| 80.95% |
With sodium carbonate; In acetone; at 30 - 40℃; for 3h; |
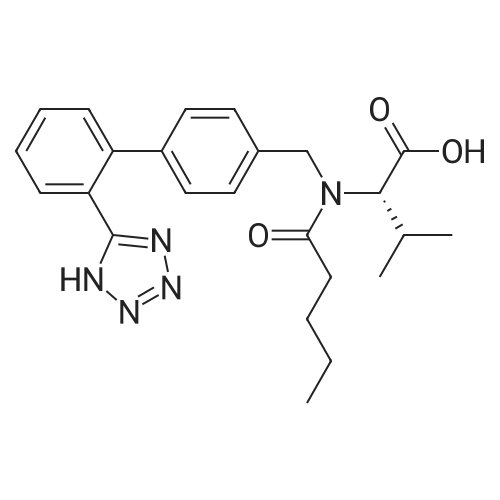
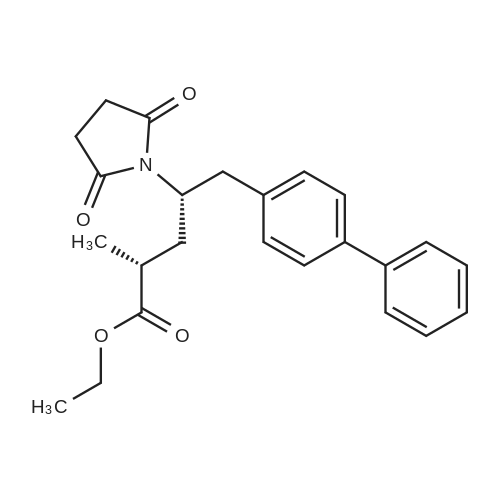
The reactor was charged with 5.0 g (12.1 mmol) of saccharide free acid and 35 ml of acetone and stirred.3.2 ml (6.1 mmol) of 20% sodium carbonate aqueous solution was added to the reaction mixture, followed by stirring at 30 for 2 hours.The reaction mixture was concentrated in vacuo at 40, and the gas was removed at 40 for 1 hour.60 ml of isopropyl acetate was then added to the reaction mixture and stirred at 30 overnight. The resulting solid was filtered and washed with isopropyl acetate,And dried under vacuum at 50 for 4 hours to obtain 4.25 g (yield: 80.95%) of the desired compound. |
|
With sodium hydroxide; In tetrahydrofuran; ethanol; water; at 20℃; for 17h; |
At room temperature, 330 mg of sakubicin was dissolved in 20 mL of a mixed solvent of ethanol and tetrahydrofuran (1: 1), 5 mL of a 1 N aqueous solution of sodium hydroxide was slowly added dropwise thereto, and the mixture was further stirred at room temperature for 17 hours,, Diluted with water and washed with ether. The aqueous phase was acidified with 1N hydrochloric acid and extracted with ethyl acetateThe organic phase was dried over magnesium sulfate and concentrated. The resulting solid was washed with ether to give the sodium salt of sulbucate. |
|
With sodium hydroxide; In Isopropyl acetate; |
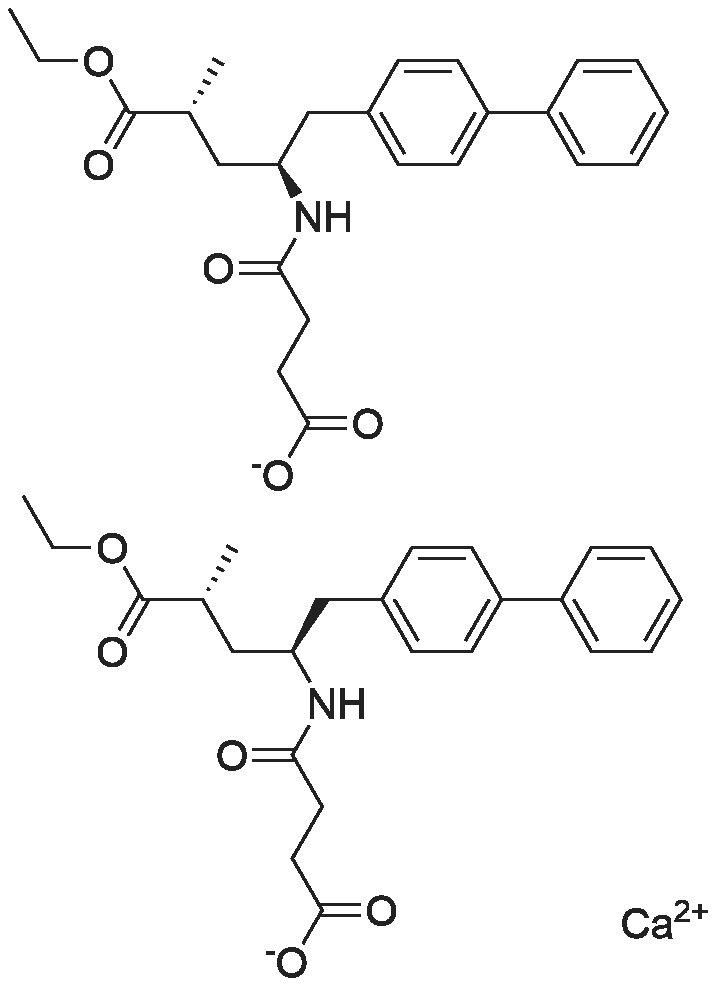
To a mixture of AHU377 free acid (17.7 g) in isopropyl acetate (190 mL), solid sodium hydroxide (1.9 g) is added, thus yielding AHU377 sodium salt which is directly used for manufacture of the calcium salt. The mixture is stirred at 25 C for 70 mm to give a turbidmixture. Addition of solid calcium chloride (2.4 g) is followed by seeding with AHU377 Modification B (obtainable e.g. as described in Example 2) at room temperature. The white suspension is stirred at 25 C for 20 h, then filtered and washed with isopropyl acetate to give solid AHU377 calcium salt after drying at 50 C under reduced pressure. |
|
With sodium hydroxide; In ethanol; at 40 - 60℃; |
Sodium hydroxide 0.10 (2.19 mmol) was dissolved in 2 ml of absolute ethanol at 40 to 45 C; At room temperature, 1.00 g (2.43 mmol) of Sacubitril was dissolved in 10 ml of absolute ethanol.the sodium hydroxide dissolved in ethanol solution added dropwise to the above mentioned Sacubitril dissolved in absolute ethanol solution with stirring to obtain a clear solution. At 55 C - 60 C, methyl tert-butyl ether was added dropwise to a solid precipitate and cooled. Filtration carried out , filter cake was washed by methyl tert-butyl ether and dried at 105 ~ 110 C under reduced pressure to obtain Sacubitril Crystalline Form A. |
|
With sodium hydroxide; In Isopropyl acetate; at 40℃; for 2h; |
Compound 1 Calcium salt1.05 equivalents of an aqueous solution of sodium hydroxide (1 mol / L) was added dropwise to the organic phase obtained in Example 1 at room temperature,40 for 2 h, separated, the water phase with 600mL × 4 IPAC extraction after pressure steaming in addition to IPAC, to get A-4 containing aqueous solution; |
| 19.9 g |
With sodium hydroxide; In ethanol; ethyl acetate; toluene; at 20℃; for 0.5h; |
Sacubitril free acid (20.16 g, 0.049 mol) is dissolved in a mixture of 55 ml of toluene and 35 ml of ethyl acetate. The solution of 1.93 g of sodium hydroxide (0.048 mol) in 43 ml of ethanol is added to the stirred solution of the acid during 30 min and at the temperature of 20C. The mixture is concentrated in vacuum by evaporation of approx. 100 ml of the solvents. The amount of 50 ml of toluene is added and the mixture is concentrated again by evaporation of approx. 50 ml of the solvents. The concentrated residue is slowly diluted by addition of 230 ml of ethyl acetate. The mixture is stirred for at least 2.5 hours. During this time period the crystalline product is separated. The obtained suspension is gradually cooled down to 15 - 18C and after approximately 30 minutes of stirring the product is filtered off and washed with about 25 ml of ethyl acetate. The isolated product is dried in vacuum at 50C until a constant weight is achieved. The amount of 19.9 g of the crystalline product (yield 94%) was obtained, chemical purity according to HPLC 99.9%, the melting point determined by DSC is 167C (Fig. 7). 1H NMR (DMSO-D6): 1.05 (d, 3H), 1.12 (t, 3H), 1.36 (t, 1H), 1.74 (t, 1H), 2.06 (m, 2H), 2.16 (m, 2H), 2.63-2.72 (m, 3H), 3.89 (m, 1H), 3.99 (q, 2H), 7.27 (d, 2H), 7.36 (t, 1H), 7.45 (t, 2H), 7.57 (d, 2H), 7.65 (d, 2H), 8.65 (d, 1H). The crystalline form was characterized by means of XRPD (Fig. 2, Table 2.). |
| 116 g |
With sodium hydroxide; In methanol; water; at 5 - 10℃; for 2h; |
Sacubitnl (130.0 g) was added to methanol (520 mL) and the reaction mixture was cooled to 5C to 10C. A solution of sodium hydroxide (12.26 g in 30 mL of water) was added to the reaction mixture at 5C to 10C and the reaction mixture was stirred for 2 hours. Methanol and water (530 mL) were recovered from the reaction mixture under reduced 200 mm to 10 mm of Hg pressure at 40C to 45C. Acetone (390 mL) was added to the reaction mixture at 40C to 45C and the mixture was stirred for 15 minutes.Acetone (390 mL) was recovered from the reaction mixture under reduced 300 mm to 10 mm of Hg pressure at 40C to 45C. Cyclohexane (390 mL) was added to the reaction mixture and stirred for 30 minutes at 20C to 25C. The reaction mixture was filtered and washed with cyclohexane (130 mL) to obtain the solid material (135 g) and dried under vacuum at 650 mm to 680 mm of Hg at 40C to 45C for 20 hours to obtain the titlecompound.Yield: 116.Og |
|
With sodium hydroxide; In water; ethyl acetate; at 20℃; for 2h;pH 6 - 9; |
Weigh 82.2g (0.2mol) AHU377 free acid placed in 3000ml round bottom flask, add 800ml of ethyl acetate, stirring to dissolve, and then dropping 30% aqueous sodium hydroxide solution ρΗ = 6.0-9.0 at room temperature The reaction was stirred for 2 hours, then concentrated to precipitate a solid, and then added 800ml of ethyl acetate and stirred for 2 hours, then filtered, the filter cake was dried in vacuo at 50 C for 16h to give AHU377 sodium salt, the X-ray powder diffraction pattern shown in Figure 1. |
| 49 g |
With sodium 2-ethylhexanoic acid; In tert-butyl methyl ether; at 55℃; for 14.25h; |
(e) Sacubitril (50 g) was added to methyl t-butyl ether (500 mL) at 20C to 25C and the mixture was stirred for 15 minutes. Sodium-2-ethyl hexonoate (21.6 g) was added to the mixture. The reaction mixture was heated to 50C to 55C for 14 hours. The reaction mixture was cooled to 35C to 40C. The reaction mixture was filtered under nitrogen atmosphere. The reaction mixture was washed with methyl t-butyl ether (400 mL) undernitrogen atmosphere. The product obtained was dried under 650 mm Hg to 680 mm Hg of pressure for 16 hours at 40C to 45C to obtain the title compound.Yield: 49 g. |
|
With sodium hydroxide; In ethanol; n-heptane; for 0.5h; |
Dissolve 112 mg of Sacubitril in 2 mL of ethanol.Add 0.1 mL of ethanol solution containing 0.19 mg of sodium hydroxide per 0.1 mL.Stir for 0.5 hours,Concentrated under reduced pressure,The residue obtained was suspended and stirred in ethanol/n-heptane (1/19, by volume) 300 mL overnight.filter,Dry at 40 C under vacuum,A solid was obtained.The solid was added to 10 mL of isopropanol.After stirring for 72 hours,Centrifuge the supernatant,The obtained solid was dried in an oven at 40 C.Obtained a white solid,That is, type A Sacubitril sodium salt. |
| 1.55 g |
With sodium 2-ethylhexanoic acid; In ethyl acetate; at 20℃; for 1h; |
Sacubitril (425 g) was added to the reaction flask.Ethyl acetate (3833.5 g) was added and stirred until fully dissolved.Sodium 2-ethylhexanoate (343.4 g) was dissolved in ethyl acetate (3097 g).After completely dissolving, pour into the reaction.The reaction was carried out for 1 hour at room temperature. Pure water (4250 g) was added, and after stirring for 0.5 hour, the layers were separated, and the aqueous layer was collected.The aqueous layer was washed with ethyl acetate (3,840 g), stirred for 0.5 hr and then layered, and the aqueous layer was collected. Isopropanol (3340.5 g) was added, concentrated under reduced pressure, and this procedure was repeated three times.A total of 647 grams of white foamed solid was obtained.HPLC analysis of crude <strong>[149709-62-6]sacubitril</strong> sodium salt with a purity of 99.82%,The impurity diacid was 0.02%, and the impurity in the decylamine was 0.04%. <strong>[149709-62-6]sacubitril</strong> sodium salt recrystallizationThe crude <strong>[149709-62-6]sacubitril</strong> sodium salt(647 g) was added to the reaction flask. Isopropanol (1002 g) was added and stirred until fully dissolved.Add n-heptane (8670 g) and mix well. <strong>[149709-62-6]sacubitril</strong> sodium salt seed crystals (43.0 g) were added. The reaction was carried out at room temperature (20-30 C) for 24 hours.The filter cake was washed with n-heptane (867 g).Dry at 55 C under vacuum (48 hours).A total of 269.93 g of a white solid was obtained after subtracting the weight of the seed crystal.The purity of the product was 99.85% by HPLC analysis. Impure diacid 0.03%, indoleamine 0.00%.The total yield was 60.3%. The crystal form of the obtained product was measured by X powder diffraction method, as shown in FIG. Add <strong>[149709-62-6]sacubitril</strong> crystal salt (1.5 g) to the reaction flask (HPLC analysis product purity is 99.85%,Impure diacid 0.04%, impure indole amine 0.00%).Add isopropanol (15 ml) and pure water (0.1 ml) and stir until fully dissolved.The liquid was concentrated to a foamed solid using a reduced pressure concentrator. Dry using an oil-free pump for 19 hours.A total of 1.55 g of a white solid was obtained. The purity of the product was 99.89% by HPLC and 0.04% of the impurity diacid.The indoleamine is 0.00%. The crystal form of the obtained product was determined by X powder diffraction method, as shown in FIG. 2It is an amorphous <strong>[149709-62-6]sacubitril</strong> sodium salt. |
| 22 g |
With sodium hydroxide; In water; acetonitrile; at 20 - 30℃; for 1h; |
Sacubitril free acid (24 gms) and acetonitrile (48 ml) were added in to a round bottom at 20- 30C and stirred for 20 min at 20-30C. Sodium hydroxide solution (2.3 gms dissolved in 7.2 ml water) was added to the resulting reaction solution at 20-30C and stirred for 1 hr, distilled out the solvent under vacuum at 40C and degassed for 2 hrs under vacuum at 40C to obtain a solid. Methanol (100 ml) was added to the resulting solid at 25-35C and stirred for 20 min. The solvent was distilled out completely under vacuum at 35-45C, degassed for 1 hr under vacuum at 35-45C and dried the solid under vacuum at 35-45C for 16 hrs to obtain <strong>[149709-62-6]sacubitril</strong> sodium salt. Yield: 22gms. |
|
With sodium hydroxide; In acetone; at 0 - 10℃; for 1h; |
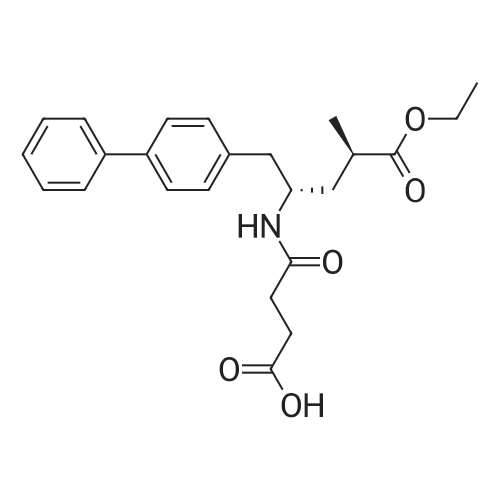
A crude product of ethyl (2R,4S)-5-(biphenyl-4-yl)-4-[(3-carboxypropionyl)amino]-2-methylpentanoate (52.4 g, 0.125 mol) and acetone (629 ml) were added to a 1 L three-necked flask equipped with a mechanical stirrer, stirred and dissolved at room temperature; cooled to 0-10 C. for 5-10 min and added dropwise with sodium hydroxide solution (25 ml, 5 mol/L), after the addition was completed, kept stirring for 1 h, concentrated to dryness, added with methanol (125 ml) or ethanol (125 ml), concentrated to dryness to give sodium 4-(((2S,4R)-1-([1,1'-biphenyl]-4-yl)-5-ethoxy-4-methyl-5-oxopentan-2-yl)amino)-4-oxobutyrate (<strong>[149709-62-6]AHU-377</strong> sodium salt). The product also does not exhibit a good solid form, and is highly hygroscopic despite less hygroscopic than potassium salt. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping