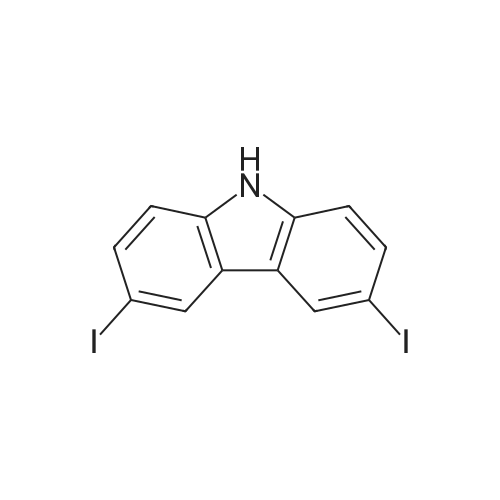
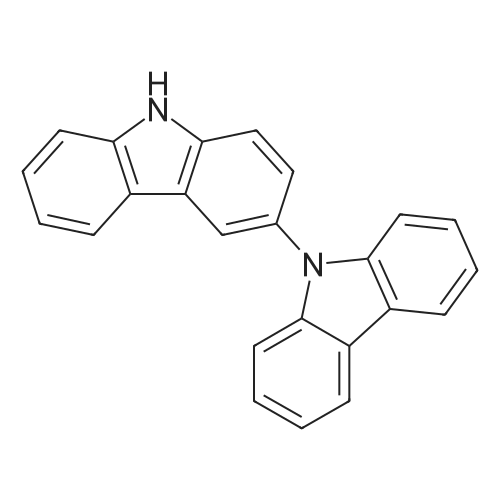
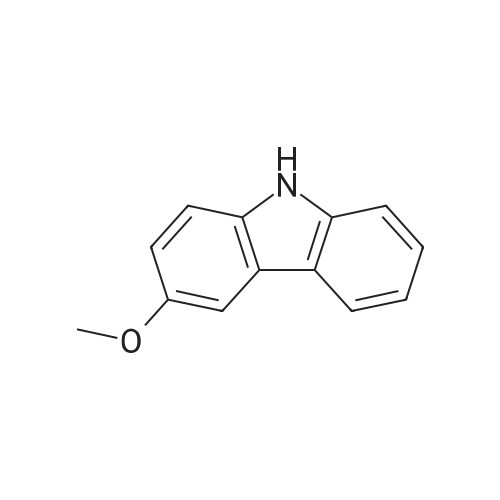
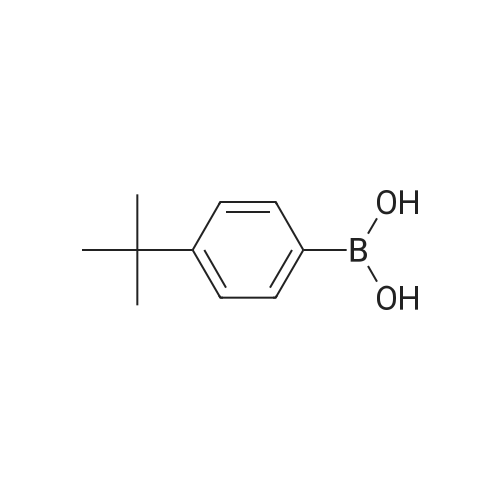
| 86% |
With N-iodo-succinimide In acetic acid at 20℃; for 1h; |
14.1 [0166] Step 1. Compound 14a-1 . 3-Iodo-9H-carbazole.
To a solution of 9H-carbazole (500 mg, 2.99 mmol) in acetic acid (15 mL) was added N-iodosuccinimide (740 mg, 3.29 mmol) and the reaction mixture was stirred at room temperature for 1 h. The mixture was evaporated and the residue was neutralized by addition of 10% aqueous potassium carbonate, and the resulting solid was collected. The crude product was purified by column chromatography eluting with hexane:ethyl acetate (33: 1) to give the title compound (750 mg, 2.56 mmol, 86%) as a pale yellow oil. LCMS: 81%, Rt 1.900, ESMS m/z 292 (M-H)~. |
| 79% |
With diphenyliodonium tetrafluoroborate; copper(II) sulfate In acetonitrile at 65℃; for 0.166667h; Inert atmosphere; Schlenk technique; |
|
| 79% |
With bis(pyridine)iodonium tetrafluoroborate; copper(II) sulfate In acetonitrile at 65℃; for 0.166667h; Inert atmosphere; |
|
| 72% |
With potassium iodate; potassium carbonate; acetic acid; potassium iodide at 80℃; for 5h; |
2.1
Potassium iodide (2.3 g, 0.020 mol) and 50 mL of glacial acetic acid were placed in a 100 mL round-bottomed flask; the reaction was carried out using a mixture of potassium carbonate (2.5 g, 0.015 mol), potassium iodate (2.5 g, 0.0115 mol) The mixture was cooled and filtered through a Buchner funnel to give a solid crude product which was washed with 200 mL of a solution of 5% (wt%) NaHSOj (wt%) in water, and the reaction was stirred at 80 ° C water bath until the color of the resulting iodine disappeared (about 5 h) The crude product was dried, recrystallized from ethanol / THF, decolorized with a small amount of activated charcoal, and finally a white crystal was obtained (6.4 g; yield: 72%). |
| 72.7% |
Stage #1: 9H-carbazole With acetic acid; potassium iodide at 110℃;
Stage #2: With potassium iodate at 100℃; for 0.166667h; |
2.2.4.1 Synthesis of 3-iodo-9H-carbazole (C1): Iodination [1]
Potassium iodide (22.2mmol, 3.68g) and 9H-carbazole (33.3mmol, 5.57g) were added into acetic acid (100mL), followed by stirring at 110°C for 1hrs. Second, after stirring the mixture for 1h, the temperature was lowered to 80°C. Finally, potassium iodate (16.7mmol, 3.57g) was slowly added, the temperature was increased to 100°C, and the reaction was allowed to continue for 10min. When the reaction was completed, the solution was cooled to room temperature, and water was added. The formed crystals were filtered under reduced pressure to obtain crystals. These crystals were recrystallized using methyl chloride, affording the final product in 72.7% yield. 1H NMR (400MHz, DMSO-d6): 7.20 (d, J=8.3Hz, 1H, ArH), 7.35 (d, J=8.0Hz, 1H, ArH), 7.46 (d, J=8.1Hz, 1H, ArH), 7.50 (d, J=1.8Hz, 1H, ArH), 7.63 (d, J=8.1Hz, 1H, ArH), 7.93 (s, 1H, ArH), 8.19 (s, 1H, ArH), 11.66 (s, 1H, NH); HRMS (m/z, MALDI): [M + H]+ calculated for C12H8IN: 293.1019; found 293.1051; Elemental analyses, found C, 49.15; H, 2.69; I, 43.23; N, 4.71; ‘C12H8IN’ requires C, 49.17; H, 2.75; I, 43.30; N, 4.78. |
| 70% |
With iodine; silver nitrate; acetic acid at 20℃; |
|
| 70% |
Stage #1: 9H-carbazole With potassium iodide In acetic acid at 100℃; for 1h;
Stage #2: With potassium iodate In acetic acid at 100℃; for 2h; |
1
Synthesis of 3-iodo-9H-carbazole. To a solution of 9H-carbazole (5.57g, 33.3 mmol) and KI (3.68g, 22.2 mmol) in AcOH (92 mL) was heated to 100 °C for 1 h. KI03 (3.57g, 16.7 mmol) was added in portions to the solution, and the resulting mixture was stirred for another 2 h at 100°C. The mixture was poured into water (500 mL) and the precipitation was collected by filtration and washed with hot water. Recrystallization was made in DCM to afford 6.8 g (70 %) of product as a white solid. |
| 68% |
With iron(III) chloride; sodium iodide In acetonitrile for 8h; |
|
| 62% |
Stage #1: 9H-carbazole With acetic acid; potassium iodide at 85℃; Reflux;
Stage #2: With potassium iodate at 85℃; for 0.166667h; |
|
| 49% |
With sodium periodate; sulfuric acid; iodine In ethanol at 60℃; for 2h; |
1.1 (1) Synthesis of Compound 2
Carbazole (Compound 1, 4.0 g, 23.6 mmol) was dissolved in ethanol (800 mL) in a 1-L three-necked flask, and then iodine (3.0 g) and NaIO4 (1.28 g) were added to the flask. After adding a solution of concentrated sulfuric acid/ethanol (2.5 mL/100 mL), the mixture was stirred at 60° C. for 2 hours. After confirming the completion of the reaction with TLC (hexane:ethyl acetate, 4:1), neutralization was performed with an ethanol solution of sodium hydroxide, and the solvent was removed. The residue was dissolved in chloroform and then washed with water/saturated sodium hydrogen carbonate solution/saturated sodium chloride solution. Then the solvent was removed with an evaporator. The residue was dissolved in ethanol, and recrystallized to obtain Compound 2. (yield: 3.4 g, yield %: 49%) (0178) 1H-NMR (400 MHz, DMSO-d6): δ 11.40 (s, 1H, NH), 8.51 (d, 1H, J=1.7 Hz, H-4), 8.14 (d, 1H, J=7.8 Hz, H-5), 7.62 (dd, 1H, J=8.5, 1.7 Hz, H-2), 7.51 (d, 1H, J=8.1 Hz, H-8), 7.40 (m, 1H, H-7), 7.36 (d, 1H, J=8.5 Hz, H-1), 7.19 (m, 1H, H-6) |
| 48% |
With sodium periodate; sulfuric acid; iodine In isopropyl alcohol at 65℃; for 6h; |
|
| 47% |
With potassium iodate; potassium iodide In acetic acid at 45℃; |
4.2.7. 3-Iodocarbazole (5) [61].
Carbazole 1 (16.7 g, 101mmol) was dissolved in boiling glacial acetic acid (250 mL), and KI (11.73 g, 135mmol) was added. The solution was cooled, ground potassium iodate (23.42 g, 150mmol) was added,and the mixture was boiled until it acquired a clear strawcoloredtint (10 min).The hot solution was decanted fromtheundissolved potassium iodate, and it was allowed to cool to45°C. The faintly brown plates were rapidly filtered off andrecrystallized from alcohol, and the solution was allowed tocool to 45°C. The faintly brown plates were rapidly filtered offand recrystallized from ethanol; the solution was allowed tocool to 45°Candfiltered, yielding 9.73 g (47%) of 5 as a brownsolid; mp 202°C (mplit. 202°C), 1H NMR (600MHz, CDCl3) δppm 8.41 (s, 1 H), 8.11 (br. s., 1 H), 8.04 (d, J = 7.70 Hz,1 H), 7.68 (dd, J = 8.53, 1.74Hz, 1 H), 7.42-7.49 (m, 2 H),7.22-7.28 (m, 2 H); 13C NMR (100MHz, CDCl3) δ ppm 139.5,138.6, 134.1, 129.2, 126.6, 125.9, 122.1, 120.5, 119.9, 112.6, 110.7.Maldi-Tof MS: m/z calcd for [M+] = C12H8IN 292.970; found 292.971. Anal. calcd. for C12H8IN: C, 49.17; H, 2.75; I, 43.30;N, 4.78. Found: C, 49.10; H, 2.71; I, 43.25; N, 4.73. |
| 47% |
Stage #1: 9H-carbazole With ammonium iodide In methanol for 0.0333333h;
Stage #2: In methanol at 75℃; |
|
| 45% |
With potassium iodate; potassium iodide In acetic acid for 0.166667h; Reflux; |
|
| 45% |
With potassium iodate; acetic acid; potassium iodide for 0.166667h; Reflux; |
|
| 44.3% |
With potassium iodate; acetic acid; potassium iodide Heating; |
|
| 44% |
With N-iodo-succinimide; acetic acid at 20℃; for 5h; Inert atmosphere; |
7.a
a) 76.9 g (0.460 mo) carbazoe and 104 g (0.460 mo) 1iodopyrroNdine2,5dione (NS) in lOOm m acetic acid are stirred under nitrogen at 20 °C. After 5 h the product is filtered off. The product is crystailzed from 900 m ethano using 2 g charcoaL The ethano soution is filtered hot. The ethano soution is cooed to 20 °C and the product is filtered off (yied: 59.5g (44 %)). |
| 44% |
With N-iodo-succinimide; acetic acid at 20℃; for 5h; Inert atmosphere; |
3.a
76.9 g (0.460 mol) carbazole and 104 g (0.460 mol) 1-iodopyrrolidine-2,5-dione (NIS) in 100m ml acetic acid are stirred under nitrogen at 20 °C. After 5 h the product is filtered off and crystalized from 900 ml ethanol using 2 g charcoal. The ethanol solution is filtered hot and cooled to 20 °C. The product is filtered off (yield: 59.5 g (44 %)). |
| 44% |
With N-iodo-succinimide; acetic acid at 20℃; for 5h; Inert atmosphere; |
1.a Synthesis Example 1
Synthesis Example 1 )benzimidazolo[1 ,2-a]benzimidazole a) 76.9 g (0.460 mol) of carbazole and 104 g (0.460 mol) of 1-iodopyrrolidine-2,5-dione (NIS) in 100m ml of acetic acid are stirred under nitrogen at 20 °C. After 5 h the product is filtered off. The product is dissolved in 900 ml of ethanol at reflux, then 2 g of charcoal are added. The ethanol solution is filtered hot and cooled to 20 °C. The product is filtered off to yield 59.5 g (44 %) of 3-iodo-9H-carbazole. |
| 44% |
With N-iodo-succinimide; acetic acid at 20℃; for 5h; Inert atmosphere; |
2.a
a) 76.9 g (0.460 mol) carbazole and 104 g (0.460 mol) 1-iodopyrrolidine-2,5-dione (NIS) in 100m ml acetic acid are stirred under nitrogen at 20 °C. After 5 h the product is filtered off. The product is crystalized from 900 ml ethanol using 2 g charcoal. The ethanol solution is filtered hot. The ethanol solution is cooled to 20 °C and the product is filtered off (yield: 59.5 (44 %)). |
| 44% |
With N-iodo-succinimide; acetic acid at 20℃; for 5h; Inert atmosphere; |
1.a
a) 76.9 g (0.460 mol) carbazole and 104 g (0.460 mol) 1-iodopyrrolidine-2,5-dione (N IS) in1 00m ml acetic acid are stirred under nitrogen at 20 00. After 5 h the product is filtered off. The product is crystalized from 900 ml ethanol using 2 g charcoal. The ethanol solution is filtered hot. The ethanol solution is cooled to 20 00 and the product is filtered off (yield: 59.5 g (44 %)). |
| 44% |
With N-iodo-succinimide; acetic acid at 20℃; for 5h; Inert atmosphere; |
3.b Example 3
b) 76.9 g (0.460 mol) carbazole and 104 g (0.460 mol) 1-iodopyrrolidine-2,5-dione (NIS) in 100m ml acetic acid are stirred under nitrogen at 20 °C. After 5 h the product is filtered off. The product is crystalized from 900 ml ethanol using 2 g charcoal. The ethanol solution is filtered hot. The ethanol solution is cooled to 20 °C and the product is filtered off (yield: 59.5 g (44 %)). |
| 40% |
With potassium iodate; acetic acid; potassium iodide at 100℃; for 2h; |
|
| 39% |
With potassium iodate; acetic acid; potassium iodide at 85℃; for 0.166667h; |
|
| 18.8% |
With periodic acid dihydrate; sulfuric acid; iodine In ethanol at 20℃; for 4h; |
3 Synthesis of Intermediate 3-1
Carbazole (15 g, 92.6 mmol) was added to ethanol (70 mL). Sulfuric acid (6 mL), water (3 mL), -HIO4.2H2O (8.2 g, 35.9 mmol) and -I2 (9.1 g, 35.9 mmol) were added thereto and stirred at room temperature for four hours. Water was added to the reaction solution and a precipitated solid was separated by filtration. Then, the obtained solid was washed with methanol. By dissolving the obtained solid in heated toluene for recrystallization, an intermediate 3-1 (5.1 g, a yield rate 18.8%) was obtained. |
| 15% |
With potassium iodate; acetic acid; potassium iodide at 80℃; for 1h; |
|
|
With potassium iodate; potassium iodide |
|
|
With sulfuric acid; iodine; periodic acid In water at 70℃; for 1h; |
|
|
With potassium iodate; potassium iodide |
|
|
With potassium iodate; potassium iodide In acetic acid |
|
|
With sulfuric acid; iodine; periodic acid In water; acetic acid at 70℃; for 1h; |
|
|
With potassium iodate; potassium iodide In acetic acid at 50℃; for 2h; |
6
Bis(3-(9-(4-diphenylaminophenyl)carbazolyl)phenylamine (F) was synthesised according to the following scheme. 15 g of carbazole, 9.5g of potassium iodide and 6.3 g of potassium iodate were dissolved in acetic acid, and were reacted at a temperature of 50°C for two hours. An aqueous solution of sodium thiosulfate was added to the resulting mixture to reduce iodine therein, and the resulting organic layer was concentrated and subjected to crystallization to provide 17.7 g of 3-iodocarbazole. |
|
With potassium iodate; acetic acid; potassium iodide |
|
|
With Iodine monochloride In acetonitrile at 0 - 20℃; for 4h; Inert atmosphere; |
|
|
With potassium iodate; acetic acid; potassium iodide |
|
|
With potassium iodate; potassium iodide |
|
|
With Iodine monochloride at 0 - 20℃; for 4h; |
|
|
With potassium iodate; potassium iodide |
|
|
With Iodine monochloride In acetonitrile at 0 - 20℃; for 4h; Inert atmosphere; |
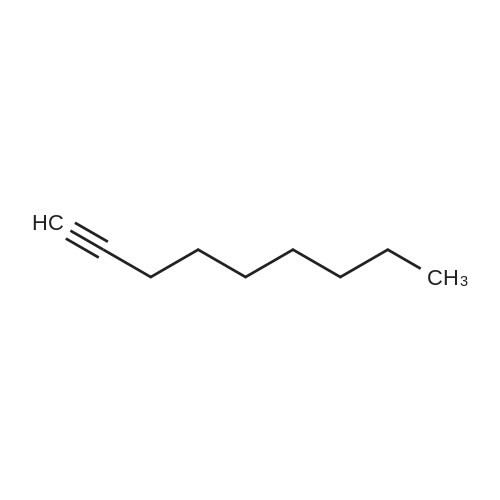
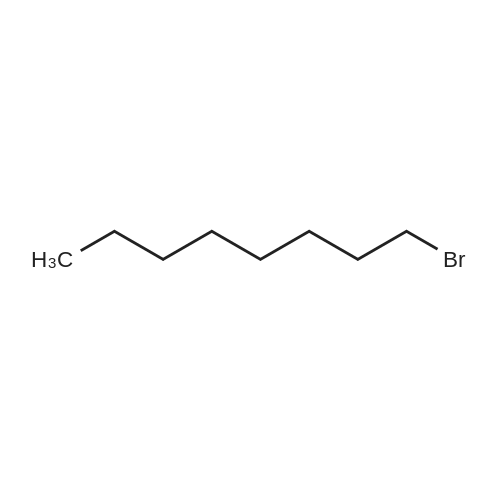
To a solution of carbazole (5.00 g, 30.0 mmol) in CH3CN (250 mL) was slowly added ICl (1.88 mL, 36 mmol) at 0 °C. The mixture was stirred at 0 °C for 2 h, then slowly warmed up to room temperature and was stirred for another 2 h. The reaction was quenched with saturated aqueous Na2S03 solution. The product was extracted with CH2Cl2 (80 mLx3). The organic extracts were combined and the volatiles were removed. The crude product of 3-iodocarbazole (la) was collected as a white solid. (-60 % yield was determined by crude 1H NMR spectra analysis.) Without further purification, the crude 3-iodocarbazole (la) was dissolved in DMF (100 mL). NaH (1.80 g, 45 mmol, 60% dispersion in mineral oil) was added to the above solution and stirred for 5 mins at room temperature. Then 1-bromohexadecane (13.74 g, 45 mmol) was added. After stirring for 4 h at room temperature, the solvent was removed. 1 M HCl (100 mL) was added to the residue, and the mixture was extracted with CH2Cl2 (3 x 100 mL). The combined organic extracts were washed with water (100 mL), and brine (100 mL), dried over anhydrous Na2S04, and concentrated to give the crude product. Purification by flash column chromatography (CH2Cl2 : Hexane, 1 : 3 v/v) gave the product (lb) together with N-hexadecyl-3,6-diiodo-carbazole. To a mixture of DMF (47 mL, 600 mmol) and 1,2-dichloroethane (50 mL) was added POCl3 (47.5 mL, 510 mmol) dropwise at 0 °C. The mixture was warmed up to 35 °C and N- hexadecyl-3-iodo-carbazole (lb) was added. After heating at 90 °C for 24 h, the mixture was cooled to ambient temperature and poured into water (500 mL). The product was extracted with chloroform (150 mL x 3). The combined organic extracts were washed with water (200 mL), and brine (200 mL), dried over anhydrous MgS04 andconcentrated. The residue was purified via flash column chromatography (CH2Cl2 : Hexane, 1 : 1 v/v) to provide pure product 1 as a white solid (7.85 g, 48 % in three steps). |
|
With potassium iodate; acetic acid; potassium iodide Reflux; |
|
| 5.1 g |
With periodic acid dihydrate; sulfuric acid; iodine In ethanol; water for 4h; |
1 Synthesis of Intermediate 1
After adding 70 mL of ethanol to 15 g of carbazole, 6 mL of sulfuric acid, 3 mL of water, 8.2 g of periodic aciddihydrate, and 9.1 g of iodine were further added at room temperature. The resultant mixture was stirred for 4 h. Theprecipitate generated by adding water to the reaction product liquid was collected by filtration and washed with methanol. The obtained solid was dissolved in hot toluene and recrystallized. The purified solid was vacuum-dried to obtain 5.1 gof white solid, which was identified as Intermediate 1 shown below by FD-MS analysis. |
|
With potassium iodate; potassium iodide In acetic acid for 0.333333h; Reflux; |
|
|
With potassium iodate; potassium iodide |
|
|
With potassium iodate; acetic acid; potassium iodide at 120℃; for 0.166667h; |
|
|
With potassium iodate; potassium iodide at 130℃; |
|
|
With sodium periodate; sulfuric acid; iodine In ethanol |
|
|
With sodium periodate; sulfuric acid; iodine In ethanol |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping