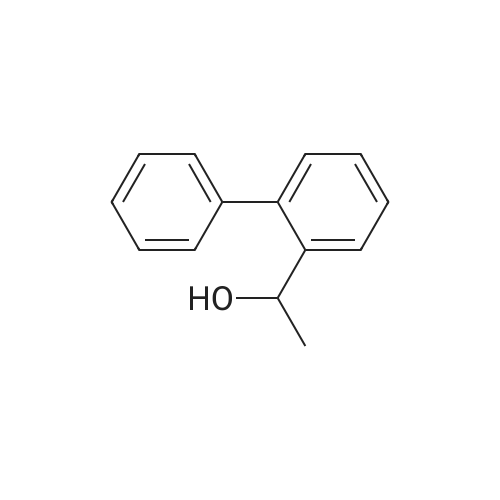
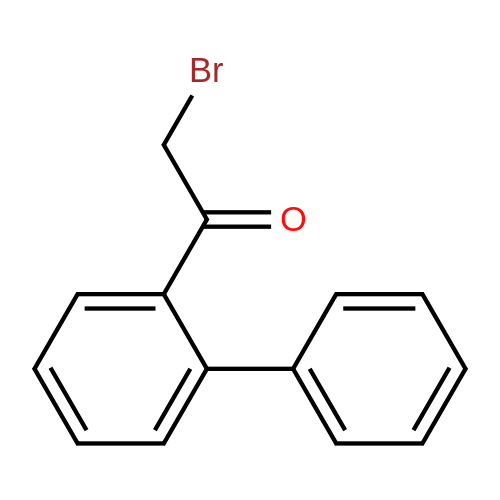
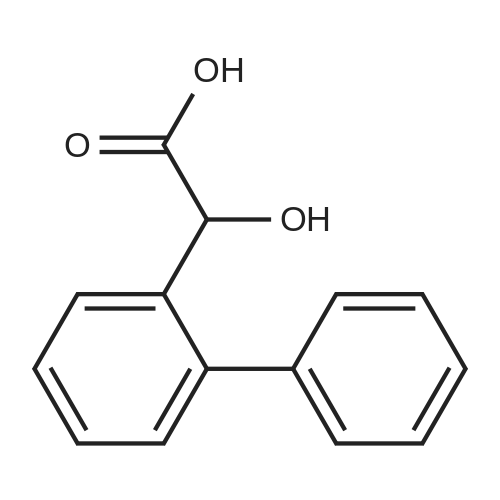
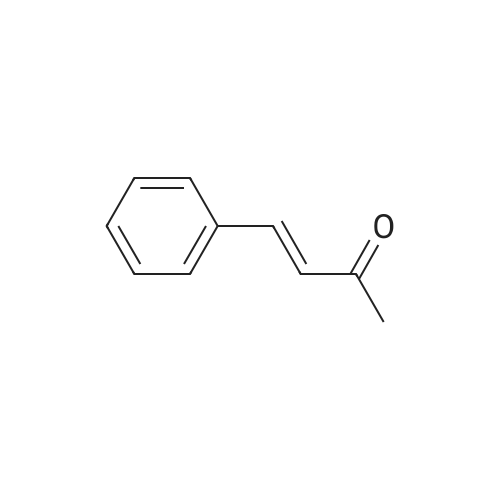
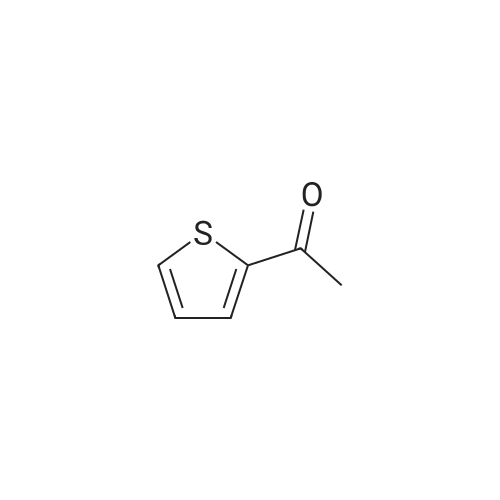
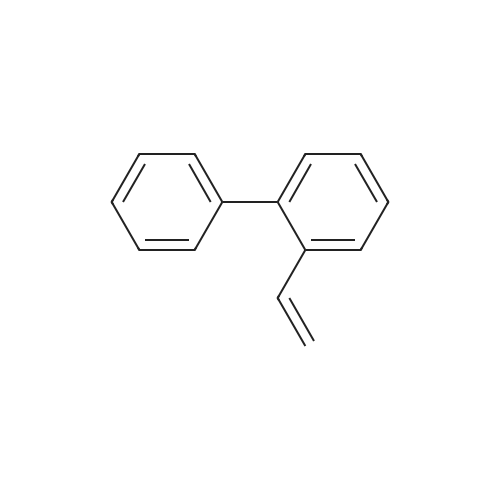
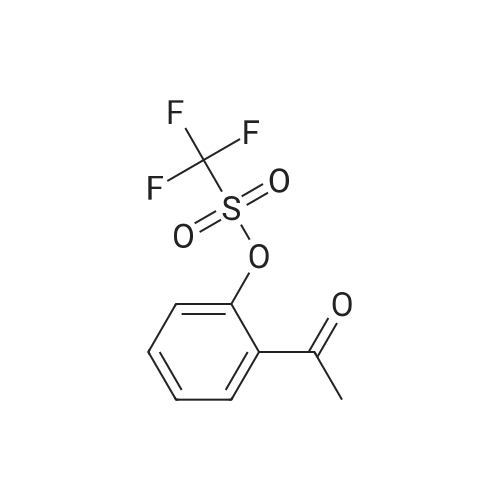
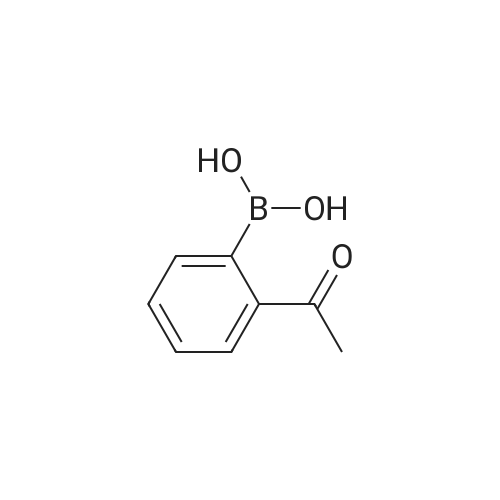
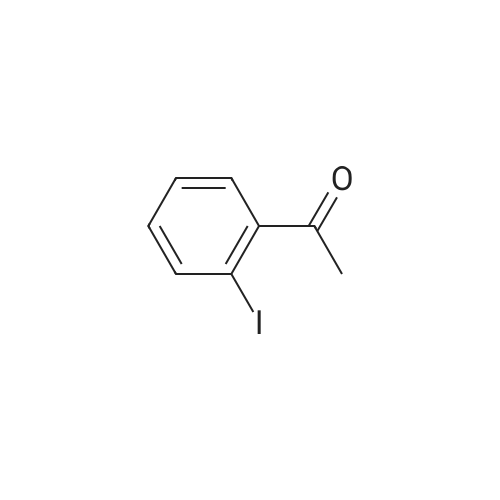
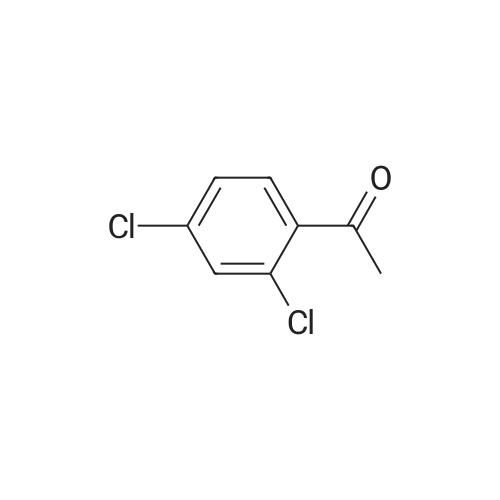
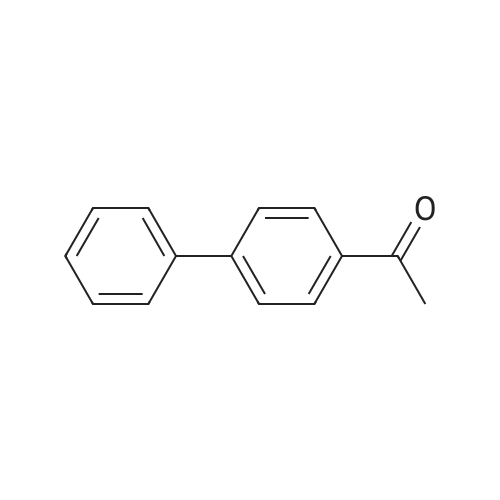
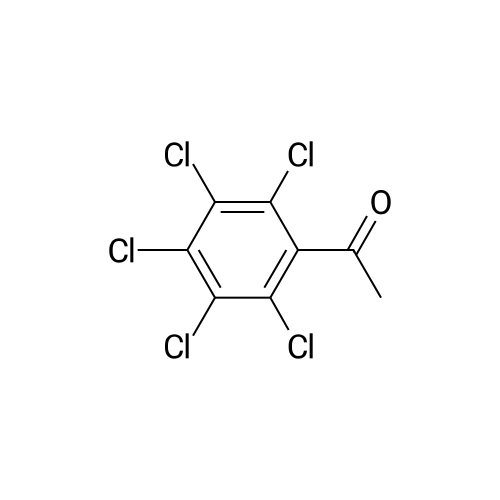
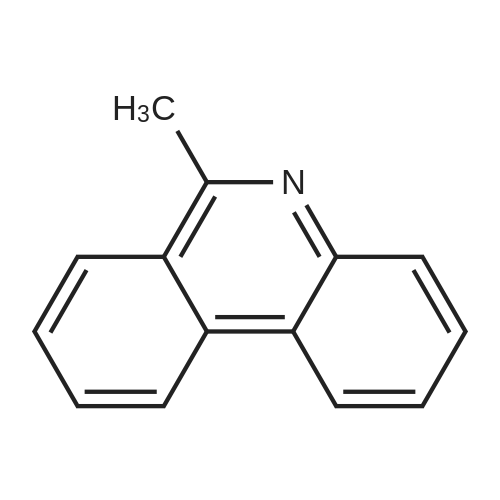
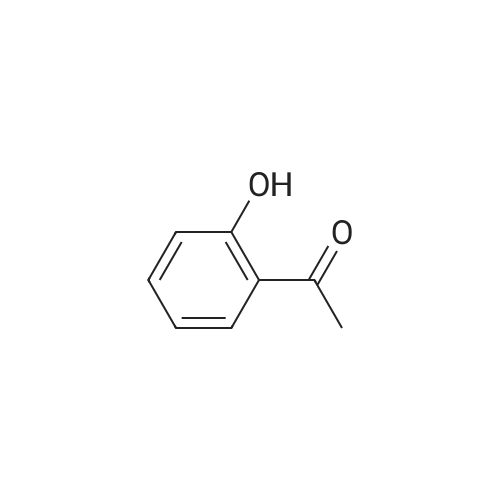
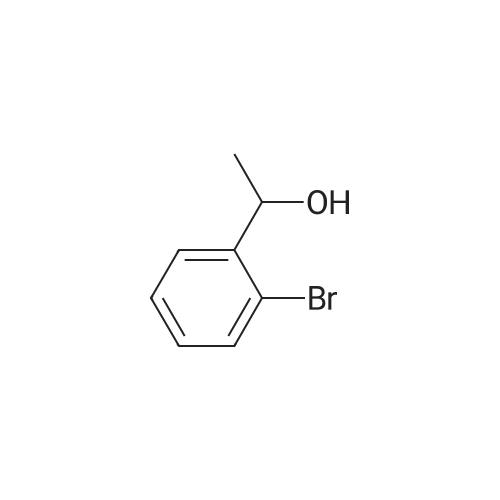
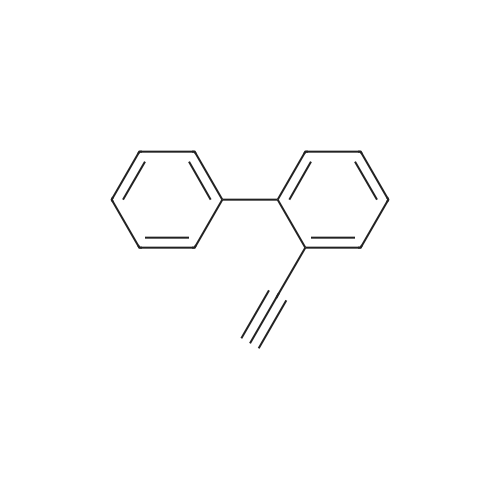
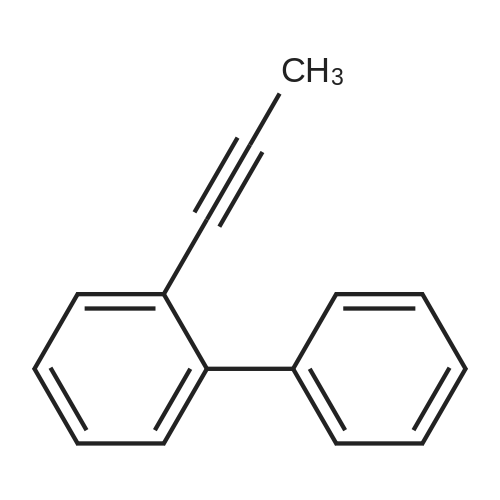
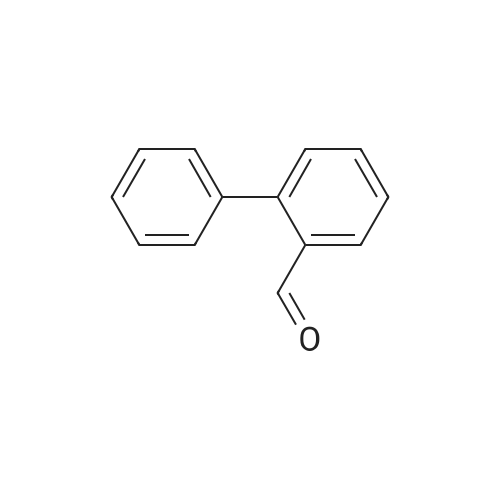
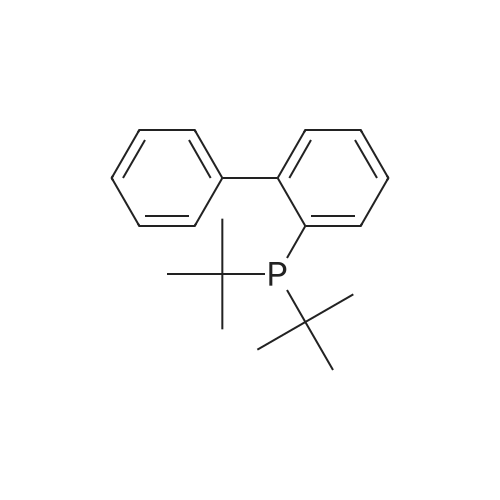
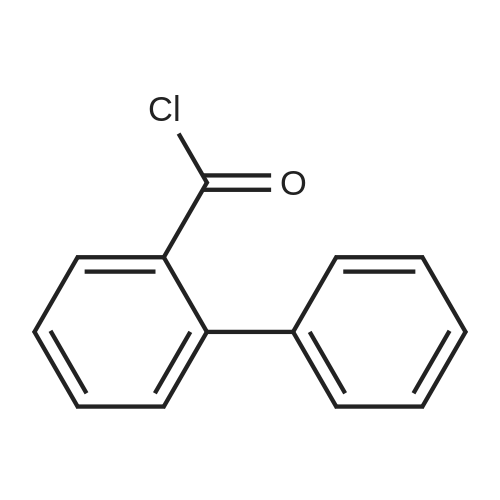
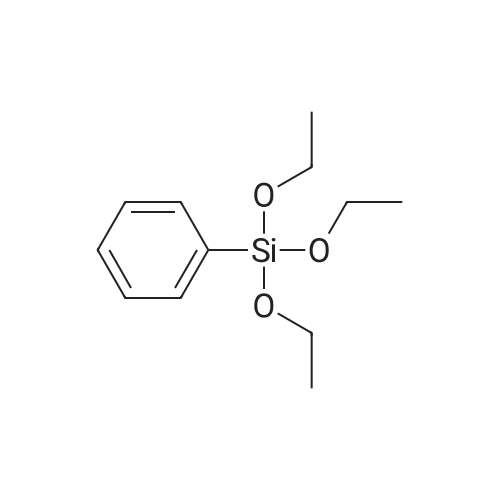
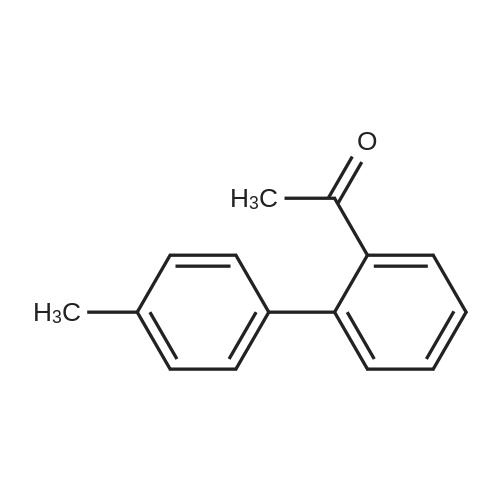
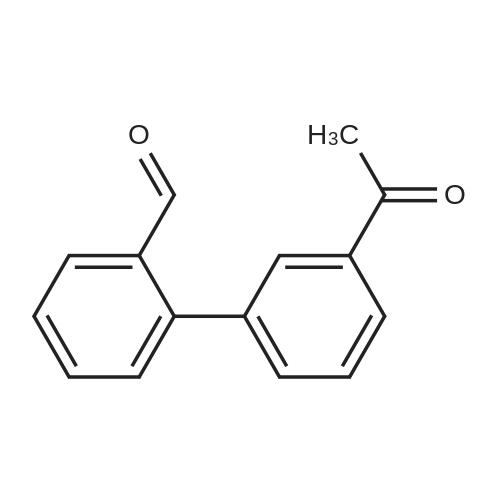
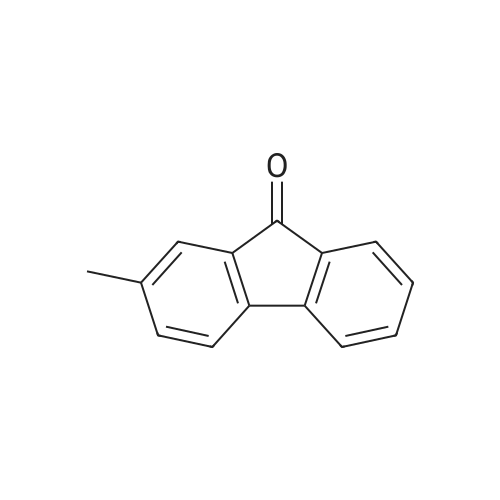
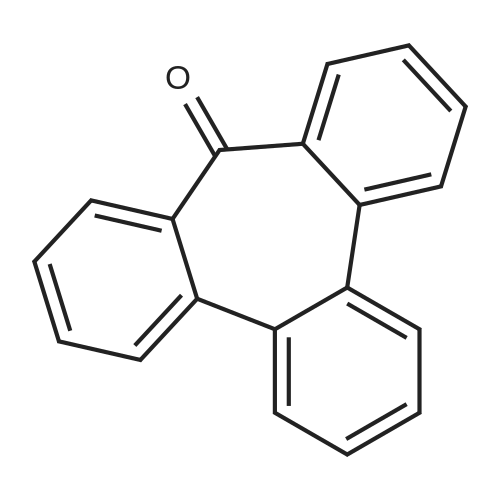
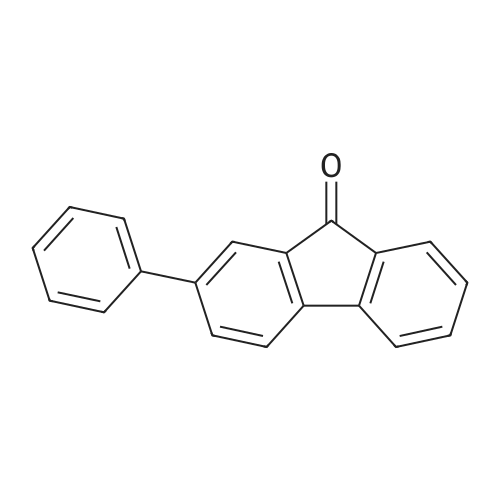
| 31% |
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,2-dimethoxyethane; water; for 48h;Reflux; |
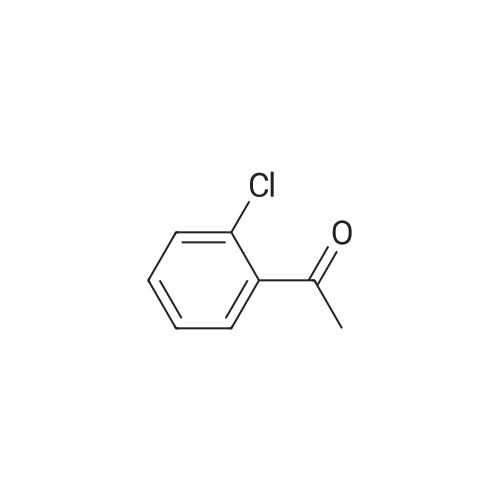
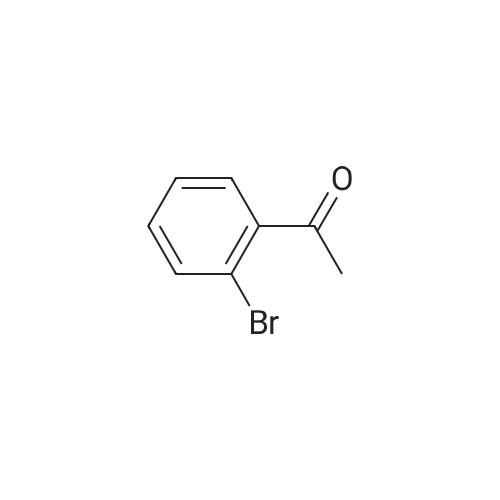
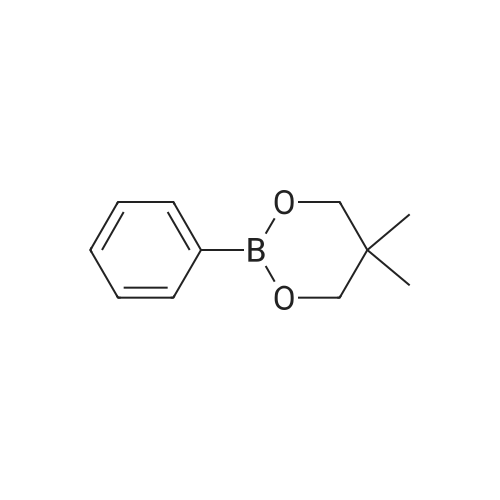
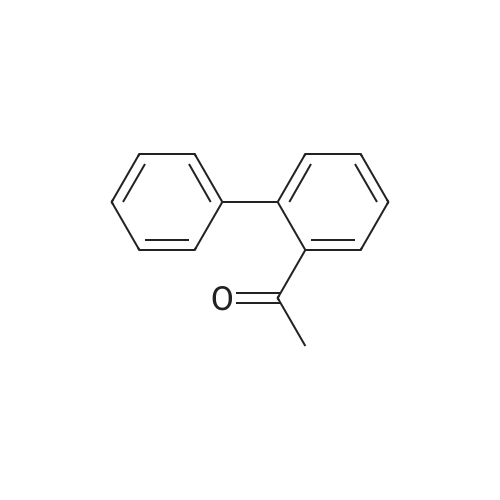
General procedure: 2'- or 3'-Bromoacetophenone (10.05 mmol, 1 equiv) and phenylboronic acid (12.04 mmol, 1.2 equiv) were dissolved in a mixture of DME (30 mL) and H2O (30 mL). Then, K2CO3 (15.08 mmol, 1.5 equiv) and tetrakistriphenylphosphinepalladium (0.05 mol, 0.005 equiv) were added and the mixture was refluxed for 48 h. After cooling to room temperature, the suspension was filtered off and the filtrate was extracted with CH2Cl2. The resulting organic layer was dried over MgSO4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (cyclohexane/AcOEt, 6:4) to afford the corresponding derivatives 1 and 2. |
| 12% |
With dichloro{1-(2-(bis(3,5-dimethylphenyl)phosphinyl)phenyl)-N-(tert-butyl)methanamine}-palladium(II); potassium carbonate; In water; N,N-dimethyl-formamide; at 80℃; for 12h;Schlenk technique; Inert atmosphere; |
General procedure: In a typical experiment, a sealed tube was charged with aryl bromide (0.10 mmol), phenylboronic acid (0.12 mmol), base (0.12 mmol), organic solvent-H2O (3:3 mL) and palladium catalyst (0.2 mol%), and the mixture was stirred at appropriate temperature. After the required reaction time, the mixture was cooled and poured out in CHCl3 (20 mL) and washed with saturated ammonium chloride and brine solution. Then, the organic phase was dried over anhydrous sodium sulfate, and the solvent was evaporated. The crude product was chromatographed on silica gel and the isolated biphenyl product was characterized by 1H,13C NMR, and GC. |
|
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; toluene;Heating / reflux; |
Biphenyl thiosemicarbazones la-lc were prepared in high yield (80-90%) from the biphenylacetophenones, which were in turn prepared in good yield (60-70%) (Scheme I) via a Suzuki cross coupling procedure from the corresponding bromoacetophenones and phenylboronic acid. Scheme I MyllH, sur er x I (i) Pd (Ph3P) 4K2C03 (aq), PhMe, reflux, phenylboronicacid. (ii) la=2'-phenyl thiosemicarbazide, MeoH, Ac OH, Reflux lb=3'-phenyl lc=4'-phenyl. |
| 89%Chromat. |
With bis-[1-(2,4,6-diisopropylpheny)-3,4,5,6-tetrahydropyrimidine]dibromopalladium; potassium tert-butylate; In water; N,N-dimethyl-formamide; at 100℃; for 3h;Inert atmosphere; |
General procedure: A Schlenk tube was charged with the appropriate aryl halide, arylboronic acid and base under nitrogen. The required amount of the catalyst stock solution and additional solvent were added to obtain a total volume of 3 mL. The reaction mixture was heated at 100 C for specified time, and then allowed to cool. The reactionmixture was extracted three times with diethyl ether, and the combined organic extracts werewashed withwater, dried (MgSO4), and evaporated to dryness. Internal standard was added to the residue and the yield was then measured by GC. The products were isolated by flash chromatography on silica gel, and characterized by m.p. and 1H NMR. |
|
With potassium phosphate; In N,N-dimethyl-formamide; at 100℃; |
General procedure: Unless otherwise stated, a mixture of 4'-bromoacetophenone(0.2149 g, 1.08 mmol), phenylboronic acid (0.1975 g, 1.62mmol), K3PO4 (0.8628 g, 3.24 mmol), and n-hexadecane (0.12ml) as the internal standard in dimethylformamide (5 ml) wereadded to a round-bottom flask containing the required amountof the immobilized palladium catalyst. The flask was heated atthe required temperature with magnetic stirring. Reaction conversions were monitored by withdrawing aliquots (0.1 ml)from the reaction mixture at different time intervals andquenching with water (1 ml). The organic components wereextracted into diethylether (2 ml 2), dried over Na2SO4, andanalyzed by GC with n-hexadecane as the reference. Theproduct identity was also further confirmed by GC-MS. |
|
With C28H44Cl2N4O2Pd; potassium hydroxide; In water; isopropyl alcohol; at 60℃; for 3h;Schlenk technique; Sealed tube; |
General procedure: The Suzuki-Miyaura reactions were carried out in a Schlenk tube. Weighed amounts of the solid reactants: phenylboronic acid (135 mg; 1.1 mmol), KOH (112.0 mg; 2.0 mmol), catalyst (20.0 mg), 2-Br-toluene (0.118 mL; 1 mmol) and 5 mL of solvent (2-propanol/water mixture) were introduced to the Schlenk tube. Next, the Schlenk tube was sealed with a rubber septum and introduced into an oil bath preheated to 80 C. The reaction mixture was magnetically stirred at the given temperature for 6 h and after this time left for several minutes to cool down. Next, the organic products were separated by extraction with 10 mL of DEE. The extracts (10 mL) were GC-FID analyzed with dodecane (0.050 mL) as an internal standard to determine the conversion of aryl bromide.The products of the reaction were determined by GC-MS. |
|
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; toluene; at 110℃; for 11h;Inert atmosphere; |
adding tetrakis(triphenylphosphine)palladium (0.3 mmol, 346.7 mg) to anhydrous three-necked flask(9 mmol, 1.24 g), after sealing, the air in the bottle was exhausted and filled with nitrogen, and repeated once. Add to the three-necked flask in turn2'-Bromoacetophenone (3 mmol, 405 muL), a mixture of phenylboric acid (3.6 mmol, 435 mg) dissolved in 3 mL of ethanol, saturated with nitrogenAnd 12 mL of toluene. The reaction mixture is refluxed at 110 C for 11 hours to obtain an intermediate product of biphenyl B.ketone. Almost completely converted. Purification by silica gel column (petroleum ether: ethyl acetate = 100:1). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping