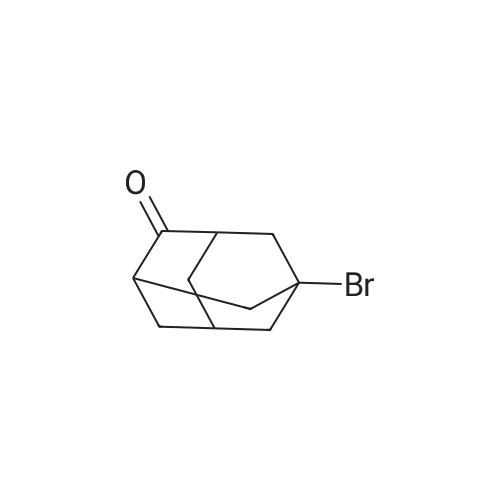
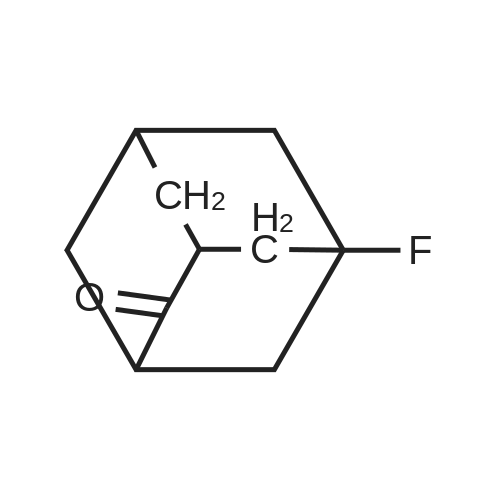
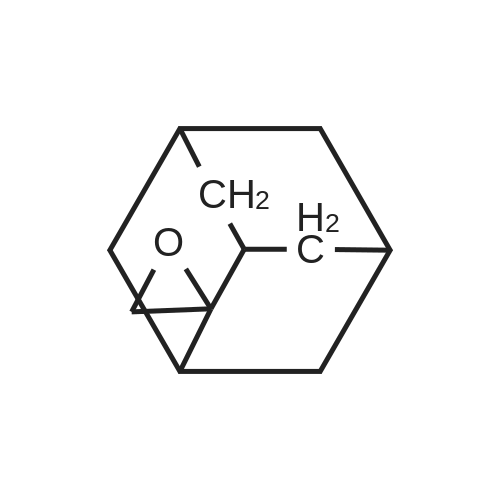
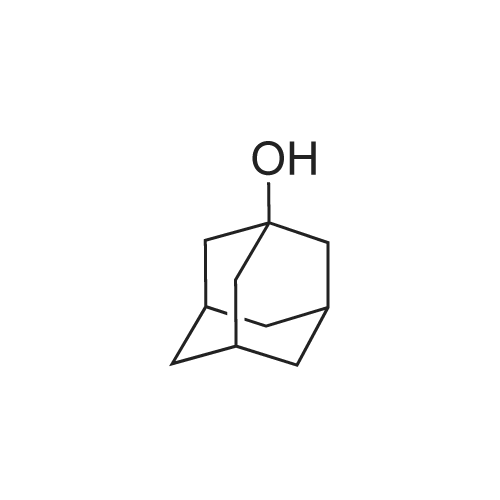
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: at 60℃; for 74.25 h; Cooling with ice Stage #2: for 1 h; Heating
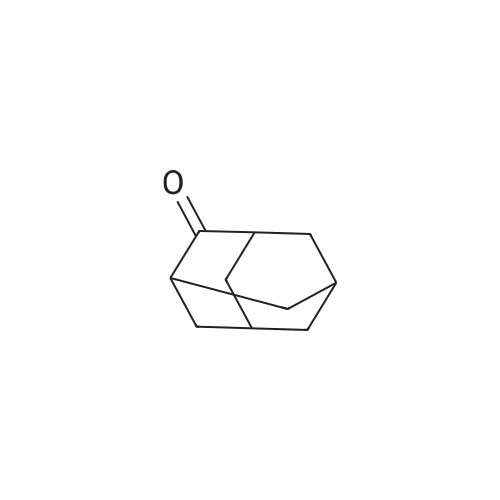
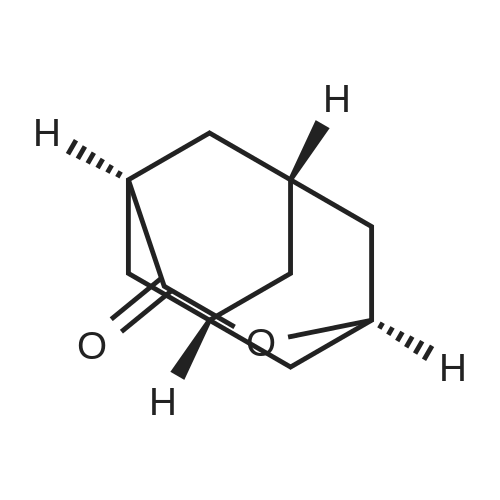
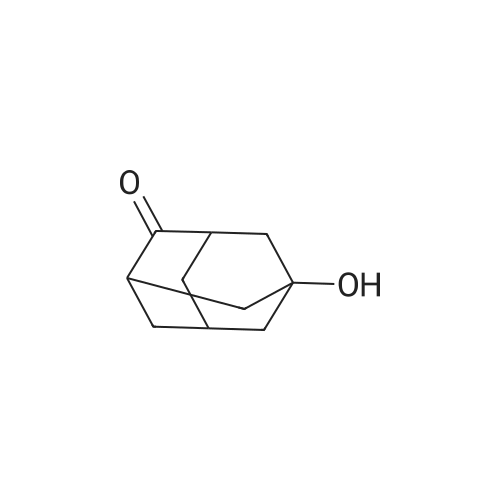
Step (i): Preparation of compound of formula (11)Adamantanone (50 grams, 333 mmol) was added with stirring to nitric acid (98percent, 440 mL) at ice bath temperature over a period of 15 minutes. The reaction mixture was stirred at room temperature for 72 hours and then heated at 60 °C, for 2 hours until most of the nitrogen dioxide evaporated. Excess nitric acid was distilled off under reduced pressure. The light yellow oil solidified upon cooling. The reaction mixture was diluted with water (200 mL) and concentrated sulphuric acid (75 mL). The resultant clear yellow solution was heated on the steam bath in a hood for 1 hour. The reaction mixture was neutralized with 30percent aqueous sodiumhydroxide solution, and while warm, extracted with chloroform. The extracts were combined, washed with brine solution and concentrated in vacuum. The crude product was dissolved in dichloromethane ( 15 mL) and hexane was added until no more precipitate was formed. The solid material was isolated by filtration and dried under vacuum to obtain compound of formula (11) (40.9 grams). Yield: 74 percent. Melting Range: 278.8-300 °C; -NMR (CDCI3): δ 2.69 (bs, 2H), 2.36-2.32 (m, 2H), 2.12-2.02 (m, 2H), 2.02-1 :88 (m, 6H), 1.80- 1.68 (m, 1H).IR: 3410, 2929, 2855, 2645, 1725, 1539, 1452, 1351, 1288, 1 1 16, 1055, 927, 900, 797;Mass (m/z): 167 [Μ+Η+].
70%
Stage #1: With nitric acid In water at 0 - 60℃; for 74.25 h; Stage #2: With sulfuric acid In water for 1 h; Heating / reflux
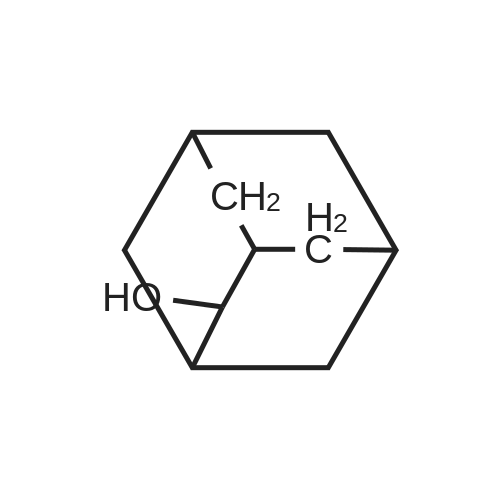
Adamantanone (12 g, 80 mmol) was added under stirring to nitric acid (98percent, 100 mL) at ice bath temperature over a period of 15 minutes. The reaction mixture was stirred at room temperature for 72 h and then heated to 60 °C, for 2 h until most of the nitrogen dioxide evaporated. Excess nitric acid was distilled off under reduced pressure. The light yellow oil solidified upon cooling (NO3 adduct of the hydroxyketone). Water (40 mL) and cone. H2SO4 (98percent, 15 mL) were added. The resulting clear yellow solution was heated on the steam bath in a hood (nitrous fumes) for 1 h. The solution was then cooled and extracted with a 2:1 mixture of n-hexane and diethylether to remove unreacted adamantanone (1.0 g). The acid layer was neutralized with 30percent aq. NaOH solution, and while warm, extracted with chloroform. The extracts were combined, washed with brine solution, and concentrated in vacuum. The crude product was dissolved in CH2Cl2 (15 mL) and hexane was added until no more precipitate was formed. The solid material was isolated by filtration and dried to get 5-hydroxy-adamantan-2-one. Yield: 9.0 g (70percent). Solid; M.R: 278.8-300 °C (decomposes) m/z (M+l) 167; 1H NMR (CDCl3) 300 MHz δ 2.70-2.55 (m, 2H)5 2.36-2.32 (m5 IH), 2.11-1.93 (m, 10H). 13C NMR (CDCl3) 75 MHz δ 217.0, 66.7, 46.7, 46.6, 44.7 (2C), 43.8, 37.9 (2C), 29.5.
Reference:
[1] Russian Journal of Organic Chemistry, 2009, vol. 45, # 8, p. 1137 - 1142
[2] Journal of Organic Chemistry, 1983, vol. 48, # 7, p. 1099 - 1101
[3] Patent: WO2011/30349, 2011, A1, . Location in patent: Page/Page column 23-24
[4] Patent: WO2007/113634, 2007, A1, . Location in patent: Page/Page column 49
[5] Chemical Communications, 2014, vol. 50, # 2, p. 186 - 188
[6] Synthesis, 1972, p. 374 - 375
[7] Organic Letters, 2003, vol. 5, # 16, p. 2943 - 2946
[8] Synthetic Communications, 1984, vol. 14, # 1, p. 65 - 68
[9] Russian Journal of Organic Chemistry, 2002, vol. 38, # 8, p. 1125 - 1129
[10] Patent: US6461876, 2002, B1,
[11] Patent: US2004/77018, 2004, A1,
[12] Patent: US7422908, 2008, B2,
[13] Patent: EP2048140, 2009, A1, . Location in patent: Page/Page column 13
[14] Journal of Molecular Catalysis B: Enzymatic, 2013, vol. 94, p. 111 - 118
3
[ 700-58-3 ]
[ 20098-14-0 ]
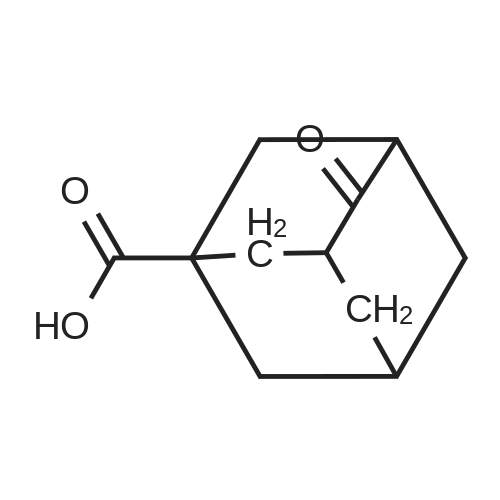
[ 73346-81-3 ]
Reference:
[1] Journal of Molecular Catalysis B: Enzymatic, 2013, vol. 94, p. 111 - 118
With sulfuric acid; sulfur trioxide; at 50 - 90℃; for 7h;
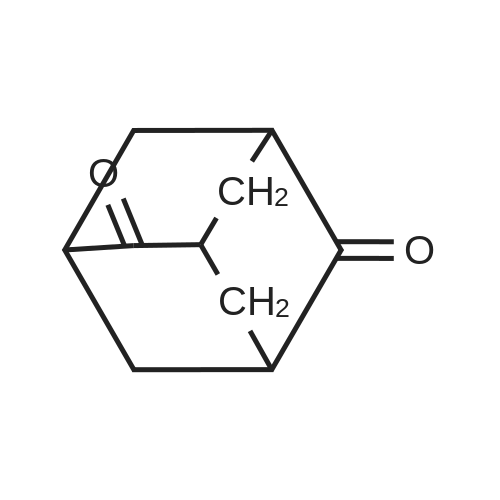
A 5 L 4-neck flask equipped with N2 inlet/bubbler with H2O trap, overhead stirring, and an addition funnel was charged with 30% oleum (10.5 volumes, 2.2 L, 8×500 g bottles+100 mL), and heated to 50 C. under a slight N2 flow. 5-Hydroxy-2-adamantanone (220 g, 81 wt % purity, 1.07 mol) was dissolved in 5 volumes HCO2H (98%, 1.10 L) and added drop-wise to the warm oleum solution over 5 hours. The addition rate was adjusted to maintain the internal temperature between 70-90 C. After stirring an additional 2 hours at 70 C. The reaction solution was cooled to 10 C. in an ice bath. 20 volumes of 10% NaCl aq (4 L) were cooled to <10 C., the crude reaction mixture was quenched into the brine solution in batches, maintaining an internal temperature <70 C. The quenched reaction solution was combined with a second identical reaction mixture for isolation. The combined product solutions were extracted 3×5 volumes with CH2Cl2 (3×2.2 L) and the combined CH2Cl2 layers were then washed 1×2 volumes with 10% NaCl (1 L). The CH2Cl2 solution was then extracted 3×5 volumes with 10% Na2CO3 (3×2.2 L). The combined Na2CO3 extracts were washed with 1×2 volumes with CH2Cl2 (1 L). The Na2CO3 layer was then adjusted to pH 1-2 with concentrated HCl (2 volumes, product precipitates out of solution). The acidic solution was then extracted 3×5 volumes with CH2Cl2 (3×2.2 L), and the organic layer was washed 1×2 volumes with 10% NaCl. The organic solution was then dried over Na2SO4, filtered, concentrated to ¼ volume, then chase distilled with 2 volumes EtOAc (1 L). Nucleation occurred during this distillation. The suspension was then chase distilled 2×5 volumes (2×2 L) with heptane and cooled to room temperature. The suspension was then filtered, and the liquors were recirculated 2× to wash the wet cake. The resultant material was dried overnight at 50 C., 20 mm Hg to afford the title compound.
With sulfuric acid; sulfur trioxide; at 50 - 90℃; for 7h;
A 5L 4-neck flask equipped with N2 inlet/bubbler with H2O trap, overhead stirring, and an addition funnel was charged with 30% oleum (10.5 volumes, 2.2 L, 8×500 g bottles+100 mL), and heated to 50 C. under a slight N2 flow. 5-Hydroxy-2-adamantanone (220 g, 81 wt % purity, 1.07 mol) was dissolved in 5 volumes HCO2H (98%, 1.10 L) and added drop-wise to the warm oleum solution over 5 hours. The addition rate was adjusted to maintain the internal temperature between 70-90 C. After stirring an additional 2 hours at 70 C. The reaction solution was cooled to 10 C. in an ice bath. 20 volumes of 10% NaCl aq (4 L) were cooled to <10 C., the crude reaction mixture was quenched into the brine solution in batches, maintaining an internal temperature <70 C. The quenched reaction solution was combined with a second identical reaction mixture for isolation. The combined product solutions were extracted 3×5 volumes with CH2Cl2 (3×2.2 L) and the combined CH2Cl2 layers were then washed 1×2 volumes with 10% NaCl (1 L). The CH2Cl2 solution was then extracted 3x5 volumes with 10% Na2CO3 (3×2.2L). The combined Na2CO3 extracts were washed with 1×2 volumes with CH2Cl2 (1 L). The Na2CO3 layer was then adjusted to pH 1-2 with concentrated HCl (2 volumes, product precipitates out of solution). The acidic solution was then extracted 3×5 volumes with CH2Cl2 (3×2.2 L), and the organic layer was washed 1×2 volumes with 10% NaCl. The organic solution was then dried over Na2SO4, filtered, concentrated to ¼ volume, then chase distilled with 2 volumes EtOAc (1 L). Nucleation occurred during this distillation. The suspension was then chase distilled 2×5 volumes (2×2 L) with heptane and cooled to room temperature. The suspension was then filtered, and the liquors were recirculated 2× to wash the wet cake. The product was dried overnight at 50 C., 20 mm Hg to afford 397.81 g product as a white crystalline solid.
With sulfuric acid; at 20 - 80℃;
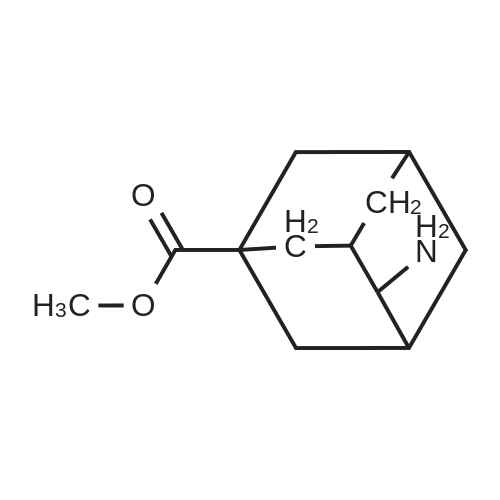
To 60% fuming sulfuric acid (135 ml) stirred under heating at 80C, a solution of 4-oxoadamantan-1-ol (15.69 g) in formic acid (250 ml) was added dropwise over 2 hours. After completion of the dropwise addition, the reaction mixture was stirred at 80C for 3 hours, to which formic acid (100 ml) was then added at room temperature and stirred overnight at room temperature. After addition of additional formic acid (50 ml), the reaction mixture was stirred at 80C for 12 hours and then stirred overnight at room temperature. The reaction mixture was poured into ice-cold water, filtered to remove insoluble materials and then extracted three times with toluene. After the combined organic layers were extracted with 4M aqueous sodium hydroxide, the aqueous layer was washed three times with chloroform. The aqueous layer was adjusted to pH 1 to 2 by addition of 12M aqueous hydrochloric acid and then extracted with chloroform. The organic layer was washed with brine and then dried over anhydrous sodium sulfate. After the desiccant was filtered off, the solvent was distilled off under reduced pressure to give 4-oxoadamantane-1-carboxylic acid (9.84 g) as a colorless powder. To a solution of 4-oxoadamantane-1-carboxylic acid thus obtained (1.50 g) in 8M ammonia/methanol (30 ml), 10% palladium on activated carbon (150 mg) was added and stirred overnight under a hydrogen atmosphere at room temperature. After addition of water (10 ml), the reaction mixture was filtered through celite and washed with methanol, and the filtrate was concentrated under reduced pressure. To the resulting crystal, acetonitrile (30 ml) was added and stirred at room temperature for 3 hours. The crystal was collected by filtration to give 4-aminoadamantane-1-carboxylic acid (1.28 g) as a colorless powder. To a solution of 4-aminoadamantane-1-carboxylic acid thus obtained (1.28 g) in methanol (13 ml), acetyl chloride (2.34 ml) was added under ice cooling and stirred at room temperature for 20 minutes and then at 45C for 8 hours. The reaction mixture was stirred overnight at room temperature and then further stirred at 45C for 4 hours. After the reaction mixture was cooled to room temperature and then concentrated, the resulting crystal was dissolved in chloroform and filtered through celite. The filtrate was concentrated under reduced pressure, and the residue was converted into a powder form by addition of acetonitrile (10 ml). The crystal was collected by filtration and washed sequentially with acetonitrile and n-hexane to give the titled compound, i.e., methyl 4-aminoadamantane-1-carboxylate (1.22 g) as a colorless powder. 1H NMR (300 MHz, CHLOROFORM-D) delta 1.58-2.40 (m, 15 H), 3.49 (brs, 1 H), 3.67 (s, 3 H).
Step 1. 4-(S-1-Phenyl-ethylamino)-adamantan-1-ol Palladium hydroxide on carbon (2.54 g, 50% water by weight, 20 wt. % Pd dry basis, 4.8 mmol) was added to a Parr bottle (350 mL) under a nitrogen atmosphere. 5-Hydroxy-2-adamantanone (15 g, 90.2 mmol), aluminum isopropoxide (18.43 g, 90.2 mmol), (S)-(-)-1-phenyl-ethylamine (99+%, 99% ee, 10.94 g, 90.3 mmol) and toluene (150 mL) were added sequentially under a nitrogen atmosphere. The mixture was well shaken for a few minutes and then was subjected to hydrogenation with H2 (55 psi) for 5 h at room temperature. 1N NaOH solution (200 mL) was then added and the reaction mixture was mixed well. After filtration through celite and washing with MeOH, the filtrate was concentrated to remove organic solvents. The remaining basic aqueous mixture was extracted with dichloromethane (3*200 mL). The combined organic phases were concentrated under house vacuum and then under high vacuum to give 4-(S-1-phenyl-ethylamino)-adamantan-1-ol (21.65 g, 88%) as an oil. A trans to cis ratio of 100:5 was determined from the crude 1H NMR. Mass spectrum: m/z: 272.2 (M+1).
With trifluorormethanesulfonic acid; at 20℃; for 4.58333h;Heating / reflux;
To a stirred solution of compound 5-hydroxyadamantan-2-one (10.0 g, 60.2 mmol) in benzene (180 mL) was added trifluoromethanesulfonic acid (5.3 mL, 60.2 mmol) over a period of 30 minutes at r.t. After stirring the reaction mixture for 5 minutes at r.t, it was refluxed for 4 h. The reaction mixture was cooled to 0C and sat. aq. NaHCO3 (76 mL) was added over a period of 30 minutes. Two layers were separated, the aqueous layer was <n="51"/>extracted with ether and the combined layer was washed with water and brine, dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure to obtain 5- Phenyladamantan-2-one (10.5 g) as a white solid in 80% yield. M.R: 53.8-60.9 C. m/z (M+l) 227; IR (cm'1): 2921,2853, 1717, 1446, 1060, 758, 698. 1H NMR (CDCl3) 300 MHz delta 7.37-7.30 (m, 4H), 7.26-7.19 (m, IH), 2.70-2.63 (m, 2H), 2.30-2.0 (m, HH). 13C NMR (CDCl3) 75 MHz delta 217.8, 147.8, 128.3 (2C), 126.1,124.6 (2C), 46.9 (2C), 44.3 (2C), 41.9, 35.9, 38.4 (2C), 28.0.
78%
With trifluorormethanesulfonic acid; at 20℃; for 4.75h;Reflux;
Step (ii): Preparation of compound of formula (12)To a stirred solution of compound of formula (11) (20.0 grams, 120.3 mmol) in benzene (365 mL) was added trifluoromethanesulfonic acid ( 10.7 mL, 60.2 mmol) over a period of 40 minutes at room temperature. After stirring the reaction mixture for 5 minutes at room temperature it was refluxed for 4 hours. The reaction mixture was cooled to 0 C and saturated aqueous sodium bicarbonate (150 mL) were added over a period of 30 minutes. Two layers were separated, the aqueous layer was extracted with ether and the combined organic layer was washed with water, brine, dried over anhydrous sodium sulphate and the solvent was evaporated under reduced pressure to obtain compound of formula (12) as a white solid (21 .2 grams). Yield: 78 %.Melting Range: 53.8-60.9 C;-NMR (CDCI3): delta 7.37-7.22 (m, 4H), 7.17-7.10 (m, 1 H), 2.67 (bs, 2H), 2.37-2.25 (m, 2H), 2.25- 2.15 (m, 4H), 2.15-2.0 (m, 5H).IR icm'1): 2912, 2850, 1716, 1597, 1495, 1444, 1294, 1059, 960, 758, 701.Mass (m/z): 227 [M+LT]. .
55%
With trifluorormethanesulfonic acid; at 80℃; for 16h;
To a solution of 5-hydroxy-2-adamantanone (2 g, 12 mmol) in benzene (30 mL) was added trifluoromethanesulfonic acid (1.1 mL, 12 mmol) dropwise.The reaction was stirred at 80 C for 16 hours.The solvent was removed under reduced pressure and the residue was subjected to silica gel column chromatography (petroleum ether/ethyl acetate = 100/1 to 4/1) to give 5-phenyl-2-adamantanone (1.48 g, yield: 55%) as White solid.
55%
With trifluorormethanesulfonic acid; at 80℃; for 16h;
5-Hydroxy-2-adamantanone (2 g, 12 mmol) in benzene (30 mL),Trifluoromethanesulfonic acid (1.1 mL, 12 mmol) was added dropwise.The system was stirred at 80 C for 16 hours.The solvent was removed by rotary evaporation under reduced pressure, and the residue was subjected to silica gel column chromatography (petroleum ether:ethyl acetate=100:1 to 4:1) to isolate Compound 1.1 (1.48 g, yield: 55%) as a white solid.
26.9 g
With trifluorormethanesulfonic acid;Reflux;
5-Phenyladamantan-2-one WO2007/113634A1 discloses the chemical reaction, but the workup (crystallization) was optimized. The reaction thus gives the product in greater purity at comparable yield. (3R,7R)-5-Hydroxyadamantan-2-one (25 g) was dissolved in benzene (200 ml). Trifluoromethanesulfonic acid (13.1 ml) was added in such a way that the reaction stirred at room temperature. Subsequently, the reaction was stirred at reflux. After again adding trifluoromethanesulfonic acid (2 ml), the reaction was stirred again at reflux. After adding tert-butyl methyl ether, the mixture was washed repeatedly with water. After filtration, the resulting solution was fully concentrated. The residue was taken up in n-heptane, hot-filtered and then crystallized. This gave 5-phenyladamantan-2-one (26.9 g) as a white solid.
26.9 g
With trifluorormethanesulfonic acid; at 20℃;Reflux;
5-Phenyladamantan-2-one WO20071113634A1 discloses the chemical reaction, but the workup (crystallization) was optimized here. The reaction thus gives the product in greater purity at comparable yield. (3R,7R)-5-Hydroxyadamantan-2-one (25 g) was dissolved in benzene (200 ml). Trifluormethanesulfonic acid (13.1 ml) was added in such a way that the reaction stirred at room temperature. Subsequently, the reaction was stirred at reflux. After again adding trifluoromethanesulfonic acid (2 ml), the reaction was stirred again at reflux. After adding tert-butyl methyl ether, the mixture was washed repeatedly with water. After filtration, the resulting solution was fully concentrated. The residue was taken up in n-heptane, hot-filtered and then crystallized. This gave 5-phenyladamantan-2-one (26.9 g) as a white solid.
3.7 g
With trifluorormethanesulfonic acid;Reflux;
To 5-hydroxyadamantan-2-one (5 g, 150 mmol) was added benzene (20 mL) and triflic acid (3 mL). The heterogenous mixture was heated to reflux, at which time all starting materials dissolved, and reflux was continued overnight. The mixture was cooled to room temperature and partitioned between water and tert-butylmethyl ether (50 mL), and the organic phase was washed 3 times with water, followed by saturated NaHCO3 and brine. The organics were dried over Na2SO4 and evaporated to give 4.7 g of crude product. Purification with flash chromatography on silica gel using 8:2 hexanes:ethyl acetate eluent gave 3.7 g of pure product as a white solid. 1H NMR (300 MHz, CDCl3) delta: 7.41-7.32 (m, 4H), 7.27-7.21 (m, 1H), 2.70 (br:s, 2H), 2.37-2.03 (m, 11H).
triphenylsulfonium 2-hydroxy-1,1,3,3,3-pentafluoropropane-1-sulfonate[ No CAS ]
[ 79-37-8 ]
[ 108-10-1 ]
[ 675200-37-0 ]
[ 56674-87-4 ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; formic acid; N,N-dimethylamino-pyridine; sulfuric acid; triethylamine; In dichloromethane; water; acetonitrile;
Synthesis Example 43 Synthesis of triphenylsulfonium 1-(difluorosulfomethyl)-2,2,22-trifluoroethyl 4-adamantanone-1-carboxylate (PAG-Y) 4-adamantanone-1-carboxylic acid was synthesised by reaction of 5-hydroxy-2-adamantanone with sulfuric acid and formic acid. Using oxalyl chloride, it was converted into a corresponding carboxylic acid chloride. In 20 g of acetonitrile were dissolved 4.9 g (0.01 mole) of triphenylsulfonium 2-hydroxy-1,1,3,3,3-pentafluoropropane-1-sulfonate of Synthesis Example 11, 1.0 g (0.01 mole) of triethylamine, and 0.24 g (0.002 mole) of N,N-dimethylaminopyridine. This was ice cooled. 2.6 g (0.012 mole) of 4-adamantanone-1-carbonyl chloride, prepared above, was added to the solution at a temperature below 5 C. The solution was stirred for 1 hour at room temperature. A dilute hydrochloric acid solution prepared from 1 g of 12N hydrochloric acid and 10 g of water was added, after which acetonitrile was distilled off in vacuum. 50 g of dichloromethane, 50 g of methyl isobutyl ketone and 20 g of water were added to the residue, whereupon the organic layer was separated. The organic layer was then washed with 20 g of water, after which the solvents were distilled off in vacuum.
With ammonia;palladium on activated charcoal; In methanol; water; under 10343.2 - 12929 Torr; for 18h;Product distribution / selectivity;
Step 1: 4-Amino-adamantan-1-ol 5-Hydroxy-adamantan-2-one (15 g, 90.24 mmol, International Specialty) and Pd/C (1.498 g, Degussa 19985880, 5%, 50% water) were added to a Parr reactor, followed by ammonia in methanol solution (7N, 300.4 ml, 2.1 mol). The reactor was pressurized with hydrogen at 200-250 psi for 18 h. The mixture was then filtered over a pad of celite and concentrated in vacuo to give 4-amino-adamantan-1-ol (15.15 g, 4/1=trans/cis by NMR-D2O) as a white solid. This material is used directly without further purification.
With ammonium formate;palladium 10% on activated carbon; In methanol; for 1h;Heating / reflux;
Ammonium formate (10 g, 0.15 mol) was added to a solution of 5-hydroxyadamantan-2-one (4.5 g, 0.027 mol, prepared as described in Tetrahedron 1968, 24, 5369) in MeOH (50ml). Then 10% Pd-C (500 mg) was added carefully and the solution heated under reflux for Ih. It was then filtered through celite and to this filtrate at O0C was added triethylamine (11.2 ml, 0.081 mol) and Boc anhydride (7.06 g, 0.0324 mol). The solution was stirred for 4 h at 20 0C and then concentrated under reduced pressure. The residue was diluted with water and extracted with EtOAc. The organic layer was dried and concentrated to give (5-hydroxy- adamantan-2-yl)carbamic acid tert-butyl ester (7 g, 96%). LC-MS (m/z) : 168 (M+l). 1HNMR (300 MHz, DMSO-d6) : delta 6.8 (d, IH), 6.7 (brs, IH), 3.45 (d, IH), 2.0 (s, IH), 1.75-1.95 (m, 4H), 1.5-1.7 (m, 6H), 1.35 (s, 9H), 1.25 (t, 2H).
With ammonium formate;palladium 10% on activated carbon; In methanol; for 1h;Heating / reflux;
Ammonium formate (10 g, 0.15 mol) was added to a solution of 5-hydroxyadamantan-2-one (4.5 g, 0.027 mol, prepared as described in Tetrahedron 1968, 24, 5369) in MeOH (50ml). Then 10% Pd-C (500 mg) was added carefully and the solution heated under reflux for Ih. It was then filtered through celite and to this filtrate at O0C was added triethylamine (11.2 ml, 0.081 mol) and Boc anhydride (7.06 g, 0.0324 mol). The solution was stirred for 4 h at 20 0C and then concentrated under reduced pressure. The residue was diluted with water and extracted with EtOAc. The organic layer was dried and concentrated to give (5- <n="69"/>hydroxyadamantan-2-yl)carbamic acid tert-butyl ester (7 g, 96%). LC-MS {m/z) 168 (M+l). 1HNMR (300 MHz, DMSOd6) : delta 6.8 (d, IH), 6.7 (brs, IH), 3.45 (d, IH), 2.0 (s, IH), 1.75- 1.95 (m, 4H), 1.5-1.7 (m, 6H), 1.35 (s, 9H), 1.25 (t, 2H).
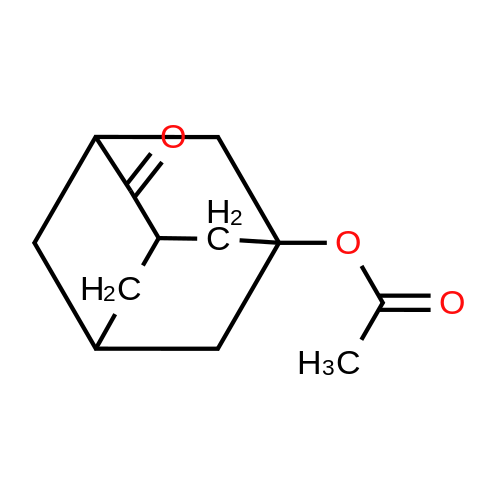
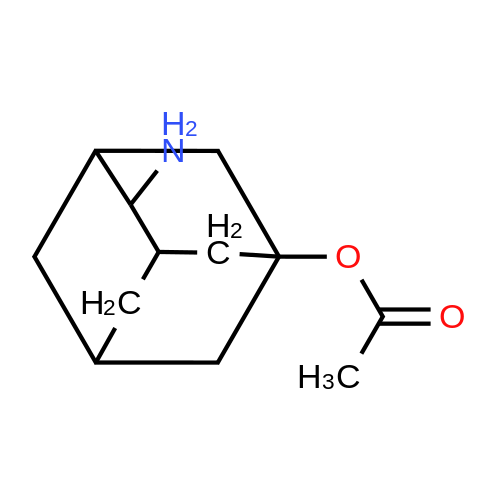
A solution of 5-hydroxy-2-adamantanone (2.6 g, 15.66 mmoles) in dichloromethane (DCM) (50 mL) was treated with dimethylaminopyridine (DMAP) (2.1 g, 17 mmoles) and acetic anhydride (2.3 mL, 23 mmoles) and stirred overnight at 50 C. The solvent was removed under reduced pressure and the residue was partitioned between water and ethyl acetate. The aqueous layer was extracted twice with ethyl acetate. Combined organic extracts were washed with water, dried (MgSO4) and filtered. The filtrate was concentrated under reduced pressure to provide the title compound as an off-white solid.
With dmap; In chloroform; at 50℃; for 2h;
To a solution of 4-oxoadamantan-1-ol (2.0 g) in chloroform (20 ml), acetic anhydride (1.3 ml) and 4-dimethylaminopyridine (1.6 g) were added and stirred at 50C for 2 hours. After distilling off the solvent under reduced pressure, the residue was diluted with water and extracted with ethyl acetate. The extracted organic layer was washed with brine and dried over anhydrous magnesium sulfate. After the desiccant was filtered off, the solvent was distilled off under reduced pressure to give 4-oxoadamantane-1-acetate (2.3 g) as a colorless powder. To a solution of 4-oxoadamantane-1-acetate thus obtained (2.3 g) in ethanol (20 ml), sodium borohydride (0.63 g) was added under ice cooling and stirred at the same temperature for 1 hour. The reaction mixture was concentrated, diluted with ethyl acetate, washed sequentially with saturated aqueous ammonium chloride and brine, and then dried over anhydrous magnesium sulfate. The desiccant was filtered off and the solvent was distilled off under reduced pressure to give the titled compound, i.e., 4-hydroxyadamantane-1-acetate (2.3 g) as a colorless powder. 1H NMR (300 MHz, CHLOROFORM-D) delta 1.22-1.50 (m, 1 H), 1.53-1.83 (m, 3 H), 1.89-2.22 (m, 12 H), 2.30-2.40 (m, 1 H), 3.75 (s, 0.4 H), 3.94 (s, 0.6 H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping