* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With chlorosulfonic acid; trifluoroacetic acid In water; toluene at 25 - 60℃; for 1 h; Stage #2: With ammonium hydroxide In isopropyl alcohol at 20 - 35℃; for 2.5 h;
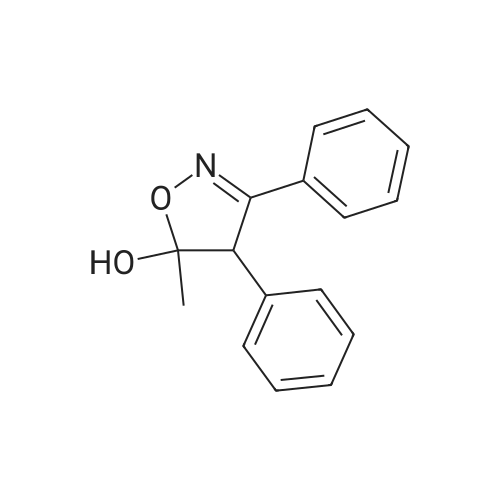
In the third step, a reaction vessel is charged with 4,5-dihydro-5-methyl-3,4diphenyl-5-isoxazolol (VI) (152 kg) and trifluoroacetic acid (TFA, 116 liters). The mixture is cooled and chlorosulfonic acid (705 kg) is added while maintaining the temperature of the reaction mixture below 25 C. After the addition is complete, the mixture is slowly heated to 60 C. and held at 60 C. for at least 1 hour. The reaction mixture is cooled and quenched by adding it to a mixture of water (456 liters) and toluene (570 liters) that is maintained below 25 C. during this addition. The reaction vessel and transfer lines are rinsed with a mixture of water (152 liters) and toluene (61 liters). The layers are separated, and the organic phase is washed with water (220 liters). The organic phase is treated with aqueous ammonium hydroxide (190 liters), and the mixture is heated to 35 C. and held at 35 C. for at least 30 minutes. An in-process check is performed to ensure that pH of the aqueous phase is not less than 9. Isopropyl alcohol (729 liters) is added, and the mixture is held at 35 C. for at least 1 hour. The mixture is cooled to 20 C. and held at 20 C. for at least 1 hour. The precipitated product is isolated and washed with isopropyl alcohol (304 liters) and then with water (at least 101 liters). The crude product is dissolved in hot methanol (709 liters). The solution is filtered to remove particulates and diluted with additional methanol (355 liters) and water (274 liters). The mixture is heated to 70 C. to dissolve the solid and then slowly cooled to initiate crystallization of the product. The mixture can be seeded if crystallization does not initiate by the time 45 C. is reached. Once crystallization occurs, the mixture is stirred at 50 C. for at least 1 hour and then slowly cooled to 5-10 C. and held at that temperature for at least 1 hour. The product is isolated and washed with a mixture of methanol and water (at least 95 liters having a 3:1 ratio of methanol to water). Alternatively, the product can be purified by recrystallization from a mixture of ethanol (1,300 liters) and water (68 liters) using the same procedure described above. The product is dried under vacuum at temperatures up to 100 C. until the amount of residual solvents by LOD or gas chromatography is not more than 0.5percent, to give 4-(5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide (VII) in a typical yield of 103 kg (62percent by weight).
53.5%
With chlorosulfonic acid; ammonium hydroxide In dichloromethane; water
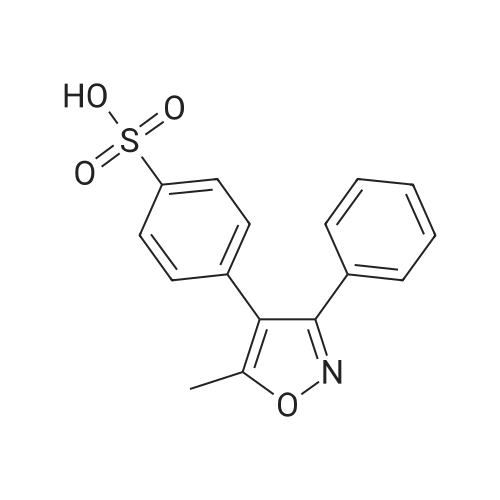
Step 3. Preparation of 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide. 5-Hydroxy-5-methyl-3,4-diphenylisoxazoline (Step 2) (142 g, 0.56 mol) was dissolved in dichloromethane (568 mL) in a 3 L roundbottom flask equipped with a heating mantle, mechanical stirrer, cold water condenser, J-KEM temperature controller and thermocouple, forming a slurry. The slurry was stirred and cooled to <10° C. Chlorosulfonic acid (335 mL, 586.3 g, 5.04 mol) was added to the slurry, keeping the temperature of the flask below 20° C. by controlling the addition. The mixture was heated to reflux (ca. 40° C.), maintained for 5 hours, then cooled to 0-5° C. The cooled reaction solution was slowly transferred to a 3 L 3-necked roundbottom flask (mechanical stirrer and thermocouple) containing water (1000 ml) previously cooled to 0-5° C., using vigorous agitation and keeping the pot temperature below 10° C. The mixture was stirred for an additional 5 minutes. The layers were separated. In a separate 3 L flask (mechanical stirrer, external ice/salt bath, thermocouple) 28percent ammonium hydroxide (700-mL) was cooled to 0-5° C. The methylene chloride solution was transferred to the stirred ammonium hydroxide solution, keeping the temperature below 10° C. The mixture was stirred at ambient temperature for 60 minutes. The resulting slurry was filtered and the solid was washed with water (200 ml) and dried, yielding the 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide as a white solid (94.3 g, 53.5percent).
Stage #1: With thionyl chloride In dichloromethane at 0 - 5℃; for 0.666667 h; Stage #2: With ammonium hydroxide In dichloromethane at 20℃; for 1 h;
5-methyl-3-phenyl-4- (4-sulfonic acid phenyl) isoxazole 11.7g (50mmol) dissolved in dichloromethaneOf thionyl chloride was added dropwise 9.3g (100mmol) at 0 ~ 5 reaction was stirred 40 minutes,Quenched with ice water, extracted with dichloromethane,Methylene chloride phase directly into the aqueous ammonia,The reaction temperature was raised to 20 deg.] C continued for 1 hourAfter the reaction, dichloromethane extraction,Washed, concentrated,Methanol to obtain 14.4 g of vediloxib,The yield was 91.4percent and the purity was 99.54percent.
Reference:
[1] Patent: CN106008387, 2016, A, . Location in patent: Paragraph 0020; 0045; 0046; 0047
3
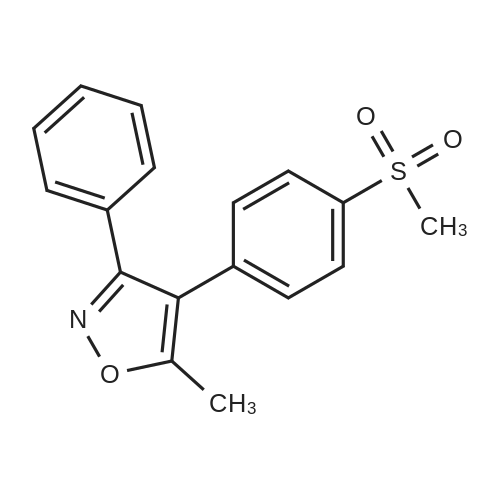
[ 37928-17-9 ]
[ 181695-72-7 ]
Yield
Reaction Conditions
Operation in experiment
93.7%
Stage #1: at 0 - 5℃; for 0.5 h; Acidic conditions Stage #2: at 0 - 20℃;
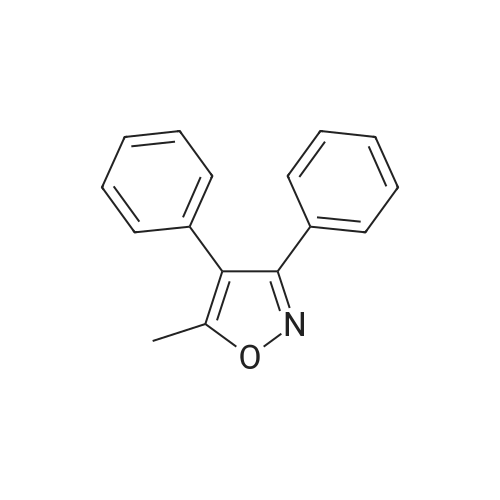
The obtained in Example 1(10 mmol) of 5-methyl-3,4-diphenylisoxazole and 2.3 g (20 mmol) of chlorosulfonic acid were stirred at 0 to 5 ° C for 30 minutes,1 g of anion exchange resin (40percent) was then added,The temperature was raised to 20 ° C,15.6 g (60 mmol) of saturated ammonium chloride was added dropwise,After completion of the reaction,Dichloromethane extraction,Washed,concentrate,Ethanol to obtain 2.95 g of valdecoxib,The yield was 93.7percentPurity 99.63percent.
92.7%
Stage #1: With chlorosulfonic acid In dichloromethane at 0 - 5℃; for 0.666667 h; Stage #2: With ammonia In dichloromethane; water at 20℃;
A solution of 11.7 g (50 mmol) of 5-methyl-3,4-diphenylisoxazole and 9.3 g (80 mmol) of chlorosulfonic acid at 0 to 5 ° C was stirred 40Minute, extracted with methylene chloride,DichloromethanephaseAmmonia was added directlyWater, the temperature rose to 20 ° C to continue the reaction, after the end of the reaction, Dichloromethane extraction, washing with water, concentration and ethanol recrystallization to obtain 14.6g of vediloxib in a yield of 92.7percent and purity of 99.69percent.
89%
Stage #1: at 20℃; Stage #2: With ammonia In ethyl acetate at 20℃; for 2 h;
After the compound 21.16g (90mmol), trifluoroacetic acid 20.52g (180mmol) added to the reaction flask, control temperature 20 dropwise chlorosulfonic acid 20.97g (180mmol), TLC monitoring completion of the reaction, the reaction was added dropwise to the above purified water, after the end of the dropwise addition, ethyl acetate was added 2 times, the combined organic phase was washed once purified, was added after the ethyl acetate solution was washed with concentrated ammonia solution, stirred at room temperature 2h, TLC monitoring of the reaction the reaction. Separated, washed twice with water purified. The combined aqueous phase was added ethyl acetate and 2 times the anti-quenching. Finally, the combined organic phase was concentrated under reduced pressure to give the crude product. Recrystallization from absolute ethanol. To give compound 25.18g (80.1mmol), yield 89.0percent, purity 99.3percent (HPLC method).
81%
Stage #1: With chlorosulfonic acid In dichloromethane for 3 h; Reflux Stage #2: With ammonium hydroxide In dichloromethane at 20℃; for 4 h;
Sequentially 10 g 5-methyl-3,4-diphenylisoxazole, was added to a 100 mL flask, 30 mL of dichloromethane, stirring, a clear solution; under ice-cooling, 20mL of chlorosulfonic acid was slowly added dropwise to the solution. After finishing the dropping, the reaction vessel was slowly warmed to reflux for about 3 hours. TLC monitored the reaction to 5-methyl-3,4-diphenylisoxazole completely disappeared, the reaction was stopped.to the 100 mL beaker by adding crushed ice, water, stirring conditions, the cold to room temperature of the reaction solution slowly into the ice water mixture. After the solution was complete, the layers were separated and the organic layer was collected. The aqueous layer was extracted twice with methylene chloride. The organic layers were combined and placed in a 250 mL flask for direct amination reaction.under stirring, 50 mL of aqueous ammonia was slowly added dropwise to the above methylene chloride solution. After the dropwise addition, the reaction was allowed to stand at room temperature for about 4 hours. TLC monitoring reaction. After completion of the reaction, the reaction solution was allowed to stand, the organic layer was separated, and the aqueous layer was extracted twice with dichloromethane. The combined organic layer was washed with saturated brine and dried over sodium sulfate. The organic layer was filtered and the filtrate was concentrated to give crude tacrolimus and the ethanol was recrystallized to give valdecoxib 10.8 g of as a white solid in 81percent yield.
60%
Stage #1: at 0 - 60℃; for 2 h; Stage #2: With ammonia In dichloromethane; water at 0 - 5℃; for 2 h;
Chlorosulfonic acid (9.6 ml, 11.5 mol) was added slowly to 3.0 g (1.0 mol) of 3,4-dipheny-5-methyl isoxazole at 0-5°C. After a clear solution was obtained, slowly the solution was heated to 50-60°C and stirred for 2 hours. The solution was poured into a mixture of 130 ml of dichloromethane and 130 ml of chilled water. The dichloromethane layer was separated and cooled to 0-5°C. Ammonium hydroxide solution (65 ml) was added to the dichloromethane layer, which was stirred for 2 hours. The dichloromethane layer was separated, and the aqueous layer was extracted with 30 ml of dichloromethane. The combined dichloromethane layer dried over 1.0 g of sodium sulfate. The dichloromethane layer was condensed to approximately one-half of its original volume, cooled to 0-5°C and stirred for 30 minutes. Solid was separated by filtration and washed with 3.0 ml of chilled dichloromethane. The filtered compound was dried at 50-55°C under vacuum. The yield of solid product is 2.4 g (60.0 percent).
Reference:
[1] Patent: CN106008386, 2016, A, . Location in patent: Paragraph 0021; 0044; 0045; 0046
[2] Patent: CN105859647, 2016, A, . Location in patent: Paragraph 0045; 0046; 0047
[3] Journal of Medicinal Chemistry, 2004, vol. 47, # 20, p. 4881 - 4890
[4] Patent: CN105418528, 2016, A, . Location in patent: Paragraph 0063; 0064
[5] Patent: CN104193694, 2016, B, . Location in patent: Paragraph 0032-0035
[6] Patent: EP1550658, 2005, A1, . Location in patent: Page/Page column 9
[7] Synthetic Communications, 2012, vol. 42, # 5, p. 639 - 649
[8] Synthetic Communications, 2012, vol. 42, # 5, p. 639 - 649
[9] Patent: CN104447600, 2016, B, . Location in patent: Paragraph 0085; 0087
[10] Patent: CN107137361, 2017, A,
4
[ 509074-26-4 ]
[ 181695-72-7 ]
Yield
Reaction Conditions
Operation in experiment
80%
Stage #1: for 0.0833333 - 0.166667 h; Stage #2: for 0.5 - 0.666667 h; Stage #3: With ammonia In water at 20 - 30℃; for 1 - 1.5 h;
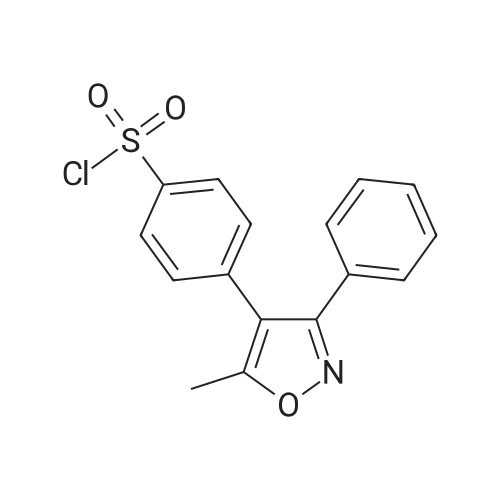
Charged 120 lit. of methylene chloride 20 kg of 4-(5-methyl-3-phenyl-4-isoxazolyl) benzene sulfonyl chloride into reactor and stirred the reaction mass for 5 to 10 minutes till the dissolution. 1 kg of carbon was charged into above solution, stirred for 30-45 minutes, filtered, and washed with 40ml of methylene chloride. The combined filtrate was charged into reactor and 90 lit. of ammonia solutions were added into the filtrate at 20-30°C for 60-90 minutes. Distilled methylene chloride at atmospheric pressure below 40°C upto 300 to 340 lit. volume left in the reactor. The reaction mass was cooled to 5-10°C and stirred for 30-45 minutes and filtered and washed with 20 lit. methylene chloride. The resultant wet cake was pressed under Nitrogen for 30-45 minutes. The wet material was charged into reactor and 100 lit. of water were added into reactor and stirred for 45-60 minutes and filtered, pressed the wet cake under nitrogen for 15-20 minutes followed by washing with 20 lit. of water. Again wet cake was pressed under nitrogen for 30-45 minutes. The resultant compound was dried under vacuum for one hours at 25-35 °C and further dried at 80-85 °C for 1 hours to afford the title compound.(Yield 15kg,80percent).
Reference:
[1] Patent: EP1550658, 2005, A1, . Location in patent: Page/Page column 7
[2] Synthetic Communications, 2012, vol. 42, # 5, p. 639 - 649
[3] Patent: EP1223167, 2002, A2, . Location in patent: Page 23
[4] Journal of Medicinal Chemistry, 2000, vol. 43, # 5, p. 775 - 777
[5] Patent: US2005/272787, 2005, A1, . Location in patent: Page/Page column 2
[6] Patent: US2005/272787, 2005, A1, . Location in patent: Page/Page column 2
[7] Patent: WO2005/123701, 2005, A1, . Location in patent: Page/Page column 38-39
[8] Patent: WO2005/85218, 2005, A1, . Location in patent: Page/Page column 10
[9] Patent: CN107137361, 2017, A, . Location in patent: Paragraph 0031-0033
5
[ 952-06-7 ]
[ 108-24-7 ]
[ 181695-72-7 ]
Yield
Reaction Conditions
Operation in experiment
30%
Stage #1: With n-butyllithium In tetrahydrofuran; hexanes at -20 - 20℃; for 2.58333 h; Stage #2: for 2 h;
A solution of desoxybenzoin keto-oxime from Step 1 (6.00 g; 28.40 mmol) in anhydrous tetrahydrofuran (THF, 80 mL) was cooled to -20° C. in an oven-dried 250 mL three-neck round-bottom flask equipped with a thermometer, nitrogen gas inlet, rubber septum and provisions for magnetic stirring. To this cold solution, n-butyllithium (1.6N in hexanes, 44.4 mL) was added, via syringe, over 35 minutes, such that the reaction temperature remained at or below -10° C. The deep red solution was stirred at -10° C. for 1 hour, warmed to room temperature, then stirred at room temperature for an additional hour. Acetic anhydride (3.2 mL, 34.1 mmol) was added in one portion, and the resulting suspension was stirred without temperature control for 2 hours. Water (100 mL) was added, and the solution was poured into 1 N HCl (100 mL) and extracted with ethyl acetate (2.x.200 mL). The combined organic solution was washed with hydrochloric acid (1 N HCl, 10 mL) and brine (100 mL), dried over magnesium sulfate and filtered. The resulting solution was evaporated under reduced pressure to yield a crude oil. The oil was applied to a column of silica gel and eluted with ethyl acetate/hexane (10-50percent ethyl acetate) to yield, upon concentration of the appropriate fractions, 5.0 g of 3,4-diphenyl4-hydrido-5-hydroxy-5-methylisoxazole. The solid was cooled to 0° C., then dissolved in cold chlorosulfonic acid (15 mL). The brown solution was stirred at 0° C. for 2 hours, then added dropwise to a stirring suspension of ice (200 mL) and dichloromethane (200 mL). The layers were separated, and the organic phase was added directly to a saturated ammonium hydroxide solution (100 mL) at 0° C. This biphasic solution was vigorously stirred at 0° C. for 2 hours, the layers were separated, and the aqueous phase was washed with dichloromethane (50 mL). The combined organic solution was dried over magnesium sulfate, filtered and evaporated under reduced pressure to approximately one-half of its original volume. Crystals formed. The stirred suspension was cooled to 0° C. and held for 30 minutes. The crystals were filtered, washed with cold dichloromethane and dried to yield 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide (2.7 g, 30percent): mp 155°-157° C. 1H NMR (CD3CN/500 MHz) δ 7.86 (d, J=8.39 Hz, 2H), 7.45 (m, 1H), 7.39 (s, 4H, 7.37 (d, J=8.39 Hz, 2H), 5.70 (s, 2H), 2.46 (s, 3H Mass Spectrum, MH+=315.
3.1 g (10 mmol) of valdecoxib was mixed with 3.5 g (35 mmol) of triethylamine and 2.6 g (20 mmol) of propionic anhydride at 20 ° CDichloromethane, after the reaction, pour into water, dichloromethane extraction, concentration, ethanol dissolved, 5 ~ 10 crystallization, Filtering and drying to obtain 3.4g of parecoxib, the yield is 92.5percent and the purity is 99.77percent
92.7%
With triethylamine In dichloromethane at 20℃;
The valdecoxib 3.1g (10mmol) and triethylamine 3.5g (35mmol) and propionic anhydride 2.6g (20mmol) were mixed in 20 Methylene chloride, after completion of the reaction, poured into water and extracted with methylene chloride, concentrated, dissolved in ethanol, 5 ~ 10 crystallization,Filtration, drying parecoxib 3.4g, yield 92.7percent, purity of 99.61percent.
92.4%
With triethylamine In dichloromethane at 20℃;
3.1 g (10 mmol) of valdecoxib was mixed with 3 g (30 mmol) of triethylamine,And propionic anhydride (3.3 g, 25 mmol) were mixed in dichloromethane at 20 ° C,After completion of the reaction,Dumped into the water,Dichloromethane extraction,concentrate,Ethanol dissolved,5 ~ 10 crystallization,filter,Drying was 3.4g of parecoxib,The yield was 92.4percentPurity 99.81percent.
83.2%
at 50 - 60℃; for 3 h;
The raw material valdecoxib III (5.0g) was added to propionic anhydride (18.6g), and stirred to dissolve.Benzene sulfonic acid (0.25g) was added, and the temperature was raised to 50 to 60 ° C and reacted for about 3 hours.TLC monitoring reaction was complete, cooled to about 0 , incubated for about 1 hour.Filter, the filter cake with methyl tert-butyl ether (10ml) was rinsed in a vacuum oven, -0.08MPa ~ -0.1MPa, vacuum dried at 50 8 to 12 hours to obtain 4.90g parecoxib sodium intermediate I, yield 83.2percent.HPLC purity of 99.93percent, the largest single miscellaneous 0.05percent, the raw material valdecoxib III
75%
at 50 - 80℃; for 0.333333 h;
Example 17: Preparation of N-((4-(5-methvl-3-Dhenvl-4-isoxazolvl)DhenvllsulfonvllDroDanamide (31, parecoxib).; 4- (5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide 19 (10.0 g, 0.032 mol) and propionic anhydride (40 mL, 0.31 mol) were charged to the 500 mL reactor. The slurry was stirred and heated to 50°C. Sulfuric acid (40 No.L, 0.8 mmol) was added in one portion. All the solids dissolved and the mixture warmed to 55.5°C within a 10 minute period after the addition was completed. The reaction mixture was then heated to 80°C and held for approximately 10 minutes. Heating was discontinued, and the mixture was allowed to cool to 50°C and held for about 60 minutes; solid started to crystallize from the reaction mixture at about 65°C. The mixture was slowly cooled to 0°C and was held at 0°C for about 60 minutes. The solid was collected by vacuum filtration. The wet cake was washed with two 45-mL portions of methyl ten-butyl ether and pulled dry at ambient temperature for about 15 minutes. The solid was further dried in a vacuum oven with a nitrogen bleed at 60°C for 18 hours to give the solid product (8.72 g 75percent yield of 31). DSC maximum endotherm for the high melting point parecoxib is 168.95. DSC maximum endotherm for the low melting point parecoxib is 147.44.
69.6%
With sulfuric acid In water at 50 - 80℃; for 0.333333 h;
4-(5-methyl-3-phenyl-4-isoxazolyl) benzenesulfonamide (25.0 g) and propionic anhydride (100 mL) were charged to the clean and dry round bottom flask and heated to 50 °C. Sulfuric acid (100.00ml) was added slowly and the mixture warmed to 55.5°C. within a 10 minute period after the addition was completed. The reaction mixture was then heated to 80°C and held for approximately 10 minutes. Heating was discontinued, and the mixture was allowed to cool to 25-35°C.Ice water was charged into another round bottom flask and was slowly cooled to 0-5 °C followed by stirring at 0-5°C for 30-45 minutes. The solid was filtered and washed with water 2-butanol and suck dried perfectly. The wet solid was charged into another round bottom flask followed by acetone and stirred for 10-15 minute till the clear solution. DM water (81 mL) was added to the reaction mixture at 25-35°C and stirred at 25-35°C for 30-45 minutes. The resultant solid was filtered and washed with DM water (1250 mL) followed by petroleum ether. The solid was further dried in a vacuum at 60°C to give the titled solid product (20.5 g 69.6percent yield).
8.9g
at 80℃; for 0.5 h;
Successively 10 g of valdecoxib, 10 mL of propionic anhydride, 0.2 mL of sulfuric acid was added to a 100 mL reaction flask, stirring slowly warmed to 80°C, the solids dissolved. The reaction was stirred for 30 minutes incubation. TLC monitoring reaction to valdecoxib disappear, stop the reaction. Slowly cooled to 10°C , the precipitated solid was filtered, washed with water to give a white solid about 8.9g.
2.1 kg
With sulfuric acid In dichloromethane; water at 20 - 30℃; for 2 h; Large scale
Will 2.2kg Valsartan and 7.3kg dichloromethane was added to the reaction flask 30L, At room temperature (20 ~ 30 ° C) was added with stirring 52.8g concentrated sulfuric acid, Then 1.83 kg propionic anhydride was slowly added dropwise,The reaction was stirred at room temperature for 2h,The reaction mixture was evaporated under reduced pressure at 20 ~ 30 ° C dichloromethane, and then slowly added under stirring 4. lkg methyl tert-butyl ether,Slowly cooled to 0 ~ 5 ° C stirring crystallization 3 ~ 5h, suction filtration, the material was washed with 2.3kg methyl tert-butyl ether, 60 ° C under reduced pressure drying 5 ~ 10h, to give 2.1 kg parecoxib.
Reference:
[1] Patent: CN105859647, 2016, A, . Location in patent: Paragraph 0048; 0049; 0050
[2] Patent: CN106008387, 2016, A, . Location in patent: Paragraph 0020; 0048; 0049; 0050
[3] Patent: CN106008386, 2016, A, . Location in patent: Paragraph 0021; 0056; 0057; 0058
[4] Journal of Medicinal Chemistry, 2000, vol. 43, # 9, p. 1661 - 1663
[5] Patent: CN106674142, 2017, A, . Location in patent: Paragraph 0038-0048
[6] Patent: WO2005/123701, 2005, A1, . Location in patent: Page/Page column 39
[7] Patent: EP1550658, 2005, A1, . Location in patent: Page/Page column 9
[8] Patent: US2003/232871, 2003, A1, . Location in patent: Page 5
[9] Patent: CN104447600, 2016, B, . Location in patent: Paragraph 0085; 0088
[10] Patent: CN104193694, 2016, B, . Location in patent: Paragraph 0036; 0037
[11] Patent: CN107137361, 2017, A, . Location in patent: Paragraph 0034-0035
35
[ 181695-72-7 ]
[ 198470-84-7 ]
Yield
Reaction Conditions
Operation in experiment
75%
With sulfuric acid In nitrogen; propionic acid anhydride
EXAMPLE 2 Preparation of N-[[4-(5-methyl-3-phenyl-4-isoxazolyl)phenyl]sulfonyl]propanamide (parecoxib, 1a). 4-(5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide (10.0 g, 0.032 mol) and propionic anhydride (40 mL, 0.31 mol) were charged to the 500 mL reactor. The slurry was stirred and heated to 50° C. Sulfuric acid (40 .box.L, 0.8 mmol) was added in one portion. All the solids dissolved and the mixture warmed to 55.5° C. within a 10 minute period after the addition was completed. The reaction mixture was then heated to 80° C. and held for approximately 10 minutes. Heating was discontinued, and the mixture was allowed to cool to 50° C. and held for about 60 minutes; solid started to crystallize from the reaction mixture at about 65° C. The mixture was slowly cooled to 0° C. and was held at 0° C. for about 60 minutes. The solid was collected by vacuum filtration. The wet cake was washed with two 45-mL portions of methyl tert-butyl ether and pulled dry at ambient temperature for about 15 minutes. The solid was further dried in a vacuum oven with a nitrogen bleed at 60° C. for 18 hours to give the solid product (8.72 g 75percent yield). DSC maximum endotherm for the high melting point parecoxib is 168.95. DSC maximum endotherm for the low melting point parecoxib is 147.44.
With ammonia; In ethyl acetate; at 20 - 25℃; for 2h;
Add 80mL of dichloromethane to the reaction bottle, add compound I11.76g, add formic acid 4.60g, add chlorosulfonic acid 58.26g dropwise at a temperature control of 10-20 C, dropwise finish after 15min, then stir for 15min, add potassium carbonate 27.64g, add Sodium iodide 1.18g, heated to 35-45 C, and the reaction was completed after 1h by TLC (developing agent n-heptane: ethyl acetate = 4: 1, compound I Rf = 0.9, compound II Rf = 0.7). Add the reaction dropwise to the purified water at 0 5 , and control the temperature to be below 20 during the dropwise addition. After the dropwise addition, add 60mL of ethyl acetate and stir for 10min, let stand for 30min, separate the liquid, and wash the organic phase once with purified water. A solution of compound II in ethyl acetate was obtained, and 32.93 g of 25% -28% concentrated ammonia solution was added. The temperature was controlled at 20-25 C and stirred for 2 h. The reaction was monitored by TLC (developing agent n-heptane: ethyl acetate = 2: 1, (Rf of compound II = 0.9, Rf of compound III = 0.4), stand for 30 minutes, separate the layers, wash the organic phase twice with purified water, evaporate the solvent under reduced pressure at 50 C, and add 80 mL of absolute ethanol-methanol (10 : 1), heated to 60 , stirred until dissolved, slowly cooled to 0 ~ 5 , stirred and crystallized for 1h, suction filtered, vacuum dried at 55-60 for 8h, to obtain 14.59g compound III, yield 92.8% , HPLC detection purity 99.8%, the largest single impurity 0.08%.
80%
Charged 120 lit. of methylene chloride 20 kg of 4-(5-methyl-3-phenyl-4-isoxazolyl) benzene sulfonyl chloride into reactor and stirred the reaction mass for 5 to 10 minutes till the dissolution. 1 kg of carbon was charged into above solution, stirred for 30-45 minutes, filtered, and washed with 40ml of methylene chloride. The combined filtrate was charged into reactor and 90 lit. of ammonia solutions were added into the filtrate at 20-30C for 60-90 minutes. Distilled methylene chloride at atmospheric pressure below 40C upto 300 to 340 lit. volume left in the reactor. The reaction mass was cooled to 5-10C and stirred for 30-45 minutes and filtered and washed with 20 lit. methylene chloride. The resultant wet cake was pressed under Nitrogen for 30-45 minutes. The wet material was charged into reactor and 100 lit. of water were added into reactor and stirred for 45-60 minutes and filtered, pressed the wet cake under nitrogen for 15-20 minutes followed by washing with 20 lit. of water. Again wet cake was pressed under nitrogen for 30-45 minutes. The resultant compound was dried under vacuum for one hours at 25-35 C and further dried at 80-85 C for 1 hours to afford the title compound.(Yield 15kg,80%).
77%
With ammonium hydroxide; In dichloromethane; at 10 - 50℃;Cooling with ice;
The system was added dropwise to 1200 ml of ice water.Temperature control is about 10 degrees,Liquid separation extraction,The organic phase is washed twice with water.The organic phase is stirred with ammonia (700 ml) at room temperature.The dot plate shows that the reaction is complete,(PE: EA=4:1),The organic phase is removed by a 50 C rotary steaming system.The remaining system was added with 2 L of ethyl acetate.1L of water is stirred until it is dissolved.Liquid separation extraction,The aqueous phase was back extracted with ethyl acetate.Combine the organic phase,Wash with purified water,The organic phase was removed by rotary evaporation to give a crude material.Add 1L of ethanol,Heat to dissolve,Recrystallization,Filtered to give a crude product (350g).The final product 310 g (986.13 mmol) was obtained (HPLC > 99.5%).White solid, yield 77%.
With ammonia; In water; at 20 - 60℃;Product distribution / selectivity;
EXAMPLE 2 25 ml of 19.5% w/v aqueous ammonia solution were added slowly to a solution of 4-(5-methyl-3-phenyl-4-isoxazolyl) benzene sulfonyl chloride (5 grams) in water (25 ml) at 25-35 C. The reaction mass was heated to 55-60 C. and stirred at this temperature until completion of the reaction. The reaction mass was cooled to about 20-30 C. The solid was filtered and washed with water (30 ml). The compound was suction dried under reduced pressure followed by drying at 25-35 C. under reduced pressure to obtain the desired crystalline Form A of valdecoxib. The yield of the compound was 4.2 grams.
With ammonia; In dichloromethane; water; at 25 - 30℃; for 0.583333 - 0.75h;Product distribution / selectivity;
EXAMPLE 1; 25 ml of 19.5% w/v aqueous ammonia solution were added slowly to a solution of 4-(5-methyl-3-phenyl-4-isoxazolyl) benzene sulfonyl chloride (5 grams) in dichloromethane (50 ml) at 25-30 C. 0.5 grams of crystalline Form A of valdecoxib (as seeding material) was charged to the reaction mass at 25-30 C. and the reaction mass was stirred for 35-45 minutes. The separated solid was filtered and washed with dichloromethane (5 ml). The compound was suction dried under reduced pressure followed by drying at 35-40 C. under reduced pressure to obtain the desired crystalline Form A of valdecoxib. The yield of the compound was 4 grams and the X-ray diffraction pattern for the product is shown in FIG. 1.
With ammonia; In water; at 0℃; for 1h;Product distribution / selectivity;
Example 16: 4-f3-Methyl-5-phenyl-4-isoxazolyllbenzenesulfonamide (valdecoxib, 19).; A solution of 5-methyl-3,4-diphenyl isoxazole 18 (250 mg, 1.1 mmol) in chlorosulfonic acid (1 ml) was stirred at 0C for 3 hours. The reaction was cautiously added to concentrated ammonium hydroxide (6 ml) in the cold (0C). The resultant reaction mixture was stirred at 0C for 1 hour. The reaction was cautiously diluted with water and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered, and the filtrate concentrated in vacuo to give the crude product. This material was chromatographed on silica gel using 25% ethyl acetate in toluene as the eluent to give the desired sulfonamide as a crystalline solid (110 mg, 40% yield of 19) : mp 85 - 87C. Anal. Calc'd. for C16H14N2O3 S: C, 61.13; H, 4.49; N, 8.91; S, 10.20. Found: C, 60.88; H, 4.61; N, 8.55; S, 10.40.
With ammonia; In dichloromethane; at 0℃; for 1h;
EXAMPLE 4 4- (5-Methyl-3-phenyl-4-isoxazolyl) benzenesulfonamide (step 3) Charged 400 ml of dichloromethane in a dry 1L four neck round bottom flask, into this 7.8 g of sulfonyl chloride from example 3 (0.02296 mol) was added at room temperature and stirred well. Reaction mass cooled to 0C and ammonia gas was purged for an hour, during this white solid was formed in the reaction mass. After one hour analysed by HPLC showed completion of reaction. Reaction mass allowed to come to room temperature and dichloromethane layer is washed thrice with water (3 x 100 ml). Dried over anhydrous sodium sulfate and filtered. Dichloromethane is distilled to obtain a one fourth of the initial volume and then cooled to 0 to 5C to get the product crystallised. The product is then filtered and washed with pre-cooled dichloromethane (2 x 10 ml) and the product dried under vacuum at 60 to 70C to obtain a crystalline product (5.6 g, HPLC purity = 99. 7%).
With ammonium hydroxide; In dichloromethane; at 20 - 30℃;Large scale;
The 3.0 kg 5 - methyl - 3, 4 - diphenyl isoxazole added to the 30 L glass reaction flask, then adding 9.9 kg methylene chloride, stirring to dissolve, cooling the solution to - 5 - 0 C, to the reaction solution slowly dropping 15.0 kg chloro sulfonic acid and temperature control ? 20 C, after dropping, water bath heating to 35 C insulation reaction 4 - 5 of H, then the reaction liquid cooling to 0 - 5 C.The 15.9 kg methylene chloride and 12.0 kg purified water by adding another 50 L in the reaction bottle, cooling to 0 - 5 C, slowly dropping has cooling of the reaction fluid and temperature control ? 20 C, after dropping stirring 0.5 h; layered, for methylene chloride level (the upper layer) 12.0 kg (25% w/w) saturated sodium chloride solution agitation washing, the delaminated, taking methylene chloride level (lower) spare; to the 50 L reaction flask add 5.4 kg concentrated ammonia water, then slowly adding the dichloromethane layer solution, at room temperature (20 - 30 C) stirring the reaction 2 - 3 H of the; added to the reaction solution after 11.7 kg isopropanol, 35 C thermal insulation to continue stirring 1.5 h, then cooled to 15 - 20 C, stirring crystallization 6 h, then adding, for material 2.4 kg of cold isopropanol washing, 60 C drying under atmospheric pressure 5 - 10 the H, get-cutting to past cloth thick 3.1 kg; will-cutting to past cloth thick 3.1 kg and 7.36 kg methanol by adding 30 L glass reaction flask, stirring under heating to 60 - 70 C reflux, filtered to remove insoluble matter, then adding 3.1 kg purified water, the temperature slowly drops to 15 - 20 C, stirring crystallization 6 h, filtered, in order to 3.1 kg cold methanol/water (V/V=3:1, W/W=2.37: 1) in the solution, 60 C drying under atmospheric pressure 5 - 10 the H, get-cutting to [...] 2.2 kg.
Weighed 15g (47.7mmol, 1e.q) of <strong>[181695-72-7]valdecoxib</strong> (compound of formula 3),Add to 22.5ml of propionic anhydride (238.5mmol, 5e.q), stir and heat to raise the temperature to 50 C,Add 0.076 g (4.8 mmol, 0.1 e.q) benzenesulfonic acid, react for 120 minutes, and cool to room temperature (15-30 C).Crystallization, filtration, vacuum drying at 55±5C, vacuum degree -0.070-0.082Mpa for 4-6h,17.27 g of parecoxib was obtained, the yield was 97.7%, and the purity was 99.9% by HPLC.
92.5%
With triethylamine; In dichloromethane; at 20℃;
3.1 g (10 mmol) of <strong>[181695-72-7]valdecoxib</strong> was mixed with 3.5 g (35 mmol) of triethylamine and 2.6 g (20 mmol) of propionic anhydride at 20 CDichloromethane, after the reaction, pour into water, dichloromethane extraction, concentration, ethanol dissolved, 5 ~ 10 crystallization, Filtering and drying to obtain 3.4g of parecoxib, the yield is 92.5% and the purity is 99.77%
92.7%
With triethylamine; In dichloromethane; at 20℃;
The <strong>[181695-72-7]valdecoxib</strong> 3.1g (10mmol) and triethylamine 3.5g (35mmol) and propionic anhydride 2.6g (20mmol) were mixed in 20 Methylene chloride, after completion of the reaction, poured into water and extracted with methylene chloride, concentrated, dissolved in ethanol, 5 ~ 10 crystallization,Filtration, drying parecoxib 3.4g, yield 92.7%, purity of 99.61%.
92.4%
With triethylamine; In dichloromethane; at 20℃;
3.1 g (10 mmol) of <strong>[181695-72-7]valdecoxib</strong> was mixed with 3 g (30 mmol) of triethylamine,And propionic anhydride (3.3 g, 25 mmol) were mixed in dichloromethane at 20 C,After completion of the reaction,Dumped into the water,Dichloromethane extraction,concentrate,Ethanol dissolved,5 ~ 10 crystallization,filter,Drying was 3.4g of parecoxib,The yield was 92.4%Purity 99.81%.
90.2%
With 4-pyrrolidin-1-ylpyridine; triethylamine; In dichloromethane; acetonitrile; at 20 - 25℃; for 2h;
In the reaction bottle, 60 mL (3: 1, V / V) of dichloromethane-acetonitrile mixed solvent was added, 12.57 g of compound III was added, the temperature was controlled at 20-25 C., 7.11 g of 4-pyrrolidinopyridine was added, and 10.41 g of propionic anhydride was added. , 16.19g of triethylamine was added, and the reaction was controlled at a temperature of 20-25 C. After 2h, the reaction was monitored by TLC (developing agent n-heptane: ethyl acetate = 2: 1, compound III Rf = 0.4, compound IV Rf = 0.1 ), 80mL of purified water was added, stirred for 20min, and allowed to stand for liquid separation. The aqueous phase was added with dichloromethane for extraction. The organic phase was evaporated to oil under reduced pressure at 35 C, 60mL of acetone was added, heated to reflux to dissolve, and slowly cooled to 0-5 At , stirring and crystallizing for 2h, suction filtration and vacuum drying to obtain 13.36g of compound IV, yield 90.2%, purity by HPLC detection 99.8%, maximum single impurity 0.06%.
87.6%
With triethylamine; In dichloromethane; at 20℃;
Compound (PCB-2) (310 g, 986.13 mmol) was added to a 5 L three-necked flask.Add 3L DCM to dissolve,Propionic anhydride (256.7 g, 1972.34 mmol) was added.Triethylamine (400 g, 3529.96 mmol) was added.Reactive at room temperature for 5-6 hours,The TLC dot plate shows the completion of the reaction (PE: EA = 3:1),Add 1L of water,Liquid separation extraction,The organic phase was washed three times with 400 ml of water.The solvent was dried to give a crude product.Add 1800 ml of acetone to reflux to dissolve.Slow cooling and crystallization,Filtering to obtain 340g of product,The product was further added with 1.5 L of acetone and heated to reflux to dissolve.Hot filtration,The mother liquor is cooled and stirred for crystallization.A white solid powder (320 g 863.8 mmol) was obtained (HPLC > 99.5%,Single miscellaneous <0.1%),The yield was 87.6%.
83.2%
With benzenesulfonic acid; at 50 - 60℃; for 3h;
The raw material <strong>[181695-72-7]valdecoxib</strong> III (5.0g) was added to propionic anhydride (18.6g), and stirred to dissolve.Benzene sulfonic acid (0.25g) was added, and the temperature was raised to 50 to 60 C and reacted for about 3 hours.TLC monitoring reaction was complete, cooled to about 0 , incubated for about 1 hour.Filter, the filter cake with methyl tert-butyl ether (10ml) was rinsed in a vacuum oven, -0.08MPa ~ -0.1MPa, vacuum dried at 50 8 to 12 hours to obtain 4.90g parecoxib sodium intermediate I, yield 83.2%.HPLC purity of 99.93%, the largest single miscellaneous 0.05%, the raw material <strong>[181695-72-7]valdecoxib</strong> III
75%
sulfuric acid; at 50 - 80℃; for 0.333333h;
Example 17: Preparation of N-((4-(5-methvl-3-Dhenvl-4-isoxazolvl)DhenvllsulfonvllDroDanamide (31, parecoxib).; 4- (5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide 19 (10.0 g, 0.032 mol) and propionic anhydride (40 mL, 0.31 mol) were charged to the 500 mL reactor. The slurry was stirred and heated to 50C. Sulfuric acid (40 No.L, 0.8 mmol) was added in one portion. All the solids dissolved and the mixture warmed to 55.5C within a 10 minute period after the addition was completed. The reaction mixture was then heated to 80C and held for approximately 10 minutes. Heating was discontinued, and the mixture was allowed to cool to 50C and held for about 60 minutes; solid started to crystallize from the reaction mixture at about 65C. The mixture was slowly cooled to 0C and was held at 0C for about 60 minutes. The solid was collected by vacuum filtration. The wet cake was washed with two 45-mL portions of methyl ten-butyl ether and pulled dry at ambient temperature for about 15 minutes. The solid was further dried in a vacuum oven with a nitrogen bleed at 60C for 18 hours to give the solid product (8.72 g 75% yield of 31). DSC maximum endotherm for the high melting point parecoxib is 168.95. DSC maximum endotherm for the low melting point parecoxib is 147.44.
69.6%
With sulfuric acid; In water; at 50 - 80℃; for 0.333333h;
4-(5-methyl-3-phenyl-4-isoxazolyl) benzenesulfonamide (25.0 g) and propionic anhydride (100 mL) were charged to the clean and dry round bottom flask and heated to 50 C. Sulfuric acid (100.00ml) was added slowly and the mixture warmed to 55.5C. within a 10 minute period after the addition was completed. The reaction mixture was then heated to 80C and held for approximately 10 minutes. Heating was discontinued, and the mixture was allowed to cool to 25-35C.Ice water was charged into another round bottom flask and was slowly cooled to 0-5 C followed by stirring at 0-5C for 30-45 minutes. The solid was filtered and washed with water 2-butanol and suck dried perfectly. The wet solid was charged into another round bottom flask followed by acetone and stirred for 10-15 minute till the clear solution. DM water (81 mL) was added to the reaction mixture at 25-35C and stirred at 25-35C for 30-45 minutes. The resultant solid was filtered and washed with DM water (1250 mL) followed by petroleum ether. The solid was further dried in a vacuum at 60C to give the titled solid product (20.5 g 69.6% yield).
sulfuric acid; at 50 - 80℃; for 2h;
In the fourth step, a reaction vessel is charged with <strong>[181695-72-7]4-(5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide</strong> (VII) (21 kg) and propionic anhydride (86 kg). The resulting suspension is warmed to 50 C., and sulfuric acid (21 ml) is added. The reaction mixture is warmed to 80 C. and held for at least 30 minutes. The mixture is slowly cooled to 50 C. to initiate crystallization of the product. The mixture is held at 50 C. for at least 30 minutes after crystallization is initiated. The mixture can be seeded if crystallization does not initiate at 50 C. The mixture is slowly cooled to 0 C. and held at 0 C. for at least 1 hour to complete the crystallization. The product was isolated, washed with methyl tert-butyl ether (80 liters), and partially dried on the filter until an in-process check indicates that LOD is not more than 5%, to give n-[[4-(5-methyl-3-phenyl-4-isoxazolyl)phenyl]sulfonyl]propanamide (VIII) as a wet cake that is carried directly into the fifth step without further purification or drying.
With dmap; triethylamine; In tetrahydrofuran; at 40℃; for 3h;
To 20L glass reactor was added <strong>[181695-72-7]valdecoxib</strong> (2.092kg), tetrahydrofuran 11.17kg, was added 4-dimethylaminopyridine 0.084kg, propionic anhydride 1.569kg, triethylamine 0.837kg.Plus Albert, 40 reaction 3h.The reaction was monitored by TLC.Completion of the reaction, the reaction solution was cooled to 10 ~ 20 , stirring crystallization 1 ~ 2h.Filtration, drained, and the resulting cake was 40 ~ 60 blast drying 12h, parecoxib was crude.Parecoxib crude product was purified three times the amount of tetrahydrofuran to give parecoxib.
8.9g
With sulfuric acid; at 80℃; for 0.5h;
Successively 10 g of <strong>[181695-72-7]valdecoxib</strong>, 10 mL of propionic anhydride, 0.2 mL of sulfuric acid was added to a 100 mL reaction flask, stirring slowly warmed to 80C, the solids dissolved. The reaction was stirred for 30 minutes incubation. TLC monitoring reaction to <strong>[181695-72-7]valdecoxib</strong> disappear, stop the reaction. Slowly cooled to 10C , the precipitated solid was filtered, washed with water to give a white solid about 8.9g.
2.1 kg
With sulfuric acid; In dichloromethane; water; at 20 - 30℃; for 2h;Large scale;
Will 2.2kg Valsartan and 7.3kg dichloromethane was added to the reaction flask 30L, At room temperature (20 ~ 30 C) was added with stirring 52.8g concentrated sulfuric acid, Then 1.83 kg propionic anhydride was slowly added dropwise,The reaction was stirred at room temperature for 2h,The reaction mixture was evaporated under reduced pressure at 20 ~ 30 C dichloromethane, and then slowly added under stirring 4. lkg methyl tert-butyl ether,Slowly cooled to 0 ~ 5 C stirring crystallization 3 ~ 5h, suction filtration, the material was washed with 2.3kg methyl tert-butyl ether, 60 C under reduced pressure drying 5 ~ 10h, to give 2.1 kg parecoxib.
With dmap; triethylamine; In dichloromethane; at 25℃; for 1h;Large scale;
(1) Add 4000 g of dichloromethane to the reaction kettle, and add 1000 g of compound B, 19.4 g of 4-dimethylaminopyridine, and 418 g of triethylamine to the reaction kettle in order under stirring, and then jacket the jacket with cooling water. The internal temperature was controlled at 25 ± 10 C, and 497 g of propionic anhydride was added dropwise.(2) After the addition is completed, the reaction is kept at 25 C with stirring and stirring, and the time and temperature are recorded every 0.5 hours; the temperature is monitored for 1 hour by TLC, and then every 0.5 hours for sampling and monitoring. PE = 1: 1) until the intermediate B disappears as the end point of the reaction, the reaction time is 1 hour, and the reaction liquid is obtained after the reaction is completed.(3) The reactor was jacketed with 45 ± 5 C hot water, and the reaction solution was concentrated under reduced pressure until the condensate did not drip.(4) Add 5000 g of ethyl acetate, and the resulting ethyl acetate solution is stirred and washed three times with purified water: the amount of purified water is 4 times the volume of the ethyl acetate solution, washed and stirred for 10 minutes each, and left for 20 minutes. Take the organic phase.(5) The kettle was jacketed with 45 C hot water under reduced pressure to concentrate the organic phase obtained in step (4) until the solid A precipitated to constant weight.(6) After the concentration is completed, the solid-liquid obtained in step (5) is separated. The separated liquid is transferred to a crystallization barrel, 2000 g of n-hexane is added, and the crystallization is performed at a temperature of 10 C. The time and time are recorded every 0.5 hours Temperature, crystallization for 2 ± 0.5 hours, moderate stirring during crystallization to obtain solid B.(7) Mix solid A and solid B, centrifuge for 10 minutes, and filter. The obtained filter cake is dried in a hot air oven at 60 ± 5 C. The temperature and time are recorded every 1 hour and dried for 8 hours. A pale yellow to off-white solid was obtained, which is parecoxib, the intermediate of parecoxib sodium of the present invention.The above-mentioned "preparation method of parecoxib" was repeated three times, and the obtained parecoxib was Sample 1, Sample 2, and Sample 3, respectively.
The obtained in Example 1(10 mmol) of 5-methyl-3,4-diphenylisoxazole and 2.3 g (20 mmol) of chlorosulfonic acid were stirred at 0 to 5 C for 30 minutes,1 g of anion exchange resin (40%) was then added,The temperature was raised to 20 C,15.6 g (60 mmol) of saturated ammonium chloride was added dropwise,After completion of the reaction,Dichloromethane extraction,Washed,concentrate,Ethanol to obtain 2.95 g of valdecoxib,The yield was 93.7%Purity 99.63%.
92.7%
A solution of 11.7 g (50 mmol) of 5-methyl-3,4-diphenylisoxazole and 9.3 g (80 mmol) of chlorosulfonic acid at 0 to 5 C was stirred 40Minute, extracted with methylene chloride,DichloromethanephaseAmmonia was added directlyWater, the temperature rose to 20 C to continue the reaction, after the end of the reaction, Dichloromethane extraction, washing with water, concentration and ethanol recrystallization to obtain 14.6g of vediloxib in a yield of 92.7% and purity of 99.69%.
89%
After the compound 21.16g (90mmol), trifluoroacetic acid 20.52g (180mmol) added to the reaction flask, control temperature 20 dropwise chlorosulfonic acid 20.97g (180mmol), TLC monitoring completion of the reaction, the reaction was added dropwise to the above purified water, after the end of the dropwise addition, ethyl acetate was added 2 times, the combined organic phase was washed once purified, was added after the ethyl acetate solution was washed with concentrated ammonia solution, stirred at room temperature 2h, TLC monitoring of the reaction the reaction. Separated, washed twice with water purified. The combined aqueous phase was added ethyl acetate and 2 times the anti-quenching. Finally, the combined organic phase was concentrated under reduced pressure to give the crude product. Recrystallization from absolute ethanol. To give compound 25.18g (80.1mmol), yield 89.0%, purity 99.3% (HPLC method).
81%
Sequentially 10 g 5-methyl-3,4-diphenylisoxazole, was added to a 100 mL flask, 30 mL of dichloromethane, stirring, a clear solution; under ice-cooling, 20mL of chlorosulfonic acid was slowly added dropwise to the solution. After finishing the dropping, the reaction vessel was slowly warmed to reflux for about 3 hours. TLC monitored the reaction to 5-methyl-3,4-diphenylisoxazole completely disappeared, the reaction was stopped.to the 100 mL beaker by adding crushed ice, water, stirring conditions, the cold to room temperature of the reaction solution slowly into the ice water mixture. After the solution was complete, the layers were separated and the organic layer was collected. The aqueous layer was extracted twice with methylene chloride. The organic layers were combined and placed in a 250 mL flask for direct amination reaction.under stirring, 50 mL of aqueous ammonia was slowly added dropwise to the above methylene chloride solution. After the dropwise addition, the reaction was allowed to stand at room temperature for about 4 hours. TLC monitoring reaction. After completion of the reaction, the reaction solution was allowed to stand, the organic layer was separated, and the aqueous layer was extracted twice with dichloromethane. The combined organic layer was washed with saturated brine and dried over sodium sulfate. The organic layer was filtered and the filtrate was concentrated to give crude tacrolimus and the ethanol was recrystallized to give valdecoxib 10.8 g of as a white solid in 81% yield.
60%
Chlorosulfonic acid (9.6 ml, 11.5 mol) was added slowly to 3.0 g (1.0 mol) of 3,4-dipheny-5-methyl isoxazole at 0-5C. After a clear solution was obtained, slowly the solution was heated to 50-60C and stirred for 2 hours. The solution was poured into a mixture of 130 ml of dichloromethane and 130 ml of chilled water. The dichloromethane layer was separated and cooled to 0-5C. Ammonium hydroxide solution (65 ml) was added to the dichloromethane layer, which was stirred for 2 hours. The dichloromethane layer was separated, and the aqueous layer was extracted with 30 ml of dichloromethane. The combined dichloromethane layer dried over 1.0 g of sodium sulfate. The dichloromethane layer was condensed to approximately one-half of its original volume, cooled to 0-5C and stirred for 30 minutes. Solid was separated by filtration and washed with 3.0 ml of chilled dichloromethane. The filtered compound was dried at 50-55C under vacuum. The yield of solid product is 2.4 g (60.0 %).
Added to the 20L glass reactor of 5-methyl-3,4-diphenyl-isoxazole 2.2kg and trifluoroacetic acid 6.75kg, chlorosulfonic acid 7.64kg.Was allowed to react at 60 3 ~ 6h, TLC monitoring of the reaction, the reaction is complete, the system under ice water was slowly added to quench the reaction 22kg, end quenched toluene 7kg extracted twice, the combined organic phases.The organic phase was added 25% aqueous ammonia 3kg, 35 reaction 1 ~ 2h.The reaction was monitored by TLC.Completion of the reaction, the filter cake washed with 17kg beating purified water, at 40 ~ 60 cake was blow dried overnight to yield crude celecoxib valdecoxib.Valdecoxib crude recrystallized celecoxib valdecoxib with ethanol / water.
In the third step, a reaction vessel is charged with 4,5-dihydro-5-methyl-3,4diphenyl-5-isoxazolol (VI) (152 kg) and trifluoroacetic acid (TFA, 116 liters). The mixture is cooled and chlorosulfonic acid (705 kg) is added while maintaining the temperature of the reaction mixture below 25 C. After the addition is complete, the mixture is slowly heated to 60 C. and held at 60 C. for at least 1 hour. The reaction mixture is cooled and quenched by adding it to a mixture of water (456 liters) and toluene (570 liters) that is maintained below 25 C. during this addition. The reaction vessel and transfer lines are rinsed with a mixture of water (152 liters) and toluene (61 liters). The layers are separated, and the organic phase is washed with water (220 liters). The organic phase is treated with aqueous ammonium hydroxide (190 liters), and the mixture is heated to 35 C. and held at 35 C. for at least 30 minutes. An in-process check is performed to ensure that pH of the aqueous phase is not less than 9. Isopropyl alcohol (729 liters) is added, and the mixture is held at 35 C. for at least 1 hour. The mixture is cooled to 20 C. and held at 20 C. for at least 1 hour. The precipitated product is isolated and washed with isopropyl alcohol (304 liters) and then with water (at least 101 liters). The crude product is dissolved in hot methanol (709 liters). The solution is filtered to remove particulates and diluted with additional methanol (355 liters) and water (274 liters). The mixture is heated to 70 C. to dissolve the solid and then slowly cooled to initiate crystallization of the product. The mixture can be seeded if crystallization does not initiate by the time 45 C. is reached. Once crystallization occurs, the mixture is stirred at 50 C. for at least 1 hour and then slowly cooled to 5-10 C. and held at that temperature for at least 1 hour. The product is isolated and washed with a mixture of methanol and water (at least 95 liters having a 3:1 ratio of methanol to water). Alternatively, the product can be purified by recrystallization from a mixture of ethanol (1,300 liters) and water (68 liters) using the same procedure described above. The product is dried under vacuum at temperatures up to 100 C. until the amount of residual solvents by LOD or gas chromatography is not more than 0.5%, to give 4-(5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide (VII) in a typical yield of 103 kg (62% by weight).
53.5%
With chlorosulfonic acid; ammonium hydroxide; In dichloromethane; water;
Step 3. Preparation of 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide. 5-Hydroxy-5-methyl-3,4-diphenylisoxazoline (Step 2) (142 g, 0.56 mol) was dissolved in dichloromethane (568 mL) in a 3 L roundbottom flask equipped with a heating mantle, mechanical stirrer, cold water condenser, J-KEM temperature controller and thermocouple, forming a slurry. The slurry was stirred and cooled to <10 C. Chlorosulfonic acid (335 mL, 586.3 g, 5.04 mol) was added to the slurry, keeping the temperature of the flask below 20 C. by controlling the addition. The mixture was heated to reflux (ca. 40 C.), maintained for 5 hours, then cooled to 0-5 C. The cooled reaction solution was slowly transferred to a 3 L 3-necked roundbottom flask (mechanical stirrer and thermocouple) containing water (1000 ml) previously cooled to 0-5 C., using vigorous agitation and keeping the pot temperature below 10 C. The mixture was stirred for an additional 5 minutes. The layers were separated. In a separate 3 L flask (mechanical stirrer, external ice/salt bath, thermocouple) 28% ammonium hydroxide (700-mL) was cooled to 0-5 C. The methylene chloride solution was transferred to the stirred ammonium hydroxide solution, keeping the temperature below 10 C. The mixture was stirred at ambient temperature for 60 minutes. The resulting slurry was filtered and the solid was washed with water (200 ml) and dried, yielding the 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide as a white solid (94.3 g, 53.5%).
The solid was cooled to 0C., then dissolved in cold CHLOROSULFONIC acid (15 mL). The brown solution was stirred at 0C. for 2 hours, then added dropwise to a stirring suspension of ice (200 mL) and DICHLOROMETHANE (200 mL). The layers were separated, and the organic phase was added directly to a saturated ammonium hydroxide solution (100 mL) at 0C. This biphasic solution was vigorously stirred at 0C. for 2 hours, the layers were separated, and the aqueous phase was washed with dichloromethane (50 mL). The combined organic solution was dried over magnesium sulfate, filtered and evaporated under reduced pressure to approximately one-half of its original volume. Crystals formed. The stirred suspension was cooled to 0C. and held for 30 minutes. The crystals were filtered, washed with COLD DICHLOROMETHANE and dried to yield 4- [5-METHYL-3-PHENYLISOXAZOL- 4-yl] benzenesulfonamide (2.7 g, 30%): mp 155. DEGREE.-157. DEGREE. C. 1H NMR (CD3 CN/500 MHz) A 7.86 (d, J=8.39 Hz, 2H), 7.45 (m, 1 H), 7.39 (s, 4H, 7.37 (d, J=8. 39 Hz, 2H), 5.70 (s, 2H), 2.46 (s, 3H Mass Spectrum, MH+=315).
With dmap; triethylamine; In tetrahydrofuran; at 20℃; for 72h;
TEA (24 mg, 0.236 mmol) and DMAP (13 mg) were added to a mixture of <strong>[181695-72-7]valdecoxib</strong> (62 mg, 0.195 mmol) and the product obtained from step 1 above in THF (1 mL) and stirred at RT for 3 d. The mixture was concentrated and the residue dissolved in EtOAc. After usual aqueous work-up and chromatographic purification, 53 mg (52%) of I-A3-PD3a obtained. 1H- NMR (300 MHz, CDCl3): delta 2.51 (s, 3H), 2.97 (t, 2H, J=6.0 Hz), 3.48 (s, 2H), 3.76 (s, 3H), 4.37 (t, 2H, J=6.0 Hz), 7.33-7.40 (m, 7H), 8.03-8.12 (m, 2H). MS (m/z): 521 [M-H]-.
With 2-(Dimethylamino)pyridine; triethylamine; In dichloromethane; ethyl acetate;
Step 1 Preparation of [[4-(5-methyl-3-phenylisoxazol-4-yl)phenyl]sulfonyl]-carbamic acid, 1,1-dimethylethylester To a stirred suspension of 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide (Example 1) (10.42 g, 33.1 mmol) in dichloromethane (100 mL) was added di-tert-butyldicarbonate (7.59 g, 34.80 mmol), dimethylaminopyridine (0.202 g, 1.66 mmol) and triethylamine (5.07 mL, 3.68 g, 36.4 mmol). The resulting homogeneous solution was stirred overnight. The reaction was diluted with ethyl acetate and dichloromethane, washed with KHSO4 solution (0.25M), brine, dried over MgSO4, filtered and concentrated in vacuo yielding a white powder. The powder was dissolved in hot ethyl acetate and diluted with isooctane, yielding [[4-(5-methyl-3-phenylisoxazol-4-yl)phenyl]sulfonyl]-carbamic acid, 1,1-dimethylethylester as a fine white powder (9.94 g, 72%): mp 167.6-170.5 C. 1 H NMR (CDCl3) 8.01 (d, J=8.66 Hz, 2H), 7.51 (s, 1H), 7.46-7.30 (m, 7H), 2.50 (s, 3H), 1.40 (s, 9H). LRMS M+H obs at m/z 415. Anal. Calc'd. for C21 H22 N2 O5 S: C, 60.86; H, 5.35; N, 6.76. Found: C, 60.79; H, 5.40; N, 6.75.
4-[4-(2,5-dimethylpyrrole-1-sulfonyl)phenyl]-5-methyl-3-phenylisoxazole[ No CAS ]
[ 181695-72-7 ]
Yield
Reaction Conditions
Operation in experiment
With water; trifluoroacetic acid; at 20 - 90℃; for 48h;Product distribution / selectivity;
8c. Valdecoxib (19).; 4-[4-(2,5-Dimethylpyrrole-1-sulfonyl)phenyl]-5-methyl-3-phenylisoxazole (30,470 mg 1.2 mmol) was mixed with trifluoroacetic acid (1 mL) and water (1 mL) and heated in a 90 C oil bath. The reaction was sampled and analyzed by TLC (silica gel 1 B-F 2.5 x 78.5 cm slide, J. T. Baker, Phillipsburg, NJ) developed with dichloromethane or ether. Compound 30 was approximately 50 % consumed. The reaction was left at ambient for 2 days (i. e. over the weekend) and then heating at 90 C was resumed for an additional 2 hours and 45 minutes. The TLC at this point indicated that no more than a trace of the starting 30 remained. The reaction was concentrated to dryness under vacuum. The brown solid residue was crystallized from 2 mL of 50% aqueous methanol. The brown solid recovered was dried at ambient temperature, 100 Torr, with a nitrogen sweep. The nmr (CD30D) was identical to the nmr of a reference sample of valdecoxib.
Step 2. Preparation of 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide. A solution of desoxybenzoin keto-oxime from Step 1 (6.00 g; 28.40 mmol) in anhydrous tetrahydrofuran (THF, 80 mL) was cooled to -20 C. To this cold solution, n-butyllithium (1.6N in hexanes, 44.4 mL) was added, via syringe, over 35 minutes, such that the reaction temperature remained at or below -10 C. The deep red solution was stirred at -10 C. for 1 hour, warmed to room temperature, then stirred at room temperature for an additional hour. Acetic anhydride (3.2 mL, 34.1 mmol) was added in one portion, and the resulting suspension was stirred without temperature control for 2 hours. Water (100 mL) was added, the solution was poured into 1N HCl (100 mL) and extracted with ethyl acetate (2*200 mL). The combined organic solution was washed with hydrochloric acid (1N HCl, 100 mL) and brine (100 mL), dried over magnesium sulfate and filtered. The resulting solution was evaporated under reduced pressure to yield a crude oil. The oil was applied to a column of silica gel and eluted with ethyl acetate/hexane (10-50% ethyl acetate) to yield, upon concentration of the appropriate fractions, 5.0 g of 3,4-diphenyl-4-hydrido-5-hydroxy-5-methylisoxazole. The solid was cooled to 0 C., then dissolved in cold chlorosulfonic acid (15 mL). The brown solution was stirred at 0 C. for 2 hours, then added dropwise to a stirred suspension of ice (200 mL) and dichloromethane (200 mL). The layers were separated, and the organic phase was added directly to a saturated ammonium hydroxide solution (100 mL) at 0 C. This biphasic solution was vigorously stirred at 0 C. for 2 hours, the layers were separated, and the aqueous phase was washed with dichloromethane (50 mL). The combined organic solution was dried over magnesium sulfate, filtered and evaporated under reduced pressure to approximately one-half of its original volume. Crystals formed. The stirred suspension was cooled to 0 C. and held for 30 minutes. The crystals were filtered, washed with cold dichloromethane and dried to yield 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide (2.7 g, 30%): mp 155-57 C. 1 H NMR (CD3 CN/500 MHz) delta 7.86 (d, J=8.39 Hz, 2H), 7.45 (m, 1H), 7.39 (s, 4H), 7.37 (d, J=8.39 Hz, 2H), 5.70 (s, 2H), 2.46 (s, 3H). Mass Spectrum, MH+=315.
30%
Step 2. Preparation of 4-[5-methyl-3-phenylisoxazol-4-yl]benzensulfonamide. A solution of desoxybenzoin keto-oxime from Step 1 (6.00 g; 28.40 mmol) in anhydrous tetrahydrofuran (THF, 80 mL) was cooled to -20 C. in an oven-dried 250 mL three-neck round-bottom flask equipped with a thermometer, nitrogen gas inlet, rubber septum and provisions for magnetic stirring. To this cold solution, n-butyllithium (1.6N in hexanes, 44.4 mL) was added, via syringe, over 35 minutes, such that the reaction temperature remained at or below -10 C. The deep red solution was stirred at -10 C. for 1 hour, warmed to room temperature, then stirred at room temperature for an additional hour. Acetic anhydride (3.2 mL, 34.1 mmol) was added in one portion, and the resulting suspension was stirred without temperature control for 2 hours. Water (100 mL) was added, and the solution was poured into 1N HCl (100 mL) and extracted with ethyl acetate (2*200 mL). The combined organic solution was washed with hydrochloric acid (1N HCl, 100 mL) and brine (100 mL), dried over magnesium sulfate and filtered. The resulting solution was evaporated under reduced pressure to yield a crude oil. The oil was applied to a column of silica gel and eluted with ethyl acetate/hexane (10-50% ethyl acetate) to yield, upon concentration of the appropriate fractions, 5.0 g of 3,4-diphenyl-4-hydrido-5-hydroxy-5-methylisoxazole. The solid was cooled to 0 C., then dissolved in cold chlorosulfonic acid (15 mL). The brown solution was stirred at 0 C. for 2 hours, then added dropwise to a stirring suspension of ice (200 mL) and dichloromethane (200 mL). The layers were separated, and the organic phase was added directly to a saturated ammonium hydroxide solution (100 mL) at 0 C. This biphasic solution was vigorously stirred at 0 C. for 2 hours, the layers were separated, and the aqueous phase was washed with dichloromethane (50 mL). The combined organic solution was dried over magnesium sulfate, filtered and evaporated under reduced pressure to approximately one-half of its original volume. Crystals formed. The stirred suspension was cooled to 0 C. and held for 30 minutes. The crystals were filtered, washed with cold dichloromethane and dried to yield 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide (2.7 g, 30%): mp 155-157 C. 1 H NMR (CD3 CN/500 MHz) delta 7.86 (d, J=8.39 Hz, 2H), 7.45 (m, 1H), 7.39 (s, 4H, 7.37 (d, J=8.39 Hz, 2H), 5.70 (s, 2H), 2.46 (s, 3H Mass Spectrum, MH+=315.
Additional specific compounds of particular interest within Formula I include each of the compounds and pharmaceutically-acceptable salts thereof as follows: ... 4-(4-cyclohexyl-2-methyloxazol-5-yl)-2-fluorobenzenesulfonamide, 5-chloro-3-(4-(methylsulfonyl)phenyl)-2-(methyl-5-pyridinyl)pyridine, 2-(3,5-difluorophenyl)-3-4-(methylsulfonyl)phenyl2-cyclopenten-1-one, 4-(4-(methylsulfonyl)phenyl]-3-phenyl-2(5H)-furanone, 4-[5-methyl-3-phenyl-isoxazol-4-yl]benzenesulfonamide, and N-[[4-(5-methyl-3-phenylisoxazol-4-yl]phenyl]sulfonyl]propanamide.
wherein the cyclooxygenase-2 selective inhibiting compound is selected from the group consisting of: celecoxib; deracoxib; valdecoxib; parecoxib; rofecoxib; etoricoxib; meloxicam; lumiracoxib; 4-(4-cyclohexyl-2-methyloxazol-5-yl)-2-fluorobenzenesulfonamide; ...
Step 4. Recrystallization of 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide The 4-[(5-methyl-3-phenyl)-4-isoxazolyl] benzenesulfonamide from Step 3 was dissolved in 300 mL of boiling methyl ethyl ketone (2-butanone) and diluted with 10% aqueous isopropyl alcohol (300 mL, (270 mL anhydrous isopropyl alcohol and 30 mL of water)). The material was cooled to room temperature, whereupon crystals formed. The crystals were isolated by filtration and dried in a vacuum drying oven (10 mm Hg, 100 C.) to afford pure Form B (112.95 g, 65%): mp 172-173 C.
3e. 4-[5-(Methyl)-3-phenylisoxazol-4-yl]benzenesulfonamide This compound was synthesised as described in patent application WO 96/25405, Example 1, the disclosure of which is incorporated by reference herein in its entirety. mp 170 C. 1H NMR (300 MHz, CD3CN) delta7.90 (d, J=8.4 Hz, 2 H), 7.39 - 7.49 (mult, 7 H), 5.48 (s, 2 H), 2.50 (s, 2 H); mass spectrum (API-TIS), m/z 315 (MH+).
Particularly preferred compounds of formula (I) include: ... 5-difluoromethyl-4-(4-(methylsulfonyl)phenyl)-3-phenylisoxazole; 4-(3-ethyl-5-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-difluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-hydroxymethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide (valdecoxib); N-propanoyl-4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 1-(2-(4-fluorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzenee; 1-(2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; ...
MK-966 (Merck & Co); L-752,860 (Merck & Co); L-783003 (Merck & Co); T-614 (Toyama); D-1367 (Chiroscience); L-748731 (Merck & Co); L-745337 (Merck & Co); and compounds of Formula I ... 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl) -1H-pyrazol-1-yl]benzenesulfonamide; 3-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-oxazolyl]benzenesulfonamide; 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; and ...
...cted from compounds, and their pharmaceutically acceptable salts, of the group consisting of meloxicam (Boehringer Ingelheim); nimesulide (Helsinn); MK-966 (Merck & Co); L-783003 (Merck & Co); T-614 (Toyama); D-1367 (Chiroscience); L-748731 (Merck & Co); L-745337 (Merck & Co); ... 5-difluoromethyl-4-(4-methylsulfonylphenyl)-3-phenylisoxazole; 4-[3-ethyl-5-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-difluoromethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-methyl-3-phenyl-isoxazol-4-yl]benzenesulfonamide; 1-[2-(4-fluorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; 1-[2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; 1-[2-(4-chlorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; ...
wherein the selective COX-2 inhibiting agent is selected from the group consisting of rofecoxib, celecoxib, valdecoxib, deracoxib, etoricoxib, 4-(4-cyclohexyl-2-methyloxazol-5-yl)-2-fluorobenzenesulfonamide, 5-chloro-3-(4-(methylsulfonyl)phenyl)-2-(methyl-5-pyridinyl)pyridine, 2-(3,5-difluorophenyl)-3-4-(methylsulfonyl)phenyl)-2-cyclopenten-1-one, N-[[4-(5-methyl-3-phenylisoxazol-4yl]phenyl]sulfonyl]propanamide, ...
A family of specific compounds of particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide; 3-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-triflouromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl) -4-(trifluoromethyl) -1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxyethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-oxazolyl]benzenesulfonamide; 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; and ...
A family of specific compounds of particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide; 3-[1-(4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxyethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-oxazolyl]benzenesulfonamide; 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; and ...
The method of claim 18wherein Formula I represents a compound selected from the group consisting of 4-(4-methylsulfonylphenyl)-3-phenyl-2,5-dihydrofuran-2-one (Compound C); 4-(4-sulfamoylphenyl)-5-methyl-3-phenyl-isoxazole (Compound D); and 4-[4-(N-propionylsulfamoyl)-phenyl]-5-methyl-3-phenyl-isoxazole (Compound E).
Step 3. Preparation of 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide. Chlorosulfonic acid (355 mL, 587 g, 5.04 mol) was cooled in an ice/salt bath and then 5-hydroxy-5-methyl-3,4-diphenylisoxazoline (142.2 g, 0.56 mol) was added at such a rate that the temperature was maintained between 10-15 C. (ca. 35 minutes). The ice bath was removed and the solution stirred at room temperature for 16 hours. The mixture was then diluted with 250 mL of dichloromethane and the solution was slowly added to ice water with stirring. The mixture was diluted with an additional 1 L portion of dichloromethane and the phases were separated. The dichloromethane solution was dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 144.1 g of white solid. This solid was dissolved in 600 mL of dichloromethane, cooled to 5 C. and was then treated with 600 mL of conc. NH4 OH and stirred at 15 C. for 15 minutes. The mixture was extracted with dichloromethane, dried over anhydrous MgSO4, filtered and concentrated in vacuo to afford 131 g of a white solid. The crude solid was dissolved in 300 mL of boiling methyl ethyl ketone (2-butanone) and diluted with 300 mL of 10% aqueous isopropyl alcohol (prepared from 270 mL anhydrous isopropyl alcohol and 30 mL of water) and allowed to cool to room temperature whereupon crystals of pure 4-[(5-methyl-3-phenyl)-4-isoxazolyl]benzenesulfonamide formed which were isolated by filtration and dried in a vacuum drying oven at 10 mm Hg, 100 C. to afford 112.95 g, 65% of pure product, mp 172-173 C.
A family of specific compounds of more particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide; 3-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-oxazolyl]benzenesulfonamide; 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; ...
A family of specific compounds of particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 5-difluoromethyl-4-(4-methylsulfonylphenyl)-3-phenylisoxazole; 4-[3-ethyl-5-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-difluoromethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-methyl-3-phenyl-isoxazol-4-yl]benzenesulfonamide; 1-[2-(4-fluorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; 1-[2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; 1-[2-(4-chlorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; ...
Particularly preferred compounds of formula (I) include: ... 5-difluoromethyl-4-(4-(methylsulfonyl)phenyl)-3-phenylisoxazole; 4-(3-ethyl-5-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-difluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-hydroxymethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 1-(2-(4-fluorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; 1-(2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; 1-(2-(4-chlorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; ...
Particularly preferred compounds of formula (I) include: ... 5-difluoromethyl-4-(4-(methylsulfonyl)phenyl)-3-phenylisoxazole; 4-(3-ethyl-5-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-difluoromethyl-3-phenylisoxazol-4yl)benzenesulfonamide; 4-(5-hydroxymethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 1-(2-(4-fluorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; 1-(2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; 1-(2-(4-chlorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; ...
A family of specific compounds of particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 5-difluoromethyl-4-(4-methylsulfonylphenyl)-3-phenylisoxazole; 4-[3-ethyl-5-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-difluoromethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-methyl-3-phenyl-isoxazol-4-yl]benzenesulfonamide; 1-[2-(4-fluorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; 1-[2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; 1-[2-(4-chlorophenyl)cyclopenten-2-yl]-4-(methylsulfonyl)benzene; ...
A family of specific compounds of more particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide; 3-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-oxazolyl]benzenesulfonamide; 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; and ...
Use according to Claim 16 wherein the compound is selected from compounds, and their pharmaceutically acceptable salts, of the group consisting of ... 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; ...
Use according to Claim 15 wherein the compound is selected from compounds, and their pharmaceutically acceptable salts, of the group consisting of ... 4-[5-difluoromethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxymethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-methyl-3-phenyl-isoxazol-4-yl]benzenesulfonamide; 1-[2-(4-fluorophenyl)cyclopenten-1-yl]-4-(methylsulfonyl)benzene; ...
A family of specific compounds of particular interest within Formula I consists of compounds and pharmaceutically-acceptable salts thereof as follows: ... 4-[5-(3-fluoro-4-methoxyphenyl)-3-(difluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide; 3-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 2-methyl-5-[1-[4-(methylsulfonyl)phenyl]-4-trifluoromethyl-1H-imidazol-2-yl]pyridine; 4-[2-(5-methylpyridin-3-yl)-4-(trifluoromethyl)-1H-imidazol-1-yl]benzenesulfonamide; 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide; 4-[5-hydroxyethyl-3-phenylisoxazol-4-yl]benzenesulfonamide; [2-trifluoromethyl-5-(3,4-difluorophenyl)-4-oxazolyl]benzenesulfonamide; 4-[2-methyl-4-phenyl-5-oxazolyl]benzenesulfonamide; and ...
Particularly preferred compounds of formula (I) include: ... 5-difluoromethyl-4-(4-(methylsulfonyl)phenyl)-3-phenylisoxazole; 4-(3-ethyl-5-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-difluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-hydyoxymethyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide; 1-(2-(4-fluorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; 1-(2-(4-fluoro-2-methylphenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; 1-(2-(4-chlorophenyl)cyclopenten-1-yl)-4-(methylsulfonyl)benzene; ...
In a further embodiment of the co-crystal according to the invention, the NSAID being a coxib is selected from: ... Etoricoxib, Lumiracoxib, Parecoxib, Rofecoxib, Valdecoxib, or Cimicoxib.
claim 1 , wherein the coxib is selected from: ... Etoricoxib, Lumiracoxib, Parecoxib, Rofecoxib, Valdecoxib, or Cimicoxib.
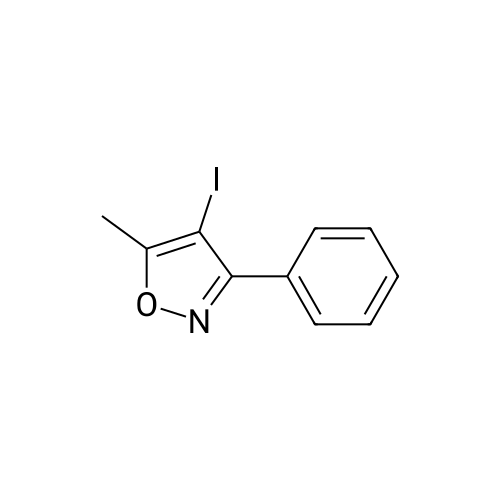
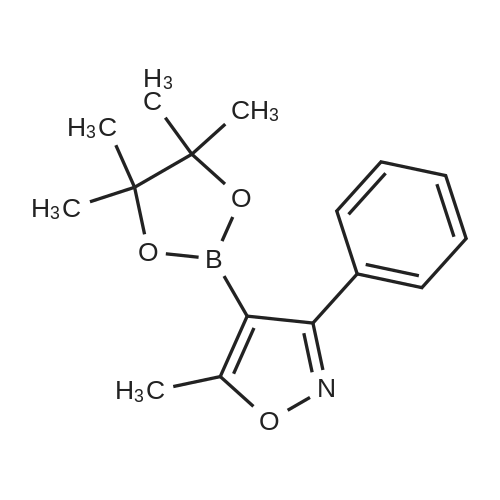
21
[ 1336-21-6 ]
[ 181696-73-1 ]
[ 181695-72-7 ]
Yield
Reaction Conditions
Operation in experiment
With chlorosulfonic acid; trifluoroacetic acid; In methanol; water; isopropyl alcohol; toluene;
Step 3 Preparation of 4-(5-Methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide (valdecoxib, 1). 4,5-Dihydro-5-methyl-3,4-diphenyl-5-isoxazolol (50.0 g, 0.197 mol) was charged to a 500 mL reactor, which had been cooled to 5 C. Trifluoroacetic acid (38.3 mL, 0.496 mol) was charged with stirring to the reactor and the 35 C. solution was cooled to ~5 C. Chlorosulfonic acid (232 g, 1.99 mol) was added slowly to control evolution of hydrogen chloride (HCl) and maintain <25 C. during the addition. The reaction solution was then heated to 60 C. and held at 60 C. for 2.5 hours. After cooling the reaction solution to 0 C. it was added slowly to a stirred 2 to 25 C. mixture of toluene (172 mL) and water (150 mL). The reactor was rinsed with a mixture of toluene (18.4 mL) and water (50 mL), which was then added to the quench mixture. The toluene layer was extracted with water (50 mL) and cooled to 0.2 C. Concentrated ammonium hydroxide (62 mL, 1.60 mol) was added slowly with cooling to maintain ~10 to 15 C. during the addition. The mixture was warmed slowly to 35 C. and held there for ~40 minutes. Isopropanol (240 mL) was added, and the reaction mixture was reheated to 35 C. and held at 35 C. for 90 minutes. The crystalline mixture was slowly cooled to 20 C. and the crude product was filtered, washed with isopropanol (100 mL) and water (100 mL). The wet cake was transferred to a 500 mL crystallizer and dissolved in methanol (350 mL) at ~58 C. Water (92 mL) was added to the methanol solution and the solution was heated to ~70 C. This solution was slowly cooled to 50 C., held for 60 minutes and then cooled to 5 C. After one hour at 5 C. the crystalline product was collected by filtration, the cake washed with 75% methanol-water (100 mL) and dried under vacuum at ~70 C. A differential scanning calorimetry (DSC) melting point of 171 to 174 deg C. (determined at 10 degrees C./minute) was found.
With sulfuric acid; In nitrogen; propionic acid anhydride;
EXAMPLE 2 Preparation of N-[[4-(5-methyl-3-phenyl-4-isoxazolyl)phenyl]sulfonyl]propanamide (parecoxib, 1a). <strong>[181695-72-7]4-(5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide</strong> (10.0 g, 0.032 mol) and propionic anhydride (40 mL, 0.31 mol) were charged to the 500 mL reactor. The slurry was stirred and heated to 50 C. Sulfuric acid (40 L, 0.8 mmol) was added in one portion. All the solids dissolved and the mixture warmed to 55.5 C. within a 10 minute period after the addition was completed. The reaction mixture was then heated to 80 C. and held for approximately 10 minutes. Heating was discontinued, and the mixture was allowed to cool to 50 C. and held for about 60 minutes; solid started to crystallize from the reaction mixture at about 65 C. The mixture was slowly cooled to 0 C. and was held at 0 C. for about 60 minutes. The solid was collected by vacuum filtration. The wet cake was washed with two 45-mL portions of methyl tert-butyl ether and pulled dry at ambient temperature for about 15 minutes. The solid was further dried in a vacuum oven with a nitrogen bleed at 60 C. for 18 hours to give the solid product (8.72 g 75% yield). DSC maximum endotherm for the high melting point parecoxib is 168.95. DSC maximum endotherm for the low melting point parecoxib is 147.44.
With potassium carbonate; In water; ethyl acetate; N,N-dimethyl-formamide;
EXAMPLE 2 N,N-bis(4-methoxybenzyl)-4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide A mixture of <strong>[181695-72-7]4-(5-methyl-3-phenyl-4-isoxazolyl)benzenesulfonamide</strong> (3.14 g, 0.01 mol) and potassium carbonate (5.53 g, 0.04 mol) in DMF (50 mL) was stirred at room temperature for 5 hours. 4-methylbenzylchloride (3.45 g, 0.022 mol) was then added and the resulting mixture was stirred overnight. Ethyl acetate (50 mL) and sat. water (100 mL) were then added and the layers were separated. The organic layer was washed with NaHCO3 (50 mL), brine, 1 N HCl (25 mL) and water (2*25 mL). Then the organic layer was then dried over MgSO4, filtered and concentrated under vacuum. The resulting product was crystallized from boiling ethanol to afford 5.2 g of the product as a white solid: mp, 133.8-135.2 C.
1,8-Diazabicyclo[5.4.0]undec-7-ene [DBU][ No CAS ]
[ 79-22-1 ]
[ 181695-72-7 ]
[ 198471-65-7 ]
Yield
Reaction Conditions
Operation in experiment
25%
In tetrahydrofuran; chloroform;
EXAMPLE 70 STR91 Methyl N-[[4-(5-methyl-3-phenylisoxazol-4-yl)phenyl]sulfonyl]carbamate, sodium salt A solution of 4-[5-methyl-3-(phenyl)isoxazol-4-yl]benzenesulfonamide (1.920 g, 6.11 mmol) in 40 mL of THF was treated with methyl chloroformate (1.16 mL, 1.38 g, 14.60 mmol) and then 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (2.80 mL, 2.79 g, 18.33 mmol) at room temperature. After 48 hours, the resulting mixture was partitioned between ethyl acetate and KHSO4 solution. The organic phase was washed with brine, dried over MgSO4, filtered and concentrated in vacuo yielding a pale yellow clear oil. This oil was purified by running two flash chromatographic columns (1st eluant 1:1; hexane:ethyl acetate; 2nd eluant CH2 Cl2 with THF) yielding the crude compound which was suitable for use without further purification. The crude compound was dissolved in 8 mL of chloroform and treated with 2 mL of saturated aqueous NaHCO3. The product separated as a crystalline solid and was collected by filtration to afford pure salt as white needles (0.607 g, 25%): mp 267.4-275.0 C. 1 H NMR (D2 O/300 MHz) 7.68 (d, 2H, J=8.5 Hz), 7.39-7.12 (m, 7H), 3.37 (s, 3H), 2.34 (s, 3H). FABLRMS m/z 401 (M+Li). FABHRMS m/z 395.0675 (M+ H, Calc'd 395.0678). Anal. Calc'd for C18 H15 N2 O5 SNa.3.66 H2 O: C, 46.96; H, 4.89, N, 6.09; Found: C, 46.91, H, 4.40, N, 6.00.
Step 1 Preparation of 1,1-dimethylethyl N-[2-[[[4-(5-methyl-3-phenylisoxazol-4-yl)phenyl]sulfonyl]amino]-2-oxoethyl]carbamate A mixture of 4-[5-methyl-3-(phenyl)isoxazol-4-yl]benzenesulfonamide (15.0 g, 47.7 mmol), N-t-boc-glycine N-hydroxysuccinimide ester (13.0 g, 47.7 mmol) and 1,8-diazobicyclo[4.3.0]undec-7-ene (14.5 g, 95.4 mmol) were mixed together in tetrahydrofuran for 1 hour at room temperature. Additional N-t-boc-glycine N-hydroxysuccinimide ester (1.3 g, 4.7 mmol) was added and the solution was stirred an additional 2 hours. The solvent was removed at reduced pressure and residue was taken up in ethyl acetate. The ethyl acetate was washed with 10% aqueous HCl, sat. aq. NaHCO3, dried over anhydrous Na2 SO4, filtered and concentrated in vacuo to afford the desired amide as a clear glassy solid (6.5 g, 75%): mp 160.2-162.0 C. 1 H NMR (CDCl3 /300 MHz) 8.04 (d, 2H, J=8.4 Hz), 7.44-7.33 (m, 5H), 7.28 (d, 2H, J=8.4 Hz), 5.24 (brs, 1H), 3.85 (m, 2H), 2.50 (s, 3H), 1.43 (s, 9H). FABLRMS m/z 472 (M+H). Anal. Calc'd for C23 H25 N3 O5 S.0.18 H2 O: C, 58.19; H, 5.38; N, 8.85. Found: C, 58.22; H, 5.73; N, 8.92.
Step 1 Preparation of [4-[4-(aminosulfonyl)phenyl]-3-phenylisoxazol-5-yl]acetic acid [4-[4-(Aminosulfonyl)phenyl]-3-phenylisoxazol-5-yl]acetic acid was prepared similar to the method of Example 55 but starting with 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide (Example 1): mp 201-202.
This mutual prodrug was synthesized using succinic anhydride and <strong>[181695-72-7]valdecoxib</strong> according to method B described in Scheme 13 with appropriate modifications. This double prodrug I-AA-MPDSa was obtained as an off-white solid. 1H-NMR (300 MHz, CDCl3): delta 2.46 (s, 6H), 2.58 (s, 4H), 7.25-7.37 (m, 16H), 7.95 (d, 2H, J=9.0 Hz). MS (m/z): 709 [M-H]-.
This mutual prodrug was synthesized using glutaric anhydride and <strong>[181695-72-7]valdecoxib</strong> according to method B described in Scheme 13 with appropriate modifications. This double prodrug I-AA-MPD8b was obtained as a colorless gum. 1H-NMR (300MHz, CDCl3+CD3OD): delta 1.68-1.74 (m, 2H), 2.15 (t, 4H, J=4.5 Hz), 2.38 (s, 6H), 7.01 (bs, 1H), 7.17-7.30 (m, 14H), 7.50 (bs, 1H), 7.88 (d, 4H, J=8.58 Hz). MS (m/z): 723 [M-H]-.
With triethylamine;dmap; In tetrahydrofuran; at 20℃; for 120h;
TEA (0.194 mL, 1.39 mmol) and DMAP (73 mg, 0.6 mmol) were added to a solution of LI-10 (1.33 g, 3.24 mmol) and <strong>[181695-72-7]valdecoxib</strong> (364 mg, 1.16 mmol) in THF (6 mL) and stirred at RT for 5 d. The mixture was concentrated and the residue was taken up in DCM. After usual aqueous work-up and chromatographic purification, 177 mg (12%) of LI-10 were obtained. 1H-NMR (300 MHz, CDCl3): delta 2.46 (s, 3H), 2.85-2.95 (m, 10H), 3.28 (t, 2H, J=6.0 Hz), 3.65 (q, 2H, J=3.0 Hz), 4.22-4.28 (m, 4H), 7.22-7.41 (m, 7H), 7.94 (d, 2H, J=9.0 Hz). MS (m/z): 609 [M+H]+. This product was converted to water-soluble hydrochloride salt form using standard methods.
With dmap; triethylamine; In tetrahydrofuran; at 20℃; for 108h;
A mixture of <strong>[181695-72-7]valdecoxib</strong> (280 mg, 0.892 mmol), DMAP (56 mg, 0.5 mmol) and TEA (150 muL, 1.06 mmol) in THF (5 mL) was stirred at RT for 4.5 d. The mixture was concentrated and the residue dissolved in EtOAc. After usual aqueous work-up and chromatographic purification, 354 mg (54%) of I-S13-PD1 were obtained. 1H-NMR (300 MHz, CDCl3): delta 2.47 (s, 3H), 3.32-3.41 (m, 4H), 4.28 (t, 2H, J=6.0 Hz), 4.47 (t, 2H, J=6.0 Hz), 7.20-7.61 (m, 22H), 8.00 (d, 2H, J=9.0 Hz). MS (m/z): 736 [M-H]-.
This double prodrug was synthesized by reacting I-A3-PD1 and <strong>[181695-72-7]valdecoxib</strong> using the method B described in Scheme 13. The double prodrug I-AA-MPD5 was obtained as an off white solid. 1H-NMR (300 MHz, CDCl3): delta 2.40 (s, 6H), 2.82 (bs, 4H), 4.20 (bs, 4H), 7.20-7.35 (m, 14H), 7.97 (d, 4H, J=9.0 Hz). MS (m/z): 833[M-H]-.
A solution of desoxybenzoin keto-oxime from Step 1 (6.00 g; 28.40 mmol) in anhydrous tetrahydrofuran (THF, 80 mL) was cooled to -20 C. in an oven-dried 250 mL three-neck round-bottom flask equipped with a thermometer, nitrogen gas inlet, rubber septum and provisions for magnetic stirring. To this cold solution, n-butyllithium (1.6N in hexanes, 44.4 mL) was added, via syringe, over 35 minutes, such that the reaction temperature remained at or below -10 C. The deep red solution was stirred at -10 C. for 1 hour, warmed to room temperature, then stirred at room temperature for an additional hour. Acetic anhydride (3.2 mL, 34.1 mmol) was added in one portion, and the resulting suspension was stirred without temperature control for 2 hours. Water (100 mL) was added, and the solution was poured into 1 N HCl (100 mL) and extracted with ethyl acetate (2×200 mL). The combined organic solution was washed with hydrochloric acid (1 N HCl, 10 mL) and brine (100 mL), dried over magnesium sulfate and filtered. The resulting solution was evaporated under reduced pressure to yield a crude oil. The oil was applied to a column of silica gel and eluted with ethyl acetate/hexane (10-50% ethyl acetate) to yield, upon concentration of the appropriate fractions, 5.0 g of 3,4-diphenyl4-hydrido-5-hydroxy-5-methylisoxazole. The solid was cooled to 0 C., then dissolved in cold chlorosulfonic acid (15 mL). The brown solution was stirred at 0 C. for 2 hours, then added dropwise to a stirring suspension of ice (200 mL) and dichloromethane (200 mL). The layers were separated, and the organic phase was added directly to a saturated ammonium hydroxide solution (100 mL) at 0 C. This biphasic solution was vigorously stirred at 0 C. for 2 hours, the layers were separated, and the aqueous phase was washed with dichloromethane (50 mL). The combined organic solution was dried over magnesium sulfate, filtered and evaporated under reduced pressure to approximately one-half of its original volume. Crystals formed. The stirred suspension was cooled to 0 C. and held for 30 minutes. The crystals were filtered, washed with cold dichloromethane and dried to yield 4-[5-methyl-3-phenylisoxazol-4-yl]benzenesulfonamide (2.7 g, 30%): mp 155-157 C. 1H NMR (CD3CN/500 MHz) delta 7.86 (d, J=8.39 Hz, 2H), 7.45 (m, 1H), 7.39 (s, 4H, 7.37 (d, J=8.39 Hz, 2H), 5.70 (s, 2H), 2.46 (s, 3H Mass Spectrum, MH+=315.
N-[[4-(5-methyl-3-phenyl-4-isoxazolyl)phenyl]sulfonyl]propanamide sodium salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
Compound 25.15g (80.0mmol) added to the reaction flask, dichloromethane 200ml, the temperature control 25 , propionic anhydride was added 20.82g (160.0mmol), triethylamine 32.33g (320mmol). 25 reaction temperature, TLC monitoring, after the reaction was added purified water 200ml, liquid separation. Aqueous phase was extracted with dichloromethane, and the combined organic phase was washed with purified water. The organic phase was removed by rotary evaporation under reduced pressure to give a solid organic solvent is an oil, acetone was added, heated under reflux until clear solution was slowly cooled to 10 , crystallization was stirred 2h, suction filtered, the filter cake was added anhydrous ethanol, stirred and dissolved at 70 adding sodium hydroxide, temperature 70 stirred 60min. Hot filtered and the filtrate cooled solid precipitation, raising grain, filtration to give the product parecoxib sodium 28.25g (72.0mmol), yield 90.0%, purity 99.5% (HPLC method)
5-methyl-3-phenyl-4- (4-sulfonic acid phenyl) isoxazole 11.7g (50mmol) dissolved in dichloromethaneOf thionyl chloride was added dropwise 9.3g (100mmol) at 0 ~ 5 reaction was stirred 40 minutes,Quenched with ice water, extracted with dichloromethane,Methylene chloride phase directly into the aqueous ammonia,The reaction temperature was raised to 20 deg.] C continued for 1 hourAfter the reaction, dichloromethane extraction,Washed, concentrated,Methanol to obtain 14.4 g of vediloxib,The yield was 91.4% and the purity was 99.54%.
72.6%
With ammonium hydroxide; In ethyl acetate; at 20℃; for 0.5h;
The compound of the formula 2 obtained by the filtration in the previous step was added to 50 ml of ethyl acetate and stirred to dissolve;After dissolution, add 48 ml (705.9 mmol, 10e.q) of ammonia water.After reacting at room temperature for 30 min, the aqueous layer was separated, and the organic layer was concentrated and evaporated.Add 25 ml of absolute ethanol to reflux for 30 minutes, cool to room temperature (15 ~ 30 C), and let stand for 10-15 h.Filter and collect the filter cake. Vacuum drying at 55±5C, vacuum degree -0.070-0.082Mpa for 4-6h,That is, 16.0 g of valdecoxib (formula 3 compound) was obtained, and the molar yield was 72.6%, and the purity was 99.9% by HPLC.
12 g of the compound of the formula II and 100 mL of ethyl acetate were successively added to the reaction flask.Irradiation at 254 nm for 4 days.The progress of the reaction was monitored by HPLC. After the reaction was terminated, the reaction solution was concentrated.Purification and purification to obtain the compound of formula I (R=H)A class of white solids, about 0.28 g, HPLC purity: 95.6%.
2) Dissolve 30.6 g of phosphoryl chloride in 200 ml of triethylamine, and then slowly add 119.3 g of chloroform under ice bath conditions to form a mixed solution. 207g of 1-nitroethylbenzene and 400ml of triethylamine are sequentially added to step S1, stir in ice bath for 1 hour, then remove the ice bath, the temperature was raised to 25 C, and 256.8 g of p-toluenesulfonamide was added. Continue stirring for 5 hours to form a reaction solution; 3) Pour the reaction solution into a separating funnel, wash with 1 L of water, then wash the chloroform layer with 6 N hydrochloric acid, and finally wash with a 5% aqueous sodium hydroxide solution; 4) drying over anhydrous magnesium sulfate and filtering; and 5) Remove the solvent with a rotary evaporator and distill the product under vacuum, get the vardecoxib. The mass of vardecoxib obtained in this group of experiments was 302.18g. The purity was 99.538% and the yield was 95.7%.
With dmap In N,N-dimethyl-formamide at 20℃; for 40h;
9 Example 9 General procedure for preparing TCO-valdecoxib.
Valdecoxib (157 mg, in DMF (4 mL)To a solution of 0.5 mmol), TCO-PNB ester (129 mg, 0.44 mmol) and DMAP (106 mg, 0.88 mmol) were added. The mixture is stirred at rt for 40 h,Dilute with ethyl acetate (100 mL),Washed with brine (40 mL), dried over sodium sulfate,Evaporated in vacuo. The product was purified by flash chromatography on silica gel eluting with DCM followed by MeOH-DCM (5%) to give compound TCO-valdecoxib (201 mg, 97%) as a white solid.
With dmap In N,N-dimethyl-formamide at 20℃; for 40h;
A3 General Procedure for the Preparation of TCO-valdecoxib .
To a solution of Valdecoxib (157 mg, 0.5 mmol) in DMF (4 mL) was added TCO-PNB ester (129 mg, 0.4 4 mmol), DMAP (106 mg, 0.88 mmol). The mixture was stirred at rt for 40 h, and diluted with ethyl acetate (100 mL), washed with brine (40 mL), dried over sodium sulfate, and evaporated in vacuo. The product was purified by flash chromatography on silica gel eluting with DCM followed by MeOH-DCM (5%) to give compound TCO-valdecoxib (201 mg, 97%) as white solid. LC-MS: 467 [M+H]+. 1 NMR (300 MHz, CDCh) d 8.03 (d, J= 8.7 Hz, 2 H), 7.65 (m, 1 H), 7.43-7.32 (m, 7 H), 5.73 (m, 1 H), 5.64 (d, J= 16.5 Hz, 1 H), 5.33 (s, 1 H), 2.50 (s, 3 H), 2.43 (m, 1 H), 2.09-0.77 (m, 9 H).
1-3
Put Intermediate IV (25.1g/0.08mol) and propionic anhydride (104.1g/0.8mol) into the reaction flask, raise the temperature to 50°C, add 0.25g of concentrated sulfuric acid dropwise, raise the temperature to 7580°C and stir for 2 hours; lower the temperature to 05, stirring and crystallization for 2 hours, suction filtration; the filter cake is slurried with 100g methyl tert-butyl ether and filtered with suction; then the filter cake is rinsed with 50g methyl tert-butyl ether, and the filter cake is dried by blowing Drying in a box at 60° C. for 4 hours with blowing air yields Intermediate V (26.9 g) with a yield of 90.7%.
Stage #1: valdecoxib With (4,4'-di-tert-butyl-2,2'-dipyridyl)-bis-(2-phenylpyridine(-1H))-iridium(III) hexafluorophosphate; N-ethyl-N,N-diisopropylamine In water; acetonitrile at 20℃; for 16h; Inert atmosphere; Sealed tube; Irradiation;
Stage #2: methyl iodide With sodium carbonate In N,N-dimethyl-formamide at 20℃; for 2h; Inert atmosphere;
With toluene-4-sulfonic acid In toluene at 100℃; for 0.5h;
General procedure A for the preparation of sulfonyl pyrroles 2 from sulfonamides 1 using catalytic p-TsOH at 100 °C (Scheme S1)
General procedure: A flask was charged with p-TsOH•H2O (5.0 mol %) and sulfonamide 1 (1.0 equiv). Then toluene (0.25 M) and 2,5-dimethoxytetrahydrofuran (1.2 equiv) were added. The mixture was stirred at 100 °C for the given time (generally between 30 min and 60 min). The reaction was cooled to room temperature then quenched with saturated aqueous NaHCO3. The resulting biphasic solution was extracted three times with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by trituration with hexane, recrystallization from toluene or column chromatography to provide sulfonyl pyrrole 2.
80%
With p-toluenesulfonic acid monohydrate In dichloromethane at 20 - 40℃; for 16h; Inert atmosphere;
With N,N-dimethyl-4-aminopyridine; N-[3-(N,N-dimethylamino)-propyl]-N'-ethyl-carbodiimide hydrochloride In dichloromethane at 20℃; for 16h; Schlenk technique; Inert atmosphere;
Stage #1: 2-(4-methoxyphenoxy)-2-methylpropanoic acid With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane at 20℃;
Stage #2: valdecoxib In dichloromethane at 20℃;
With iron(III) trifluoromethanesulfonate; sodium phosphate In dichloromethane at 20℃; Inert atmosphere; Sealed tube; Irradiation;
General Procedure D
General procedure: In a nitrogen-filled glovebox, an oven-dried 6-mL vial equipped with a stir bar is charged withFeCl3 (0.097 g, 3.0 equiv, 0.60 mmol), Na3PO4 (0.098 g, 3.0 equiv, 0.60 mmol), the carboxylicacid (1.0 equiv, 0.20 mmol), the sulfonamide (3.0 equiv, 0.60 mmol), and methylene chloride (2.0mL, 0.10 M). The vial is sealed, removed from the glovebox, and irradiated at 427 nm with two 40W Kessil Lamp PR160 lamps at a distance of 10 cm with stirring at 1000 rpm. A fan is used tomaintain the vial at room temperature. After 24 h, the crude reaction mixture is diluted with 1.5mL EtOAc and adsorbed directly on diatomaceous earth (Celite). The product is purified by flashchromatography on silica gel, eluting with mixtures of ethyl acetate in hexanes.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping