* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With lithium tri-butoxyaluminohydride In 1,2-dimethoxyethane at -78℃; for 6.5 h;
In a 11 3-neck flask under argon was placed Quinoxaline-6-carbonyl chloride in [600ML] of [
DME. TO THIS SOLUTION WAS ADDED LITHIUM TRI-TERT-BUTOXYALUMINOHYDRIDE (1 EQ. ) AT-78°C] over 1.5 h. The reaction was kept at that temperature for [5H.] Then ice was added, and the reaction was diluted with AcOEt and filtrated over celite. The two layers were separated and the organic phase was washed with [NAHC03] sat. Quinoxaline-6-carbaldehyde was obtained upon evaporating the solvent in 73percent yield as yellowish solid. HPLC : 1.49 min. LC-MS: M/Z ESI: 0.81 min, 159.37 [(M+1). 1H NMR (CDCL3) 6] 10.28 (s, 1H), 8.97 (s, 2H), 8.61 (s, [1H),] 8.27 (q, 6Hz, 9Hz, 2H).
With selenium(IV) oxide In 1,4-dioxane at 200℃; for 3 h; Microwave irradiation
b) Quinoxaline-6-carbaldehyde. A suspension of 6-methylquinoxaline (8.0 g; 0.055 mol.) and selenium dioxide (6.77 g; 0.061 mol.) in 1,4-dioxane (5.0 mL) was irradiated at 200° C. for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated five further times and the combined, cooled reaction mixtures were dissolved in CH2Cl2, filtered through a plug of celite, and concentrated in vacuo. Purification via flash column chromatography (silica gel, 20-50percent ethyl acetate in hexanes) followed by crystallization from CH2Cl2 provided quinoxaline-6-carbaldehyde (40.0 g, 91percent) as a white solid. 1H NMR (400 MHz, CDCl3) δ ppm 10.25 (s, 1H) 8.95 (s, 2H) 8.57 (d, J=1.3 Hz, 1H) 8.24 (dd, J=8.6, 1.5 Hz, 1H) 8.20 (d, J=8.6 Hz, 1H). MS(ES+) m/e 159 [M+H]+.
91%
With selenium(IV) oxide In 1,4-dioxane at 220℃; for 3 h; Microwave heating
A suspension of 3,4-diaminotoluene (50.0 g; 0.409 mol.) and glyoxal (40percent aq. soln.; 52.0 mL; 0.450 mol.) in water (150 mL) and CH3CN (20.0 mL) was heated to 60 0C for 1 h. Heating was then discontinued and brine (100 mL) was added. The solution was extracted with EtOAc (3 x 150 mL) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification via distillation under reduced pressure (1200C, 10 torr) provided 6-methylquinoxaline (48.0 g, 81 percent) as a clear, colorless oil. 1 H NMR (400 MHz, CDCl3) δ ppm 2.61 (s, 3 H) 7.61 (dd, J=8.59, 1.77 Hz, 1 H) 7.88 (s, 1 H) 8.00 (d, J=8.59 Hz, 1 H) 8.79 (dd, J=9.85, 1.77 Hz, 2 H) MS(ES+) m/e 145 [M+H]+. A suspension of 6-methylquinoxaline (8.O g; 0.055 mol.) and selenium dioxide (6.77 g; 0.061 mol.) in 1 ,4-dioxane (5.0 mL) was irradiated at 2000C for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated five further times and the combined, cooled reaction mixtures were dissolved in CH2CI2, filtered through a plug of celite, and concentrated in vacuo. Purification via flash column chromatography (silica gel,20-50percent ethyl acetate in hexanes) followed by crystallization from CH2CI2 provided quinoxaline-6-carbaldehyde (40.0 g, 91percent) as a white solid. 1H NMR EPO <DP n="114"/>(400 MHz, CDCI3) δ ppm 10.25 (s, 1 H) 8.95 (s, 2 H) 8.57 (d, J=1.3 Hz, 1 H) 8.24 (dd, J=8.6, 1.5 Hz, 1 H) 8.20 (d, J=8.6 Hz, 1 H). MS(ES+) m/e 159 [M+H]+.
91%
With selenium(IV) oxide In 1,4-dioxane at 200℃; for 0.5 h; Microwave irradiation
A suspension of 6-methylquinoxaline (8.0 g; 0.055 mol.) and selenium dioxide (6.77 g; 0.061 mol.) in 1 ,4-dioxane (5.0 mL) was irradiated at 200 0C for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated five further times and the combined, cooled reaction mixtures were dissolved in CH2CI2, filtered through a plug of celite, and concentrated in vacuo. Purification via flash column chromatography (silica gel, 20-50percent ethyl acetate in hexanes) followed by crystallization from CH2CI2 provided quinoxaline-6-carbaldehyde (40.0 g, 91 percent) as a white solid. 1H NMR (400 MHz, CDCI3) δ ppm 10.25 (s, 1 H) 8.95 (s, 2 H) 8.57 (d, J=1.3 Hz, 1 H) 8.24 (dd, J=8.6, 1.5 Hz, 1 H) 8.20 (d, J=8.6 Hz, 1 H). MS(ES+) m/e 159 [M+H]+.
64%
With selenium(IV) oxide In ethyl acetate at 160℃; for 7 h;
Example 242 : Synthesis of (1E,6E)-1-(4-hydroxyphenyl)-7-(quinoxalin-6-yl)hepta-1,6-diene-3,5-dione (CU348); (1) Synthesis of quinoxaline-6-carboxaldehyde; 6-Methylquinoxaline (500 mg, 3.47 mmol) and selenium dioxide (423 mg, 3.81 mmol) in a sealed vessel were stirred at 160°C for 7 h. After cooled to room temperature, the reaction mixture was dissolved in ethyl acetate. The solution was washed with brine twice, and dried over MgSO4. After filtration, the filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (hexane/ethyl acetate = 80/20 to 60/40) to obtain the title compound as a pale brown solid (355 mg, 64percent).
10%
at 160℃; for 72 h;
6-Methylquinaxoline (100 g, 0.69 mol) was heated in a sealed tube to 160 0C and was then added selenium dioxide (100 g, 0.90 mol). The sealed tube was then stirred at 160 0C for 3 days, then allowed to cool to room temperature. The contents solidified and were dissolved in dichloromethane. Solids were filtered through a celite/silica gel cake. The cake was washed with dichloromethane and washes were combined and concentrated to give a pinkish solid, which was washed with hexane and then dried under vacuum to give quinoxaline-6- carbaldehyde as a white solid (50.5 g, contained ca. 10percent of 6-methylquinaxoline).
With sodium periodate In 1,4-dioxane; water; <i>tert</i>-butyl alcohol for 3 h;
Quinoxaline-6-carbaldehyde: To a mixture of 6-Vinyl-quinoxaline (0.68 g, 4.35 mmol) in dioxane (44 mL) and water (35 mL) was added osmium tetraoxide (2.5percent wt in t-BuOH, 2.18 mL, 0.17 mmol) followed by sodium periodate (2.79 g, 13.1 mmol) and the reaction stirred for 3 h. The solution was diluted with ethyl acetate (150 mL) and then washed with water (3.x.75 mL) followed by brine (75 mL). The organics were dried with magnesium sulfate, filtered, and concentrated in vacuo. Purification by flash column chromatography on silica gel using 10-20percent ethyl acetate in dichloromethane as eluant yielded 470 mg (68percent yield) of Quinoxaline-6-carbaldehyde as a white solid. 1H NMR (DMSO-d6): 10.30 (s,1H), 9.11 (d,2H,J=1.9 Hz), 8.74 (d,1H,J=1.4 Hz), 8.26 (m,2H). LC/MS (Method A): r.t.=0.69 min., purity=94.8percent, calculated mass=158, [M+H]+=159.
With diisobutylaluminium hydride In tetrahydrofuran at -78℃; for 0.5 h;
[0185] To a solution of N-methoxy-N-methylquinoxaline-6-carboxamide (4.34 g, 20 mmol, 1.0 eq.) in THF (40 mL) cooled at -78 °C was added DiBAl-H (40 mL, 40 mmol, 2.0 eq.) dropwise. The resulted mixture was stirred at -78 °C for 30 min, then quenched by the addition of aqueous NH4C1 solution. The mixture was adjusted to pH 7 with HCl (IN, 60 mL) and extracted with EA (50 mL X 3). The organic layers were combined, dried over anhydrous Na2S04 and concentrated. The resulting residue was purified via flash column chromatography (PE/EA=4/l,v/v) to afford quinoxaline-6- carbaldehyde
93.7%
With diisobutylaluminium hydride In tetrahydrofuran at -78℃; for 0.5 h;
To a solution of N-methoxy-N-methylquinoxaline-6-carboxamide (4.34 g, 20 mmol, 1.0 eq.) in THF (40 mL) cooled at -78 °C was added DiBAl-H (40 mL, 40 mmol, 2.0 eq.) dropwise. The resulted mixture was stirred at -78 °C for 30 min, then quenched by the addition of aqueous NH4C1 solution. The mixture was adjusted to pH 7 with HC1 (1 N, 60 mL) and extracted with EA (50 mL X 3). The organic layers were combined, dried over anhydrous Na2S04 and concentrated. The resulting residue was purified via flash column chromatography (PE/EA=4/l,v/v) to afford quinoxaline-6- carbaldehyde as yellow solid (2.96 g, 93.7percent)
82%
With diisobutylaluminium hydride In tetrahydrofuran; toluene at -10℃;
Stage 3 A solution of Compound (vi) in THF is treated with a solution of DIBAL-H in toluene at approximately -10° C. The mixture is quenched into aqueous hydrochloric acid and warmed to 20° C. The mixture is diluted with brine. The aqueous phase is removed and backextracted with ethyl acetate. The combined organics are washed with brine, partially concentrated, diluted with heptane, partially distilled again, diluted with heptane, cooled, and filtered. The resulting Compound (vii) wetcake is washed with heptane and dried under vacuum at 50° C.
With lithium tri-butoxyaluminohydride; In 1,2-dimethoxyethane; at -78℃; for 6.5h;
In a 11 3-neck flask under argon was placed Quinoxaline-6-carbonyl chloride in [600ML] of [ DME. TO THIS SOLUTION WAS ADDED LITHIUM TRI-TERT-BUTOXYALUMINOHYDRIDE (1 EQ. ) AT-78C] over 1.5 h. The reaction was kept at that temperature for [5H.] Then ice was added, and the reaction was diluted with AcOEt and filtrated over celite. The two layers were separated and the organic phase was washed with [NAHC03] sat. Quinoxaline-6-carbaldehyde was obtained upon evaporating the solvent in 73% yield as yellowish solid. HPLC : 1.49 min. LC-MS: M/Z ESI: 0.81 min, 159.37 [(M+1). 1H NMR (CDCL3) 6] 10.28 (s, 1H), 8.97 (s, 2H), 8.61 (s, [1H),] 8.27 (q, 6Hz, 9Hz, 2H).
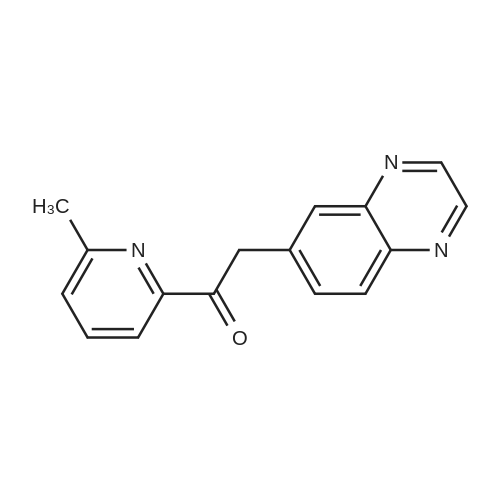
To a solution of <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (2.0 g, 0.0126 mol) in THF (40 mL) and iPrOH (10 mL), was added (phenylamino-(2-methyl-pyrimidin-4-yl)-methyl)-phosphonic acid diphenyl ester (5.46 g, 0.0126 mol) and Cs2CO3 (5.39 g, 0.0164 mol). The mixture was stirred at room temperature for 20 hours and then treated with 3N HCl (10 mL) for 1 hour. The reaction mixture was then diluted with methyl t-butyl ether and extracted with IN HCl twice. The combined aqueous layers were neutralized with 30% aqueous KOH to a pH of ca. 8, then extracted with ethyl acetate (3x). Organic layers were dried over MgSO4 and concentrated to yield a dark orange oil, which was purified on silica gel column with ethyl acetate/hexane (4:1) to give l-(2-methyl-pyrimidin-4-yl)-2-quinoxalin-6-yl-ethanone (2.02 g, 60%) as a yellow solid. LC-MS/ES+: M+l: 265.16. 1H NMR (300 MHz, CDCl3), delta 8.91 (d, IH), 8.85 (m, 2H), 8.1 (d, 2H), 7.75 (d, 2H), 4.71 (s, 2H), 2.81 (s, 3H).
To a solution of <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (0.96 g, 6.1 mol; see Example 4(a) above) in a mixed solvent of THF (40 mL) and iPrOH (10 mL), was added (phenylamino-(2- trifluoromethyl-pyrimidin-4-yl)-methyl)-phosphonic acid diphenyl ester (3.0 g, 6.1 mmol; see Example 3(b) above) and Cs2CO3 (2.64 g, 8 mmol). It was stirred at room temperature overnight and then treated with 3N HCl (10 mL) for 1 hour. The reaction mixture was then diluted with methyl t-butyl ether and extracted with IN HCl twice. The combined aqueous layers were neutralized with 30% aqueous KOH to pH of ca. 8, then extracted with ethyl acetate (3x). Organic layers were dried over MgSO4 and concentrated to yield a dark orange oil, which was purified on silica gel column with EtOAc/hexane (4:1) to give 2-quinoxalin-6-yl-l-(2- trifluoromethyl-pyrimidin-4-yl)-ethanone (1.67 g) as a yellow solid. LC-MS/ES+: M+l: 319.39. 1H NMR (300 MHz, CDCl3), delta 9.08 (d, IH), 8.75 (m, 2H), 8.03-7.96 (m, 3H), 7.67 (dd, IH), 4.71 (s, 2H).
With selenium(IV) oxide; In 1,4-dioxane; at 200℃; for 3.0h;Microwave irradiation;Product distribution / selectivity;
b) Quinoxaline-6-carbaldehyde. A suspension of 6-methylquinoxaline (8.0 g; 0.055 mol.) and selenium dioxide (6.77 g; 0.061 mol.) in 1,4-dioxane (5.0 mL) was irradiated at 200 C. for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated five further times and the combined, cooled reaction mixtures were dissolved in CH2Cl2, filtered through a plug of celite, and concentrated in vacuo. Purification via flash column chromatography (silica gel, 20-50% ethyl acetate in hexanes) followed by crystallization from CH2Cl2 provided quinoxaline-6-carbaldehyde (40.0 g, 91%) as a white solid. 1H NMR (400 MHz, CDCl3) delta ppm 10.25 (s, 1H) 8.95 (s, 2H) 8.57 (d, J=1.3 Hz, 1H) 8.24 (dd, J=8.6, 1.5 Hz, 1H) 8.20 (d, J=8.6 Hz, 1H). MS(ES+) m/e 159 [M+H]+.
91%
With selenium(IV) oxide; In 1,4-dioxane; at 220℃; for 3.0h;Microwave heating;
A suspension of 3,4-diaminotoluene (50.0 g; 0.409 mol.) and glyoxal (40% aq. soln.; 52.0 mL; 0.450 mol.) in water (150 mL) and CH3CN (20.0 mL) was heated to 60 0C for 1 h. Heating was then discontinued and brine (100 mL) was added. The solution was extracted with EtOAc (3 x 150 mL) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification via distillation under reduced pressure (1200C, 10 torr) provided 6-methylquinoxaline (48.0 g, 81 %) as a clear, colorless oil. 1 H NMR (400 MHz, CDCl3) delta ppm 2.61 (s, 3 H) 7.61 (dd, J=8.59, 1.77 Hz, 1 H) 7.88 (s, 1 H) 8.00 (d, J=8.59 Hz, 1 H) 8.79 (dd, J=9.85, 1.77 Hz, 2 H) MS(ES+) m/e 145 [M+H]+. A suspension of 6-methylquinoxaline (8.O g; 0.055 mol.) and selenium dioxide (6.77 g; 0.061 mol.) in 1 ,4-dioxane (5.0 mL) was irradiated at 2000C for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated five further times and the combined, cooled reaction mixtures were dissolved in CH2CI2, filtered through a plug of celite, and concentrated in vacuo. Purification via flash column chromatography (silica gel,20-50% ethyl acetate in hexanes) followed by crystallization from CH2CI2 provided quinoxaline-6-carbaldehyde (40.0 g, 91%) as a white solid. 1H NMR EPO <DP n="114"/>(400 MHz, CDCI3) delta ppm 10.25 (s, 1 H) 8.95 (s, 2 H) 8.57 (d, J=1.3 Hz, 1 H) 8.24 (dd, J=8.6, 1.5 Hz, 1 H) 8.20 (d, J=8.6 Hz, 1 H). MS(ES+) m/e 159 [M+H]+.
91%
With selenium(IV) oxide; In 1,4-dioxane; at 200℃; for 0.5h;Microwave irradiation;
A suspension of 6-methylquinoxaline (8.0 g; 0.055 mol.) and selenium dioxide (6.77 g; 0.061 mol.) in 1 ,4-dioxane (5.0 mL) was irradiated at 200 0C for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated five further times and the combined, cooled reaction mixtures were dissolved in CH2CI2, filtered through a plug of celite, and concentrated in vacuo. Purification via flash column chromatography (silica gel, 20-50% ethyl acetate in hexanes) followed by crystallization from CH2CI2 provided quinoxaline-6-carbaldehyde (40.0 g, 91 %) as a white solid. 1H NMR (400 MHz, CDCI3) delta ppm 10.25 (s, 1 H) 8.95 (s, 2 H) 8.57 (d, J=1.3 Hz, 1 H) 8.24 (dd, J=8.6, 1.5 Hz, 1 H) 8.20 (d, J=8.6 Hz, 1 H). MS(ES+) m/e 159 [M+H]+.
64%
With selenium(IV) oxide; In ethyl acetate; at 160℃; for 7.0h;
Example 242 : Synthesis of (1E,6E)-1-(4-hydroxyphenyl)-7-(quinoxalin-6-yl)hepta-1,6-diene-3,5-dione (CU348); (1) Synthesis of quinoxaline-6-carboxaldehyde; 6-Methylquinoxaline (500 mg, 3.47 mmol) and selenium dioxide (423 mg, 3.81 mmol) in a sealed vessel were stirred at 160C for 7 h. After cooled to room temperature, the reaction mixture was dissolved in ethyl acetate. The solution was washed with brine twice, and dried over MgSO4. After filtration, the filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (hexane/ethyl acetate = 80/20 to 60/40) to obtain the title compound as a pale brown solid (355 mg, 64%).
10%
With selenium(IV) oxide; at 160℃; for 72.0h;
6-Methylquinaxoline (100 g, 0.69 mol) was heated in a sealed tube to 160 0C and was then added selenium dioxide (100 g, 0.90 mol). The sealed tube was then stirred at 160 0C for 3 days, then allowed to cool to room temperature. The contents solidified and were dissolved in dichloromethane. Solids were filtered through a celite/silica gel cake. The cake was washed with dichloromethane and washes were combined and concentrated to give a pinkish solid, which was washed with hexane and then dried under vacuum to give quinoxaline-6- carbaldehyde as a white solid (50.5 g, contained ca. 10% of 6-methylquinaxoline).
With hydrogenchloride; In tetrahydrofuran; dichloromethane;
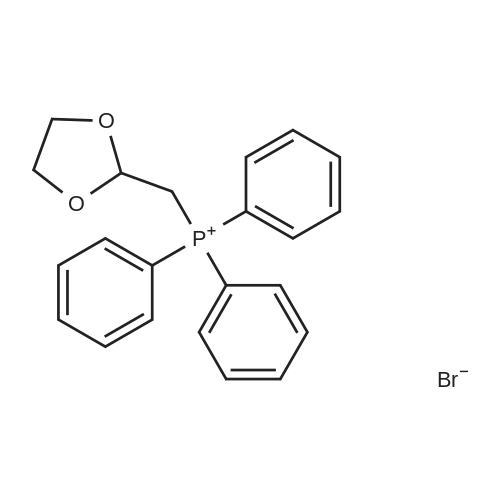
REFERENCE EXAMPLE 27 (2E)-3-(6-Quinoxalinyl)-2-propenal A mixture of <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (0.62 g, 3.92 mmol, prepared as described in Photochem. Photobiol. 1991, 54, 7), (1,3-dioxolan-2-ylmethyl)triphenylphosphonium bromide (2.50 g, 5.82 mmol), and TDA-1 (1.20 mL, 3.75 mmol) in dichloromethane (20 mL) and sat. aq. K2CO3 (20 mL) was heated to reflux for 4 h. The layers were separated and the aqueous layer was extracted with dichloromethane (2*20 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried (Na2SO4), and concentrated. THF (10 mL) and 10% HCl (10 mL) were added and the mixture was stirred for 1 h at rt. The mixture was cooled to 0 C., the precipitated solids were removed by filtration, washed with water and dried in vacuo to give 0.47 g (65%) of the title compound as a tan solid. MS 185 (M+H)+.
0.47 g (65%)
With hydrogenchloride; In tetrahydrofuran; dichloromethane;
REFERENCE EXAMPLE 28 (E)-3-(6-Quinoxalinyl)-2-propenal A mixture of <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (0.62 g, 3.92 mmol, prepared as described in Photochem. Photobiol. 1991, 54, 7), (1,3-dioxolan-2-ylmethyl)triphenylphosphonium bromide (2.50 g, 5.82 mmol), and TDA-1 (1.20 mL, 3.75 mmol) in dichloromethane (20 mL) and sat. aq. K2CO3 (20 mL) was heated to reflux for 4 h. The layers were separated and the aqueous layer was extracted with dichloromethane (2*20 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried (Na2SO4), and concentrated. THF (10 mL) and 10% HCl (10 mL) were added and the mixture was stirred for 1 h at rt. The mixture was cooled to 0 C., the precipitated solids were removed by filtration, washed with water and dried in vacuo to give 0.47 g (65%) of the title compound as a tan solid. MS 185 (M+H)+.
0.47 g (65%)
With hydrogenchloride; In tetrahydrofuran; dichloromethane;
Reference Example 27 3-(6-Quinoxalinyl)-2-propenal A mixture of <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (0.62 g, 3.92 mmol, prepared as described in Photochem. Photobiol. 1991, 54, 7), (1,3-dioxolan-2-ylmethyl)triphenylphosphonium bromide (2.50 g, 5.82 mmol), and TDA-1 (1.20 mL, 3.75 mmol) in dichloromethane (20 mL) and sat. aq. K2CO3 (20 mL) was heated to reflux for 4 h. The layers were separated and the aqueous layer was extracted with dichloromethane (2*20 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried (Na2SO4), and concentrated. THF (10 mL) and 10% HCl (10 mL) were added and the mixture was stirred for 1 h at rt. The mixture was cooled to 0 C., the precipitated solids were removed by filtration, washed with water and dried in vacuo to give 0.47 g (65%) of the title compound as a tan solid. MS 185 (M+H)+.
With sodium hydroxide; dibenzoyl peroxide; In N-Bromosuccinimide; water; chlorobenzene;
EXAMPLE 3 Catalytic Air Dehydrogenation of 6-Hydroxymethylmethyl-quinoxaline With 5% Pt/C In a 100 ml flask, 6-methyl-quinoxaline (1.25 g, 8.68 mmol) was dissolved with N-bromosuccinimide (2.32, 13.0 mmol) and benzoyl peroxide (0.15 g, 0.62 mml) in 31 g of chlorobenzene. The mixture was heated up to 85 C. to give a pale yellow solution that turned reddish with time. The solution was maintained at 85 C. for two hours and it was then cooled to -10 C. and filtered. The solution was vacuum dried to give an orange solid residue. The orange solid was then mixed with an alkaline solution prepared by dissolving 2.6 g of sodium hydroxide pellets in 50 ml of water. The mixture was gently heated to 85-95 C. to give a solution of 6-hydroxymethyl-quinoxaline that was mixed with the catalyst (5% Pt/C, 0.2 g) and maintained at that temperature with air sparging. The reaction was monitored by GCMS showing the consumption of 6-hydroxymethyl-quinoxaline and the formation of 6-quinoxalinecarboxaldehyde.
6-[di(1H-imidazol-1-yl)methyl]quinoxaline 4-methylbenzenesulfonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
7 parts (23.0%)
In acetone;
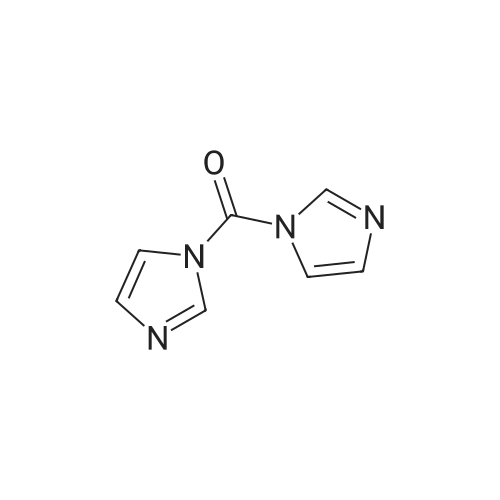
EXAMPLE 16 A mixture of 7.8 parts of <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> and 24.3 parts of 1,1'-carbonylbis-1H-imidazole was stirred for 1 hour at 100 C. After cooling, the reaction mixture was partitioned between water and ethyl acetate. The organic layer was dried, filtered and evaporated. The residue was purified by column chromatography (silica gel; CH2 Cl2 /CH3 OH/NH4 OH 85:15:1). The eluent of the desired fraction was evaporated and the residue was converted into the 4-methylbenzenesulfonate(1:2) salt in 2-propanone, yielding 7 parts (23.0%) of 6-[di(1H-imidazol-1-yl)methyl]quinoxaline 4-methylbenzenesulfonate(1:2); mp. 240.3 C. (comp. 51).
(b) To a stirred solution of 10 parts of intermediate 5, namely 6-quinoxalinemethanol in 133 parts of dichloromethane were added 20 parts of manganese(IV) oxide. After stirring for 3 hours at room temperature, the reaction mixture was filtered and the filtrate was evaporated, yielding 6.6 parts (67.3%) of 6-quinoxalinecarboxaldehyde; mp. 134 C. (interm. 6).
(5Z)-5-(6-quinoxalinylmethylidene)-2-thioxo-1,3-thiazolidin-4-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
98%
With sodium acetate; In acetic acid; for 18h;Heating / reflux;
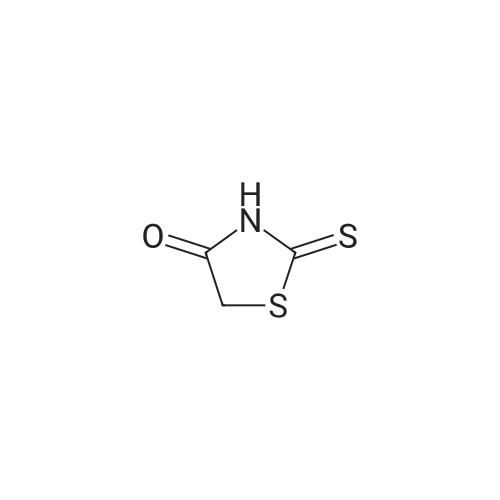
A mixture of <strong>[130345-50-5]6-quinoxalinecarbaldehyde</strong> (6.75 g, 42.7 mmoles), rhodanine (5.69 g, 42.7 mmoles) and sodium acetate (10.5 g, 128 mmoles) in acetic acid (150 mL) was heated under reflux for 18 hours. The mixture solidified during the reaction, which was triturated with water, the solid collected and washed successively with water, methanol and dichloromethane to give the title compound as a dark red solid (11.5g, 98%). 1 H NMR (400 MHz, DMSO-c/6) delta ppm 7.90 (s, 1 H) 8.03 (dd, J=8.84, 1.77 Hz, 1 H) 8.21 (d, J=8.59 Hz, 1 H) 8.31 (d, J=1.77 Hz, 1 H) 9.02 (dd, J=6.32, 1.77 Hz, 2 H) 14.01 (s, 1 H).
With piperidine; In ethanol; at 150℃; for 0.5h;Microwave irradiation;
A solution of the compound from Example 1c) (295 mg; 1.0 mmol.), <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> from 1 b) (158 mg;1.0 mmol.) and piperidine (0.11 ml 1.1 mmol.) in ethanol (2.5 ml_) was stirred and heated at 150 0C for 30 min. in a Biotage Initiator microwave synthesizer. The reaction mixture was then cooled, poured into 1 M aqueous hydrochloric acid (50.0 ml_) and filtered to give the title compound (286 mg; 66%) as a yellow powder. 1 H NMR (400 MHz, DMSOd6) delta ppm 7.81 (s, 2 H) 7.98 (dd, J=8.59, 1.52 Hz, 1 H)8.01 (s, 1 H) 8.18 (d, J=8.59 Hz, 1 H) 8.24 (d, J=1.26 Hz1 1 H) 8.99 (s, 2 H) 13.17 (s, 1 H). This material was further purified by crystallization from water (5.0 mI_)/1 M aqueous sodium hydroxide (1.0 mL)/ethanol (1.0 ml_) to afford the monosodium salt, hemihydrate of the title compound (201 mg; 46%) as bright yellow needles. C18H8N4OSCI3Na.0.5 H2O requires: %C, 46.3; %H, 1.9; %N, 12.0; found: %C, 46.3; %H, 2.1 ; %N, 11.8.
N-(4-chloro-3-[(5Z)-4-oxo-5-(6-quinoxalinylmethylidene)-4,5-dihydro-1,3-thiazol-2-yl]amino}phenyl)-2-methylpropanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
20%
With piperidine; In ethanol; at 150℃; for 0.666667h;Microwave heating;
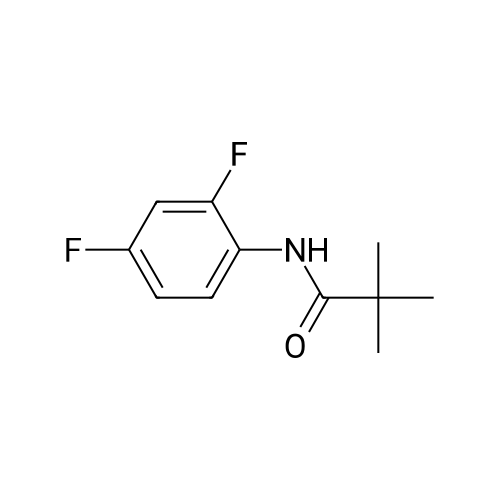
Lambda/-{4-chloro-3-[(4-oxo-4,5-dihydro-1,3-thiazol-2-yl)amino]phenyl}-2-methylpropanamide (0.165 g, 0.529 mmole) and <strong>[130345-50-5]6-quinoxalinecarbaldehyde</strong> (0.083 g, 0.532 mmole) in ethanol (2.0 ml_) was treated with piperdine (0.052 mL, 0.525 mmol.) The reaction was heated in a microwave to 1500C for 40 min. The reaction mixture was diluted with water (3.0 mL) and washed with 1 N Hydrochloric acid and then water and dried. This was reprecipitated by dissolving in basic ethanol and re-acidfied with acetic acid. The yellow solid was filtered and dried to provide (0.0482 g, 20%.) 1 H NMR (400 MHz, DMSO-Of6) delta ppm 12.36 (bs, 1 H) 10.01 (s, 1 H) 8.95 (s, 1 H) 8.18 (s, 1 H) 8.16 (d, J=8.76, 1 H) 7.99 (d, J=8.61 , 1 H) 7.83 (s, 1 H) 7.43 (s, 3H) 2.80 (m, 1 H) 1.09 (s, 3H) 1.08 (s, 3H)
20%
With piperidine; In ethanol; at 150℃; for 0.666667h;Microwave;
lambda/-{4-chloro-3-[(4- oxo-4,5-dihydro-1 ,3-thiazol-2-yl)amino]phenyl}-2-methylpropanamide (0.165 g, 0.529 mmole) and <strong>[130345-50-5]6-quinoxalinecarbaldehyde</strong> (0.083 g, 0.532 mmole) in ethanol (2.0 ml.) was treated with piperdine (0.052 ml_, 0.525 mmol.) The reaction was heated in a microwave to 1500C for 40 min. The reaction mixture was diluted with water (3.0 ml.) and washed with 1 N Hydrochloric acid and then water and dried. This was reprecipitated by dissolving in basic ethanol and re-acidfied with acetic acid. The yellow solid was filtered and dried to provide (0.0482 g, 20%.) 1 H NMR <n="47"/>(400 MHz, DMSOd6) delta ppm 12.36 (bs, 1 H) 10.01 (s, 1 H) 8.95 (s, 1 H) 8.18 (s, 1 H) 8.16 (d, J=8.76, 1 H) 7.99 (d, J=8.61 , 1 H) 7.83 (s, 1 H) 7.43 (s, 3H) 2.80 (m, 1 H) 1.09 (s, 3H) 1.08 (s, 3H)
Potassium carbonate (0.066 g, 0.478 mmol) was added to a slurry of the compound from example 31 (c) (0.058 g, 0.238 mmol) in 2-butanone (4 mL), followed by isovaleryl chloride (0.029 mL, 0.238 mmol). The mixture was stirred at room temperature for 18 h, then evaporated under reduced pressure and the residue partitioned between brine and ethyl acetate. The extracts were dried (MgSO4) and evaporated under reduced pressure to give the crude amide product. A suspension of the crude amide (0.100 g; 0.307 mmol), <strong>[130345-50-5]6-quinoxalinecarbaldehyde</strong> (0.049 g; 0.307 mmol), and piperidine (0.030 mL; 0.307 mmol) in ethanol (2.0 mL) was stirred and irradiated at 1500C for 30 min. in a Biotage Initiator microwave synthesizer. Upon cooling, the reaction mixture was acidified with 1 M aq. HCI and the resulting suspension was filtered, washed with water, and dried in vacuoto afford the title compound (0.111 g, 78%) as a yellow solid. 1H NMR (400 MHz, DMSO-Of6) delta ppm 12.9 (s, 1 H) 10.1 (s, 1 H) 8.98 (s, 2 H) 8.22 (d, J=1.5 Hz, 1 H) 8.18 (d, J=8.6 Hz, 1 H) 7.99 (dd, J=8.8, 1.8 Hz, 1 H) 7.95 (s, 1 H) 7.50 (d, J=1.5 Hz, 1 H) 7.47 (d, J=9.3 Hz, 1 H) 7.41 (dd, J=9.1 , 2.0 Hz, 1 H) 2.19 (d, J=7.1 Hz, 2 H) 2.00 - 2.10 (m, 1 H) 0.92 (d, J=6.6 Hz, 6 H). MS(ES+) m/e 466 [M+H]+.
A mixture of the compound from example 145(b) (0.300 g, 0.703 mmol), <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (0.122 g, 0.772 mmol), piperidine (0.030 ml_, 0.707 mmol) and ethanol (1 ml_) was heated in a microwave reactor at 150 C for 20 min, then cooled. 1 M aqueous HCI (1 ml_) was added, followed by water (1 ml_). The precipitate was filtered and dried, then a sample purified by rp-HPLC (ODS, 10-90% acetonitrile/water + 0.1 % trifluoroacetic acid) to give the title compound(64%) as a solid. 1 H NMR (400 MHz, DMSOd6) delta ppm 1.27 (s, 6 H) 1.35 (s, 9 H) 6.72 (s, 1 H) 7.00 (s, 1 H) 7.45 (d, J=8.84 Hz, 1 H) 7.53 (s, 1 H) 7.93 (s, 1 H) 7.98 (dd, J=8.84, 1.77 Hz, 1 H) 8.16 (d, J=8.59 Hz, 1 H) 8.21 (m, 1 H) 8.97 (s, 2 H) 9.63 (s, 1 H) 12.84 (s, 1 H)
With sodium periodate;osmium(VIII) oxide; In 1,4-dioxane; water; tert-butyl alcohol; for 3h;
Quinoxaline-6-carbaldehyde: To a mixture of 6-Vinyl-quinoxaline (0.68 g, 4.35 mmol) in dioxane (44 mL) and water (35 mL) was added osmium tetraoxide (2.5% wt in t-BuOH, 2.18 mL, 0.17 mmol) followed by sodium periodate (2.79 g, 13.1 mmol) and the reaction stirred for 3 h. The solution was diluted with ethyl acetate (150 mL) and then washed with water (3×75 mL) followed by brine (75 mL). The organics were dried with magnesium sulfate, filtered, and concentrated in vacuo. Purification by flash column chromatography on silica gel using 10-20% ethyl acetate in dichloromethane as eluant yielded 470 mg (68% yield) of Quinoxaline-6-carbaldehyde as a white solid. 1H NMR (DMSO-d6): 10.30 (s,1H), 9.11 (d,2H,J=1.9 Hz), 8.74 (d,1H,J=1.4 Hz), 8.26 (m,2H). LC/MS (Method A): r.t.=0.69 min., purity=94.8%, calculated mass=158, [M+H]+=159.
With piperidine; In ethanol; for 48h;Heating / reflux;
Compound (ii), Compound (vii), and piperidine are refluxed for 48 h and the resulting mixture is cooled to 10 C. The resulting solid is filtered solid Compound (viii) is washed with ethanol. The intermediate product is slurried in ethanol and treated with aqueous hydrochloric acid at 20 C. The resulting suspension is filtered, washed with ethanol, water, and ethanol again. Compound A (as the free acid) wetcake is dried under vacuum at 50 C.
With piperidine; In ethanol; at 150℃; for 4h;Microwavw irradiation;
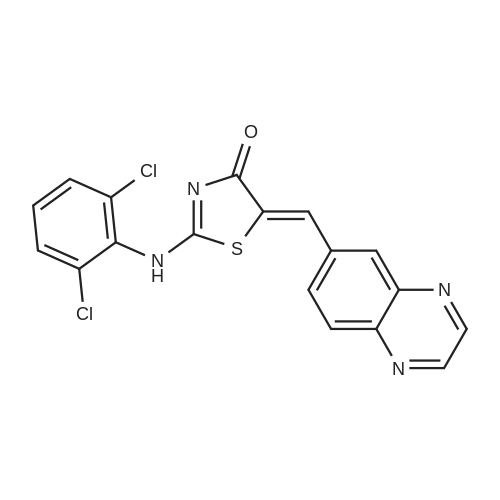
d) (5Z)-2-[(2,6-Dichlorophenyl)amino]-5-(6-quinoxalinylmethylidene)-1,3-thiazol-4(5H)-one. A suspension of 2-[(2,6-dichlorophenyl)amino]-1,3-thiazol-4(5H)-one (4.95 g; 0.019 mol.), <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (3.00 g; 0.019 mol.) and piperidine (1.88 mL; 0.019 mol.) in ethanol (10.0 mL) was stirred and irradiated at 150 C. for 30 min. in a Biotage Initiator microwave synthesizer. The above procedure was repeated seven further times and the combined, cooled reaction mixtures were poured into water (500 mL) and acidified with 1M aq. HCl (100 mL). The resulting suspension was filtered, washed with water and MeOH, and dried in vacuo to provide (5Z)-2-[(2,6-dichlorophenyl)amino]-5-(6-quinoxalinylmethylidene)-1,3-thiazol-4(5H)-one (47.0 g, 88%) as a dull brown powder. 1 H NMR (400 MHz, DMSO-d6) delta ppm 13.11 (s, 1H) 8.97 (s, 2H) 8.20 (s,1H) 8.17 (d, J=8.6 Hz, 1H) 8.00 (s,1H) 7.96 (dd, J=8.7, 1.6 Hz, 1H) 7.58 (d, J=8.1 Hz, 2H) 7.25 (t, J=8.1 Hz, 1H). MS(ES+) m/e 401 [M+H]+.
With diisobutylaluminium hydride; In tetrahydrofuran; at -78℃; for 0.5h;
[0185] To a solution of N-methoxy-N-methylquinoxaline-6-carboxamide (4.34 g, 20 mmol, 1.0 eq.) in THF (40 mL) cooled at -78 C was added DiBAl-H (40 mL, 40 mmol, 2.0 eq.) dropwise. The resulted mixture was stirred at -78 C for 30 min, then quenched by the addition of aqueous NH4C1 solution. The mixture was adjusted to pH 7 with HCl (IN, 60 mL) and extracted with EA (50 mL X 3). The organic layers were combined, dried over anhydrous Na2S04 and concentrated. The resulting residue was purified via flash column chromatography (PE/EA=4/l,v/v) to afford quinoxaline-6- carbaldehyde
93.7%
With diisobutylaluminium hydride; In tetrahydrofuran; at -78℃; for 0.5h;
To a solution of N-methoxy-N-methylquinoxaline-6-carboxamide (4.34 g, 20 mmol, 1.0 eq.) in THF (40 mL) cooled at -78 C was added DiBAl-H (40 mL, 40 mmol, 2.0 eq.) dropwise. The resulted mixture was stirred at -78 C for 30 min, then quenched by the addition of aqueous NH4C1 solution. The mixture was adjusted to pH 7 with HC1 (1 N, 60 mL) and extracted with EA (50 mL X 3). The organic layers were combined, dried over anhydrous Na2S04 and concentrated. The resulting residue was purified via flash column chromatography (PE/EA=4/l,v/v) to afford quinoxaline-6- carbaldehyde as yellow solid (2.96 g, 93.7%)
82%
With diisobutylaluminium hydride; In tetrahydrofuran; toluene; at -10℃;Product distribution / selectivity;
Stage 3 A solution of Compound (vi) in THF is treated with a solution of DIBAL-H in toluene at approximately -10 C. The mixture is quenched into aqueous hydrochloric acid and warmed to 20 C. The mixture is diluted with brine. The aqueous phase is removed and backextracted with ethyl acetate. The combined organics are washed with brine, partially concentrated, diluted with heptane, partially distilled again, diluted with heptane, cooled, and filtered. The resulting Compound (vii) wetcake is washed with heptane and dried under vacuum at 50 C.
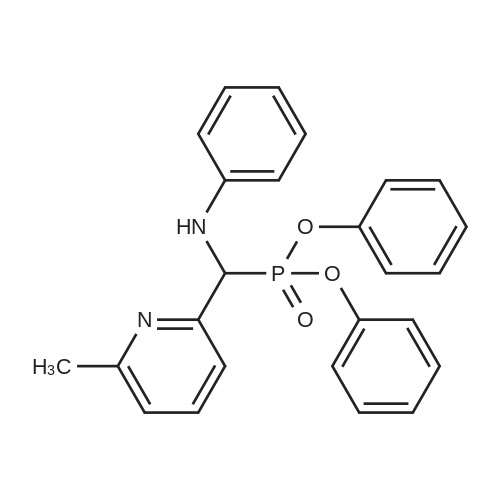
General procedure: To a stirred solution of 10a or 10b (18.97 mmol) in a mixture of THF (40 mL) and i-PrOH (10 mL), diphenyl (6-methylpyridin-2-yl)(phenylamino)methylphosphonate (18.97 mmol) and Cs2CO3 (24.65 mmol) were added. The mixture was stirred at room temperature for 16 h, and to it, 3 N HCl (25 mL) was added dropwise over a period of 5 min. The reaction mixture was diluted with MTBE (30 mL). The aqueous layer was separated, and the organic layer was extracted with 1 N HCl (2 × 50 mL). The combined aqueous layer was neutralized with 50% aqueous KOH solution (pH 78) and extracted with EtOAc (3 × 100 mL). The EtOAc solution was dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. The residue was purified by MPLC on silica gel using EtOAc/hexane as eluent to give the titled compound 11a or 11b as a light yellow solid.
With potassium hydroxide; In methanol; at 20℃; for 24h;
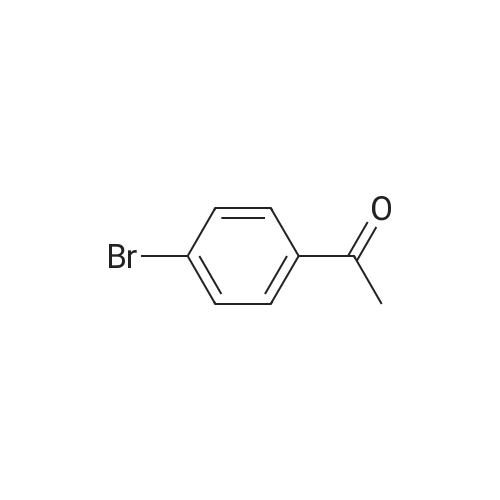
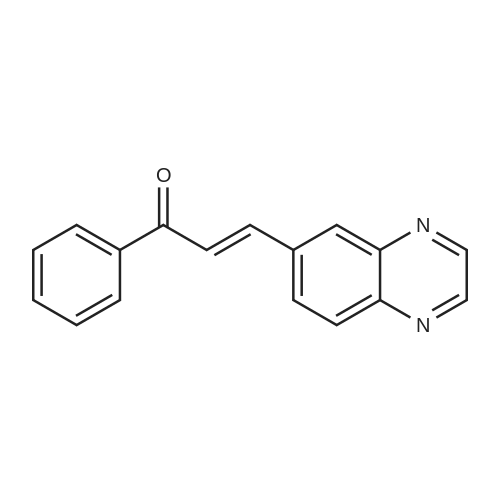
General procedure: The chalcones were prepared through an aldol condensation reaction between acetophenones (1.0 mmol) and the corresponding aldehydes (1.0 mmol), in methanol (15 mL), KOH (50 % v/v), at room temperature with magnetic stirring for 24 h.14 Distilled water and 10% hydrochloric acid were added to the reaction for total precipitation of the compounds, which were then obtained by vacuum filtration and later recrystallized from hot ethanol. The purity of the synthesized compounds was analyzed by thin-layer chromatography (TLC) using Merck silica pre-coated aluminum plates of 200 mum thickness, with several solvent systems of different polarities. Compounds were visualized with ultraviolet light (l = 254 and 360 nm) and using sulfuric anisaldehyde solution followed by heat application as the developing agent.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
With potassium hydroxide; In methanol; at 20℃; for 24h;
General procedure: The novel chalcones (1-8)were prepared by magnetic stirring with acetophenone (1 mmol), methanol (30 ml), KOH 50% w/v (5 ml) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (12) (1 mmol), at room temperature for 24 h. Distilled water and chloridric acid 10% were added in the reaction for total precipitation of the compounds. The compounds were then obtained by vacuum filtration and later recrystallized in dichloromethane/hexane. The purified chalcones were obtained with yields between 41% and 93%.
[0197] To a solution of N-(3-bromo-4-fluorophenyl)pivalamide (9.48 g, 34.6 mmol, 1.2 eq.) in THF (100 mL) cooled at -78 C was added w-BuLi (27.6 mL, 69 mmol, 2.4 eq.) dropwise. The resulting mixture was stirred at -78 C for 1 h, then a solution of <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (4.5 g, 28.8 mmol, 1.0 eq.) in THF (200 mL) was added dropwise. The mixture was stirred at -78 C for lh, then quenched by the addition of NH4C1 solution. The mixture was extracted with EA (100 mL X 3) and the combined organic layers were dried over Na2S04 and concentrated. The resulting residue was purified via flash column chromatography (PE/EA=1/1, v/v) to afford N-(4-fluoro-3- (hydroxy(quinox ivalamide as a yellow foam (5.16 g, 50.7%).
50.7%
To a solution of N-(3-bromo-4-fluorophenyl)pivalamide (9.48 g, 34.6 mmol, 1.2 eq.) in THF (100 mL) cooled at -78 C. was added n-BuLi (27.6 mL, 69 mmol, 2.4 eq.) dropwise. The resulting mixture was stirred at -78 C. for 1 h, then was added a solution of <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (4.5 g, 28.8 mmol, 1.0 eq.) in THF (200 mL) dropwise. The mixture was stirred at -78 C. for 1 h, then quenched by the addition of NH4Cl solution. The mixture was extracted with EA (100 mL×3) and the combined organic layers were dried over Na2SO4 and concentrated. The resulting residue was purified by flash column chromatography (PE/EA=1/1, v/v) to afford N-(4-fluoro-3-(hydroxy(quinoxalin-6-yl)methyl)phenyl)pivalamide as a yellow foam (5.16 g, 50.7%).
[0186] To a solution of N-(2,4-difluorophenyl)pivalamide (740 mg, 3.46 mmol, 1.2 eq.) in THF (10 mL) cooled at -78 C was added n-BuLi (2.76 mL, 6.9 mmol, 2.4 eq.) dropwise. The resulting mixture was stirred at -78 C for 1 h, then a solution of <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (450 mg, 2.88 mmol, 1.0 eq.) in THF (20 mL) was added dropwise. The mixture was stirred at -78 C for lh, then quenched by the addition of NH4C1 solution. The mixture was extracted with EA (20 mL X 3) and the combined organic layers were dried over Na2S04 and concentrated. The resulting residue was purified via flash column chromatography (PE/EA=2/1, v/v) to afford N-(2,4-difluoro-3-(hydroxy(quinoxalin- 6-yl)methyl)phenyl)pivalamide as a yellow foam (541 mg, 50.7%).
General procedure: A mixture of an aldehyde (1.0 eq) and an amine (1.0 eq) was heated in a sealed tube at 60 C for 6 h. The crude material was dried under vacuum over phosphorus(V) oxide to give quantitatively the imine which was used in the next step without further purification.
(-)-2-((3,4-trans)-4-methylpyrrolidin-3-yl)-7-(tetrahydro-2H-pyran-4-yl)imidazo[5,1-f][1,2,4]triazin-4(3H)-one[ No CAS ]
[ 130345-50-5 ]
(+)-2-((3,4-trans)-4-methyl-1-(quinoxalin-6-ylmethyl)pyrrolidin-3-yl)-7-(tetrahydro-2H-pyran-4-yl)imidazo[5,1-f][1,2,4]triazin-4(3H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
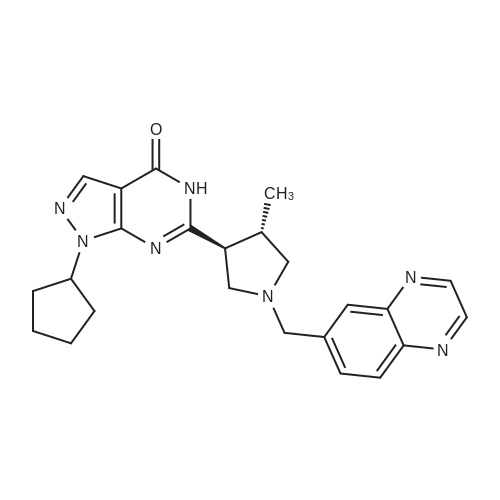
86. f+)-2-f(3.,4-trans)-4-methyl-l-f uinoxalin-6-ylmethyl)pyrroli(iin-3-yl)- 7-ftetrahvdro-2H-pyran-4-yl)imidazo[5,l- 1 [l,2,41triazin-4f3H -one [1045] To a stirred solution of (-)-2-((3,4-trans)-4-methylpyrrolidin-3-yl)-7- (tetrahydro-2H-pyran-4-yl)imidazo[5,l- J[l,2,4]triazin-4(3H)-one (125 mg, 0.41 mmol) in MeOH (10 mL) was added <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (98 mg, 0.62 mmol) at RT under an argon atmosphere. After being stirred for 2 h, NaCNBH3 (78 mg, 1.23 mmol) was added to the reaction mixture and stirring was continued for another 16 h at RT. The volatiles were evaporated under reduced pressure. The residue was diluted with ice-cold water (15 mL) and extracted with DCM (2 x 20 mL). Combined organic layer was dried over sodium sulphate, filtered and concentrated in vacuo to obtain the crude product. The crude material was purified by silica gel column chromatography to afford (+)-2-((3,4-trans)-4-methyl-l-(quinoxalin-6- ylmethyl)pyrrolidin-3 -yl)-7-(tetrahydro-2H-pyran-4-yl)imidazo [5 , 1 -f [ 1 ,2,4]triazin- 4(3H)-one (70 mg, 38%) as an off-white solid; 1H-NMR (DMSO d6, 400 MHz): delta 8.91 (d, 2H), 8.08 (d, 1H), 8.01 (s, 1H), 7.87 (d, 1H), 7.64 (s, 1H), 3.95-3.91 (m, 4H), 3.54-3.50 (m, 3H), 3.04-3.01 (m, 1H), 2.96-2.94 (m, 1H), 2.87-2.84 (m, 2H), 2.72- 2.64 (m, 1H), 2.34-2.31 (m, 1H), 1.87-1.82 (m, 4H), 1.12 (d, 3H); Mass (ESI): 446 [M++l]; LC-MS: 98.47%; 446 [M++l]; (column; X-bridge C-18, (50x3.0 mm, 3.5mu); RT 2.72 min. 5mM NH4OAc in water: ACN; 0.8 ml/min); UPLC (purity): 96.41%; (column; Acquity BEH C-18, (50x2.1 mm, 1.7mu); RT 1.38 min. 0.025% TFA (Aq): ACN; 0.50 ml/min; Chiral HPLC: 99.37%, R, = 23.23 min (Chiralpak IA, 250 x 4.6mm, 5mu; mobile phase (A) 0.1% TEA in n-Hexane (B) Ethanol (A: B : 75 :25); flow Rate: 1.00 mL/min); Optical rotation [a]D2: +8.65 (c = 0.25, DCM); TLC: 10% MeOH/DCM (Rf: 0.6).
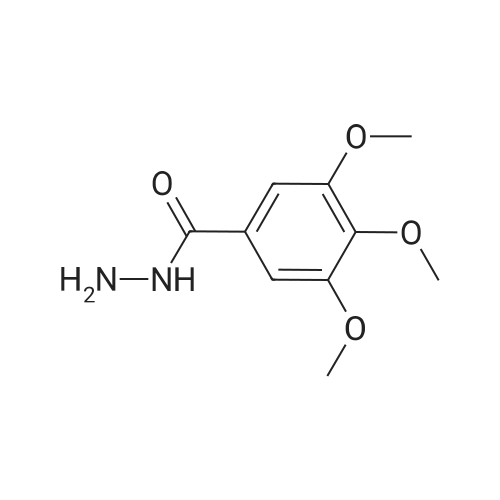
(E)-3,4,5-trimethoxy-N'-(quinoxalin-6-ylmethylene)benzohydrazide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
In methanol; for 2h;Reflux;
General procedure: The hydrazones were synthesized from the 3,4,5-trimethoxybenzohydrazide (2 mmol) or benzohydrazide(2 mmol) and the appropriate aldehydes (2 mmol) in methanol(15 mL) and refluxed for 2 h. After cooling, the crude product wascollected by filtration, washed and recrystallized from hot ethanolto give white solids [11]. Hydrazones 10, 22, 26 and 34 are novelcompounds, which were recently patented by our group [25].
2-(quinoxalin-6-ylmethylene)malononitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
With piperidine; In ethanol; at 20℃; for 1h;
Piperidine (4.7 pL, 0.047 mmol) was added to a solution of malononitrile (34 mg, 0.52 mmol) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (75 mg, 0.47 mmol) in EtOH (1 mL). The mixture was stirred at ambient temperature for 1 h. The resulting precipitate was collected and washed with cold EtOH to give the intermediate as a light brown solid (66 mg, 68%). ?H NMR (400 MHz, CDC13) 8.97 (s, 2H), 8.55 (d, J= 2.1 Hz, 1H), 8.37 (dd, J= 8.9, 2.1 Hz, 1H), 8.27 (d, J = 8.9 Hz, 1H), 8.01 (s, 1H).
68%
With piperidine; In ethanol; at 20℃; for 1h;
Piperidine (4.7 muIota_, 0.047 mmol) was added to a solution of malononitrile (34 mg, 0.52mmol) and <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (75 mg, 0.47 mmol) in EtOH (1 mL). The mixture was stirred at ambient temperature for 1 h. The resulting precipitate was collected and washed with cold EtOH to give the intermediate as a light brown solid (66 mg, 68%). 1 H NMR (400 MHz, CDCl3) delta 8.97 (s, 2H), 8.55 (d, J = 2.1 Hz, 1 H), 8.37 (dd, J = 8.9, 2.1 Hz, 1 H), 8.27 (d, J = 8.9 Hz, 1 H), 8.01 (s, 1 H).
Acetic acid (0.023 mL, 0.4 mmol) was added to a stirred suspension of intermediate 17 (40 mg, 0.19 mmol), <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (CAS: 130345-50-5; 40 mg, 0.25mmol) in MeOH (1 mL) at rt and under N2 atmosphere. The mixture was further stirred at rt for 1 h and then sodium cyanoborohydride (25 mg, 0.4 mmol) was added. The mixture was further stirred at rt for 16 h. The reaction mixture was quenched with Na2CO3 (aq. sat. soltn.) and diluted with DCM. The organic layer was separated, dried over MgSO4, filtered and the filtrate was evaporated in vacuo. The residue thusobtained was purified by flash column chromatography (SiO2 amino functionalized, EtOAc in heptane, 0/100 to 100/0). The desired fractions were concentrated in vacuo to yield product 10 as yellow oil (11 mg, 16% yield).
Acetic acid (0.020 mL, 0.35 mmol) was added to a stirred suspension of intermediate 19 (34 mg, 0.17 mmol), <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (CAS: 130345-50-5; 37 mg, 0.23 mmol) in MeOH (1 mL) at rt and under N2 atmosphere. The mixture was further stirredat rt for 2.5 h and then sodium cyanoborohydride (34 mg, 0.54 mmol) was added. The mixture was further stirred at rt for 60 h. The reaction mixture was quenched with NaHCO3 (aq. sat. soltn.) and diluted with DCM. The organic layer was separated, dried over Mg504, filtered and the filtrate was evaporated in vacuo. The residue thus obtained was purified by reverse phase HPLC (Stationary phase: C18 XBridge 30 x100 mm 5 jim, mobile phase: gradient from 81% lOmIVI NH4CO3H pH 9 solution in water, 19% CH3CN to 64% 10mM NH4CO3H pH 9 solution in water, 36% CH3CN). The desired fractions were collected and concentrated in vacuo to yield product 12 as yellow oil (12.4 mg, 22% yield).
Diisopropylethylamine (0.177 mL, 1.03 mmol) was added to a stirred solution of intermediate 2a (50 mg, 0.21 mmol, HC1 salt) in DCM (1.1 mL) at rt and the mixture was stirred at rt for 5 mi <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (CAS: 130345-50-5; 39 mg, 0.24 mmol) and sodium triacetoxyborohydride (65.5 mg, 0.31 mmol) were added and the mixture was stirred at rt for 16 h. The reaction mixture was quenched withNaHCO3. The organic layer was separated, dried over MgSO4, filtered and the filtrate was evaporated in vacuo. The residue thus obtained was purified by flash column chromatography (silica gel, MeOH in DCM, 0/100 to 10/90). The desired fractions were concentrated in vacuo to yield product 18 as a colorless sticky solid (33 mg, 46% yield).46
With sodium cyanoborohydride; acetic acid; In methanol;
General procedure: The following compounds were prepared following a reductive amination procedurelike the one described for the preparation of product 20 starting from the corresponding amine and aldehyde intermediates using sodium triacetoxyborohydride in DCM.Changes of solvent, reductant are mentioned in the Table below. In the case a base or acid was used this is also noted in the Table A below.
Triethylamine (0.29 mL, 2.01 mmol) was added to a stirred solution of intermediate 27 (150 mg, 0.52 mmol, bis hydrochloric acid salt) in DCM (2.5 mL) and the mixture was stirred at rt for 2 min. Then <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (CAS: 130345-50-5; 82.3 mg, 0.52 mmol) followed by sodium cyanoborohydride (46 mg, 0.73 mmol) were added at rt. The mixture was further stirred at rt for 15 h. The reaction mixture was diluted with NaHC03 (aq. sat. soltn.) and DCM. The organic layer was separated, dried over MgS04, filtered and the filtrate was evaporated in vacuo. The residue was purified by RP HPLC (Stationary phase: CI 8 XBridge 30 x 100 mm 5 mupiiota), Mobile phase: Gradient from 81 % 10 mM NH4CO3H pH 9 solution in water, 19% CH3CN to 64% 10 mM NH4CO3H pH 9 solution in water, 36% CH3CN), to yield product 41 (82 mg, 44%)) as a white solid. (0594) Product 41 (111 mg) was separated into enantiomers via chiral SFC [Stationary phase: CHIRALCEL OJ-H 5muiotaeta 250x20mm, Mobile phase: 83% C02, 17% MeOH (0.3% iPrNH2)] yielding product 42 (46 mg) and product 43 (47 mg).
Acetic acid (0.03 mL, 0.52 mmol) was added to a stirred solution of Intermediate 3a (55 g, 0.24 mmol) and <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (CAS: 130345-50-5; 49 mg, 0.31 mmol) in MeOH (1 mL) at rt. The solution was stirred for at rt for 2.5 h. Then sodium cyanoborohydride (37 mg, 0.59 mmol) was added and the mixture was further stirred at rt for 60 h. Then, NaHC03 (aq. sat. soltn.) and DCM were added to the mixture. The organic layer was separated, dried over MgS04, filtered and the filtrate was evaporated in vacuo. The crude product was purified by RP HPLC (Stationary phase: CI 8 XBridge 30 x 100 mm 5 muiotaeta), mobile phase: gradient from 81% lOmM NH4CO3H pH 9 solution in water, 19% CH3CN to 64% lOmM NH4C03H pH 9 solution in water, 36% CH3CN). The desired fractions were collected and extracted with EtOAc and DCM/2-PrOH (9/1). The desired fractions were collected and concentrated in vacuo. The crude product was purified by ion exchange chromatography (ISOLUTE SCX-2, MeOH and then 7N solution of NH3 in MeOH). The desired fractions were collected and concentrated in vacuo to yield to yield product 4 (20.3 mg, 23% yield) as yellow oil.
Acetic acid (0.03 mL, 0.52 mmol) was added to a stirred solution of Intermediate 3b (54 mg, 0.23 mmol) and <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (CAS: 130345-50-5; 53 mg, 0.33 mmol) in MeOH (1 mL) at rt. The solution was stirred for at rt for 2.5 h. Then sodium cyanoborohydride (43 mg, 0.68 mmol) was added and the mixture was further stirred at rt for 60 h. Then, NaHC03 (aq. sat. soltn.) and DCM were added to the mixture. The organic layer was separated, dried over MgS04, filtered and the filtrate was evaporated in vacuo. The crude product was purified by RP HPLC (Stationary phase: CI 8 XBridge 30 x 100 mm 5 muiotaeta), mobile phase: gradient from 81% lOmM NH4CO3H pH 9 solution in water, 19% CH3CN to 64% lOmM NH4CO3H pH 9 solution in water, 36%> CH3CN). The desired fractions were collected and extracted with EtOAc and DCM/2-PrOH (9/1). The desired fractions were collected and concentrated in vacuo. The crude product was purified by ion exchange chromatography (ISOLUTE SCX-2, MeOH and then 7N solution of NH3 in MeOH). The desired fractions were collected and concentrated in vacuo to yield to yield product 5 (16.5 mg, 19%) yield) as yellow oil.
General procedure: Titanium tetraisopropoxide (0.09 mL, 0.31 mmol) and <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (CAS: 130345-50-5; 49 mg, 0.31 mmol) were added to a stirred mixture of (0350) Intermediate 8 (50 mg, 0.2 mmol) in DCM (0.63 mL) at rt. The mixture was stirred at rt for 18 h. Then, the reaction mixture was cooled to 0 C and methylmagnesium bromide (0.73 mL, 1.02 mmol; 1.4 M solution in THF) was added followed by THF (0.6 mL). The mixture was stirred at 0 C for 5 min and then at rt for 3 h. The reaction mixture was quenched with NH4C1 (aq. sat. soltn.) and extracted with DCM. The organic layer was separated, dried over Na2S04, filtered and the filtrate was evaporated in vacuo. The residue thus obtained was purified by flash column chromatography (silica gel, MeOH in DCM, 0/100 to 10/90). The desired fractions were concentrated in vacuo to yield product 15 (20 mg, 24% yield) as a brown sticky solid.
Diisopropylethylamine (0.17 mL, 0.98 mmol) was added to a stirred suspension of Intermediate 8 (70 mg, 0.19 mmol) in DCM (1 mL) at rt and the mixture was stirred at rt for 5 min. Then, <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (CAS: 130345-50-5; 38 mg, 0.22 mmol) and sodium triacetoxyborohydride (62 mg, 0.29 mmol) were added and the mixture was further stirred at rt for 16 h. The reaction mixture was quenched with NaHCC"3 (aq. sat. soltn.). The organic layer was separated, dried over MgS04, filtered and the filtrate was evaporated in vacuo. The residue thus obtained was purified by flash column chromatography (silica gel, MeOH in DCM, 0/100 to 10/90). The desired fractions were concentrated in vacuo to yield product 14 (43 mg, 57% yield) as a white solid.
Titanium tetraisopropoxide (0.09 mL, 0.31 mmol) and <strong>[130345-50-5]6-quinoxalinecarboxaldehyde</strong> (CAS: 130345-50-5; 49 mg, 0.31 mmol) were added to a stirred mixture of (0350) Intermediate 8 (50 mg, 0.2 mmol) in DCM (0.63 mL) at rt. The mixture was stirred at rt for 18 h. Then, the reaction mixture was cooled to 0 C and methylmagnesium bromide (0.73 mL, 1.02 mmol; 1.4 M solution in THF) was added followed by THF (0.6 mL). The mixture was stirred at 0 C for 5 min and then at rt for 3 h. The reaction mixture was quenched with NH4C1 (aq. sat. soltn.) and extracted with DCM. The organic layer was separated, dried over Na2S04, filtered and the filtrate was evaporated in vacuo. The residue thus obtained was purified by flash column chromatography (silica gel, MeOH in DCM, 0/100 to 10/90). The desired fractions were concentrated in vacuo to yield product 15 (20 mg, 24% yield) as a brown sticky solid.
With Ag NPs decorated reduced graphene oxide; In methanol; at 20℃; for 0.8h;Sonication;
General procedure: An equimolar mixture of aromatic aldehyde (2 mmol), methyl acetoacetate (1 mmol), aromatic amines (2 mmol), and 5 wt% Ag NPs/rGO nanocomposite in 5 mL methanol was introduced in a 20 mL heavy walled pear-shaped twonecked flask with non-standard tapered outer joint. The flask was attached to a 12 mm tip diameter probe and the reaction mixture was sonicated at an ambient temperature for the specified period at 50% power of the processor and in a 4 s pulse mode till a solid product separates out. When the reaction was completed, the resulting solid precipitate was filtered and dried along with the catalyst. Then, the solid precipitate was dissolved in acetone and the catalyst was recovered by filtration. This solution was concentrated at room temperature to generate the crude product. The crude product was purified by crystallization from ethanol.
Diisopropylethylamine (0.18 mL, 1.05 mmol) was added to a suspension of intermediate 86 (50 mg, 0.21 mmol, hydrochloric acid salt) in DCM (1.1 mL) and the mixture was stirred at rt for 5 minutes. Then <strong>[130345-50-5]quinoxaline-6-carbaldehyde</strong> (CAS: 130345-50-5; 40 mg, 0.25 mmol) and sodium triacetoxyborohydride (89 mg, 0.42 mmol) were added and the mixture was stirred at rt for 17 h. Then aqueous NaHC03 (aq. sat. soltn.) was added and the organic layer was separated, dried (MgS04), filtered and concentrated in vacuo. The crude product was purified by RP HPLC (Stationary phase: C18 XBridge 30 x 100 mm 5 muiotaeta, Mobile phase: Gradient from 81 % 10 mM NH4CO3H pH 9 solution in water, 19 % CH3CN to 64 % 1 OmM NH4CO3H pH 9 solution in water, 36 % CH3CN). The desired fractions were collected and concentrated in vacuo affording product 58 as a yellow oil (18 mg, 25 % yield).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping