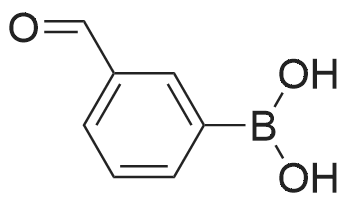
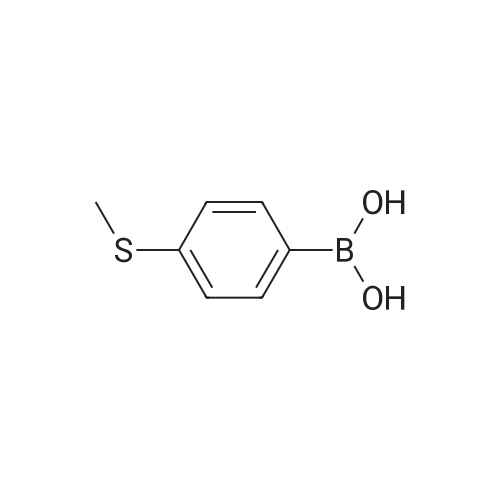
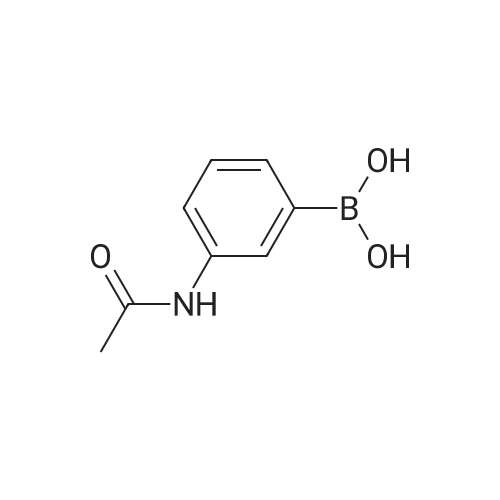
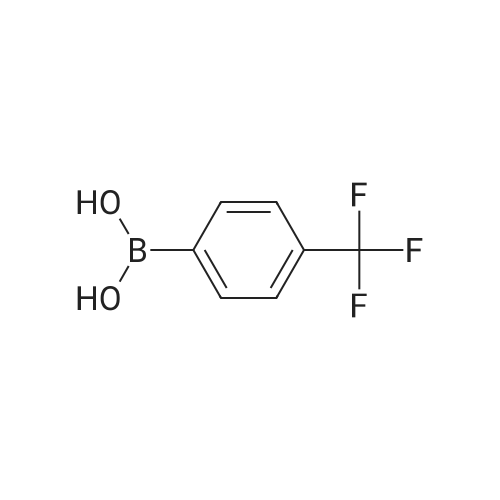
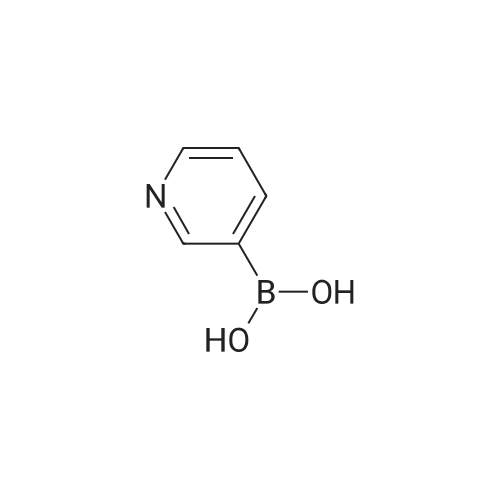
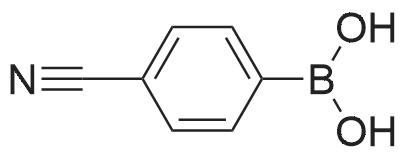
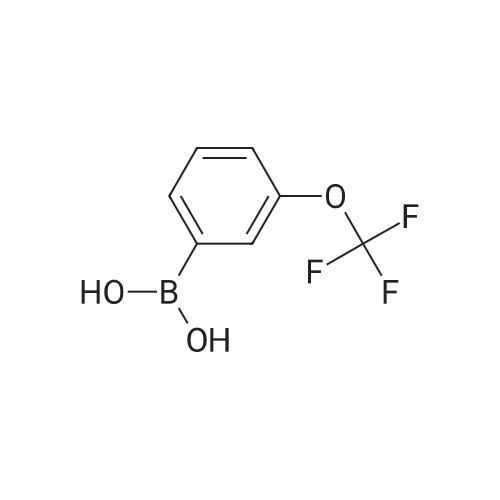
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With dihydrogen peroxide In ethanol; water at 20℃;
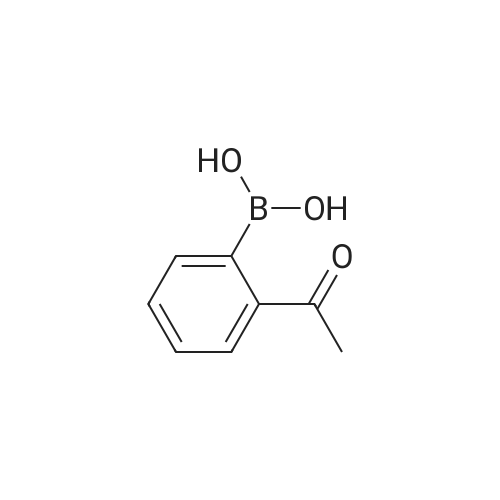
General procedure: In a 50 mL round-bottomed flask, a mixture of arylboronic acid(1 mmol), H2O2 (30percent aq, 0.2 mL), ZnO nanocatalyst (5 molpercent; sampleZnO-1) and 2 mL of water were stirred at room temperature under aerobic condition. The progress of the reaction was monitored by thin layer chromatography (TLC). After completion of the reaction, the reaction mixture was diluted with 20 mL of water and extracted with (3×20) mL of diethyl ether. The combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed in a rotary evaporator under reduced pressure. The crude product was purified by column chromatography (hexane/ ethylacetate, 9:1) on silica (100–200mesh) to get the desired product. The products were identified by 1HNMR and 13C NMR.
89%
With air; Ru(at)imine-nanoSiO2 catalyst In water; isopropyl alcohol at 30℃; for 0.8 h; Green chemistry
General procedure: A 50mL round bottom flask was charged with arylboronic acid (1mmol), 5mg Ru catalyst and 4ml iPrOH:H2O (1:1). The reaction mixture was stirred at room temperature under aerobic condition. The progress of the reaction was monitored by TLC. After the completion of the reaction, the mixture was diluted with 20mL of water and extracted with diethyl ether. The combined organic layers were washed with brine and dried over by anhydrous Na2SO4 and evaporated in a rotary evaporator under reduced pressure. The crude was purified by column chromatography on silica gel (hexane:ethyl acetate, 9:1) to afford the desired product. The purity of the compound was confirmed by 1H NMR, 13C NMR.
88%
With dihydrogen peroxide In water at 20℃; for 0.166667 h; Green chemistry
General procedure: In a 50mL round-bottomed flask, a mixture of arylboronic acid (1mmol), H2O2 (30percent aq, 0.2mL), bio-silica (5mg) and 2mL of water was added and stirred at room temperature in aerobic condition. The reaction was monitored by TLC. After completion of the reaction the reaction mixture was diluted with 20mL of water and extracted with (3×20) mL of diethylether and the combined organic layer was washed with brine and dried over by Na2SO4 and evaporated in a rotary evaporator under reduced pressure. The crude was purified by column chromatography (hexane/ethylacetate, 9:1) on mesh silica (100–200) to get the desired product. The products were confirmed by 1H NMR, 13C NMR, FT-IR spectroscopy and mass spectrometry.
Reference:
[1] Journal of Organic Chemistry, 2017, vol. 82, # 10, p. 5236 - 5241
[2] Advanced Synthesis and Catalysis, 2018, vol. 360, # 10, p. 2013 - 2019
[3] Applied Catalysis A: General, 2018, vol. 562, p. 58 - 66
[4] Tetrahedron Letters, 2016, vol. 57, # 36, p. 4050 - 4052
[5] Tetrahedron Letters, 2015, vol. 56, # 14, p. 1780 - 1783
[6] Chemistry Letters, 2016, vol. 45, # 3, p. 268 - 270
[7] ACS Catalysis, 2017, vol. 8, # 6, p. 5313 - 5322
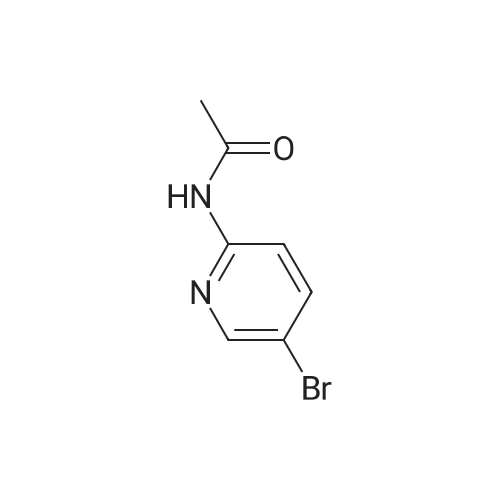
2
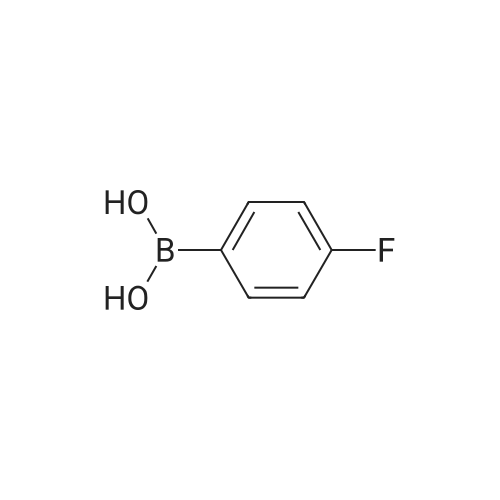
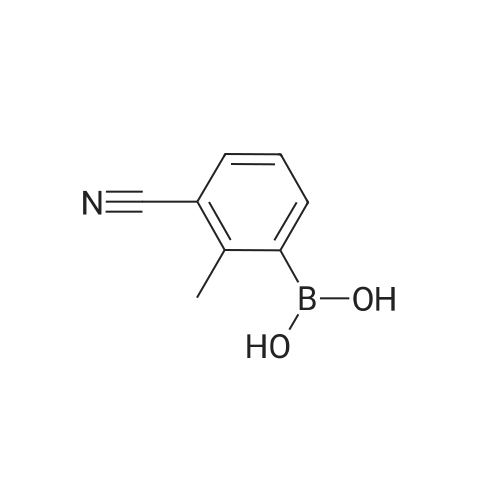
[ 150255-96-2 ]
[ 25487-66-5 ]
Yield
Reaction Conditions
Operation in experiment
89%
Stage #1: With potassium hydroxide In ethylene glycol at 175℃; for 3 h; Stage #2: With hydrogenchloride In water; ethylene glycol
1. Preparation of 3-carboxyphenylboronic acid 10 g (68 mmol) of 3-cyanophenylboronic acid and 15.26 g (272 mmol, 4 eq.) of potassium hydroxide powder were suspended in 40 ml of ethylene glycol and heated to 175° C. After three hours, the reaction mixture was allowed to cool and was diluted with 60 ml of water. The pH was adjusted to 2-3 with 32percent hydrochloric acid, which precipitated the 3-carboxyphenylboronic acid in colorless crystalline form, which was isolated by filtering it off with suction. The crystals were washed with water and dried under a gentle vacuum at 35° C. The yield was 10.04 g (60.5 mmol, 89percent).
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at 20℃; for 0.5 h; Stage #2: Heating / reflux
20 g of 3-bromobenzonitrile was dissolved in 100 ml of dry THF, and then mixed with 37.6 ml of triisopropoxyborane in the atmosphere of nitrogen. The solution was cooled at -78° C., and then 98.3 ml of a 1.6M n-butyl lithium hexane solution was dropwisely added to the cooled solution for about 30 minutes with stirring. The mixture was stirred at room temperature for 30 minutes, cooled at 0° C. and mixed with 220 ml of 4M sulfuric acid. The solution was heated and refluxed overnight, again cooled at 0° C., mixed with 340 ml of a 5M aqueous solution of sodium hydroxide, and then extracted with 200 ml of diethyl ether. The aqueous phase was separated, mixed with 6M hydrochloric acid until to give pH 2, and then twice extracted with 300 ml of ethyl acetate. The obtained ethyl acetate layer was dried over MgSO4, and the solvent was distilled away. The obtained crude product was recrystallized from DMF-water to obtain 11.6 g (72percent) of the title compound as needle-like pale yellow crystals. 1H NMR (270 MHz): δ (DMSO-d6) 8.5(brs, 2H), 8.3-7.6(m, 4H).
62%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78 - 20℃; for 1 h; Inert atmosphere Stage #2: With sulfuric acid In tetrahydrofuran; diethyl ether; hexane at 0℃; Reflux
3-bromobenzonitrile (20 g, 110 mmol) was dissolved in 100 mL of dry THF, and then mixed with triisopropoxyborane (71 ml, 309 mmol) in the atmosphere of nitrogen. The solution was cooled at -78° C., and then n-butyl lithium (76 mL, 121 mmol, 1.6M in hexane) was dropwisely added to the cooled solution for about 30 minutes with stirring. The mixture was stirred at room temperature for 30 min, cooled at 0° C. and mixed with 220 mL of 4M sulfuric acid. The solution was heated and refluxed overnight, again cooled at 0° C., mixed with 340 mL of a 5M aqueous solution of sodium hydroxide, and then extracted with 200 mL of diethyl ether. The aqueous phase was separated, mixed with 6M hydrochloric acid until to give pH 2, and then twice extracted with 300 mL of with 6M hydrochloric acid until to give pH4 2, and then twice extracted with 300 mL of ethyl acetate. The obtained ethyl acetate layer was dried over Na2SO4, and the solvent was distilled away. The obtained crude product was recrystallized from DMF-water to obtain (3-cyanophenyl)boronic acid (10.12 g, 62percent) as a solid.1H NMR (400 MHz, DMSO-d6): δ 8.39 (brs, 2H), 8.13 (s, 1H), 8.07 (d, J=7.6 Hz, 1H), 7.86 (d, J=7.6 Hz, 1H), 7.56 (t, J=7.6 Hz, 1H).
72%
With hydrogenchloride; sodium hydroxide; n-butyllithium; sulfuric acid In tetrahydrofuran; hexane
Productive Example 1 Preparation of 3-cyanophenylboronic acid In 100 ml of anhydrous tetrahydrofuran, was dissolved 20 g of 3-bromobenzonitrile. To the resulting solution, was added 37.6 ml of triisopropoxyborane under a nitrogen atmosphere. The formed solution was cooled to -78° C., and 98.3 ml of a 1.6 M solution of n-butyllithium in hexane was dropped thereinto under stirring for about 30 minutes. After stirring the resulting mixture at room temperature for 30 minutes, the mixture was then cooled to 0° C., and 220 ml of a 4 M sulfuric acid was added. The prepared solution was refluxed overnight and subsequently recooled to 0C. To the cooled solution, was added 340 ml of a 5 M aqueous solution of sodium hydroxide. The resulting solution was extracted with 200 ml of diethyl ether, and an aqueous layer was separated. To the aqueous layer, was added a 6 M hydrochloric acid until pH attained 2. The resulting mixture was extracted with 300 ml of ethyl acetate twice. The extract was dried over magnesium sulfate, and the solvent was then evaporated. The resulting crude product was recrystallized from dimethylformamide-water to provide 11.6 g (72percent) of the title compound as a needlelike light-yellow crystal. 1H-NMR (270 MHz, DMSO-d6): δ 7.6-8.3 (m, 4H), 8.5 (brs, 2H).
Stage #1: With diisopropopylaminoborane; triethylamine; triphenylphosphine; palladium dichloride In tetrahydrofuran at 65℃; for 12 h; Inert atmosphere Stage #2: With methanol In tetrahydrofuran at 0℃; Inert atmosphere
General procedure: Triphenylphosphene (0.131 g, 0.5 mmol, 20 mol percent), p-iodoanisol (0.585 g, 2.5 mmol), and triethylamine (1.78 mL, 12.5 mmol) were added to a 50 mL round-bottomed flask equipped with a sidearm, condenser, and stir bar. This solution was then degassed by alternating vacuum and argon three times. Palladium dichloride (0.023 g, 0.13 mmol, 5 mol percent) was then added under positive argon pressure. After stirring at room temperature for 15 min, diisopropylaminoborane (5 mL, 1 M solution in THF, 5 mmol) was added and the reaction mixture was degassed again by alternating vacuum and argon three times. The reaction solution was then heated to reflux. After 12 h of reflux the reaction was cooled to 0 °C and 6 mL of methanol was added through the condenser slowly (Caution: exothermic reaction with evolution of hydrogen). After 15 min of stirring all the solvent was removed under reduced pressure to yield a black solid. This solid was dissolved with sodium hydroxide (3 M, 8 mL) and subsequently washed with hexanes (3.x.10 mL). The aqueous layer was then cooled to 0 °C (ice bath) and acidified to pH <=1 with concentrated HCl, with the boronic acid usually precipitating out as a white solid. The aqueous fraction was then extracted with diethyl ether (3.x.10 mL). The organic fractions were combined, dried with magnesium sulfate and filtered. The solvent was then removed under reduced pressure yielding a white solid.
Stage #1: for 8 h; Heating / reflux Stage #2: at 20℃; for 12 h;
In a three-necked flask with a capacity 3 liters, 123.4g of the 3-formylphenylboronic acid prepared in Production Example 2, 68.6g of hydroxylamine hydrochloride, 1050 ml of formic acid and 112.1g of sodium formate are placed and mixed with each other. The resultant mixture liquid was heated for 8 hours while refluxing. The resultant reaction mixture liquid was left to stand in the ambient atmosphere for one night. Thereafter, in the case where a precipitate was generated in the reaction mixture liquid, this mixture liquid was agitated while cooling with ice and, in the case where no precipitate was generated in the reaction mixture liquid, the mixture liquid was mixed with a small amount of precipitation seed particles and agitated. From the resultant reaction mixture liquid, the solid precipitate was collected, by filtration and dried. The target compound, 3-cyanophenylboronic acid was obtained in an amount of 82.0g. The yield thereof was 68percent. The results of the 1H-NMR measurement of the target compound (200 MHz, δ ppm, CDCl3) were shown below. 7.47 (t, J = 7.7 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.9 - 8.0 (br. d, 1H), 8.0 - 8.1 (br. s, 1H)
Part C. Preparation of 3-cyanophenylboronic Acid. 3-Bromobenzonitrile (10.0 g, 55 mmol) was dissolved in dry THF (100 mL) and cooled to -100° C. (Et2 O/N2). n-Butyllithium (24.2 mL, 2.5 M in hexane) was added over 30 minutes, maintaining the internal temp under -90°. After 20 minutes, triisopropylborate (18.0 mL) was added over 15 minutes, again maintaining the internal temperature. After the addition was complete, the reaction was allowed to warm slowly to room temperature over 1.5 hours. The reaction was stirred at room temp for 16 hours, then cooled to 15° C., after which 6 M HCl (25 mL) was added. After stirring vigorously for 3.5 hours, the reaction was partitioned between water and EtOAc. After extracting a second time with EtOAc, the combined organics were washed with 2 M NaOH. The aqueous extract was neutralized with 6 M HCl. The white precipitate was filtered, yielding the desired product (4.80 g, 60percent). 1 H NMR (DMSO-d6): δ 8.37 (s, 2H), 8.10 (s, 1H), 8.03 (dt, 1H, J=7.3, J'=1.1), 7.83 (dt, 1H, J=7.6, J'=1.4), 7.53 (t, 1H, J=7.7).
Reference:
[1] Patent: US6060491, 2000, A,
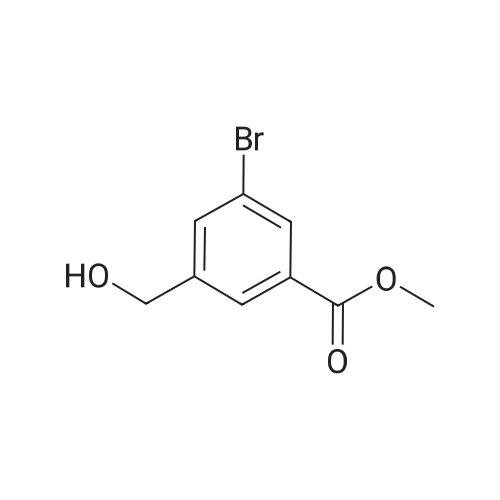
7
[ 6952-59-6 ]
[ 150255-96-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2005, vol. 13, # 8, p. 2859 - 2872
[2] Journal of the American Chemical Society, 2012, vol. 134, # 28, p. 11667 - 11673
[3] Organic Letters, 2012, vol. 14, # 18, p. 4814 - 4817,4
[4] Journal of Organic Chemistry, 2013, vol. 78, # 13, p. 6427 - 6439
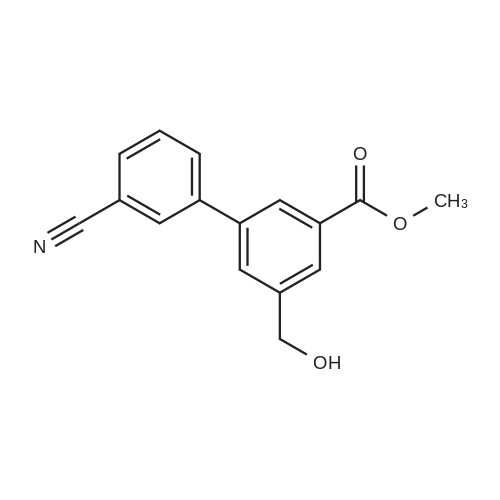
8
[ 5419-55-6 ]
[ 6952-59-6 ]
[ 150255-96-2 ]
Reference:
[1] Patent: EP1043310, 2000, A1,
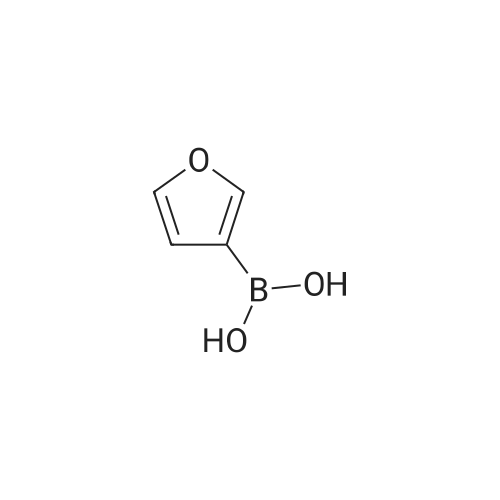
9
[ 13675-18-8 ]
[ 69113-59-3 ]
[ 150255-96-2 ]
Reference:
[1] Journal of Organic Chemistry, 2018, vol. 83, # 4, p. 1842 - 1851
10
[ 13675-18-8 ]
[ 6952-59-6 ]
[ 150255-96-2 ]
Reference:
[1] Journal of Organic Chemistry, 2018, vol. 83, # 4, p. 1842 - 1851
11
[ 938443-32-4 ]
[ 150255-96-2 ]
Reference:
[1] Synlett, 2011, # 15, p. 2223 - 2227
12
[ 76-09-5 ]
[ 150255-96-2 ]
[ 214360-46-0 ]
Yield
Reaction Conditions
Operation in experiment
4.72 g
With sodium sulfate In tetrahydrofuran at 25℃; for 12 h; Inert atmosphere
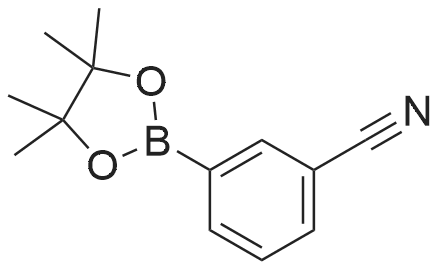
To a solution of 3-cyanophenyl boronic acid (2.5 g, CAS: 150255-96-2) in anhydrous THF (150 ml) under an atmosphere of nitrogen were added pinacol (2.95 g) and Na2SO4 (10 g) at 25°C.The reaction mixture was stirred for 12 h at 25°C under an atmosphere of nitrogen. Then, Na2SO4was filtered off and all volatiles were evaporated. The residue was partitioned between ethylacetate and water. The organic layer was then separated, dried over Na2SO4 and concentrated to afford the title compound (4.72 g) as off white solid which was directly used in the next reaction step without further characterization.
Reference:
[1] Journal of the American Chemical Society, 2013, vol. 135, # 7, p. 2552 - 2559
[2] Patent: WO2017/37146, 2017, A1, . Location in patent: Page/Page column 195; 196
[3] Journal of Organic Chemistry, 2018, vol. 83, # 4, p. 1842 - 1851
13
[ 150255-96-2 ]
[ 370864-73-6 ]
Reference:
[1] Angewandte Chemie - International Edition, 2016, vol. 55, # 33, p. 9676 - 9679[2] Angew. Chem., 2016, vol. 128, # 33, p. 9828 - 9831,4
14
[ 150255-96-2 ]
[ 1000016-93-2 ]
Reference:
[1] Patent: WO2007/149031, 2007, A1,
15
[ 150255-96-2 ]
[ 1527473-33-1 ]
Reference:
[1] Journal of Medicinal Chemistry, 2015, vol. 58, # 1, p. 419 - 432
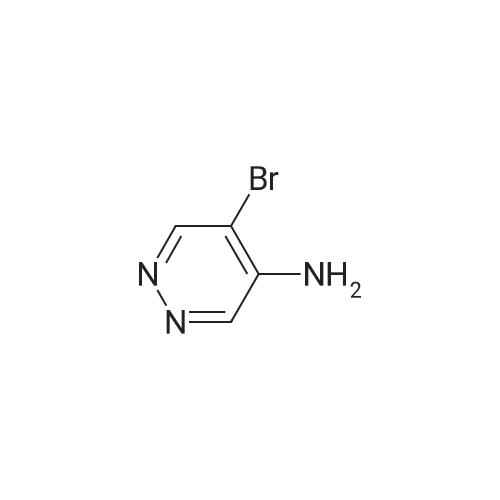
With sodium carbonate; triphenylphosphine;tris-(dibenzylideneacetone)dipalladium(0); In tetrahydrofuran; water; at 80℃; for 16h;
To a mixture of THF (80 ml) and 2N sodium carbonate (34 ml, aqueous) is added 6-amino- 8-BROM-1, 7-naphthyridine (4.007 g), triphenylphosphine (0.37 g) and 3-cyanophenyl- boronic acid (3.23 g). The mixture is degassed under argon three times and then bis (di- benzylidene-acetone) palladium (0) (0.4 g) is added and the mixture is degassed under argon three more times. The mixture is heated at 80C under argon for 16 hours then cooled and filtered. The mixture is diluted with ethyl acetate and washed with 2N sodium hydroxide then brine. After drying over sodium sulfate the organic layer is evaporated and suspended in ether. The solid precipitate thus obtained is the title compound, which is filtered off.
With tetrabutylammomium bromide; sodium carbonate;bis(triphenylphosphine)palladium(II)-chloride; In benzene;
Part D. Preparation of methyl 2-(3'-cyanophenyl)nicotinate. Methyl 2-bromonicotinate (2.0 g, 9.3 mmol) and 3-cyanophenylboronic acid (2.7 g, 18.4 mmol) were combined in 190 mL benzene. Sodium carbonate (19 mL of a 2 M aqueous solution), tetrabutylammonium bromide (152 mg, 0.5 mmol), and bis(triphenylphosphine)palladium(II) chloride (325 mg, 0.5 mmol) were added. The entire mixture was evacuated to remove dissolved gasses, then placed under argon. The reaction was refluxed for 14 hours, diluted with water and EtOAc, separated, dried over Na2 SO4, filtered, and evaporated. The resulting yellow solid was chromatographed on silica gel (30% EtOAc/hexanes) to yield a light yellow solid (1.70 g, 77%). 1 H NMR (CDCl3): delta 8.81 (dd, 1H, J=4.8, J'=1.8), 8.23 (dd, 1H, J=8.0, J'=1.9), 7.85 (s, 1H), 7.73 (m, 2H), 7.55 (t, 1H, J=7.7), 7.43 (m, 1H), 3.76 (s, 3H).
General procedure: Triphenylphosphene (0.131 g, 0.5 mmol, 20 mol %), p-iodoanisol (0.585 g, 2.5 mmol), and triethylamine (1.78 mL, 12.5 mmol) were added to a 50 mL round-bottomed flask equipped with a sidearm, condenser, and stir bar. This solution was then degassed by alternating vacuum and argon three times. Palladium dichloride (0.023 g, 0.13 mmol, 5 mol %) was then added under positive argon pressure. After stirring at room temperature for 15 min, diisopropylaminoborane (5 mL, 1 M solution in THF, 5 mmol) was added and the reaction mixture was degassed again by alternating vacuum and argon three times. The reaction solution was then heated to reflux. After 12 h of reflux the reaction was cooled to 0 C and 6 mL of methanol was added through the condenser slowly (Caution: exothermic reaction with evolution of hydrogen). After 15 min of stirring all the solvent was removed under reduced pressure to yield a black solid. This solid was dissolved with sodium hydroxide (3 M, 8 mL) and subsequently washed with hexanes (3×10 mL). The aqueous layer was then cooled to 0 C (ice bath) and acidified to pH ?1 with concentrated HCl, with the boronic acid usually precipitating out as a white solid. The aqueous fraction was then extracted with diethyl ether (3×10 mL). The organic fractions were combined, dried with magnesium sulfate and filtered. The solvent was then removed under reduced pressure yielding a white solid.
3-(4-amino-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
41%
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; ethanol; water; at 80℃;
Synthesis of 3-(4-amino-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzonitrile (BA39); A solution of 3-Cyanophenylboronic acid (23 mg, 0.15 mmol) in EtOH (3.3 ml) was added to a solution of <strong>[862730-04-9]3-iodo-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine</strong> (40 mg, 0.13 mmol) in DME (12 ml). Pd(PPh3)4 (30 mg, 0.03 mmol) and saturated Na2CO3 (1.9 ml) were added and the reaction was heated to 80 C. under an argon atmosphere overnight. After cooling, the reaction was extracted with saturated NaCl and CH2Cl2. Organic phases were combined, concentrated in vacuo and purified by RP-HPLC (MeCN:H2O:0.1% TFA) to yield BA39 (14.9 mg, 41% yield). ESI-MS (M+H)+ m/z calcd 279.1, found 279.0.
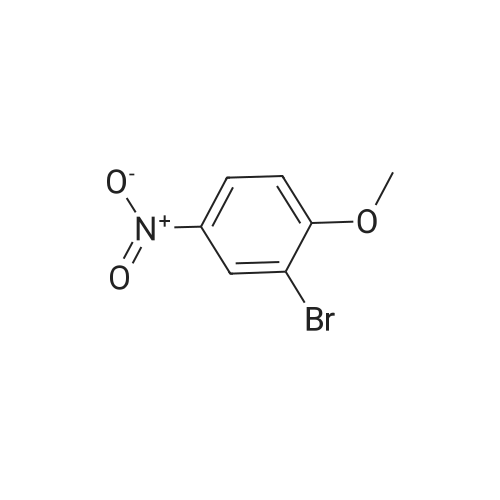
With caesium carbonate;dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; In ethylene glycol dimethylether; water; at 90℃; for 4h;Heating / reflux;
[1,1'Bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane(1:1) (1.43 g) was added to a stirred mixture of 3-cyanophenylboronic acid (10.0 g, 61.8 mmol) and <strong>[5197-28-4]2-bromo-4-nitroanisole</strong> (14.35 g, 62 mmol) in 2.0N cesium carbonate (92.7 ML, 185.4 mmol) and ethylene glycol dimethylether (200 ML).The flask was purged with nitrogen and heated at 90 C. (oil bath) for 4 hours.The mixture was allowed to cool to room temperature overnight, during which time the product precipitated from solution.The solid was collected on a Buchner funnel, washed with water and dried under reduced pressure to give 2-(3-cyanophenyl)-4-nitroanisole (15.7 g).
With sodium hydrogencarbonate;palladium diacetate; In DMF (N,N-dimethyl-formamide); water; at 80℃; for 6.5h;
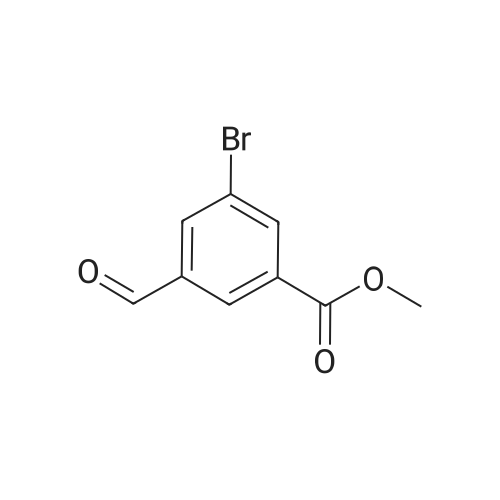
In a three-necked flask with a capacity of 2 liters, 3-cyanophenylboronic acid in an amount of 67.65g and sodium hydrogen carbonate in an amount of 116.0g were placed and then a solution of 111.9g of <strong>[377734-27-5]methyl 5-bromo-3-formylbenzoate</strong> prepared in step (A) in 142 ml of dimethyl formamide (DMF) was placed. The resultant mixture liquid in the flask was mixed with 592 ml of DMF and 149 ml of water. The flask was gas-tightly sealed, the air inside the flask was replaced by an argon gas and then 0.2231g of palladium acetate was fed into the flask. The resultant reaction mixture liquid in the flask was heated to a temperature of 80C and agitated at this temperature for 6.5 hours. Thereafter, the resultant reaction mixture liquid was subjected to a hot-filtration to remove an insoluble fraction from the reaction mixture liquid, and the resultant filtrate was heated to a temperatures of 80C and agitated. The heated and agitated filtrate was gradually added with 585 ml of water, and the resultant filtrate mixture was left to cool to room temperature. The precipitate generated in the filtrate mixture was collected by filtration, and the collected precipitate was washed with 590 ml of water and then dried. A crude product of the target compound, methyl 5-(3-cyanophenyl)-3-formylbenzoate was obtained in an amount of 103.15g which corresponded to a yield of 84.5%. The crude product was subjected to a purification procedure as follows. The dried crude product in an amount of 50g was placed in a three-necked flask with a capacity of 2 liters and mixed with 150 ml of hydrous THF having a water content of 3%. The resultant mixture was heated to a temperature of 80C to provide a solution of the crude product. The solution was subjected to a hot-filtration. The filtrate was again heated to a temperature of 80C and mixed with 750 ml of 2-propyl alcohol. The resultant mixture liquid was left to cool to room temperature, to recrystallize the target compound. After cooling, the resultant precipitate generated in the cooled mixture liquid was collected by filtration. A purified product of the target compound, methyl 5-(3-cyanophenyl)-3-formylbenzoate was obtained in an amount of 45.19g which corresponded to a recrystallization yield of 90%. The result of the 1H-NMR (200 MHz, delta ppm, CDCl3) of the purified product was as follows. 4.02 (s, 3H), 7.5 - 7.8 (m, 2H), 7.8 - 8.0 (m, 2H), 8.2 - 8.3 (s, 1H), 8.4 - 8.6 (m, 2H), 10.2 (s, 1H)
3-(2,6-dimethyl-pyridin-4-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium phosphate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; at 90℃; for 18h;
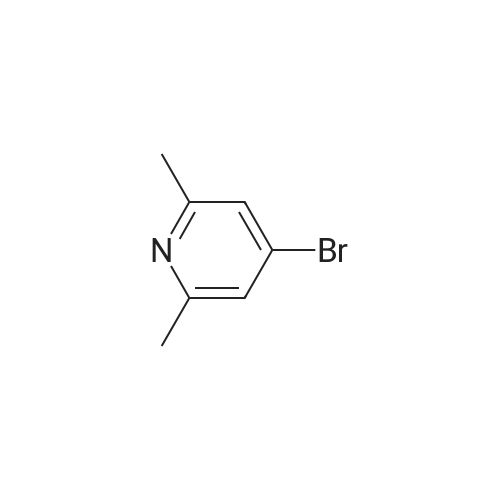
Compound K5 was obtained from 3- (2, 6-dimethyl-pyridin-4-yl) -benzonitrile [prepared by the following procedure: A mixture of 3-cyanophenylboronic acid [CAS-No. 150255- 96-2] (4.74 g, 32.25 mmol), <strong>[5093-70-9]4-bromo-2,6-dimethylpyridine</strong> [Chem. Pharm. Bull. 38: 2446 (1990) and J. Org. Chem. 27: 1665 (1962) ] (5.00 g, 26.87 mmol) and K3P04 (8.56 g, 35.78 mmol) in dioxane (126 mL) was degassed and Pd (Ph3P) 4 (1.53 g, 1.37 mmol) was added. The reaction mixture was stirred at 90°C for 18 h. Evaporated to dryness, taken up in EtOAc, washed with sat. NAHCO3-SOLUTION and brine, dried over NA2SO4. Removal of the solvent in vacuum left a brown solid, which was purified by silica gel column chromato- graphy with cyclohexane/EtOAc. ] (2.2 g, 10.6 mmol) by treatment with tert-butyl bromoacetate and activated zinc, followed by hydrolysis with 10percent HCl according to general procedure K (method d).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In methanol; toluene;Heating / reflux;
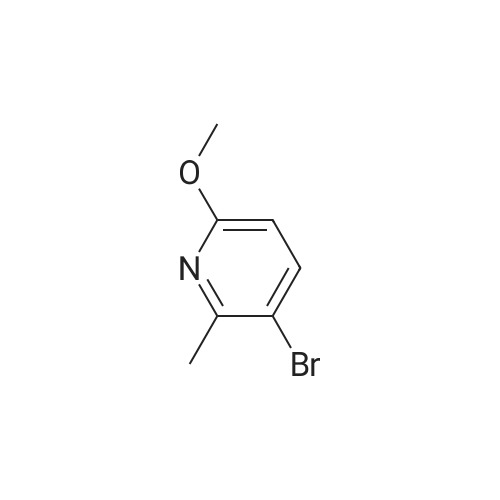
Step 2: 3-(6-Methoxy-2-meth(at)lpyridin-3-(at))benzonitrile: To a nitrogen flushed mixture of 3- cyanophenylboronic acid (5.0 g, 34 mmol), <strong>[126717-59-7]3-bromo-6-methoxy-2-methylpyridine</strong> (4.6 g, 22.7 mmol) and palladium tetrakis (triphenylphosphine) mg, 0.68 mmol) is added toluene (150 mL), methanol (12 mL) and sodium carbonate (22.7 mmol) and the mixture is heated overnight at reflux. The cooled reaction is concentrated and the residue is purified by chromatography eluting with cyclohexane-3.2 to 9% ethyl acetate. Product containing fractions are combined and concentrated to afford 3-(6-methoxy-2-methylpyridin-3- yl) benzonitrile (3.09g, 61% yield) as a white solid. LC/MS: MS m/e = 225 (M + H) ; RT 3.58 min; ¹H NMR (CDCl3, 8 ppm) 7.65 (1H, m), 7.60 (1H, m), 7.56 (2H, m), 7.40 (1H, d), 6.63 (1H, d), 3.98 (3H, s), 2.40 (3H, s).
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 80℃; for 14h;
Compound K7 was obtained from 3- (6-cyclopropyl-pyridin-3-yl)-benzonitrile [prepared by the following procedure: A mixture of 3-cyanophenylboronic acid [CAS-No. 150255- 96-2] (8.82 g, 60 mmol), crude <strong>[579475-29-9]5-bromo-2-cyclopropylpyridine</strong> {prepared by the following procedure: A mixture of 2, 5-dibromopyridine (11.85 g, 50 mmol), cyclopropyl zinc chloride (0.4 M in THF, 160 mL, 64 mmol), Pd (PPH3) 4 (578 mg, 1 mol%) in THF (55 mL) was stirred under Argon atmosphere at 70C for 1.5 h. Cooled to RT, poured into sat. NAHCO3-SOLUTION, extracted with ether, washed with brine, dried over NA2S04. Removal of the solvent in vacuum left a brown oil (12.36 G).} (ca. 60 mmol), Pd (PPH3) 4 (1.733 g, 3 mol%) and K2CO3 (13.82 g, 100 mmol) in toluene (250 mL), EtOH (22 mL) and H20 (50 mL) was stirred at 80C for 14 h. Cooled to RT, diluted with EtOAc, washed with sat. NAHCO3-SOLUTION and brine, dried over NA2SO4. Removal of the solvent in vacuum left a brown solid, which was purified by silica gel column chromatography with cyclohexane/EtOAc. ] (9.88 g, 44.87 mmol) by treatment with tert-butyl bromoacetate and activated zinc, followed by hydrolysis with 10% HCl according to general procedure K (method d).
With bis(((1Z,3Z)-3-((1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)imino)-1,3-diphenylprop-1-en-1-yl)oxy)palladium; potassium carbonate; In water; N,N-dimethyl-formamide; at 80℃; for 4.5h;Catalytic behavior;
General procedure: A mixture of aryl halide (1mmol), olefin (1mmol), K2CO3 (1mmol), and the Pd-Schiff base complex (2.1mg, 0.2mol %) in DMF-H2O (1:1) was stirred at 80C for 3-4h. The reaction progress was monitored periodically by TLC. After completion of the reaction, it was cooled to room temperature and the product was extracted with ethyl acetate (3×10mL) from aqueous phase. The combined organic fractions were dried over Na2SO4, the solvent removed under reduced pressure to afford a crude product. The residue was purified by short column chromatography on silica gel eluted with petroleum ether/ethyl acetate afforded the desired coupled products up to 98% yield. The products were confirmed by 1H and 13C NMR.General procedure for the Suzuki-Miyaura reaction: A mixture of aryl halide (1mmol), arylboronic acid (1mmol), K2CO3 (1mmol), and the Pd- Schiff base complex (2.1mg, 0.2mol %) in DMF-H2O (1:1) was stirred at 80C for 3-5h. The progress of reaction was monitored by TLC until the complete consumption of the aryl halide. After the reaction, the mixture was cooled down to room temperature and repeatedly extracted with ethyl acetate. The combined organic layer was separated, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel to give the corresponding coupling products in up to 95% isolated yield. The products were confirmed by 1H and 13C NMR.
83%
With potassium carbonate; In water; at 70℃; for 0.5h;Inert atmosphere;
General procedure: In a typical run, h-BN(at)Fur(at)Pd(OAc)2 (0.05 mmol) was added to a mixture of arylboronic acid 1 (1.0 mmol), aryl bromide 2 (1.5 mmol) and K2CO3 (1.5 mmol) in water (1 mL). The resulting mixture was stirred at 70 C under Ar protection, and the progress of the reaction was monitored by TLC. After completion of the reaction, ethyl acetate was added to the reaction mixture and the catalyst was separated. The organic phase was washed with water, dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. Finally, the residue was isolated by chromatography on a column of silica gel to afford the corresponding product 3.
80%
With potassium phosphate; [bis{2-(3-(2,4,6-trimethylbenzyl)imidazolin-2-yliden-1-yl)-4-methylphenyl}amido]chloronickel; In 1,4-dioxane;Inert atmosphere; Schlenk technique; Reflux;
General procedure: In a typical reaction, to a 25-mL Schlenk tube equipped with amagnetic stirring bar were added nickel(II) catalyst 2a (0.005mmol, 1.0mol %), aryl halides (0.5mmol), phenylboronic acids (0.75mmol), and anhydrous K3PO4 (2.0mmol). The tube was then evacuated (3×5min) under vacuum and backfilled with N2. Dried dioxane (4.0mL) was injected via a syringe, and the reaction mixture was stirred at reflux until the aryl halides had disappeared as monitored by TLC. The reaction mixture was treated with H2O (20mL), then extracted with Et2O (3×15mL). The combined organic extracts were washed with sat. aq NaCl (10mL) and dried with anhydrous MgSO4. The solvent was removed under reduced pressure and the crude material was purified by silica gel column chromatography to give the corresponding biaryl. All the coupling products are known and their structures were identified by comparing their 1H NMR and 13C NMR spectral data with those reported in literature.
67%
With caesium carbonate;tetrakis(triphenylphosphine) palladium(0); In N,N-dimethyl-formamide; for 4h;Heating / reflux;
Preparation Example 5. C-Biphenyl-3-yl-methylamine To a solution of 3-cyanophenylboronic acid (1.0g, 6.81 mmol) and bromobenzene (1.07g, 6.81mmol) in N,N-dimethylformamide (100mL) were added tetrakis(triphenylphosphine)palladium(0) (0.393g, 0.341 mmol) and cesium carbonate (2.77g, 8.51mmol) under nitrogen atmosphere, and the mixture was stirred for 4 hours under reflux. The reaction mixture was allowed to room temperature, ethyl acetate and water were added for partitioning, the organic layer was washed with water and dried over anhydrous magnesium sulfate. The solvent was evaporated, the residue was purified by silica gel column chromatography (hexane : ethyl acetate = 10 : 1), and biphenyl-3-carbonitrile (821 mg, 67%) was obtained as a yellow solid. Next, a solution of the resulting biphenyl-3-carbonitrile (821 mg, 4.58mmol) in tetrahydrofuran (5mL) was added to a solution of lithium aluminum hydride (0.435g, 11.5mmol) in tetrahydrofuran (5mL) that had been cooled on an ice bath, and the solution was stirred for 6 hours at room temperature. The reaction solution was cooled on an ice bath, a mixture solution of methanol and water (9:1) was added thereto, an aqueous solution of saturated ammonium chloride was further added, filtration was carried out through Celite pad and insoluble matter was removed. The filtrate was partitioned, the organic layer was dried over anhydrous magnesium sulfate, and the title compound (527mg, 63%) was obtained as a brown oil. This was used in the next reaction without further purification.
67%
With caesium carbonate;tetrakis(triphenylphosphine) palladium(0); In N,N-dimethyl-formamide; for 4h;Heating / reflux;
To a solution of 3-cyanophenylboronic acid (1.0g, 6.81 mmol) and bromobenzene (1.07g, 6.81mmol) in N,N-dimethylformamide (100mL) were added tetrakis(triphenylphosphine)palladium(0) (0.393g, 0.341 mmol) and cesium carbonate (2.77g, 8.51mmol) under nitrogen atmosphere, and the mixture was stirred for 4 hours under reflux. The reaction mixture was allowed to room temperature, ethyl acetate and water were added for partitioning, the organic layer was washed with water and dried over anhydrous magnesium sulfate. The solvent was evaporated, the residue was purified by silica gel column chromatography (hexane : ethyl acetate = 10 : 1), and biphenyl-3-carbonitrile (821 mg, 67%) was obtained as a yellow solid. Next, a solution of the resulting biphenyl-3-carbonitrile (821mg, 4.58mmol) in tetrahydrofuran (5mL) was added to a solution of lithium aluminum hydride (0.435g, 11.5mmol) in tetrahydrofuran (5mL) that had been cooled on an ice bath, and the solution was stirred for 6 hours at room temperature. The reaction solution was cooled on an ice bath, a mixture solution of methanol and water (9:1) was added thereto, an aqueous solution of saturated ammonium chloride was further added, filtration was carried out through Celite pad and insoluble matter was removed. The filtrate was partitioned, the organic layer was dried over anhydrous magnesium sulfate, and the title compound (527mg, 63%) was obtained as a brown oil. This was used in the next reaction without further purification.
64%
With Ti0.97Pd0.03O1.97; potassium carbonate; In water; at 100℃; for 6h;
General procedure: A mixture of aryl halide 1 (0.4mmol), arylboronic acid 2 (0.8mmol), Ti0.97Pd0.03O1.97 (nanoparticles; 10wt %), K2CO3 (110mg, 0.8mmol) in water (2mL) was stirred at 100C for 6h. After the completion of the reaction, diethyl ether (15ml x 3 times) was poured into the mixture, washed with water (15mL), extracted with diethyl ether (3×20mL), dried over anhydrous Na2SO4 and evaporated under vacuum; the residue was purified by column chromatography (petroleum ether or petroleum ether/ethyl acetate) to obtain the desired coupled product.
With potassium carbonate;Pd(PPh3)4; In tetrahydrofuran; ethanol; water; toluene;
Example K35 3-[3-(6-Ethyl-4-methyl-pyridin-3-yl)-phenyl]-3-oxo-propionic acid tert-butyl ester The title compound was obtained from 3-(6-ethyl-4-methyl-pyridin-3-yl)-benzonitrile [prepared by the following procedure: A mixture of 3-cyanophenylboronic acid [CAS-No. 150255-96-2] (3.53 g, 24 mmol), 5-bromo-2-ethyl-4-methyl-pyridine{prepared by the following procedure: A mixture of <strong>[3430-26-0]2,5-dibromo-4-methyl-pyridine</strong> [CAS-No. 3430-26-0] (5.0 g, 20 mmol), diethylzinc (1.1 M in toluene, 10.9 mL, 12 mmol), Pd(PPh3)4 (231 mg, 1 mol %) in THF (90 mL) was stirred under argon atmosphere at 70 C. for 0.5 h. Cooled to room temperature, poured into sat. NaHCO3-solution, extracted with ether, washed with brine, dried over Na2SO4. Removal of the solvent in vacuum followed by silica gel column chromatography with cyclohexane/EtOAc left a colorless oil (5.321 g).} (5.321 g, 20 mmol), Pd(PPh3)4 (693 mg, 3 mol %) and K2CO3 (5.53 g, 40 mmol) in toluene (100 mL), EtOH (9 mL) and H2O (20 mL) was stirred at 80 C. for 3.5 h. Cooled to room temperature, diluted with EtOAc, washed with sat. NaHCO3-solution and brine, dried over Na2SO4. Removal of the solvent in vacuum left a brown solid, which was purified by silica gel column chromatography with cyclohexane/EtOAc.] (2.330 g, 10.5 mmol) by treatment with tert-butyl bromoacetate and activated zinc, followed by hydrolysis with 10% HCl according to general procedure K (method d). Obtained as a brown oil (2.926 g, 82%). MS (EI) 339 (M+).
3-(3-hydroxymethyl-pyridin-4-yl)-benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; toluene;Heating / reflux;
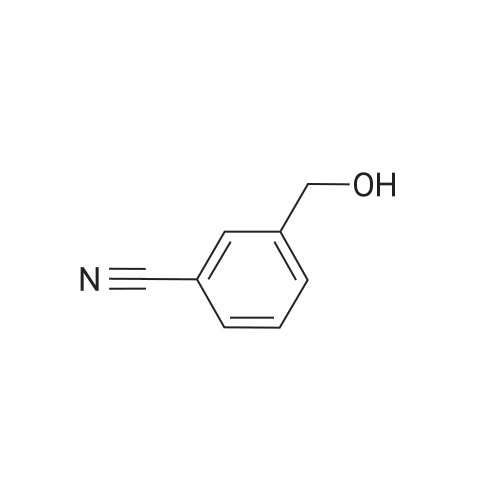
To a flask charged with (4-bromo-pyridin-3~yl)-methanol (90 mg, 0.48 mmol), 3-cyanophenylboronic acid (84.4 mg, 0.57 mmol) and tetrakis(triphenylphousphine)-palladium(0) (28 mg) was added 2 M Na2CO3 (2 mL) and toluene (3 ml_). The reaction mixture was refluxed overnight and then extracted with ethyl acetate. The organic layer was dried over sodium sulfate and purified by column chromatography to yield 3-(3-Hydroxymethyl-pyridin-4- yl)-benzonitrile as a white solid.1HNMR (CDCI3, 300 MHz) delta (ppm) 8.74 (s, 1 H), 8.60 (d, J = 4.8 Hz, 1 H), 7.9-7.2 (m, 7H), 4.62 (s, 2H), 3.34 (bs, 1 H); LCMS: 2.815 min, m/z: 211 (M + 1)
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; toluene;Heating / reflux;
To a flask charged with <strong>[197007-87-7](4-bromo-pyridin-3-yl)-methanol</strong> (90 mg, 0.48 mmol), 3-cyanophenylboronic acid (84.4 mg, 0.57 mmol) and tetrakis(triphenylphousphine)-palladium(0) (28 mg) was added 2 M Na2COe (2 ml_) and toluene (3 ml_). The reaction mixture was refluxed overnight and then extracted with ethyl acetate. The organic layer was dried over sodium sulfate and purified by column chromatography to yield 3-(3-Hydroxymethyl-pyridin-4- yl)-benzonitrile as a white solid.1HNMR (CDCI3, 300 MHz) delta (ppm) 8.74 (s, 1 H), 8.60 (d, J= 4.8 Hz, 1 H), 7.9-7.2 (m, 7H), 4.62 (s, 2H), 3.34 (bs, 1 H); LCMS: 2.815 min, m/z: 211 (M + 1).
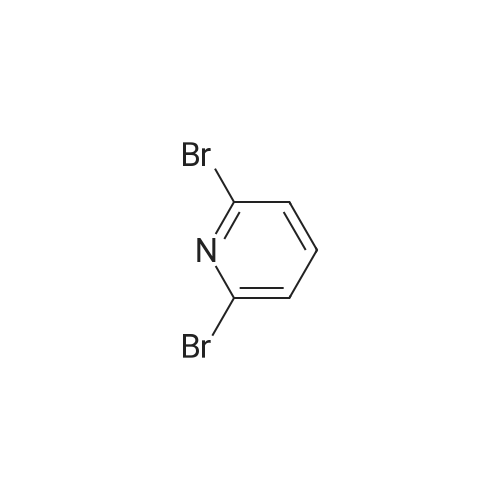
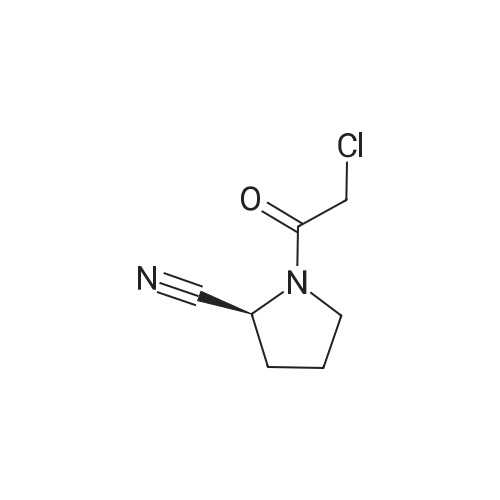
(2S)-1-({2-[6-(3-Cyano-phenyl)-pyridin-2-ylamino]-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, hydrochloride salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; In 1,4-dioxane;
Example 130 (2S)-1-({2-[6-(3-Cyano-phenyl)-pyridin-2-ylamino]-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, Hydrochloride Salt This compound was obtained in analogy to example 36, steps A] to C] starting from 2,6-dibromopyridine, 1,2-diaminoethane, 3-cyanophenylboronic acid and IIA. The residue obtained by flash chromatography was dissolved in dioxane and precipitated by treatment with HCl in dioxane yielding a white powder. MS (ISP): 375.5 (MH+).
(2S)-1-({2-[5-(3-Cyano-phenyl)-pyridin-2-ylamino]-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, hydrochloride salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran;
Example 118 (2S)-1-({2-[5-(3-Cyano-phenyl)-pyridin-2-ylamino]-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, Hydrochloride Salt This compound was obtained in analogy to example 36, steps A] to C] starting from 2,5-dibromopyridine, 1,2-diaminoethane, 3-cyanophenylboronic acid and IIA. The residue obtained by flash chromatography was dissolved in THF and precipitated by treatment with HCl in ether yielding a white powder. MS (ISP): 375.5 (MH+).
4-(3-cyanophenyl)-1-(pyridin-2-yl)-1H-imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.5mg (9%)
With hydrogenchloride; sodium carbonate;Pd(PPh3)4; In tetrahydrofuran; 1-methyl-pyrrolidin-2-one; methanol; 1,2-dimethoxyethane; dichloromethane; ethyl acetate;
4-(3-Cyanophenyl)-1-(2-pyridyl)-1H-imidazole A mixture of <strong>[87941-55-7]4-bromo-1-trityl-1H-imidazole</strong> (0.2g, 0.51 mmol), 3-cyanophenylboronic acid (0.11 g, 0.77 mmol), and Pd(PPh3)4 (0.06g, 0.05 mmol) in a solution of ethylene glycol dimethyl ether (2 mL) and 2M sodium carbonate (2 mL) was heated in a sealed vial overnight at 120 C. After cooling, the reaction mixture was diluted with dichloromethane (30 mL) and washed with water (50 mL) and brine (30 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. Silica gel chromatography of the crude residue using 2% ethyl acetate in hexanes afforded 0.07 g (34%) 4-(3-cyanophenyl)-1-trityl-1H-imidazole as white foam. A solution of 4-(3-cyanophenyl)-1-trityl-1H-imidazole (0.07 g, 0.17 mmol) in tetrahydrofuran (1.36 mL) was treated with 2N hydrochloric acid (0.68 mL) and the resulting mixture was heated at reflux for 45 minutes. After cooling the reaction mixture was concentrated in vacuo and the residue dissolved in dichloromethane (20 mL). The organic phase was successively washed with 1N sodium hydroxide (10 mL), water (20 mL) and brine (20 mL). The organic solution was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography of the residues using 3% methanol in dichloromethane afforded 0.02 g (72%) of 4-(3-cyanophenyl)imidazole as a white solid. A solution of 4-(3-cyanophenyl)imidazole (0.02 g, 0.11 mmol) in N-methylpyrrolidinone (0.5 mL) was treated with 2-bromopyridine (1.05 mL, 11.1 mmol) and the reaction mixture heated overnight at 160 C. After cooling the reaction mixture was diluted with dichloromethane (40 mL) and the organic phase was successively washed with water (10*50 mL) and brine (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. Silica gel chromatography using 30% ethyl acetate in hexanes afforded 2.5mg (9%) of 4-(3-cyanophenyl)-1-(2-pyridyl)-1H-imidazole, as an off-white solid.
With methanesulfonic acid; In tetrahydrofuran; tert-butyl methyl ether;
Example 135 (2S)-1-({2-[6-(3-Cyano-phenyl)-pyridin-2-ylamino]-1,1-dimethyl-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, Methansolfonic Acid Salt This compound was obtained in analogy to example 36, steps A] to C] starting from 2,6-dibromopyridine, 1,2-diamino-2-methlypropane, 3-cyanophenylboronic acid and IIA. The residue obtained by flash chromatography was dissolved in THF and precipitated by treatment with methanesulfonic acid in tert-butyl methyl ether and ethyl acetate yielding a white powder. MS (ISP): 403.5 (MH+).
(2S)-1-({2-[5-(3-Cyano-phenyl)-pyridin-2-ylamino]-1,1-dimethyl-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, hydrochloride salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran;
Example 117 (2S)-1-({2-[5-(3-Cyano-phenyl)-pyridin-2-ylamino]-1,1-dimethyl-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile, Hydrochloride Salt This compound was obtained in analogy to example 36, steps A] to C] starting from 2,5-dibromopyridine, 1,2-diamino-2-methlypropane, 3-cyanophenylboronic acid and IIA. The residue obtained by flash chromatography was dissolved in THF and precipitated by treatment with HCl in ether yielding a white powder. MS (ISP): 403.6 (MH+).
Example 145 (2S)-1-({2-[5-(3-Cyano-phenyl)-pyrimidin-2-ylamino]-1,1-dimethyl-ethylamino}-acetyl)-pyrrolidine-2-carbonitrile This compound was obtained in analogy to example 36, steps A] to C] starting from 5-bromo-2-iodopyrimidine, 1,2-diamino-2-methlypropane, 3-cyanophenylboronic acid and IIA. It was isolated as its free amine, as a slightly brown foam. MS (ISP): 404.5 (MH+).
3-[[1,2-dihydro-1-[2'-(methylsulfonyl)-[1,1'-biphenyl]-4-yl]-2-oxo-3-pyridinyl]amino]-benzenecarboximidamide[ No CAS ]
[ 477739-69-8 ]
[ 598-54-9 ]
[ 150255-96-2 ]
[ 477739-70-1 ]
Yield
Reaction Conditions
Operation in experiment
With pyridine; triethylamine;silica gel; In dichloromethane; ethyl acetate;
Part E. 3-[[1,2-dihydro-1-[2'-(methylsulfonyl)-[1,1'-biphenyl]-4-yl]-2-oxo-3-pyridinyl]amino]-benzonitrile: The compound of Ex. 64, Part D (100 mg, 0.294 mmol), 3-cyanophenylboronic acid (86 mg, 0.588 mmol), pyridine (0.04 mL, 0.647 mmol), triethylamine (0.09 mL, 0.647 mmol), copper acetate (106 mg, 0.588 mmol), and 4A molecular sieves were added together with CH2Cl2 (20 mL) in a round-bottom flask equipped with a drying tube. The mixture was stirred at RT overnight, filtered through Celite, washed with CH2Cl2, concentrated, and chromatographed with 2:3 EtOAc/CH2Cl2 to give 0.14 g of the desired product. 1H NMR (CDCl3): delta 8.27-8.24 (d, J=8.0 Hz, 1H), 7.71-7.51 (m, 6H), 7.43-7.27 (m, 5H), 7.22-7.19 (d, J=7.4 Hz, 1H), 7.06-7.7.03 (d, J=7.0 Hz, 1H), 6.38-6.36 (t, 1H), 2.74 (s, 1H); MS (ES+): 442.4, (M+H)+, 464.2, (M+Na)+.
3-[1-(2'-dimethylaminomethyl-biphenyl-4-yl)-2-oxo-azepan-3-ylamino]-benzamidine[ No CAS ]
3-Amino-1-(4-bromo-phenyl)-azepan-2-one[ No CAS ]
[ 598-54-9 ]
[ 150255-96-2 ]
3-[1-(4-Bromo-phenyl)-2-oxo-azepan-3-ylamino]-benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
44%
With pyridine; triethylamine;silica gel; In dichloromethane;
Part C. 3-[1-(4-Bromo-phenyl)-2-oxo-azepan-3-ylamino]-benzonitrile: A mixture of the compound of Ex. 74, Part B (0.35 mmol), 3-cyanophenylboronic acid (1.5 eq), copper acetate (2 eq), triethyl amine (2 eq), pyridine (2 eq), 4 A molecular sieves was stirred in dichloromethane for 30 min. The mixture was then filtered through a pad of silica gel eluding with ethyl acetate. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography eluding with 0-25% EtOAc/Hex to give the product as clear oil (44%). Mass spectrum, ESI (M+H) 384.2, 386.2.
With sodium hydroxide;palladium diacetate; In methanol; water;
Example 1 7-(3-Cyanophenyl)isoquinolinone (IV) A solution of sodium hydroxide (214 g, 5.35 mol) in water (10 L) was added to a stirred suspension of 3-cyanophenylboronic acid (24% water content, 1.13 kg, 5.8 mol) and 7-bromoisoquinolinone (1.0 kg, 4.46 mol) in methanol (10 L) and the mixture stirred for 1 hour. Palladium acetate (10 g, 44.5 mmol) was added and the mixture was heated at reflux under N2 for 5 hours and then cooled to room temperature and stirred overnight. The light grey solid was collected by filtration. The damp solid was reslurried in water (10 L) and heated to 80 C. for 30 minutes. The mixture was then cooled to room temperature and the solid collected by filtration, washed with water (2 L) then methanol (2 L) and dried in vacuo at 50 C. to give 7-(3-cyanophenyl)isoquinolinone (1.09 kg, 4.43 mol, 99 %) as a light grey solid. Mp >300 C. 1H (TFAD, 300MHz) delta8.78 (1H, s), 8.28 (1H, d), 8.09 (3H, m), 7.84 (1H, d), 7.74 (2H, m), 7.51 (1H, d) ppm.
With sodium carbonate;tetrakis(triphenylphosphine)palladium (0); In 1,2-dimethoxyethane; ethanol; water;
EXAMPLE 295 5-[3-(3,4,8,9-Tetrahydro-6-methoxy-3,3,8,8-tetramethylfuro[2,3-h]isoquinolin-1-yl)phenyl]-2-pyridinamine A solution of sodium carbonate (198 mg, 1.86 mmol) in water (1 mL) and tetrakis(triphenylphosphine)palladium(0) (55 mg, 0.0475 mmol) were added to a solution of <strong>[7169-97-3]N-(5-bromo-2-pyridinyl)acetamide</strong> (243 mg, 1.13 mmol) and 3-cyanophenylboronic acid (249 mg, 1.70 mmol) in 1,2-dimethoxyethane (2 mL) and ethanol (1 mL) and the mixture was stirred at 80 C for 15 hours. Water was poured into the reaction mixture, which was extracted twice with tetrahydrofuran. The combined organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resultant crystals were washed with diethyl ether to obtain N-[5-(3-cyanophenyl)-2-pyridinyl]acetamide (205 mg, yield: 77%). 1H NMR (CDCl3) delta 2.25 (3H, s), 7.54-7.92 (5H, m), 8.02 (1H, br s), 8.32 (1H, d, J = 8.0 Hz), 8.48 (1H, d, J = 2.2 Hz).
9-hydroxy-4-(4-methylsulfanylphenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
[ 622860-48-4 ]
4-(4-fluorophenyl)-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
4-(9-hydroxy-1,3-dioxo-1,2,3,6-tetrahydro-pyrrolo[3,4-c]carbazol-4-yl)-benzonitrile[ No CAS ]
3-(9-Hydroxy-1,3-dioxo-1,2,3,6-tetrahydro-pyrrolo[3,4-c]carbazol-4-yl)-benzonitrile[ No CAS ]
4-(2,5-dichlorophenyl)-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-Hydroxy-4-(3-hydroxymethyl-phenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
4-(2-Acetyl-phenyl)-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-hydroxy-4-(4-methanesulfonylphenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-hydroxy-4-(4-trifluoromethylphenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-hydroxy-4-(3-trifluoromethylphenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
4-Furan-2-yl-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-Hydroxy-4-(3-trifluoromethoxy-phenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-Hydroxy-4-(4-trifluoromethoxy-phenyl)-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
9-hydroxy-4-naphthalen-2-yl-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
4-(1,3-benzodioxol-5-yl)-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
4-(2,4-dimethoxy-pyrimidin-5-yl)-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
4-biphenyl-4-yl-9-hydroxy-6H-pyrrolo[3,4-c]carbazole-1,3-dione[ No CAS ]
N-[3-(9-hydroxy-1,3-dioxo-1,2,3,6-tetrahydro-pyrrolo[3,4-c]carbazol-4-yl)-phenyl]acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; water; at 90℃; for 4h;Combinatorial reaction / High throughput screening (HTS);Product distribution / selectivity;
EXAMPLE 36The Preparation of a series of compounds using multiple parallel synthetic techquires from 9-hydroxy-4-iodopyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione prepared as in Example 7 Combichem Procedure 1; In a 8 ml screw cap vial was added a solution of 9-Hydroxy-4-iodo-6H-pyrrolo[3,4-c]carbazole-1,3-dione, (0.1 mmol) prepared as in example 7 in dioxane (1 ml), a solution of Reagent 1 (see table) (0.1 mmol) in 1:1 dioxane/2.5 M K2CO3 (1 ml) and [1,1'Bis(diphenylphosphino)ferrocene]-dichloropalladium(II) complex with dichloromethane (0.003 g, 0.0037 mmol). The vial was capped and the reaction mixture was shaken for 4 hours at 90 C. After cooling to room temperature, the solution was was removed under vacuum. Purification was carried out via reverse-phase HPLC (3% n-propanol in acetonitrile and 3% n-propanol in water as the eluent; C-18 column). The products were characterised by mass spectral analysis (See Table 1).
N-[4-(3-cyanophenyl)-6-phenylpyridin-2-yl]-4-(3,4-diethoxyphenyl)-4-oxobutanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
tris-(dibenzylideneacetone)dipalladium(0);
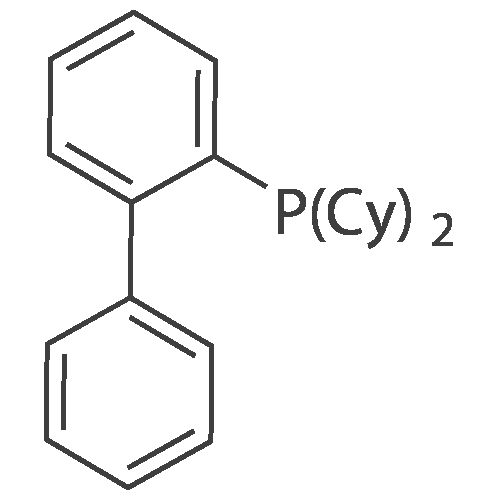
Example 132 N-[4-(3-cyanophenyl)-6-phenylpyridin-2-yl]-4-(3,4-diethoxyphenyl)-4-oxobutanamide Using N-(4-chloro-6-phenylpyridin-2-yl)-4-(3,4-diethoxyphenyl)-4-oxobutanamide obtained in Example 43-(3) (22.6 mg), tris(dibenzylideneacetone)dipalladium (1.1 mg), biphenyl-2-yl(dicyclohexyl)phosphine (1.8 mg), 1N aqueous potassium carbonate solution (0.1 mL) and (3-cyanophenyl)boronic acid (18.4 mg) as starting materials and in the same manner as in Example 119, N-[4-(3-cyanophenyl)-6-phenylpyridin-2-yl]-4-(3,4-diethoxyphenyl)-4-oxobutanamide (15.4 mg) was obtained. HPLC (220 nm) purity 90% (retention time 2.11 min) MS (ESI+, m/e) 520 (M+H)
N-[4-(3-cyanophenyl)-6-phenylpyridin-2-yl]-4-(2,3-dihydro-1-benzofuran-5-yl)-4-oxobutanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
tris-(dibenzylideneacetone)dipalladium(0);
Example 163 N-[4-(3-cyanophenyl)-6-phenylpyridin-2-yl]-4-(2,3-dihydro-1-benzofuran-5-yl)-4-oxobutanamide Using N-(4-chloro-6-phenylpyridin-2-yl)-4-(2,3-dihydro-1-benzofuran-5-yl)-4-oxobutanamide obtained in Example 114-(1) (20.3 mg), tris(dibenzylideneacetone)dipalladium (1.1 mg), biphenyl-2-yl(dicyclohexyl)phosphine (1.8 mg), 1N aqueous potassium carbonate solution (0.1 mL) and (3-cyanophenyl)boronic acid (18.4 mg) as starting materials and in the same manner as in Example 119, N-[4-(3-cyanophenyl)-6-phenylpyridin-2-yl]-4-(2,3-dihydro-1-benzofuran-5-yl)-4-oxobutanamide (10.5 mg) was obtained. HPLC (220 nm) purity 98% (retention time 2.04 min) MS (ESI+, m/e) 474 (M+H)
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 100℃; for 24.0h;
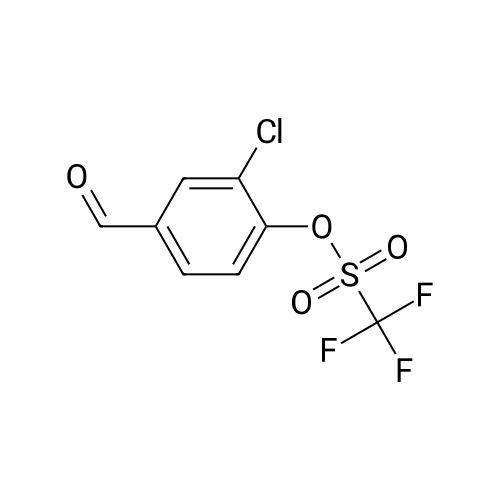
Preparation of Intermediate ?'-Chloro-A'-formyl-biphenyl-S-carbonitrile (l-6b):Trifluoro-methanesulfonic acid 2-chloro-4-formyl-phenyl ester (N6a: 10.0 g), 3-cyano-phenyl- boronic acid (5.26 g) and palladium tetrakis(triphenylphosphine) (3.00 g) were combined in 100 ml. of dimethoxyethane, then treated with 10 ml_ of a 2M aqueous sodium carbonate solution before being heated in a 100C oil bath. After 24 hours, the reaction mixture was cooled to ambient temperature, the organic phase was separated, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by chromatography on silica gel eluting with 10% ethyl acetate in heptane to afford the title compound (l-6b).1H NMR (400 MHz, Chloroform-c/) delta ppm 7.49 (d, J=7.89 Hz, 1 H), 7.59 (t, J=7.99 Hz, 1 H), 7.68 - 7.76 (m, 3 H), 7.86 (dd, J=7.79, 1.56 Hz, 1 H), 8.01 (d, J=1.45 Hz, 1 H), 10.03 (s, 1 H).
With caesium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; water; at 100℃; for 3h;
EXAMPLE 16; (2R,3R,4S,5R,6E)-N-[(3R)-9-(3-cyanophenyl)-4-oxo-2,3,4,5-tetrahydro-1,5-benzothiazepin-3-yl]-3,4,5-trihydroxy-2-methoxy-8,8-dimethylnon-6-enamide; Step 1: Preparation of 2'-fluoro-3'-nitrobiphenyl-3-carbonitrile (42); 334 mg of 3-cyanophenylboronic acid (2.273 mmol), 46.41 mg of 1,1'-bis(diphenylphosphino)ferrocenepalladium chloride (C35H30Cl4FeP2Pd, MW 816.65, 0.057 mmol) and 2.96 g of caesium carbonate (9.09 mmol) are placed in a 50 mL round-bottomed flask, with stirring and under an argon atmosphere, containing 10 mL of water, 3 mL of dioxane and 500 mg of 17 (2.273 mmol). The medium is heated at 100 C. with stirring for 3 hours. 20 mL of EtOAc are then added and the mixture is washed with twice 20 mL of water. The organic phase is dried over MgSO4, filtered and evaporated to dryness. The crude product is chromatographed on a silica cartridge (30 g) eluting with heptane/EtOAc (as an EtOAc gradient: 10% to 50%). 230 mg of expected product 42 are collected (white solid).1H NMR (500 MHz, DMSO-d6), delta (ppm): 7.59 (t, J=8.0 Hz, 1H); 7.77 (t, J=8.0 Hz, 1H); from 7.93 to 8.04 (m, 3H); 8.12 (broad s, 1H); 8.22 (dt, J=2.0 and 8.0 Hz, 1H).
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; for 6h;Reflux;
3'-Aminomethyl-biphenyl-3-carboxylic acid ethyl ester: To a stirred solution of 3-bromo-ethyl benzoate (500 mg, 2.18 mmol) in DME (20 mL) was added 3-cyano-phenyl-boronic acid (320 mg, 2.18 mmol), 2 N K2CO3 (1.9 mL) followed by tetrakistriphenylphosphine palladium (125 mg) and heated at reflux for 6 hrs. After cooling to room temperature, the reaction mixture was diluted with EtOAc (25 mL) and washed with H2O (10 mL×2). The organic layer was dried over Na2SO4, concentrated, and the residue purified by silica gel column chromatography with EtOAc and hexane as eluents to give 3'-cyano-biphenyl-3-carboxylic acid ethyl ester (383 mg, 70% yield).
With caesium carbonate;bis-triphenylphosphine-palladium(II) chloride; In 1,4-dioxane; for 0.5h;Reflux; Inert atmosphere;
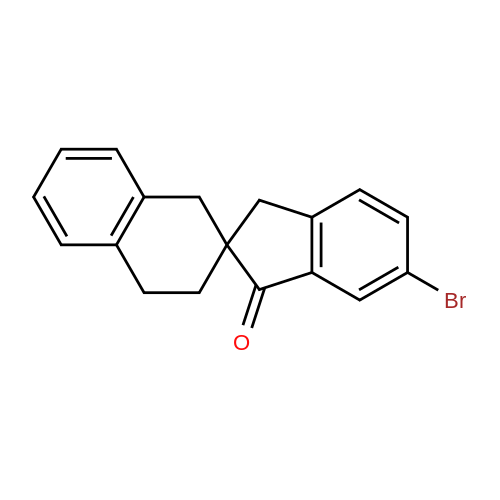
6-Bromo-3',4'-dihydro-r//-spiro[indene-2,2'-naphthalen]-l(3//)-one (163 mg, 0.5 mmol), 3-cyanophenylboronic acid (147 g, 1 mmol) in [l,4]-dioxane (12 niL), Cs2CO3 (2 N, 3.2 mL), then Pd(PPrIs)2Cl2 (5 mg, 0.01 mmol) was added under Ar2, the mixture was refluxed for 30 minutes. The reaction mixture was concentrated in vacuo to give the residue, which was purified by TLC to give 3-(l-oxo-l,3,3',4'-tetrahydro- lEta-spiro[indene-2,2'- naphthalene] -6-yl)benzonitrile (35 mg, 6%).
With sodium carbonate;bis-triphenylphosphine-palladium(II) chloride; In ethanol; water; toluene; at 80℃; for 18h;Inert atmosphere;
5.1 A solution of 10.6 g (100 mmol) of sodium carbonate in 50 ml of water is added to a solution of 7.34 g (50.0 mol) of 3-cyanobenzene-boronic acid and 12.0 g (50.0 mmol) of <strong>[135034-10-5]3-chloro-6-iodopyridazine</strong> in 100 ml of ethanol and 50 ml of toluene, and the mixture is heated to 80 C. under nitrogen. 351 mg (0.50 mmol) of bis(triphenylphosphine)palladium(II) chloride are then added. The reaction mixture is stirred at 80 C. for 18 hours. The mixture is allowed to cool to room temperature, the resultant precipitate is filtered off with suction and washed with water. The residue is recrystallised from 2-propanol : 3-(6-chloropyridazin-3-yl)benzonitrile as brown crystals; ESI 216.
With sodium carbonate;bis-triphenylphosphine-palladium(II) chloride; In water; toluene; at 80℃; for 18h;Inert atmosphere;
A solution of 212 mg (2.0 mmol) of sodium carbonate in 1 ml of water is added to a solution, kept under nitrogen, of 240 mg (1.00 mmol) of <strong>[135034-10-5]3-chloro-6-iodopyridazine</strong> in 1 ml of toluene, and the mixture is heated to 80 C. 7.0 mg (0.010 mmol) of bis(triphenylphosphine)palladium(II) chloride are added, and a solution of 147 mg (1.00 mmol) of 3-cyanobenzene-boronic acid is subsequently added dropwise. The reaction mixture is stirred at 80 C. for 18 hours. The reaction mixture is cooled to room temperature, water is added, and the solid is filtered off with suction and washed with water. The residue is dried in vacuo: 3-(6-chloropyridazin-3-yl)benzonitrile as colourless crystals; ESI 216.
With sodium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,2-dimethoxyethane; water; at 120℃; for 2h;
To a solution of l-bromo-4-(trifluoromethoxy)benzene (0.50 g, 2.07 mmol) in DME (6 mL) were added 3-cyanophenyl boronic acid (0.37 g, 2.49 mmol), 1,1 '- bis(diphenylphosphino)ferrocene)-dichloropalladium(II) (0.046 g, 0.062 mmol) and Na2C03 (2 mL of aqueous solution, 0.44 g, 4.14 mmol). The resulting mixture was stirred at 120 C for 2h. After removal of organic solvent, the resulting residue was dilited with water (10 mL) and extracted with methylene chloride (10 mL x 2). The organic layer was dried over MgS04 and concentrated in vacuo. The resulting crude residue was purified by flash column chromatography (n-hexane : ethyl acetate = 10 : 1 ratio) to give a Jl.
With sodium sulfate; In tetrahydrofuran; at 25℃; for 12h;Inert atmosphere;
To a solution of 3-cyanophenyl boronic acid (2.5 g, CAS: 150255-96-2) in anhydrous THF (150 ml) under an atmosphere of nitrogen were added pinacol (2.95 g) and Na2SO4 (10 g) at 25C.The reaction mixture was stirred for 12 h at 25C under an atmosphere of nitrogen. Then, Na2SO4was filtered off and all volatiles were evaporated. The residue was partitioned between ethylacetate and water. The organic layer was then separated, dried over Na2SO4 and concentrated to afford the title compound (4.72 g) as off white solid which was directly used in the next reaction step without further characterization.
With palladium diacetate; sodium carbonate; In water; N,N-dimethyl-formamide; at 110℃; for 48h;
<strong>[58313-23-8]Ethyl 3-iodobenzoate</strong> (939mg, 3.40mmol) was dissolved in DMF (2mL) and a solution of 3-cyanophenylboronic acid (500mg, 3.40mmol) in DMF (5mL) was added. A solution of Pd(OAc)2 (15mg, 0.068mmol) and Na2C03 (858mg, 10.2mmol) in water (2mL) was added and the reaction mixture was heated at 110C for 2d. The reaction mixture was concentrated in vacuo and partitioned between DCM (150mL) and water (50mL). The aqueous fraction was extracted with DCM (50mL) and the combined organic fractions were dried (MgSC^) and concentrated in vacuo. The residue was purified by column chromatography to give the title compound (767mg, 90%) as a pale yellow liquid. LCMS: ES+ 252.1 [MH]+.
90%
With palladium diacetate; sodium carbonate; In water; N,N-dimethyl-formamide; at 110℃; for 48h;
<strong>[58313-23-8]Ethyl 3-iodobenzoate</strong> (939mg, 3.4Ommol) was dissolved in DIVIF (2mL) and a solution of 3-cyanophenylboronic acid (500mg, 3.4Ommol) in DIVIF (5mL) was added. A solutionof Pd(OAc)2 (15mg, 0.O68mmol) and Na2CO3 (858mg, 10.2mmol) in water (2mL) wasadded and the reaction mixture was heated at 110C for 2d. The reaction mixture was concentrated in vacuo and partitioned between DCM (1 5OmL) and water (5OmL). The aqueous fraction was extracted with DCM (5OmL) and the combined organic fractions were dried (MgSO4) and concentrated in vacuo. The residue was purified by columnchromatography to give the title compound (767mg, 90%) as a pale yellow liquid.LCMS: ES 252.1 [IVIHf.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; for 5.0h;Reflux; Inert atmosphere;
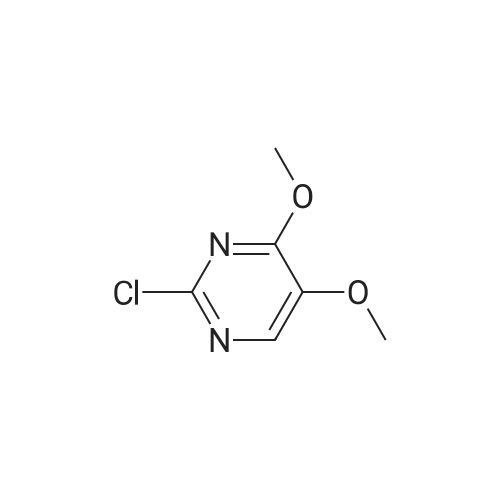
a. Preparation of Compound 3-(4,5-Dimethoxypyrimidin-2-yl)benzonitrile 2-Chloro-4,5-dimethoxypyrimidine (100 mg, 0.57 mmol), (3-cyanophenyl)boronic acid (126 mg, 0.86 mmol), Pd(PPh3)4 (66 mg, 0.06 mmol) and Na2C03(182 mg, 1.72 mmol) were dissolved in a mixture of dioxane (9 mL) and water (3 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 5 hours. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with sat. NH4CI followed by brine. The organic layer was dried over Na2S04 and concentrated in vacuo and the resulting residue was flash chromatographed on silica gel eluting with 0 to 20% EtOAc/Hexane. This afforded 3-(4,5-dimethoxypyrimidin-2-yl)benzonitrile as a white solid (138 mg, 100%); m.p.134-136 C; H NMR (400 MHz, CDC13) delta 8.61 (s, 1H), 8.54 (d, J= 8 Hz, 1H), 8.09 (s, 1H), 7.65 (d, J= 8 Hz, 1H), 7.51 (t, J= 8 Hz, 1H), 4.13 (s, 3H), 3.95 (s, 3H); 13C NMR (100 MHz, CDC13) 6 159.8, 153.6, 141.7, 138.5, 137.0, 132.8, 131.6, 131.3, 129.2, 1 18.9, 1 12.6, 56.4, 54.2
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 100℃; for 16h;
a. Preparation of Compound A mixture of 5-bromo-2,3-dimethoxypyridine (217 mg, 1.0 mmol), 3- cyanophenylboronic acid (220 mg, 1.5 mmol), Pd(PPh3)4 (173 mg, 0.15 mmol) and K2C03 (276 mg, 2.0 mmol) in 1 ,4- dioxane (5.0 ml) and H20 (1.5 ml) was degassed for 30 min. This mixture was heated to 100 C and stirred for 16 h. The reaction mixture was cooled to room temperature and partitioned between NaHC03 and EtOAc (3x), and washed with NaCl (lx). The organic phase was dried over Na2S04 and was concentrated. The resulting residue was purified by ISCO flash chromatography using 20% EtOAc in hexane to give 190 mg (79% yield) desired product as white solid. 'H NMR (300 MHz, CDC13) delta: 7.91 (d, J - 2.1 Hz, 1H), 7.79- 7.73 (m, 2H), 7.61-7.53 (m, 2H), 7.17 (d, J = 2.1 Hz, 1H), 4.05 (s, 3H), 3.94 (s, 3H).
With dihydrogen peroxide; In ethanol; water; at 20℃;
General procedure: In a 50 mL round-bottomed flask, a mixture of arylboronic acid(1 mmol), H2O2 (30% aq, 0.2 mL), ZnO nanocatalyst (5 mol%; sampleZnO-1) and 2 mL of water were stirred at room temperature under aerobic condition. The progress of the reaction was monitored by thin layer chromatography (TLC). After completion of the reaction, the reaction mixture was diluted with 20 mL of water and extracted with (3×20) mL of diethyl ether. The combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed in a rotary evaporator under reduced pressure. The crude product was purified by column chromatography (hexane/ ethylacetate, 9:1) on silica (100-200mesh) to get the desired product. The products were identified by 1HNMR and 13C NMR.
91%
With dihydrogen peroxide; In water; at 20℃; for 0.0833333h;
General procedure: A mixture of arylboronic acid (0.5 mmol), 10 mg cellulose (15 wt%) and 2 mL distilled H2O were taken in an oven dried 10 mL round bottomed flask. To this 30% aq H2O2 (0.5 mL) was added dropwise and stirred at room temperature for 5 min. After completion of the reaction (monitored by TLC), aqueous layer was centrifuged to recover the catalyst for further use. The products were extracted with EtOAc (3x10 mL), dried with anhydrous Na2SO4 and then vacuum dried. The crude product was purified by column chromatography on silica gel (EtOAc/ hexane) to obtain the desired product.
89%
With air; Ru(at)imine-nanoSiO2 catalyst; In water; isopropyl alcohol; at 30℃; for 0.8h;Green chemistry;
General procedure: A 50mL round bottom flask was charged with arylboronic acid (1mmol), 5mg Ru catalyst and 4ml iPrOH:H2O (1:1). The reaction mixture was stirred at room temperature under aerobic condition. The progress of the reaction was monitored by TLC. After the completion of the reaction, the mixture was diluted with 20mL of water and extracted with diethyl ether. The combined organic layers were washed with brine and dried over by anhydrous Na2SO4 and evaporated in a rotary evaporator under reduced pressure. The crude was purified by column chromatography on silica gel (hexane:ethyl acetate, 9:1) to afford the desired product. The purity of the compound was confirmed by 1H NMR, 13C NMR.
88%
With dihydrogen peroxide; In water; at 20℃; for 0.166667h;Green chemistry;
General procedure: In a 50mL round-bottomed flask, a mixture of arylboronic acid (1mmol), H2O2 (30% aq, 0.2mL), bio-silica (5mg) and 2mL of water was added and stirred at room temperature in aerobic condition. The reaction was monitored by TLC. After completion of the reaction the reaction mixture was diluted with 20mL of water and extracted with (3×20) mL of diethylether and the combined organic layer was washed with brine and dried over by Na2SO4 and evaporated in a rotary evaporator under reduced pressure. The crude was purified by column chromatography (hexane/ethylacetate, 9:1) on mesh silica (100-200) to get the desired product. The products were confirmed by 1H NMR, 13C NMR, FT-IR spectroscopy and mass spectrometry.
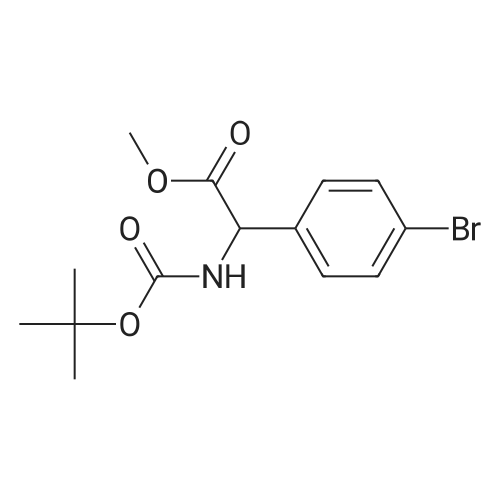
methyl 2-((tert-butoxycarbonyl)amino)-2-(3'-cyano-[1,1'-biphenyl]-4-yl)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate; In tetrahydrofuran; water; at 100.0℃; for 2.0h;Microwave irradiation; Inert atmosphere; Sealed tube;
General procedure: Aryl bromide (0.20g) and the appropriate boronic acid or boronate ester (1.5 eq) were dispersed in degassed THF (3mL) and degassed 1M Na2CO3(aq) (1mL) in a 10mL microwave vessel. A steady stream of nitrogen was bubbled through the mixture for 5min, before adding PdCl2(PPh3)2 (41mg, 0.1 eq), then immediately sealing the tube. The mixture was heated at 100C in an aluminium heating block for 2h, at which point LC-MS analysis indicated the reaction was complete. After cooling, the mixture was diluted with EtOAc (10mL) and water (10mL), and the aqueous layer discarded. The organic layer was filtered through a plug of cotton wool, before concentration and further purification by FCC (eluent DCM or MeOH/DCM 0:100 to 5:95 for more polar compounds).
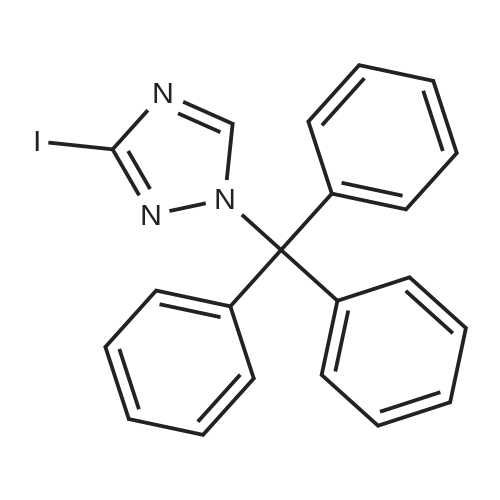
3-(1-trityl-1H-1,2,4-triazol-3-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
227 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 100℃; for 4h;
3-iodo-1-trityl -1H-1,2,4- triazole (300 mg),3-cyanophenyl boronic acid (101 mg), tetrakis triphenylphosphine palladium (79 mg), ethanol 2M aqueous sodium carbonate (1.5 mL) (1.0 mL) - toluene (1.5 mL) mixed solution of the mixture was stirred for 4 hours at 100 C. After cooling to room temperature, and separated the two layers.The aqueous layer was extracted with ethyl acetate, it was concentrated under reduced pressure and the organic layers were washed with. The residue was purified by column chromatography (silica gel cartridge, hexane: ethyl acetate = 88: 12-50: 50) to give the title compound (227 mg) as a colorless solid.
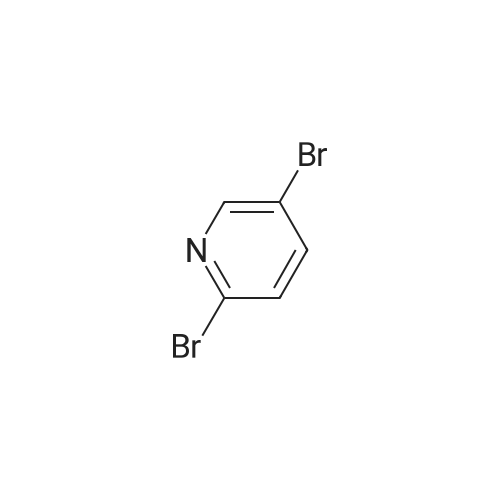
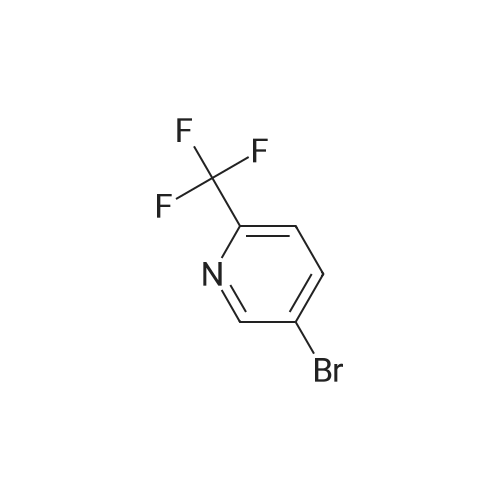
3-[6-(trifluoromethyl)-3-pyridyl]benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); sodium carbonate; In 1,4-dioxane; water; at 100℃; for 0.5h;Microwave irradiation;
To 242 3-cyanophenylboronic acid (500 mg, 3.4 mmol), 243 <strong>[436799-32-5]5-bromo-2-(trifluoromethyl)pyridine</strong> (850mg, 3.7 mmol) and 244 1,1?-bis(diphenylphosphino)ferrocenedichloropalladium(II) (250 mg, 0.34 mmol)were added 115 1,4-dioxane (5 mL) and 1 mol/L aqueous sodium carbonate solution (5 mL), and themixture was stirred with heating using a microwave reactor at 100 C. for 30 min. To the reaction mixturewas added 52 water, and the mixture was extracted with ethyl acetate. The organic layer was dried oversodium sulfate. The desiccant was filtered off, and the solvent was evaporated. The obtained residue waspurified by silica gel column chromatography (hexane/ethyl acetate) to give the 245 title compound (643 mg , 2.6 mmol, 76%). MS (ESI) m/z 249 (M+H)+
methyl 3-(3-cyanophenyl)-1H-indole-6-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With potassium fluoride; tris-(dibenzylideneacetone)dipalladium(0); tri tert-butylphosphoniumtetrafluoroborate; In tetrahydrofuran; at 40℃;Inert atmosphere;
Under N2 atmosphere, <strong>[860457-92-7]methyl 3-bromo-1H-indole-6-carboxylate</strong> (1.12 g, 4.43 mmol), (3-cyanophenyl)boronic acid (1.30 g, 8.84 mmol), tris(dibenzylideneacetone) dipalladium (0) (608 mg, 0.66 mmol), tri-tert- butylphosphonium tetrafluoroborate (385 mg, 1.32 mmol), anhydrous KF (769 mg, 13.30 mmol), and anhydrous THF (20 mL) were mixed and stirred at 40 oC overnight. The mixture was cooled down to rt, filtered through celite and washed by ethyl acetate (100 mL). The filtrate was concentrated in vacuo, and the residue was purified by flash chromatography on silica gel with hexanes and ethyl acetate (0-60 %) to give product (1.05 g, 86%).1H NMR (600 MHz, CD3OD) delta 8.20 (S, 1H), 8.02 (brs, 2H), 7.93 (d, J = 12.0 Hz, 1H), 7.84 (m, 2H), 7.62 (m, 2H), 3.95 (s, 3H).
methyl 5-(3-cyanophenyl)-6-methoxynicotinate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
308 mg
With palladium diacetate; triethylamine; triphenylphosphine; In N,N-dimethyl-formamide; at 100℃;
Methyl 5-bromo-6-methoxynicotinate (500 mg),(3-cyanophenyl) boronic acid (400 mg),Palladium (II) acetate (10 mg),Triphenylphosphine (35 mg),Triethylamine (0.9 ml) and DMF (6 ml)Was stirred at 100 C. overnight.The mixture was cooled to room temperature and extracted with ethyl acetate and water. The obtained organic layer was washed with saturated brine, dried over magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (308 mg).
methyl 2-(3-cyanophenyl)-1-methyl-1H-imidazole-5-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
239 mg
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In ethanol; toluene; at 80℃;
Methyl 2-bromo-1-methyl-1 H-imidazole-5-carboxylate (659 mg) (3-cyanophenyl) boronic acid (600 mg),(1,1'-diphenylphosphinoferrocene) dichloropalladium (200 mg),2 M aqueous potassium carbonate solution (3 ml),A mixture of toluene (10 ml) and ethanol (3 ml) was stirred at 80 C. overnight.The mixture was cooled to room temperature and extracted with ethyl acetate and water. The obtained organic layer was washed with saturated brine, dried over magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give the title compound (239 mg)
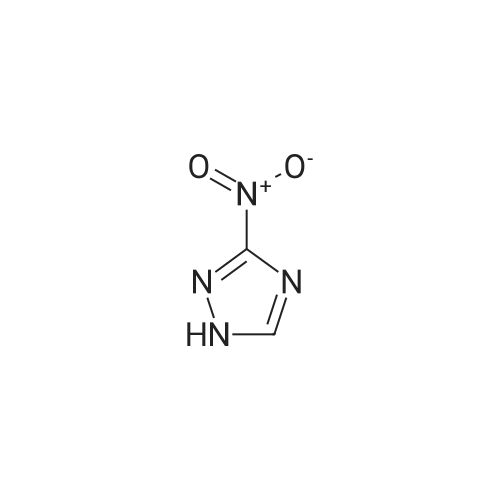
3-(3-nitro-1H-1,2,4-triazol-1-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With pyridine; copper diacetate; In dichloromethane; at 20℃; for 16h;Molecular sieve;
To a solution of 3-nitro-4H-1 ,2,4-triazole (CAS Number 24807-55-4; 1 .00 g, 8.77 mmol) and (3-cyanophenyl)boronic acid (2.57 g, 17.54 mmol) in DCM (25 ml ) was added copper (II) acetate (2.62 g, 13.1 mmol), pyridine (1.43 ml, 17.5 mmol) and molecular sieves (0.60 g) and stirred at rt for 16 h. The resulting reaction mixture was poured into water (250 ml) and extracted into DCM (2 x 200 ml). Combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude residue, which was purified by column chromatography (compound eluted at 30% EtOAc in n-hexane) to yield 3-(3-nitro- 1 H-1 ,2,4-triazol-1 -yl)benzonitrile (0.60 g, 2.79 mmol). The crude product was taken directly onto the next step.
With pyridine; copper diacetate; In dichloromethane; at 40℃; for 18h;Molecular sieve;
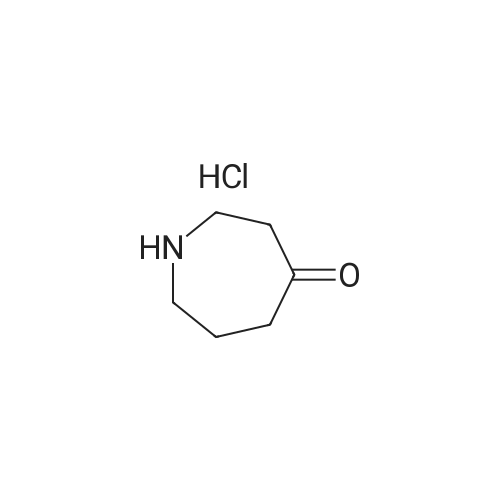
Azepan-4-one hydrochloride (125 mg, 0.84 mmol), (3-cyanophenyl)boronic acid (24 mg, 1.67 mmol), copper(II) acetate (152 mg, 0.84 mmol), pyridine (202 mul, 2.51 mmol) and 4 molecular sieves (500 mg) were suspended in anhydrous DCM (10 ml). The reaction mixture was stirred at 40 C. for 18 h. The reaction mixture was cooled to rt and filtered through Celite. The filtrate was concentrated in vacuo to give a brown oil. The oil was purified by flash column chromatography eluting with gradient from 0-100% EtOAc in heptane followed by 0-100% MeOH in EtOAc. The product containing fractions were combined and reduced in vacuo to afford the title compound as a white solid (54 mg, 30%). 1H NMR (250 MHz, Chloroform-d) delta 7.32-7.23 (m, 1H), 6.99-6.92 (m, 1H), 6.91-6.83 (m, 2H), 3.83-3.71 (m, 2H), 3.70-3.56 (m, 2H), 2.78-2.62 (m, 4H), 1.95-1.79 (m, 2H). LCMS Method 4-Tr=1.58 min (ES+) (M+H)+ 215.2.
With potassium carbonate; In acetonitrile; at 80℃; for 20h;Inert atmosphere;
The dichloropyridine (652 mg, 4.0 mmol, 1.0 equiv) and the boronic acid (588 mg, 4.0 mmol, 1.0 equiv) were dissolved in CH3CN (16 mL, 0.25 M) in a 40 mL vial equipped with a magnetic stir bar. An aqueous solution of K2C03 (2 M, 2 mL, 4.0 mmol, 1.0 equiv) wasadded and the resulting solution was degassed by bubbling N2 for 10 minutes. Pd(PPh3)4 (231 mg, 0.2 mmol, 5 mol%) was then added and the mixture was heated to 80 C for 20 hours under a nitrogen atmosphere. Upon completion, the reaction mixture was diluted with EtOAc (20 mL), filtered over Celite, and concentrated in vacuo. The crude residue was purified by flash column chromatography over silica (hexanes/EtOAc gradient 0% to 100%) to afford the product as awhite solid (211 mg, 23% yield).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In water; acetonitrile; at 80℃; for 12h;Inert atmosphere;
The dichloropyridine (656 mg, 4.0 mmol, 1.0 equiv) and the boronic acid (588 mg, 4.0 mmol, 1.0 equiv) were dissolved in CH3CN (16 mL, 0.25 M) in a 40 mL vial equipped with a magnetic stir bar. An aqueous solution of K2C03 (2 M, 2 mL, 4.0 mmol, 1.0 equiv) wasadded and the resulting solution was degassed by bubbling N2 for 10 minutes. Pd(PPh3)4 (231 mg, 0.2 mmol, 5 mol%) was then added and the mixture was heated to 80 C for 12 hours under a nitrogen atmosphere. Upon completion, the reaction mixture was diluted with EtOAc (20 mL), filtered over Celite, and concentrated in vacuo. The crude residue was purified by flash column chromatography over silica (hexanes/EtOAc gradient 0% to 100%) to afford the product as awhite solid (360.4 mg, 39% yield).
With oxygen; copper diacetate; triethylamine; In N,N-dimethyl-formamide; at 65℃; for 4h;
General procedure: To a solution of <strong>[461-89-2]6-azauracil</strong> (100 mg, 0.88 mmol) inDMF (10.0 mL) was added base (1.76 mmol) and Cu(OAc) 2(159 mg, 0.88 mmol) at room temperature. The resulting reationmixture was degassed with oxygen for 10 min and then addedarylboronic acids (0.96 mmol) at room temperature and stirredat appropriate temperature (Table-1) under oxygen atmosphere.The reaction mixture was diluted with water (15 mL) andextracted with dichloromethane (3 × 15 mL). The organic layerwashed with H 2 O (15 mL), brine solution (15 mL), dried overNa 2 SO 4 and concentrated. The obtained crude product waspurified by column chromatography (0 to 10 % CH 3 OH/CH 2 Cl 2 )to afford the title compounds.
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; potassium carbonate; In 1,2-dimethoxyethane; water; at 80℃; for 1h;Sealed tube;
To a pressure tube was added 3-cyanophenyl boronic acid (1.9 g, 13 mmol), 4- bromo-3-methylpyridine (2.5g, 12 mmol), potassium carbonate (5.0 g, 36 mmol), dichloro[l,l'-bis(diphenylphosphino)ferrocene]palladium(II) DCM adduct (490 mg, 0.60 mmol), ethylene glycol dimethyl ether (45 mL), and water (15 mL). After being degassed with nitrogen for approximately 2 minutes, the tube was sealed with a septum and heated to 80 C for 1 hour. The mixture was cooled to room temperature, poured into water, and extracted three times with EtOAc. The combined extracts were washed with brine, dried with sodium sulfate, filtered, and evaporated to dryness. Purification by silica gel chromatography (0 - 75% EtOAc/hexanes) yielded Intermediate 4B (1.7 g, 73%). LCMS (method A): m/z 195.2 (M+H) H NMR (CDC13) delta 8.56 (s, 1H), 8.52 (d, 1H), 7.73 (m, 1H), 7.65 - 7.56 (m, 3H), 7.13 (d, 1H), 2.27 (s, 3H).
With pyridine; copper diacetate; In tetrahydrofuran; dichloromethane; at 15℃; for 12h;Molecular sieve;
To a solution of compound 1A (54.56 mg, 371.34 umol, 1 eq) and compound 1 (100 mg, 371.34 umol, 1 eq) in DCM (5 mL) and THF (1 mL) was added Cu(OAc)2 (67.45 mg, 371.34 umol, 1 eq)Py (88.12 mg, 1.11 mmol, 89.92 uL, 3 eq) and 4A MS (100 mg, 371.34 umol, 1 eq). The mixture was stirred at 15C for 12 hr under oxygen atmosphere. LCMS showed the desired compound was detected. The reaction mixture was filtered. The filtrate was concentrated under reduced pressure to give a residue. The residue was purifiedby prep-TLC (SiO2, Petroleum ether : Ethyl acetate = 1:1). Compound 2 (80 mg, 215.98 umol, 58.16% yield) was obtained as a white solid. LCMS: RT = 1.404 min, MS cal.: 370.1, [M-H] - = 369.3.
3-(1-methyl-1H-imidazol-2-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium carbonate; In water; isopropyl alcohol; at 130℃; for 18h;Inert atmosphere; Sealed tube;
A mixture of (3-cyanophenyl)boronic acid (16 g, 109 mmol), <strong>[16681-59-7]2-bromo-1-methyl-1H-imidazole</strong> (intermediate 1, 15.8 g, 98 mmol) and sodium carbonate (46.2 g, 436 mmol) in IPA (120 mL) and water (120 mL) was stirred and degassed with nitrogen for 20 min, and then PdCl2(dppf)- DCM adduct (4.45 g, 5.44 mmol) was added and stirred at 130 C for 18 h in a sealed tube. After cooling, the reaction mixture was diluted with ethyl acetate (300 mL), washed with water (300 mL), dried over anhydrous sodium sulphate, filtered and evaporated under reduced pressure. The crude compound was purified by silica gel chromatography eluting with 70% ethyl acetate in pet. ether to afford the title compound. LCMS (method C): rt = 0.57, [M+H]+= 184.
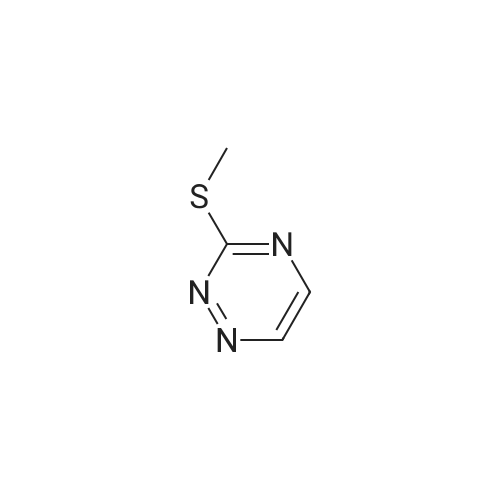
3-(6-amino-2-(methylthio)pyrimidin-4-yl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
With bis(di-tert-?butyl(4-?dimethylaminophenyl)?phosphine)?dichloropalladium(II); caesium carbonate; In water; toluene; at 115℃; for 16h;
Bis(di-/er/-butyl(4-dimethylamiiiophenyl)phosphine)dichloropalladium(II) (1.0 g, 5 mol%) ws added to a mixture of 6-chloro-2-(methylthio)pyrimidin-4-amine (Combi-Blocks, cat ST-1384) (5.0 g, 29 mmol), (3-cyanophenyl)boronic acid (8.4 g, 57 mmol) and cesium carbonate (37 g, 114 mmol) in toluene (100 mL) and water (10 mL) The mixture was purged with nitrogen, and then stirred at 115 C for 16 h. The reaction mixture was cooled to room temperature, filtered through a Celite plug with DCM and concentrated under reduced pressure. Water (200 mL) was added to the residue and the resulting solid was collected by filtration, and then dried to give the desired product (6.5 g, 94 %), which was used in the next step without further purification. LC-MS calculated for CnHi N+S (M+H)+: m/z = 243.1; found 243.2.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; for 12h;Reflux;
General procedure: 10.0 grams (g) (27.6 mmol, mmol) of 1,3-dibromo-5-iodobenzene,4.06 g (27.6 mmol) of (2-cyanophenyl) boronic acid, 1.60 g (1.38 mmol)Tetrakis (triphenylphosphine) palladium (0) (Pd (PPh3) 4), and 9.55g (69.1mmol)Potassium carbonate was added to a solution containing 60 ml (mL) of tetrahydrofuran (THF) and 30 mLMixed solvent with water, and the reaction mixture was stirred under reflux for 12 hours.After the reaction is completed, the reaction product is cooled to room temperature,And an aqueous solution layer was removed therefrom by extraction.Filtering the resultant obtained through silica gel under reduced pressure,And the filtrate was concentrated under reduced pressure.The product obtained therefrom was separated by silica gel column chromatography to provide an intermediate (A)(4.38 g, 47% yield).
41%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; for 12h;Reflux;
General procedure: 10.0 grams (g) (27.6 millimoles, mmol) of 1,3-dibromo-5-iodobenzene, 4.06 g (27.6 mmol) of (2-cyanophenyl)boronic acid, 1.60 g (1.38 mmol) of tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4), and 9.55 g (69.1 mmol) of potassium carbonate were added to a mixed solution containing 60 milliliters (mL) of tetrahydrofuran (THF) and 30 mL of water, and the reaction mixture was stirred for 12 hours under reflux. After the reaction was completed, the reaction product was cooled to room temperature, and the aqueous solution layer was removed therefrom through extraction. The resultant obtained therefrom was filtered through a silica gel under reduced pressure, and the filtrate was concentrated under reduced pressure. The product obtained therefrom was separated by silica gel column chromatography to provide Intermediate (A) (4.38 g, yield of 47%).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 120℃; for 12h;
Add 100mmol of <strong>[56962-04-0]3-bromo-5-chlorophenol</strong>, 100mmol of 3-cyanophenylboronic acid, 41.4g of potassium carbonate (300mmol), 800ml of THF and 200ml of water into the reaction flask, and add 1mol% of Pd(PPh3)4 . React at 120C for 12h. After the completion of the reaction, the reaction was stopped, the reactant was cooled to room temperature, water was added, and the organic phase was concentrated to obtain a white solid, which was filtered and washed with water. The obtained solid was recrystallized and purified with toluene to obtain a white powder M1. Among them, the amount of Pd(PPh3)4 added is 1 mol% of <strong>[56962-04-0]3-bromo-5-chlorophenol</strong>.
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; anhydrous sodium carbonate; In water monomer; N,N-dimethyl-formamide; at 100℃; for 2h;Inert atmosphere;
General procedure: Under N2, to a solution of compound 2 (1.7 g, 10.0 mmol) in a mixture of DMF(30 mL) and H2O (5 mL) were added the appropriate compound 3 (14.0 mmol), sodiumcarbonate (2.1 g, 20.0 mmol) and Pd(dppf)Cl2 (366 mg, 0.50 mmol). The mixture wasstirred at 100 C for 2 h. The mixture was diluted with water (60 mL) and extractedwith ethyl acetate (3100 mL). The organic layer was washed with brine, dried andconcentrated under reduced pressure. The residue was purified by flash chromatography(CH2Cl2:MeOH, 20:1 - 10:1) to provide the target compound.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






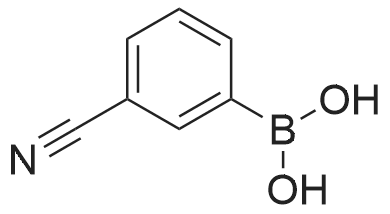

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping