| 79% |
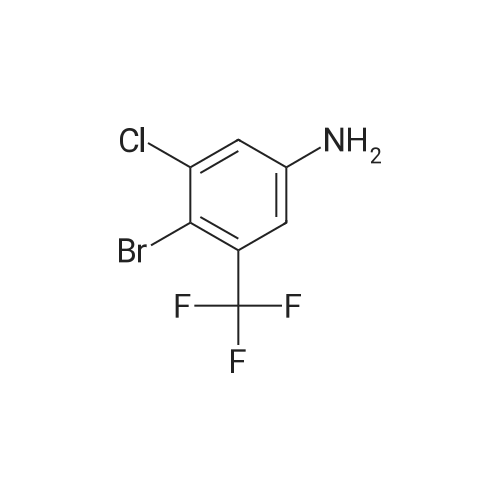
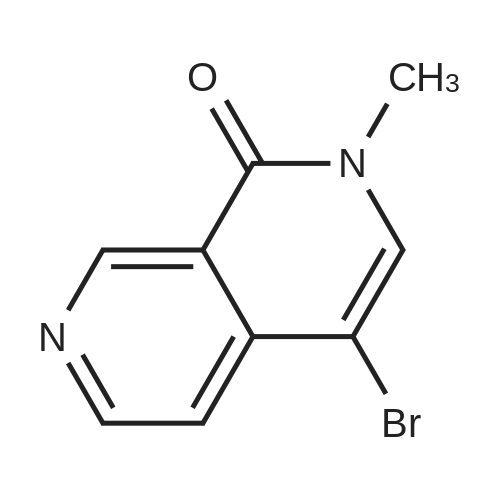
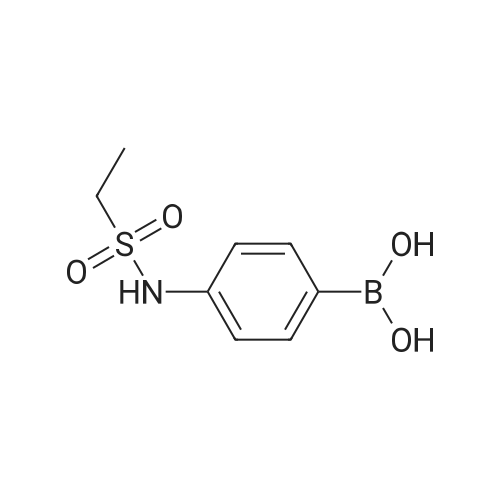
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate In 1,4-dioxane; water at 80℃; for 18h; Inert atmosphere; |
150 Example 150N- { 3’- [(5aS,6S,9aR)-8-cyano-2,6-dimethyl-7-oxo-9a-phenyl-4,5,5 a,6,7,9a-hexahydro-2H- benzo [g] indazol-3 -yl]biphenyl-3 -yl} methanesulfonamide
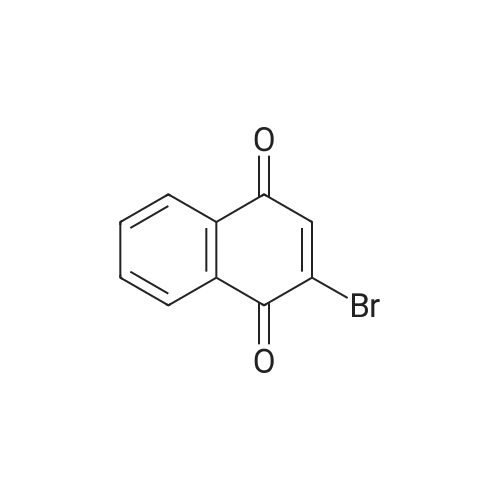
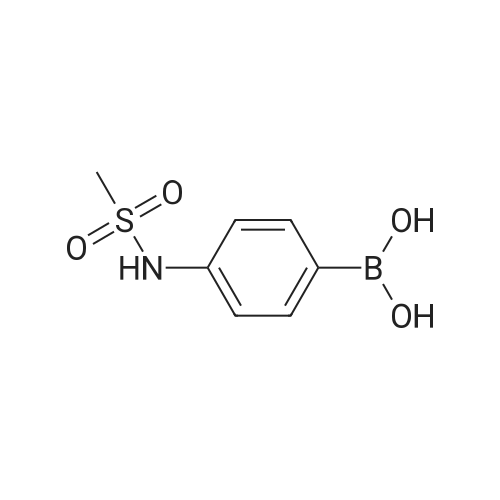
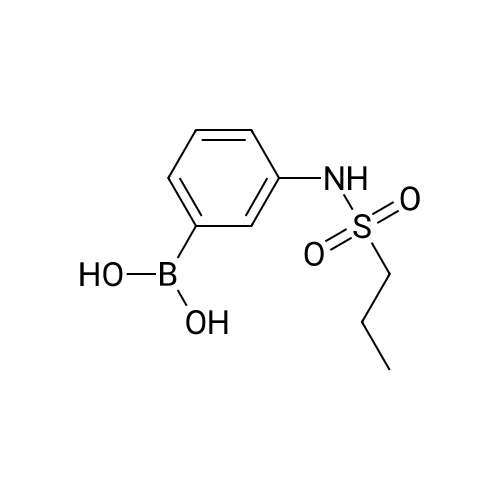
Example 150N- { 3’- [(5aS,6S,9aR)-8-cyano-2,6-dimethyl-7-oxo-9a-phenyl-4,5,5 a,6,7,9a-hexahydro-2H- benzo [g] indazol-3 -yl]biphenyl-3 -yl} methanesulfonamideExample 148D (0.05 g, 0.106 mmol), [1,1’-bis(diphenyl)phosphine)ferrocene] dichloropalladium(II) (0.0077 g, 0.0105 mmol), cesium carbonate (0.103 g, 0.318 mmol), and 3-(methylsulfonylamino)phenylboronic acid (0.068 g,0.318 mmol) were dissolved in dioxane (3 mL) and water (0.3 mL). Then nitrogen gas was bubbled through the mixture for 10 minutes followed by heating at 80 °C for 18 hours. After cooling to ambient temperature, 1 N aqueous ammonium chloride was added followed by extraction with ethyl acetate. The organic extracts were dried over anhydrous magnesium sulfate, filtered, and concentrated by rotary evaporation. The resultant residue was dissolvedin dichloromethane, and the solution was applied to a silica gel flash chromatography column eluted with 0% to 100% ethyl acetate in heptane to afford the titled compound (0.047 g, 79%). ‘HNMR(400 MHz, CDC13) ppm 8.51 (s, 1H), 7.63 (m, 3H), 7.47 (m, 4H), 7.32 (m, 3H), 7.22 (m, 1H), 6.99 (d, J=7.4 Hz, 2H), 6.47 (s, 1H), 3.87 (s, 3H), 3.08 (m, 3H), 2.79 (m, 2H), 2.43 (m, 2H), 1.83 (dd, J=13.6, 6.7 Hz, 1H), 1.57 (m, 1H), 1.17 (d, J=6.4 Hz, 3H); MS(ESI+) m/z 563 (M+H). |
| 79% |
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate In 1,4-dioxane; water at 80℃; for 18h; Inert atmosphere; |
150 N-{3'-[(5aS,6S,9aR)-8-cyano-2,6-dimethyl-7-oxo-9a-phenyl-4,5,5a,6,7,9a-hexahydro-2H-benzo[g]indazol-3-yl]biphenyl-3-yl}methanesulfonamide
Example 148D (0.05 g, 0.106 mmol), [1,1′-bis(diphenyl)phosphine)ferrocene]dichloropalladium(II) (0.0077 g, 0.0105 mmol), cesium carbonate (0.103 g, 0.318 mmol), and 3-(methylsulfonylamino)phenylboronic acid (0.068 g, 0.318 mmol) were dissolved in dioxane (3 mL) and water (0.3 mL). Then nitrogen gas was bubbled through the mixture for 10 minutes followed by heating at 80° C. for 18 hours. After cooling to ambient temperature, 1 N aqueous ammonium chloride was added followed by extraction with ethyl acetate. The organic extracts were dried over anhydrous magnesium sulfate, filtered, and concentrated by rotary evaporation. The resultant residue was dissolved in dichloromethane, and the solution was applied to a silica gel flash chromatography column eluted with 0% to 100% ethyl acetate in heptane to afford the titled compound (0.047 g, 79%). 1H NMR (400 MHz, CDCl3) δ ppm 8.51 (s, 1H), 7.63 (m, 3H), 7.47 (m, 4H), 7.32 (m, 3H), 7.22 (m, 1H), 6.99 (d, J=7.4 Hz, 2H), 6.47 (s, 1H), 3.87 (s, 3H), 3.08 (m, 3H), 2.79 (m, 2H), 2.43 (m, 2H), 1.83 (dd, J=13.6, 6.7 Hz, 1H), 1.57 (m, 1H), 1.17 (d, J=6.4 Hz, 3H); MS (ESI+) m/z 563 (M+H)+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping