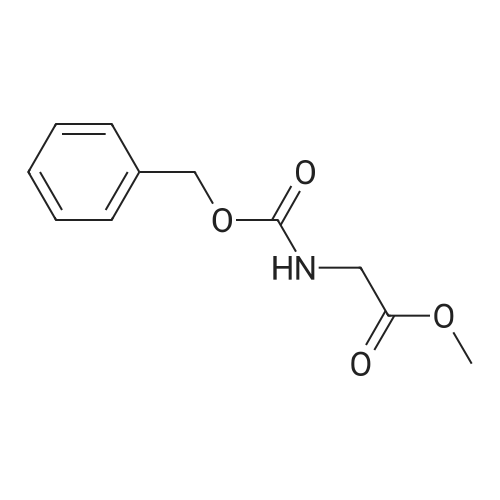
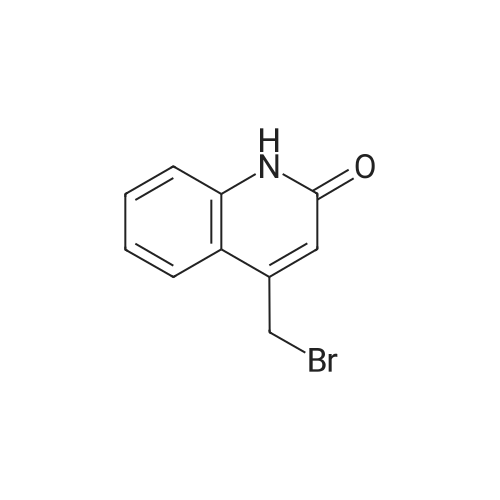
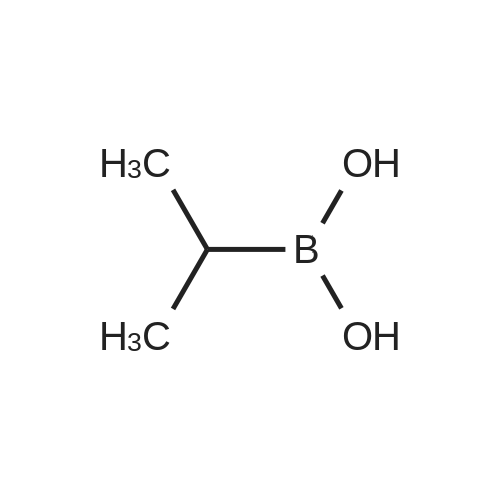
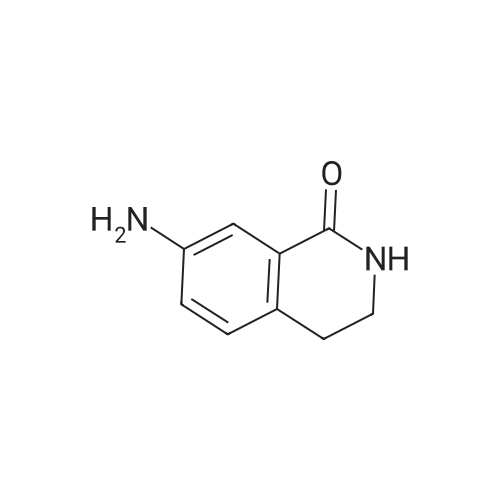
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Letters in Organic Chemistry, 2011, vol. 8, # 1, p. 48 - 52
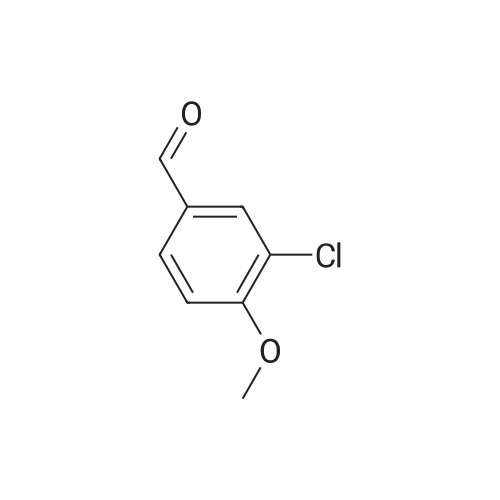
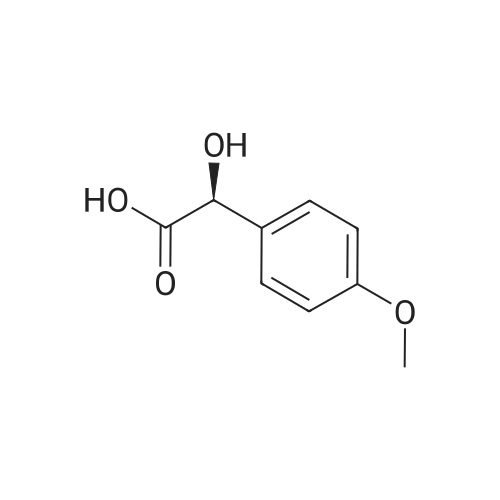
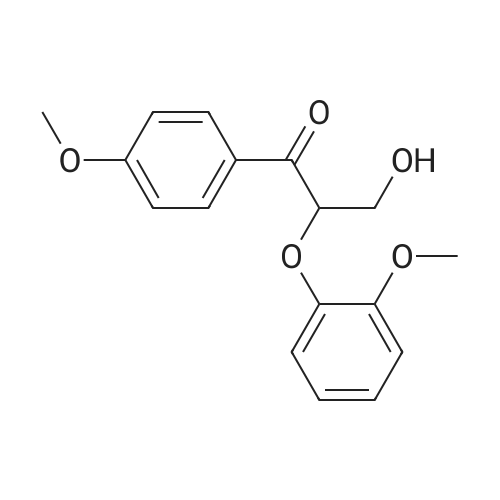
14
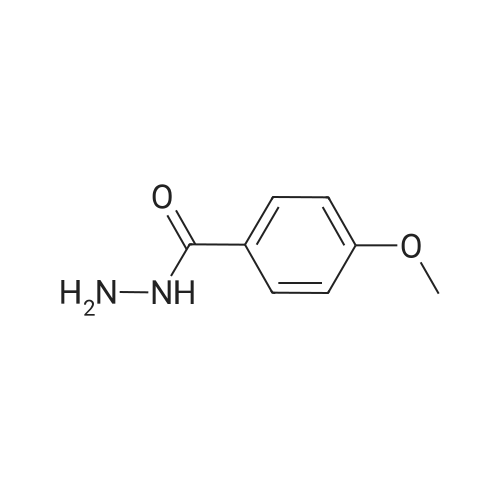
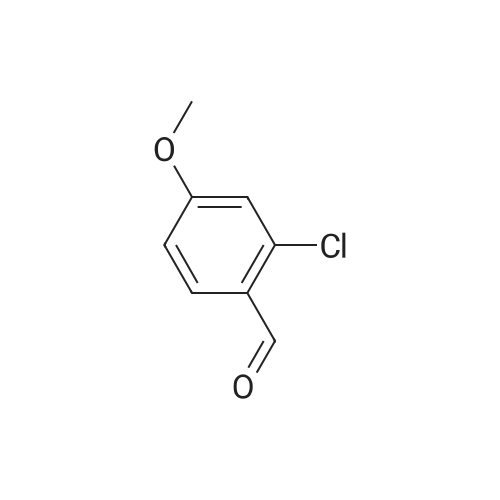
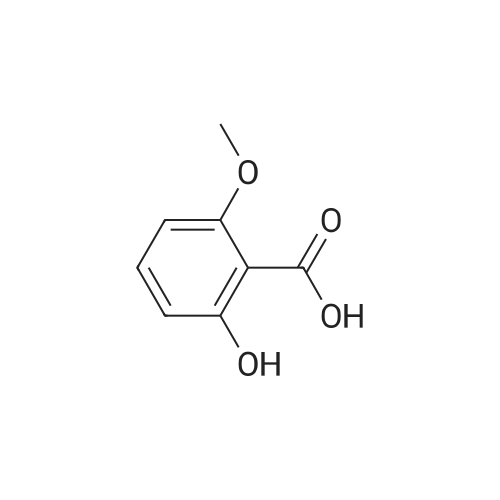
[ 123-11-5 ]
[ 78-93-3 ]
[ 104-27-8 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1927, vol. 2, p. 54,55[2] Sci. Rep. Tohoku Univ., Ser. 1: Phys., Chem., Astron., vol. 16, p. 535,537[3] Chem. Zentralbl., 1927 I,2730; II,1471,
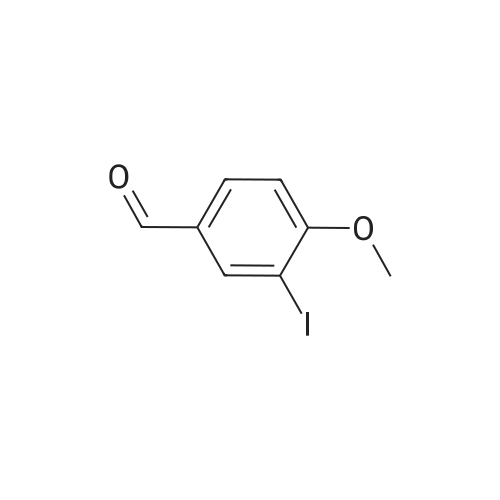
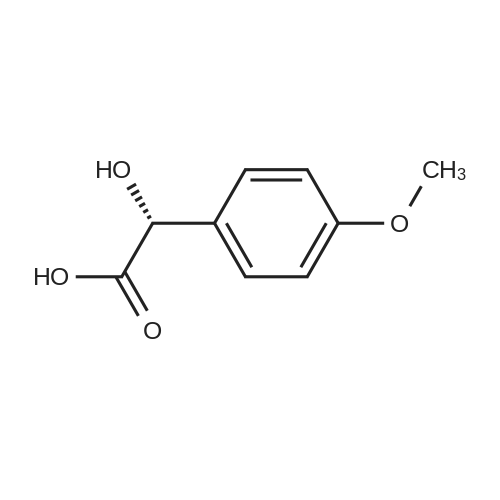
15
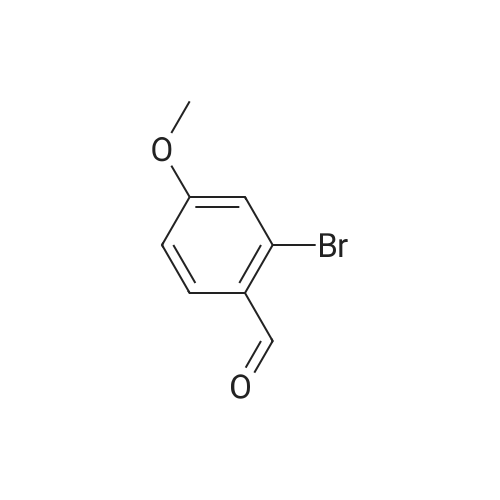
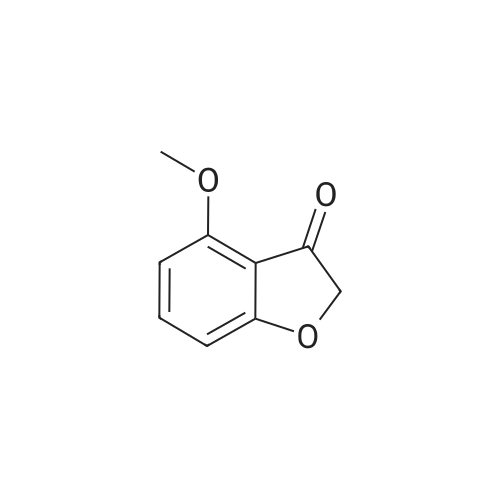
[ 105-13-5 ]
[ 78-93-3 ]
[ 5440-80-2 ]
[ 123-11-5 ]
[ 104-27-8 ]
Reference:
[1] Catalysis Science and Technology, 2017, vol. 7, # 9, p. 1928 - 1936
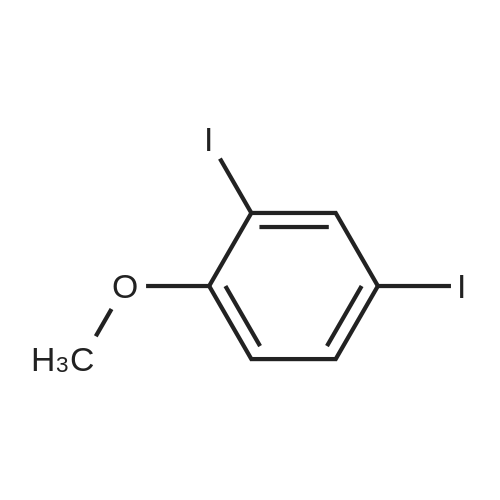
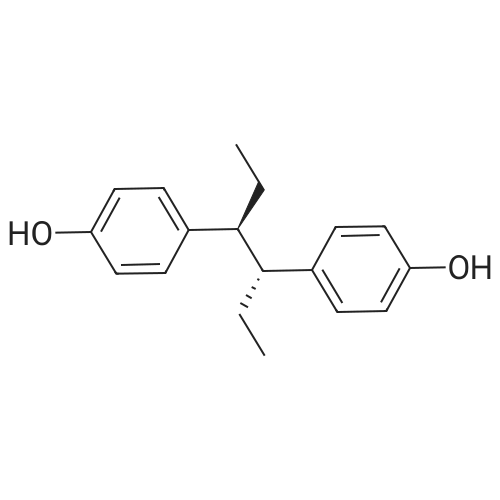
16
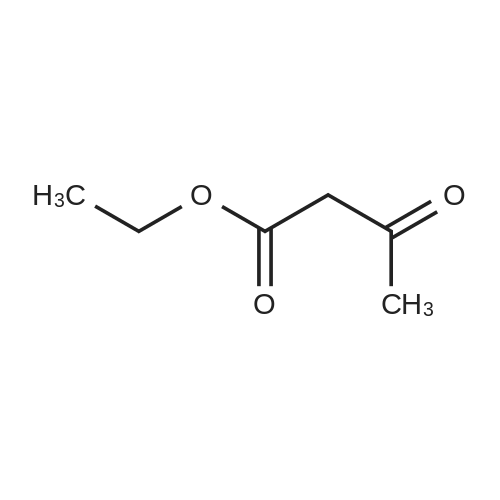
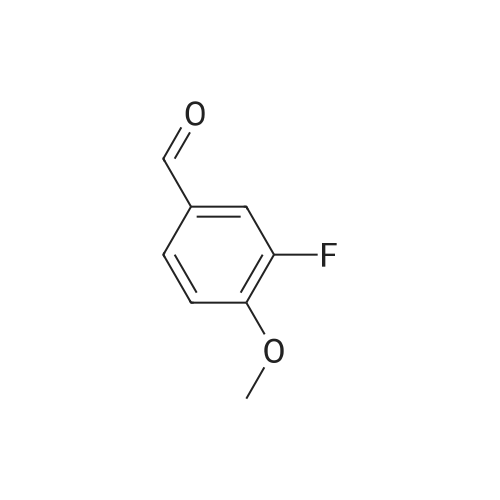
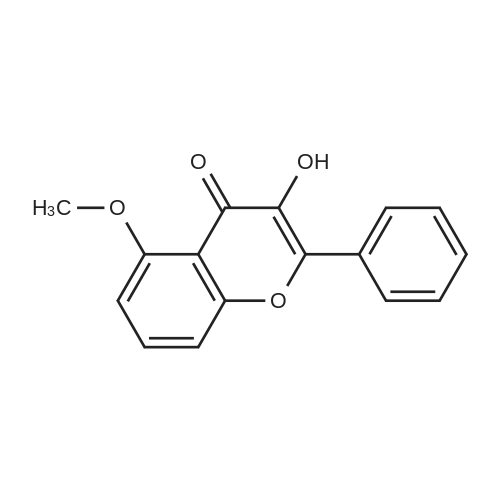
[ 123-11-5 ]
[ 107-13-1 ]
[ 3528-45-8 ]
Yield
Reaction Conditions
Operation in experiment
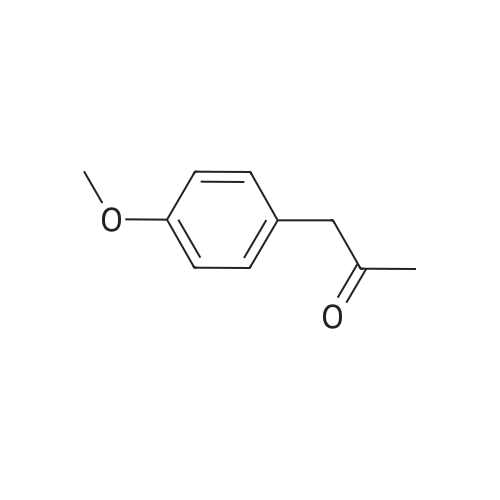
60%
Stage #1: With hydrazine In tetrahydrofuran; water at 0 - 20℃; for 2.33 h; Stage #2: for 3.25 h; Stage #3: With sodium butanolate In butan-1-ol at 25 - 120℃; for 3.33 h;
To a stirred solution of acrylonitrile (50.Og, 0.94 mol) in dry THF (200 ml_) cooled at O0C, hydrazine hydrate (49.5 g, 1.0 mol) is added over a period of about 20 min and the reaction mixture is stirred at RT for about 2 h. To the reaction mixture, p-anisaldehyde (134.7 g, 0.99 mol) is added over a period of about 15 min and stirred for about 3 h. The volatiles are evaporated under reduced pressure, then the reaction mixture is diluted with n-BuOH (200 mL). A solution of freshly prepared n-BuONa (90.1g, 0.94 mol) in n- BuOH (470 mL) is added dropwise at 250C for about 20 min and then the mixture is heated to 12O0C for about 3 h. The reaction mixture is cooled to RT and quenched by adding ice water (2 L). The crude product is extracted with Et2O (2 x 1 L). The combined organic layer is extracted with 1 N HCI (2 x 1 L). The pH of the aqueous layer is adjusted to approximately 14 with a 50percent NaOH solution and extracted with CH2CI2 (2 x 1 L). The combined organic layer is washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product is triturated with petroleum ether and the suspension is filtered. The light yellowish solid obtained is dried under vacuum to afford intermediate 1a (115 g, 60percent yield), which is used in the following step without purification.
60%
Stage #1: With hydrazine hydrate In tetrahydrofuran at 0 - 20℃; for 2 h; Stage #2: at 20℃; for 2 h; Reflux
To a solution of acrylonitrile (30 mL, 455 mmol) in THF (250 mL), NH2NH2 H20 (23.1 9 mL, 478 mmol) was added drop-wise at 0 °C After addition was complete, the mixture was stirred at rt for 2 h, then 4-methoxybenzaldehyde (55.4 mL, 455 mmol) was added drop-wise. The mixture was stirred at rt overnight, then at reflux for 2 h. After cooling to rt the mixture was quenched by addition of 300 mL of ice water. The mixture was extracted with ethyl acetate (3 x), then the combined organic layers were extracted with 1 N HCl. The aqueous layer was neutralized with aqueous 10 N NaOH solution, then extracted with ethyl acetate The organic layer was washed with H2O and brine, then dried over Na2SO4 . Filtration, concentration, and recrystrallization with Et2O gave the target compound as a white solid (50 g, 60percent).
60%
Stage #1: With hydrazine hydrate In tetrahydrofuran at 0 - 20℃; for 2 h; Stage #2: at 20℃; Reflux
To a solution of acrylonitrile (30 mL, 455 mmol) in THF (250 mL), NH2NH2 H20 (23.1 9 mL, 478 mmol) was added drop-wise at 0 °C After addition was complete, the mixture was stirred at rt for 2 h, then 4-methoxybenzaldehyde (55.4 mL, 455 mmol) was added drop-wise. The mixture was stirred at rt overnight, then at reflux for 2 h. After cooling to rt the mixture was quenched by addition of 300 mL of ice water. The mixture was extracted with ethyl acetate (3 x), then the combined organic layers were extracted with 1 N HC1. The aqueous layer was neutralized with aqueous 1 0 N NaOH solution, then extracted with ethyl acetate The organic layer was washed with H20 and brine, then dried over Na2S0 . Fi ltration, concentration, and recrystrallization with Et20 gave the target compound as a white sol id (50 g, 60percent).
60%
Stage #1: With hydrazine hydrate In tetrahydrofuran at 0 - 20℃; for 2 h; Stage #2: at 20℃; Reflux
1 H-Pyrazolo[3.4-b]pyridine-5-carboxylic acid Step 1 . 1 -(4-MethoxybenzvO- 1 H-pyrazol-5-amine. To a solution of acrylonitrile (30 mL, 455 mmol ) in THF (250 mL), NH2NH2 H20 (23. 1 mL, 478 mmol) was added drop-wise at 0 "C. After addition was complete, the mixture was stirred at rt for 2 h, then 4-methoxybenzaldehyde (55.4 mL, 455 mmol) was added drop-wise. The mixture was stirred at rt overnight, then at reflux for 2 h. After cooling to rt the mixture was quenched by addition of 300 mL of ice water. The mixture was extracted with ethyl acetate (3 x), then the combined organic layers were extracted with 1 N HC1. The aqueous layer was neutralized with aqueous 1 0 N NaOH solution, then extracted with ethyl acetate. The organic layer was washed with H20 and brine, then dried over Na2SOj. Fi ltration, concentration, and recrystrall ization with Et20 gave the target compound as a white solid (50 g, 60percent).
60%
Stage #1: With hydrazine hydrate In tetrahydrofuran at 0 - 20℃; for 2 h; Stage #2: at 20℃; Reflux
To a solution of acrylonitrile (30 mL, 455 mmol) in THF (250 mL), NH2NH2 H20 (23.1 9 mL, 478 mmol) was added drop-wise at 0 °C After addition was complete, the mixture was stirred at rt for 2 h, then 4-methoxybenzaldehyde (55.4 mL, 455 mmol) was added drop-wise. The mixture was stirred at rt overnight, then at reflux for 2 h. After cooling to rt the mixture was quenched by addition of 300 mL of ice water. The mixture was extracted with ethyl acetate (3 x), then the combined organic layers were extracted with 1 N HCl. The aqueous layer was neutralized with aqueous 10 N NaOH solution, then extracted with ethyl acetate The organic layer was washed with H2O and brine, then dried over Na2SO4 . Filtration, concentration, and recrystrallization with Et2O gave the target compound as a white solid (50 g, 60percent).
60%
Stage #1: With hydrazine hydrate In tetrahydrofuran at 0 - 20℃; for 2 h; Stage #2: at 20℃; Reflux
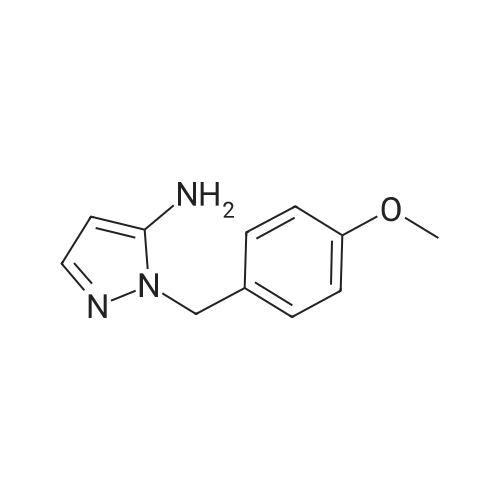
[0178] Step 1. 1-(4-Methoxybenzyl)-1H-pyrazol-5-amine. To a solution of acrylonitrile (30 mL, 455 mmol) in THF (250 mL), NH2NH2H2O (23.19 mL, 478 mmol) was added dropwise at 0 °C. After addition was complete, the mixture was stuffed at rt for 2 h, then 4- methoxybenzaldehyde (55.4 mL, 455 mmol) was added drop-wise. The mixture was stirred at rt overnight, then at reflux for 2 h. After cooling to rt the mixture was quenched by addition of 300 mL of ice water. The mixture was extracted with ethyl acetate (3 x), then the combined organic layers were extracted with 1 N HC1. The aqueous layer was neutralized with aqueous 10 N NaOH solution, then extracted with ethyl acetate. The organic layer waswashed with H20 and brine, then dried over Na2SO4. Filtration, concentration, and recrystrallization with Et20 gave the target compound as a white solid (50 g, 60percent).
60%
Stage #1: With hydrazine hydrate In tetrahydrofuran at 0 - 20℃; for 2 h; Stage #2: at 20℃; Reflux
0186] Step 1. l-(4-Methoxybenzyl)-lH-pyrazol-5-amine. To a solution of acrylonitrile (30 mL, 455 mmol) in THF (250 mL), NH2NH2 H20 (23.19 mL, 478 mmol) was added drop- wise at 0 °C. After addition was complete, the mixture was stirred at rt for 2 h, then 4- methoxybenzaldehyde (55.4 mL, 455 mmol) was added drop-wise. The mixture was stirred at rt overnight, then at reflux for 2 h. After cooling to rt the mixture was quenched by addition of 300 mL of ice water. The mixture was extracted with ethyl acetate (3 x), then the combined organic layers were extracted with 1 N HC1. The aqueous layer was neutralized with aqueous 10 N NaOH solution, then extracted with ethyl acetate. The organic layer was washed with H20 and brine, then dried over Na2S04. Filtration, concentration, and recrystrallization with Et20 gave the target compound as a white solid (50 g, 60percent).
30%
Stage #1: With hydrazine In ethanol at 20℃; for 20 h; Stage #2: at 20 - 30℃; for 60 h; Stage #3: With sodium hydroxide In ethanol; isopropyl alcohol at 120℃; for 2 h;
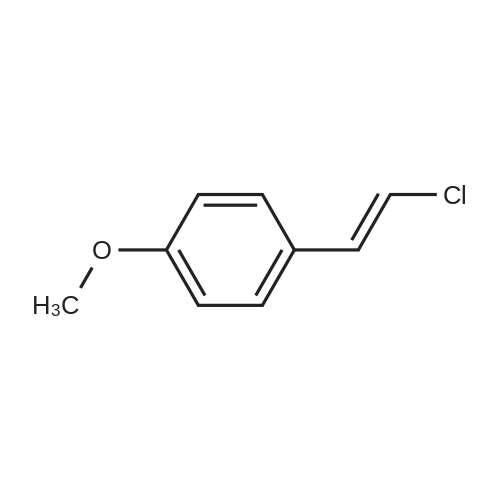
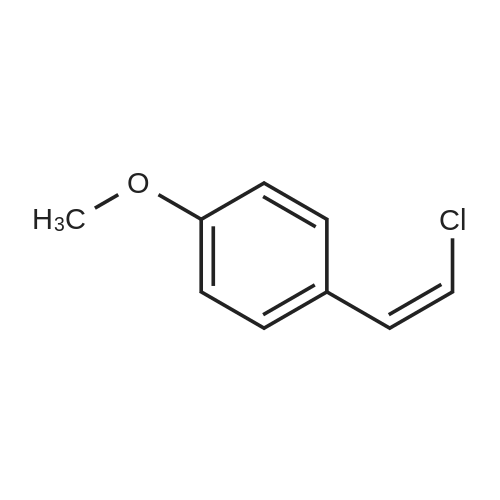
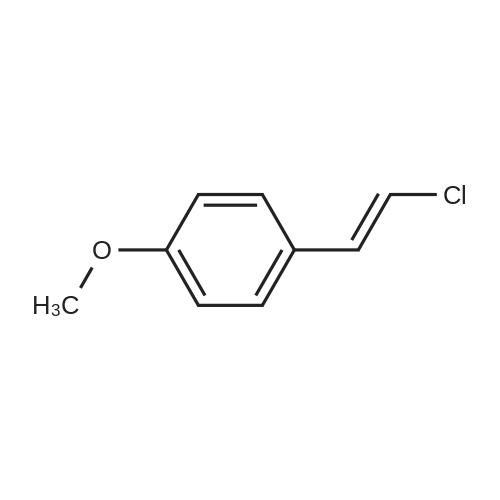
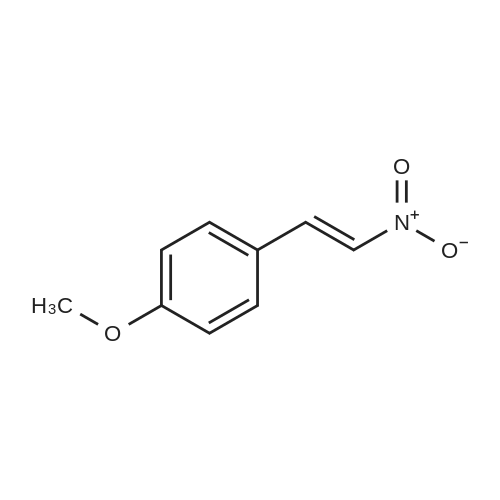
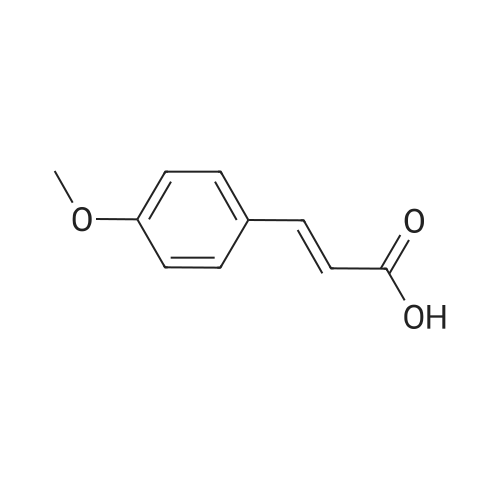
Hydrazine (10 mL, 320 mmol) was added over 10 min. to a vigorously stirred solution of 2-propenenitrile (22.3 mL, 339 mmol) in EtOH (100 mL), which was cooled in an ice-water bath to below 20° C. After stirring for 20 h, the reaction mixture was cooled in an ice-water bath, and 4-methoxybenzaldehyde (41.1 mL, 338 mmol) was added slowly. The reaction mixture was stirred at room temperature for 60 h. The solution was concentrated under reduced pressure and the residue was dissolved in isopropyl alcohol (100 mL). NaOH (7 g, 200 mmol) was added and the resulting mixture was heated at 120° C. for 2 h. The solution was concentrated under reduced pressure and the residue was diluted with water and EtOAc. The layers were separated and the aqueous layer was then extracted with further EtOAc. The combined organic extract was washed with 1 M HCl. The HCl layers were combined and the pH was adjusted to 14 using NaOH. The resulting slurry was extracted with DCM. The DCM layers were dried over Na2SO4, filtered and concentrated under reduced pressure to afford 21 g of the sub-title compound (30percent). LCMS calc. for C11H14N3O (M+H)+: m/z=204.1. Found: 204.2.
12.8%
Stage #1: With hydrazine hydrate In ethanol at 0 - 20℃; for 24 h; Inert atmosphere Stage #2: at 0 - 20℃; for 24 h; Inert atmosphere Stage #3: With sodium hydroxide In isopropyl alcohol for 2.5 h; Reflux
In a round-bottom flask, equipped with a stirrer, thermometer and reflux condenser, mix under nitrogen in the specified order: 120 mL of ethanol and 23.4 g (0.436 mol) of acrylonitrile. Cool the mixture to 0 °; add, dropwise, 21 g (0.414 mol) of hydrazine hydrate, maintaining the temperature. Stir the mixture for 24 hours at room temperature. Cool the reaction mass to 0 °, add 60 g (0.436 mol) of para-methoxybenzaldehyde, and stir at room temperature for 24 hours. After that, distill off the solvent, dissolve the residue in 130 mL of 2- propanol, add 9.2 g (0.23 mol) of NaOH, and boil for 2.5 hours. After that, concentrate the reaction mass, dissolve the residue in water, and extract with 125 mL of ethyl acetate four times. Combine the organic layers and wash with 200 mL of 2M hydrochloric acid. Separate the aqueous layer. Neutralize the aqueous layer with 2M NaOH and extract the product with 100 mL of dichloromethane four times. Wash the organic layer with water, dry with sodium sulfate, and distill off the solvent. Purify the resulting product by column chromatography, eluent hexane : ethyl acetate (1:1). Yield: 10.58 g (12.8percent).
11%
Stage #1: With hydrazine hydrate In ethanol at 20℃; for 16 h; Stage #2: at 20℃; for 16 h; Stage #3: With sodium hydroxide In butan-1-ol at 120℃; for 6 h;
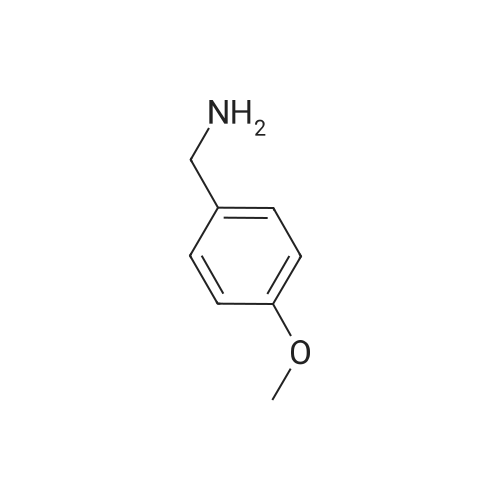
Intermediate 18l-[4-(Methyloxy)phen l]methyl}-lH-pyrazol-5-amineHydrazine hydrate (12.82 g, 400 mmol) was added dropwise to a cooled ( volume under reduced pressure, poured onto 300 mL of water, and then extracted with Et20 (2 x 200 mL). The combined ether phases were extracted with IN HC1 (3 x 100 mL). The combined HC1 extracts were combined and cooled in an ice/water bath. Added next was 6N NaOH until basic (pH > 12). The contents were extracted with Et20 (4 x 100 mL), washed with water, dried over MgS04, filtered, and concentrated under reduced pressure. The residue was purified via silica gelchromatography (eluent : 0 - 50percent EtOAc:Hex). The final product was collected as 8.94 g (11percent). 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 3.79 (s, 3 H), 5.14 (s, 2 H), 5.55 (d, J=1.77 Hz, 1 H), 6.81 - 6.94 (m, 2 H), 7.12 (d, J=8.84 Hz, 2 H), 7.31 (d, J=2.02 Hz, 1 H).
Stage #1: at 20℃; for 3 h; Stage #2: With sodium butanolate In butan-1-ol at 120℃; for 3 h; Inert atmosphere
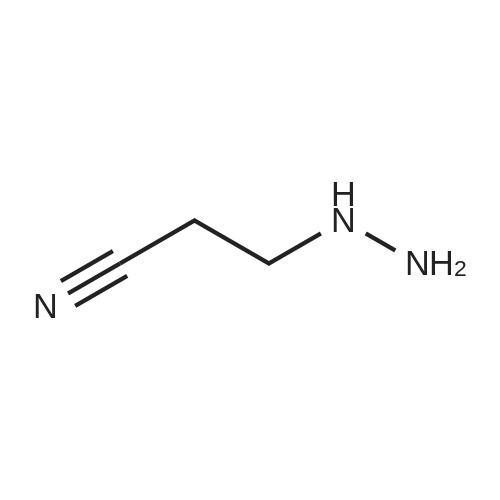
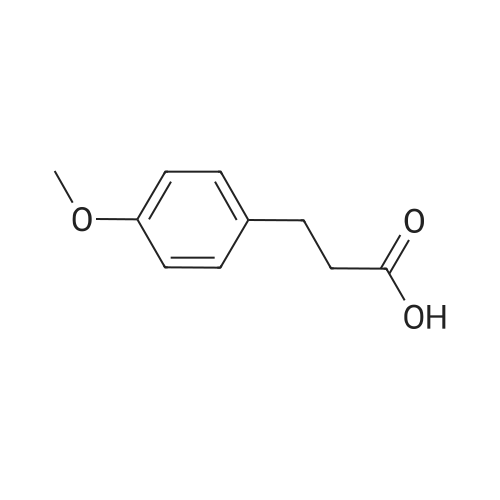
To a solution of 3-hydrazinylpropanenitrile (15.0 g, 0.176 mol) in anhydrous THF (60 mL) was added 4-methoxybenzaldehyde (25.4 g, 0.187 mol) dropwise. The resulting mixture was stirred at r.t. for 3 hrs. The volatiles were evaporated and the residue was diluted with -BuOH (38 mL). A freshly prepared solution of -BuONa (0.176 mol) in -BuOH (88 mL) was added dropwise to the resulting mixture, which was then heated to 120°C under N2 atmosphere and kept for 3 hrs. After cooling to r.t., the reaction mixture was quenched with ice water (375 mL) and extracted with DCM (375 mL). The organic phase was washed with brine, dried over Na2S04 and concentrated under reduced pressure. The crude product was purified by flash chromatography (silica gel, 0 to 50percent ethyl acetate in petroleum ether) to afford l-(4-methoxybenzyl)-lH-pyrazol-5-amine (1) (20.5 g, 57percent yield) as a yellow solid. LC-MS (ESI): m/z (M+ 1) 204.1
Stage #1: at 0 - 20℃; for 1.25 h; Stage #2: at 20℃; for 17 h; Stage #3: With potassium <i>tert</i>-butylate In butan-1-ol at 20 - 120℃; for 18.5 h; Sealed tube
Example 14Preparation of 3-(4-fluorophenylsulfonyl)-N-QH-pyrazol-3-yl)isoquinolin-l- amine[00260] Step A: To a stirred solution of acrylonitrile (7.96 g, 150 mmol) inTHF at 0 °C was added hydrazine monohydrate (9.70 g, 155 mmol) portionwise. The resulting mixture was stirred at 0 °C for 15 min, then warmed to rt and stirred for 1 h.To the mixture was added portionwise p-anisaldehyde (21.10 g, 155 mmol) and the mixture was stirred at rt for 17 h. The mixture was concentrated and n-butanol (30 mL) was added followed by a solution of sodium tert-butoxide (14.42 g, 150 mmol) in n-butanol (70 mL). The mixture was stirred at rt for 10 min, heated at 120 °C m a sealed tube for 3.5 h, then cooled to rt and stirred for 15 h. The mixture was poured into IN HC1 (600 mL) and extracted with diethyl ether (3x 100 mL). The aqueous phase was basified with IN sodium hydroxide and extracted with diethyl ether (3x 120 mL). The latter organic layers were combined, dried over MgS04, filtered, and concentrated under reduced pressure. The residue was triturated with hexane, filtered, and dried to afford l-(4-methoxybenzyl)-lH-pyrazol-5 -amine as a yellow solid (14 g, 46 percent). 1H NMR (300 MHz, CDC13) δ 7.30-7.26 (m, 1H), 7.12 (d, J= 8.5 Hz, 2H), 6.86 (d, J= 8.5 Hz, 2H), 5.55 (s, 1H), 5.15 (s, 2H), 3.78 (s, 3H), 3.37 (s, 2H); LC-MS (ESI) m/z 204 (M + H)+.
Reference:
[1] Synthesis, 2008, # 11, p. 1685 - 1687
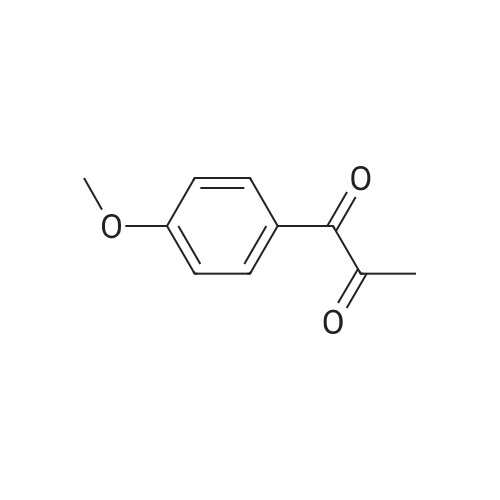
[2] Tetrahedron, 2005, vol. 61, # 4, p. 875 - 878
[3] Synthetic Communications, 2004, vol. 34, # 6, p. 1117 - 1123
[4] Tetrahedron, 1998, vol. 54, # 43, p. 13237 - 13252
[5] Journal of Medicinal Chemistry, 2012, vol. 55, # 5, p. 1831 - 1843
[6] Tetrahedron Letters, 2008, vol. 49, # 32, p. 4788 - 4791
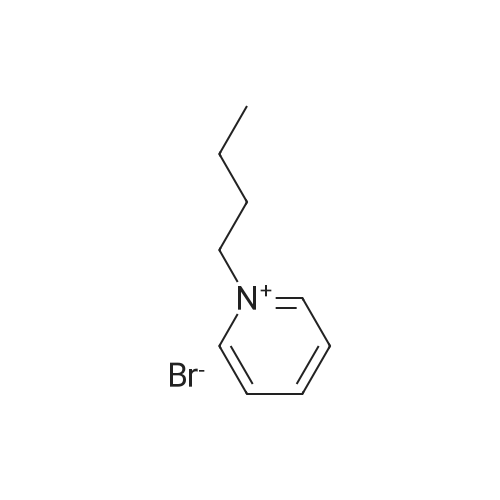
21
[ 623-73-4 ]
[ 123-11-5 ]
[ 2881-83-6 ]
Reference:
[1] Journal of Organic Chemistry, 1998, vol. 63, # 10, p. 3333 - 3336
[2] Journal of Organic Chemistry, 2004, vol. 69, # 22, p. 7599 - 7608
22
[ 123-11-5 ]
[ 105-36-2 ]
[ 2881-83-6 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2016, vol. 14, # 41, p. 9720 - 9724
23
[ 123-11-5 ]
[ 2881-83-6 ]
Reference:
[1] Synlett, 2006, # 10, p. 1547 - 1548
24
[ 123-11-5 ]
[ 2881-83-6 ]
Reference:
[1] Journal of Chemical Research, 2007, # 3, p. 160 - 161
[2] Chemistry - An Asian Journal, 2011, vol. 6, # 8, p. 2073 - 2079
[3] RSC Advances, 2013, vol. 3, # 31, p. 12616 - 12620
25
[ 123-11-5 ]
[ 13623-25-1 ]
Reference:
[1] Synthetic Communications, 1991, vol. 21, # 21, p. 2231 - 2256
[2] Synthetic Communications, 1991, vol. 21, # 21, p. 2231 - 2256
[3] Chemical Communications, 2011, vol. 47, # 22, p. 6290 - 6292
[4] Molecular Crystals and Liquid Crystals, 2011, vol. 545, p. 149 - 155
[5] Organic and Biomolecular Chemistry, 2017, vol. 15, # 35, p. 7374 - 7379
26
[ 123-11-5 ]
[ 62803-47-8 ]
Reference:
[1] Chemical Communications, 2011, vol. 47, # 22, p. 6290 - 6292
[2] Molecular Crystals and Liquid Crystals, 2011, vol. 545, p. 149 - 155
[3] Organic and Biomolecular Chemistry, 2017, vol. 15, # 35, p. 7374 - 7379
27
[ 3167-62-2 ]
[ 123-11-5 ]
[ 3167-63-3 ]
[ 477727-71-2 ]
[ 123098-29-3 ]
[ 768-60-5 ]
Reference:
[1] Canadian Journal of Chemistry, 1989, vol. 67, p. 1332 - 1343
28
[ 3167-62-2 ]
[ 123-11-5 ]
[ 3167-63-3 ]
[ 18684-79-2 ]
[ 18684-94-1 ]
[ 768-60-5 ]
Reference:
[1] Canadian Journal of Chemistry, 1989, vol. 67, p. 1332 - 1343
29
[ 123-11-5 ]
[ 34841-06-0 ]
Yield
Reaction Conditions
Operation in experiment
87%
With N-Bromosuccinimide In neat (no solvent) at 20℃; for 1 h; Milling; Green chemistry
General procedure: 1-Methoxy-3,5-dimethylbenzene(100mg, 0.73 mmol), N-Bromosuccinimide (NBS,260 mg,1.46 mmol) and one ball (5 mmdiameter, stainless steel) were transferred to a milling jar (10 mL, stainlesssteel). The ball-milling operation was performed and the progress of reaction was monitored by TLC/1H NMR.[1]After completion, the reaction mixture was transferred into 30 mL ethyl acetate and cooled at 0 °C. The product was isolated as filtrate upon paper filtration and waste succinimide as precipitate. The resulting filtrate were concentrated in vacuo to isolate 250 mg (yield: 85percent) of 2b as colourless powder. To test the efficiency in large scale, the reaction was also performed for the mono-bromination of 1-methoxy-3,5-dimethylbenzene in 1.3 g scale for 1 h and the product was isolated in 87percent yield.[1] The milling apparatus was stopped and small portion of the sample was collected from the reaction jar to study either TLC/ proton NMR. Following, the reaction was started again andthis operation time was excluded for reporting the reaction timing.
87%
With N-Bromosuccinimide In neat (no solvent) at 20℃; for 1 h; Milling; Green chemistry
General procedure: 1-Methoxy-3,5-dimethylbenzene (100mg, 0.73 mmol), N-Bromosuccinimide (NBS,260 mg,1.46 mmol) and one ball (5 mmdiameter, stainless steel) were transferred to a milling jar (10 mL, stainlesssteel). The ball-milling operation was performed and the progress of reactionwas monitored by TLC/1H NMR.[1]After completion, the reaction mixture was transferred into 30 mL ethyl acetateand cooled at 0 °C. The product was isolated as filtrate upon paper filtrationand waste succinimide as precipitate. The resulting filtrate were concentrated in vacuoto isolate 250 mg (yield: 85percent) of 2bas colourless powder. To test the efficiency in largescale, the reaction was also performed for the mono-bromination of1-methoxy-3,5-dimethylbenzene in 1.3 g scale for 1 h and the product wasisolated in 87percent yield.[1] Themilling apparatus was stopped and small portion of the sample was collectedfrom the reaction jar to study either TLC/ proton NMR. Following, the reaction was started again andthis operation time was excluded for reporting the reaction timing.
87%
With N-Bromosuccinimide; iodine In acetonitrile for 12 h; Darkness; Inert atmosphere
General procedure: To a reaction tube charged with NBS (1.5 equiv, 0.3 mmol), catalyst (10 molpercent, 0.02 mmol) and CH3CN (1.0 mL),was added para-chloroanisole 1a (0.2 mmol). After being stirred at room temperature for 12 h in dark, the reaction was quenched by saturated aq. solution of Na2S2O3 (2 mL). The resulting mixture was extracted by ethyl acetate (3 5 mL). The combined organic extracts were washed by brine (10 mL), dried over Na2SO4 and filtered through a pad of Celite. The filtrate was concentrated under reduced pressure and the residuewas purified by flash chromatography on a silica gel column with petroleum ether/dichloromethane (5:1) as the eluent to give 4.3.1. 2-Bromo-4-chloroanisole (2a)
82%
With iodine pentoxide; potassium bromide In water at 20℃; for 20 h;
General procedure: A mixture of arene (0.5 mmol), I2O5 (334 mg, 1.0 mmol), and KBr (148 mg, 1.25 mmol) was dissolved in 2mL of H2O. The reaction was complete after stirring for the indicated time at room temperature. The mixture was extracted by ethyl acetate and concentrated under reduced pressure, and the mixture was purified by flash column chromatography (silica gel) to afford the desired product.
77%
With N-Bromosuccinimide; thioacetamide In acetonitrile at 20℃; for 20 h;
General procedure: Reaction conditions: Thiourea (5.1 molpercent, 2 mg, 0.026 mmol) was added to an acetonitrile solution (10 mL) containing NBS (1.15 equiv, 104.4 mg, 0.587 mmol). Anisole (56.3 mg, 0.51 mmol) was added immediately to the resulting stirred solution and allowed to stir at room temperature for 10 min. The reaction was quenched by the addition of 10percent aqueous solution of Na2S2O3 (10 mL) and extracted with ethyl acetate (70 mL). The organic solution was then washed with additional 10percent Na2S2O3 (2 * 10 mL), followed by deionized water (3 * 15 mL) and brine (2 * 10 mL). The organic solution was then dried over anhydrous Na2SO4 and the solvent was evaporated in vacuo. The major product of each reaction was isolated by centrifugal thin-layer chromatography using a 2 mm thick silica gel 60GF254 coated plate (5percent CH2Cl2/hexanes). The products reported herein are known compounds and were characterised by GC-MS, IR, 1H and 13C NMR. Their spectroscopic data are in agreement with those reported in the literature.
55%
With lithium bromide monohydrate; [bis(acetoxy)iodo]benzene In 2,2,2-trifluoroethanol at 20℃; for 0.166667 h;
General procedure: To a solution of alkoxybenzylalcohol 1 (0.2 mmol) in CF3CH2OH (1 mL) were added LiBr·H2O (0.2 mmol) and PhI(OAc)2 (0.2 mmol) atroom temperature. After completion of the reaction as indicated by TLC monitoring, saturated aq. Na2SO3 wasadded and the mixture was extracted with CH2Cl2. The combined organic layers were washed with brine, driedover anhydrous Na2SO4 and then concentrated in vacuo. The residue was purified by silica gel columnchromatography to afford pure monobrominated compounds 2.
42.1%
With bromine In 1,2-dichloro-ethane at 0 - 60℃;
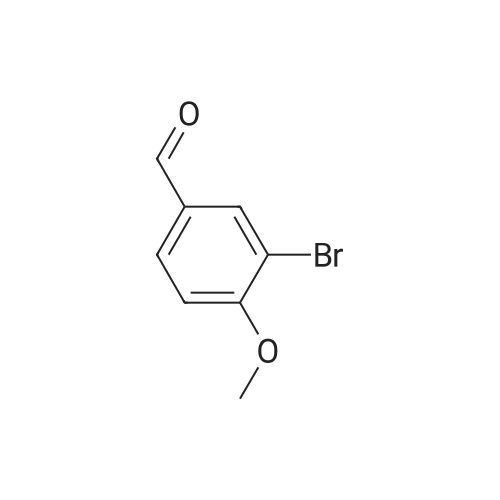
To a stirred solution of 4-methoxybenzaldehyde (30.0 g, 220.4 mmol) taken in DOE (300 mL), at 0 00 bromine (38.7 g, 242.2 mmol) was added drop wise. It was heated at 60 00 over night. The reaction mixture was quenched with ice water, and then extracted with ethyl acetate. The organic layer was washed with sodium thiosulphate,water and brine solution, was dried over anhydrous Na2SO4 and concentrated. The product was purified by column chromatography to yield the title product (20.0 g, 42.1percent) as an off white solid.
42.1%
With bromine In 1,2-dichloro-ethane at 0 - 60℃;
To a stirred solution of 4-methoxybenzaldehyde (30.0 g, 220.4 mmol) was taken inDOE (300 mL), at 0 00 was added bromine (38.7 g, 242.2 mmol) drop wise. It washeated at 60 00 for over night. The reaction mixture was quenched with ice water,and then extracted with ethyl acetate. The organic layer was washed with sodium thiosulphate, water and brine solution, over anhydrous Na2SO4 and concentrated. The product was purified by column chromatography to yield the title product (20.0 g, 42.1percent) as an off white solid.
Reference:
[1] Journal of the Iranian Chemical Society, 2011, vol. 8, # 2, p. 531 - 536
[2] Tetrahedron Letters, 2014, vol. 55, # 13, p. 2154 - 2156
[3] Tetrahedron Letters, 2015, vol. 55, # 13, p. 2154 - 2156
[4] Tetrahedron, 2017, vol. 73, # 50, p. 7105 - 7114
[5] Synthetic Communications, 2014, vol. 44, # 2, p. 181 - 187
[6] Tetrahedron Letters, 2009, vol. 50, # 2, p. 158 - 160
[7] Tetrahedron, 2017, vol. 73, # 46, p. 6564 - 6572
[8] Journal of Chemical Research, 2015, vol. 39, # 6, p. 336 - 339
[9] Chemistry of Natural Compounds, 1993, vol. 29, # 6, p. 748 - 759[10] Khimiya Prirodnykh Soedinenii, 1993, # 6, p. 839 - 853
[11] Synlett, 2018, vol. 29, # 17, p. 2275 - 2278
[12] New Journal of Chemistry, 2016, vol. 40, # 10, p. 8198 - 8201
[13] Patent: WO2014/202580, 2014, A1, . Location in patent: Page/Page column 82
[14] Patent: WO2014/202528, 2014, A1, . Location in patent: Page/Page column 69
[15] Journal of the American Chemical Society, 1917, vol. 39, p. 1711
[16] Justus Liebigs Annalen der Chemie, 1928, vol. 460, p. 135
[17] Justus Liebigs Annalen der Chemie, 1928, vol. 460, p. 135
[18] Journal of the Chemical Society, 1924, vol. 125, p. 1062
[19] Justus Liebigs Annalen der Chemie, 1845, vol. 56, p. 308[20] Annales de Chimie (Cachan, France), 1845, vol. <3> 14, p. 486
[21] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1979, p. 829 - 837
[22] Journal of the Chemical Society [Section] C: Organic, 1970, p. 2234 - 2238
[23] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1984, # 11, p. 1797 - 1802
[24] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 2, p. 463 - 466
[25] Tetrahedron Letters, 2009, vol. 50, # 7, p. 831 - 833
[26] Indian Journal of Heterocyclic Chemistry, 2012, vol. 21, # 3, p. 281 - 288
Reference:
[1] Journal of Organic Chemistry, 2000, vol. 65, # 12, p. 3880 - 3881
33
[ 123-11-5 ]
[ 54439-75-7 ]
[ 43192-31-0 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2007, vol. 5, # 1, p. 143 - 150
34
[ 123-11-5 ]
[ 351-54-2 ]
[ 654-11-5 ]
Reference:
[1] Journal of Fluorine Chemistry, 2007, vol. 128, # 1, p. 29 - 33
[2] Organic Process Research and Development, 2008, vol. 12, # 2, p. 339 - 344
35
[ 123-11-5 ]
[ 4903-09-7 ]
Reference:
[1] European Journal of Organic Chemistry, 2018, vol. 2018, # 4, p. 485 - 493
[2] Advanced Synthesis and Catalysis, 2013, vol. 355, # 14-15, p. 2936 - 2941
[3] Journal of Organic Chemistry, 2017, vol. 82, # 14, p. 7529 - 7537
[4] Tetrahedron, 1996, vol. 52, # 26, p. 8863 - 8866
[5] Journal of Medicinal Chemistry, 1998, vol. 41, # 18, p. 3367 - 3372
[6] New Journal of Chemistry, 2016, vol. 40, # 10, p. 8198 - 8201
[7] Justus Liebigs Annalen der Chemie, 1928, vol. 460, p. 135
[8] Journal of the Chemical Society, 1937, p. 1798,1804
[9] Journal of the Chemical Society, 1965, p. 954 - 973
[10] Journal of Medicinal Chemistry, 2002, vol. 45, # 24, p. 5260 - 5279
[11] Synthetic Communications, 2014, vol. 44, # 5, p. 660 - 664
36
[ 507-40-4 ]
[ 123-11-5 ]
[ 4903-09-7 ]
Reference:
[1] Journal of the American Chemical Society, 1951, vol. 73, p. 702
37
[ 473-34-7 ]
[ 123-11-5 ]
[ 4903-09-7 ]
Reference:
[1] Journal of the Chemical Society, 1956, p. 1417,1418, 1421
38
[ 507-40-4 ]
[ 123-11-5 ]
[ 64-19-7 ]
[ 4903-09-7 ]
Reference:
[1] Journal of the American Chemical Society, 1951, vol. 73, p. 702
39
[ 123-11-5 ]
[ 2314-37-6 ]
Yield
Reaction Conditions
Operation in experiment
85%
With N-iodo-succinimide; [bis(trifluoromethanesulfonyl)imidate](triphenylphosphine)gold(I) In 1,2-dichloro-ethane; toluene at 85℃; for 14 h;
General procedure: To a stirred solution of the substrate (1 mmol) in CH2Cl2 or (CH2Cl)2 (0.1 M) were added Ph3PAuNTf2 (0.025 mmol, 19 mg; complex Ph3PAuNTf2 toluene, 2:1) followed by N-iodosuccinimide (1.1 mmol, 248 mg). The resulting solution was stirred at r.t. or under reflux until complete conversion of the starting material. After removal of the solvent under reduced pressure, the crude material was purified by flash column chromatography using different gradients of hexanes and EtOAc to obtain the pure desired products.
36%
With iodine; Selectfluor In methanol at 50℃; for 21 h;
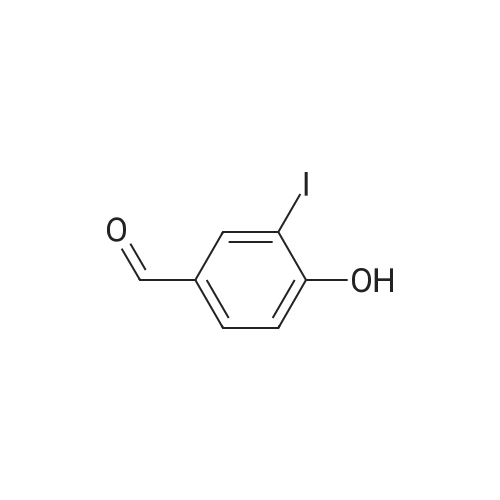
A mixture of p-anisaldehyde (1.00 g, 7.34 mmol), iodine (932 mg, 3.67 mmol) and selectfluor (1.56 g, 4.40 mmol) in 75 mL of methanol was heated at 50 °C for 21 h. The reaction was cooled and diluted with dichloromethane, washed with saturated aqueous sodium bicarbonate, dried over MgSO4 and concentrated to give a brown residue. The residue was purified by column chromatography, eluting with 20-40percent gradient EtOAc in hexanes, to give 690 mg (36percent) of 3-iodo-4-methoxybenzaldehyde as a yellow solid; MS 263.1 (M+H).
Reference:
[1] European Journal of Organic Chemistry, 2000, # 8, p. 1527 - 1533
[2] European Journal of Organic Chemistry, 2012, # 30, p. 5935 - 5942,8
[3] Synthesis, 2010, # 16, p. 2776 - 2786
[4] Organic letters, 2003, vol. 5, # 4, p. 415 - 418
[5] Tetrahedron Letters, 1997, vol. 38, # 35, p. 6305 - 6306
[6] Synlett, 2014, vol. 25, # 3, p. 399 - 402
[7] Synthetic Communications, 2007, vol. 37, # 21, p. 3855 - 3860
[8] Journal of Organic Chemistry, 2017, vol. 82, # 2, p. 1205 - 1217
[9] Organic Letters, 2018, vol. 20, # 23, p. 7726 - 7730
[10] Journal of Organic Chemistry, 2006, vol. 71, # 3, p. 1027 - 1032
[11] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 21, p. 6731 - 6734
[12] Patent: WO2009/76602, 2009, A1, . Location in patent: Page/Page column 124
[13] Journal of the Chemical Society, 1956, p. 1417,1418, 1421
[14] Journal fuer Praktische Chemie (Leipzig), 1899, vol. <2> 59, p. 143
[15] Tetrahedron Letters, 2004, vol. 45, # 43, p. 8083 - 8085
[16] Patent: WO2006/82245, 2006, A1, . Location in patent: Page/Page column 156
[17] Patent: US2005/65066, 2005, A1, . Location in patent: Page/Page column 69
[18] Patent: WO2004/80480, 2004, A1, . Location in patent: Page/Page column 150
[19] Patent: WO2006/5683, 2006, A1, . Location in patent: Page/Page column 151
[20] Patent: US2003/229120, 2003, A1, . Location in patent: Page 52
40
[ 105-13-5 ]
[ 2314-37-6 ]
[ 123-11-5 ]
Yield
Reaction Conditions
Operation in experiment
71%
With iodic acid In N,N-dimethyl-formamide at 60℃; for 2 h; Inert atmosphere
General procedure: To a solution of p-bromobenzyl alcohol I-1 (187 mg, 1.0 mmol) in DMF (2.0 mL) was added HIO3 (194 mg, 1.1 mmol). The mixture was stirred at 60 °C for 2 h under an Ar atmosphere. After the reaction, the reaction mixture was poured into aq Na2S2O3, and extracted with a mixture of Et2O: hexane=1:1 (3*10 mL). The organic layer was dried over Na2SO4. After being filtration and removal of the solvent under reduced pressure, the residue was purified by flash short column chromatography on silica gel (EtOAc-hexane, 1:4) to give p-bromobenzaldehyde II-1 in 95percent yield.
71%
With iodic acid In N,N-dimethyl-formamide at 20 - 60℃; for 2 h; Inert atmosphere
General procedure: 4-Bromobenzyl alcohol (187 mg, 1.0 mmol)Was dissolved in 2.0 mL of DMF solvent,This was mixed with iodic acid (194 mg, 1.1 mmol).The mixture was stirred for 2 hours at room temperature to 60 ° C. in an Ar atmosphere.To the reaction mixture was added aqueous sodium thiosulfate solution and extracted with diethyl ether: hexane = 1: 1 (3 × 10 mL).The extract was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure.The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give 4-bromobenzaldehyde as a final product (yield 91percent). The same treatment was carried out except that the primary alcohol or secondary alcohol was used instead of 4-bromobenzyl alcohol used in Example 1 based on the following general formula (1) to obtain a carbonyl compound shown in Table 1 Were obtained in yields shown in Table 1, respectively.
Reference:
[1] Tetrahedron, 2016, vol. 72, # 44, p. 6948 - 6954
[2] Patent: JP2018/2680, 2018, A, . Location in patent: Paragraph 0032-0035
Reference:
[1] Journal of the American Chemical Society, 1935, vol. 57, p. 909,911
47
[ 1068-55-9 ]
[ 123-11-5 ]
[ 18228-46-1 ]
Yield
Reaction Conditions
Operation in experiment
95%
Stage #1: at 8 - 20℃; for 1 h; Stage #2: With ammonium chloride In tetrahydrofuran; water; toluene
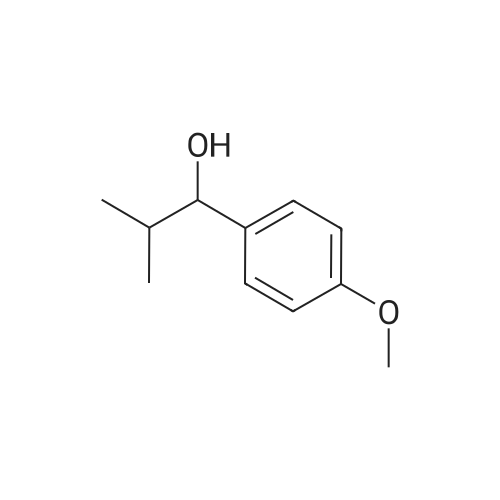
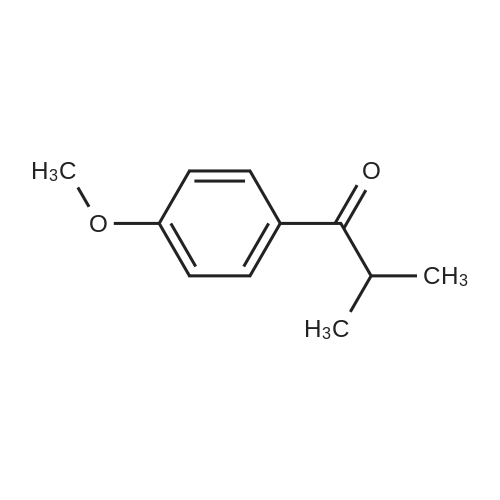
1-(4-methoxy-phenyl)-2-methyl-propan-1-ol A solution of 1.5 ml 4-methoxy-benzaldehyde in 10 ml of toluene is added dropwise at 8° C. to 7.4 ml of a 2 M solution of isopropylmagnesium chloride in tetrahydrofuran. Then the mixture is allowed to come up to ambient temperature and stirred for 1 hour. Then the reaction is stopped by the addition of semisaturated aqueous ammonium chloride solution. The phases are separated and the aqueous phase is extracted with toluene. The combined organic phases are washed with semisaturated aqueous ammonium chloride solution and dried on magnesium sulphate. The solvents are eliminated in vacuo and the product thus obtained is further reacted directly. Yield: 2.12 g (95percent of theory) HPLC (method 1): retention time=3.22 min.
Reference:
[1] Patent: US2011/269737, 2011, A1, . Location in patent: Page/Page column 63
[2] Australian Journal of Chemistry, 1986, vol. 39, # 12, p. 2095 - 2110
[3] Patent: US2597446, 1949, ,
48
[ 123-11-5 ]
[ 75-26-3 ]
[ 18228-46-1 ]
Reference:
[1] Journal of the American Chemical Society, 1980, vol. 102, # 27, p. 7926 - 7927
[2] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1973, p. 301 - 310
[3] Journal of Medicinal Chemistry, 1986, vol. 29, # 9, p. 1668 - 1674
[4] Organic Letters, 2014, vol. 16, # 17, p. 4650 - 4653
[5] Organic Letters, 2018, vol. 20, # 10, p. 2906 - 2910
49
[ 75-29-6 ]
[ 123-11-5 ]
[ 18228-46-1 ]
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 10, p. 3967 - 3969
Reference:
[1] Journal of Organic Chemistry, 2017, vol. 82, # 13, p. 7070 - 7076
55
[ 2033-24-1 ]
[ 123-11-5 ]
[ 1929-29-9 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2017, vol. 15, # 35, p. 7374 - 7379
56
[ 123-11-5 ]
[ 1929-29-9 ]
Reference:
[1] Synthetic Communications, 1991, vol. 21, # 21, p. 2231 - 2256
[2] Synthetic Communications, 1991, vol. 21, # 21, p. 2231 - 2256
[3] Journal of the Chemical Society, 1877, vol. 31, p. 409[4] Journal of the Chemical Society, 1878, vol. 33, p. 215
[5] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 10, p. 3192 - 3203
[6] Molecular Crystals and Liquid Crystals, 2011, vol. 545, p. 149 - 155
[7] Chemistry Letters, 2013, vol. 42, # 9, p. 1051 - 1052
[8] Journal of the Indian Chemical Society, 2013, vol. 90, # 10, p. 1853 - 1860
[9] Patent: CN106146450, 2016, A,
57
[ 123-11-5 ]
[ 141-78-6 ]
[ 1929-29-9 ]
Reference:
[1] Journal of the Chemical Society, 1909, vol. 95, p. 1722
58
[ 201230-82-2 ]
[ 100-66-3 ]
[ 6971-51-3 ]
[ 612-16-8 ]
[ 123-11-5 ]
[ 105-13-5 ]
Reference:
[1] Chemistry Letters, 1987, p. 1113 - 1116
59
[ 75-52-5 ]
[ 123-08-0 ]
[ 123-11-5 ]
[ 3179-08-6 ]
[ 3179-10-0 ]
Reference:
[1] Chemistry - A European Journal, 2009, vol. 15, # 29, p. 7052 - 7062
60
[ 99-91-2 ]
[ 123-11-5 ]
[ 6552-63-2 ]
Yield
Reaction Conditions
Operation in experiment
96.9%
With methanol; sodium In methanol at 20℃;
General procedure: Na (0.01 g, 0.4 mmol) was cut to pieces and added into anhydrous MeOH (15 mL) under ice bath; the fresh MeONa/MeOH solution was prepared till no gas evolved. 4-Methoxybenzaldehyde (1.36 g, 10.0 mmol) was added to the fresh, cooled MeONa/MeOH solution (15 mL), and then 10.0 mmol acetyl aromatic compound was added dropwise. The mixture was stirred at room temperaturefor2–8 h(36–54 hfor2nand2oduetothesteric hindrance of thenaphthyl andbiphenyl). Theend ofthereaction was monitored by TLC. Afterward, water (20 mL) and EtOAc (75 mL, 25 mL × 3) were added into the mixture. Theinorganic precipitatewas removed, and theorganic solution was washed with brine three times and dried. The crude product was puried by silica gel column chromatography with EtOAc/hexane as eluent to get pureα,β-unsaturated carbonyl compounds (2a–2o).
88.3%
With sodium hydroxide In ethanol at 0℃;
General procedure: The target compounds were synthesised in accordance with the reaction shown in Figures 1 and 2. Appropriate aldehyde (0.01 mol) and acetophenone derivatives (0.01 mol) were dissolved in anhydrous ethanol (15 mL). The reaction mixture was stirred at 0 °C for 8 h. Then, 10percent NaOH (5 mL) was slowly added to the above mixture under stirring until the reaction was complete. The precipitate was filtered and washed with still water. The pure compounds were obtained by re-crystallisation in acetone and water.
85%
at 20℃; for 0.666667 h; Neat (no solvent)
General procedure: To acetophenone (1 mmol) catalyst STA (0.1 mmol or 10 mol percent) was dissolved by stirring for 5-10 min. and further to this 4-chlorobenzaldehyde (1 mmol) was added and the mixture was magnetically stirred at room temperature until the completion of the reaction as confirmed by TLC. After the completion of the reaction the crude product was separated out, and then washed with water. The solid was filtered and dried. The crude product was further purified by recrystallization. All the products were characterised by their melting point, IR and 1H NMR and 13C NMR spectral analysis.
Reference:
[1] Bulletin of the Korean Chemical Society, 2016, vol. 37, # 2, p. 200 - 206
[2] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1994, vol. 33, # 5, p. 455 - 459
[3] Synthetic Communications, 2010, vol. 40, # 19, p. 2887 - 2896
[4] Medicinal Chemistry Research, 2012, vol. 21, # 6, p. 922 - 931
[5] Natural Product Research, 2015, vol. 29, # 19, p. 1804 - 1810
[6] European Journal of Medicinal Chemistry, 2010, vol. 45, # 11, p. 5292 - 5301
[7] Tetrahedron Letters, 2012, vol. 53, # 6, p. 646 - 649
[8] Acta Poloniae Pharmaceutica - Drug Research, 2012, vol. 69, # 5, p. 893 - 900
[9] Journal of the Chinese Chemical Society, 2017, vol. 64, # 1, p. 17 - 24
[10] Journal of the Indian Chemical Society, 2009, vol. 86, # 12, p. 1347 - 1351
[11] Journal of the Korean Chemical Society, 2013, vol. 57, # 2, p. 234 - 240
[12] Indian Journal of Chemistry, Section A: Inorganic, Physical, Theoretical and Analytical, 1990, vol. 29, # 1, p. 67 - 69
[13] Justus Liebigs Annalen der Chemie, 1918, vol. 415, p. 256
[14] Journal of the Indian Chemical Society, 1990, vol. 67, # 7, p. 571 - 574
[15] Australian Journal of Chemistry, 1997, vol. 50, # 4, p. 309 - 319
[16] Acta Poloniae Pharmaceutica - Drug Research, 2010, vol. 67, # 1, p. 55 - 61
[17] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 15, p. 4657 - 4660
[18] E-Journal of Chemistry, 2010, vol. 7, # 1, p. 295 - 298
[19] Journal of Heterocyclic Chemistry, 2010, vol. 47, # 6, p. 1350 - 1355
[20] Journal of Enzyme Inhibition and Medicinal Chemistry, 2011, vol. 26, # 2, p. 280 - 287
[21] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 18, p. 5409 - 5419
[22] Chemistry of Heterocyclic Compounds, 2011, vol. 47, # 5, p. 575 - 583
[23] Tetrahedron Letters, 2012, vol. 53, # 9, p. 1160 - 1162
[24] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 6, p. 2010 - 2018
[25] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 9, p. 3039 - 3043
[26] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2012, vol. 51, # 5, p. 774 - 779
[27] Asian Journal of Chemistry, 2010, vol. 22, # 7, p. 5581 - 5587
[28] Zeitschrift fur Anorganische und Allgemeine Chemie, 2012, vol. 638, # 7-8, p. 1224 - 1232
[29] European Journal of Medicinal Chemistry, 2012, vol. 55, p. 467 - 474
[30] Synlett, 2012, vol. 23, # 17, p. 2485 - 2490,6
[31] Journal of the Indian Chemical Society, 2013, vol. 90, # 2, p. 221 - 229
[32] Materials Research Bulletin, 2014, vol. 50, p. 446 - 453
[33] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2014, vol. 127, p. 261 - 267
[34] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 21, p. 5804 - 5812
[35] Chemistry and Biodiversity, 2015, vol. 12, # 1, p. 116 - 132
[36] Chinese Journal of Catalysis, 2016, vol. 37, # 4, p. 571 - 578
[37] European Journal of Medicinal Chemistry, 2016, vol. 112, p. 48 - 59
[38] New Journal of Chemistry, 2016, vol. 40, # 8, p. 6777 - 6786
[39] Helvetica Chimica Acta, 2016, vol. 99, # 8, p. 608 - 616
[40] ChemMedChem, 2016, vol. 11, # 24, p. 2649 - 2655
[41] New Journal of Chemistry, 2017, vol. 41, # 3, p. 1186 - 1192
[42] Synthetic Communications, 2017, vol. 47, # 11, p. 1102 - 1109
[43] Organic and Biomolecular Chemistry, 2018, vol. 16, # 18, p. 3389 - 3395
61
[ 58518-76-6 ]
[ 123-11-5 ]
[ 6552-63-2 ]
Reference:
[1] Journal of Organic Chemistry, 1988, vol. 53, # 5, p. 1022 - 1025
62
[ 123-11-5 ]
[ 74-88-4 ]
[ 32723-67-4 ]
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 6, p. 1078 - 1083
Reference:
[1] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 9, p. 3290 - 3298
[2] Organic Letters, 2011, vol. 13, # 4, p. 664 - 667
[3] Organic Letters, 2017, vol. 19, # 2, p. 316 - 319
69
[ 123-11-5 ]
[ 7515-17-5 ]
Reference:
[1] Journal of Organic Chemistry, 2016, vol. 81, # 15, p. 6783 - 6791
70
[ 558-13-4 ]
[ 123-11-5 ]
[ 79-22-1 ]
[ 7515-17-5 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2010, vol. 45, # 11, p. 5428 - 5437
71
[ 105-13-5 ]
[ 104-92-7 ]
[ 38493-59-3 ]
[ 123-11-5 ]
Reference:
[1] Journal of Organic Chemistry, 2000, vol. 65, # 12, p. 3880 - 3881
72
[ 1190405-04-9 ]
[ 123-11-5 ]
[ 387-89-3 ]
Reference:
[1] Zeitschrift fur Naturforschung - Section B Journal of Chemical Sciences, 2009, vol. 64, # 5, p. 541 - 550
73
[ 123-11-5 ]
[ 60032-63-5 ]
Reference:
[1] Journal of Organic Chemistry, 2017, vol. 82, # 2, p. 1205 - 1217
74
[ 123-11-5 ]
[ 22996-21-0 ]
Yield
Reaction Conditions
Operation in experiment
89%
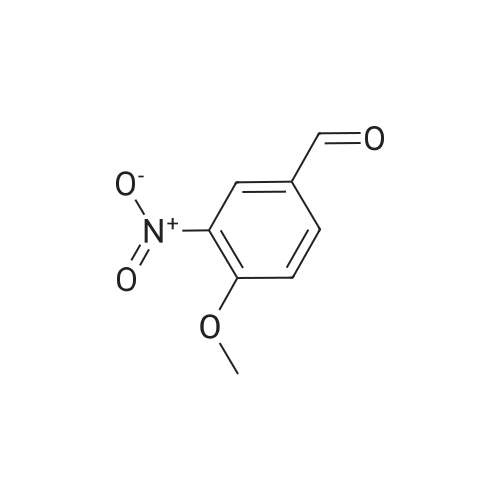
at 0 - 20℃; for 0.5 h; Inert atmosphere
General procedure: 10 mmol of the desired p-substituted benzaldehyde was dissolved in 5 mL of conc. sulfuric acid, and cooled to 0 °C, and then 1.2 equiv. of nitric acid was dissolved in 1 mL of conc. sulfuric acid, and then slowly added to the reaction mixture. The reaction was allowed to warmgradually to room temperature and stirred for 30 min at room temperature. It was then poured into 50 mL of ice-cold water. The produced precipitate was filtered and washed with cold water. The product was purified on reverse phase column chromatography with gradient increase of methanol in water containing 0.1percent of formic acid as eluent.
Reference:
[1] European Journal of Medicinal Chemistry, 2014, vol. 77, p. 361 - 377
[2] Drug Development Research, 2016, p. 241 - 250
75
[ 123-11-5 ]
[ 22996-21-0 ]
[ 31680-08-7 ]
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 19, p. 8088 - 8094
76
[ 879583-48-9 ]
[ 123-11-5 ]
[ 43212-67-5 ]
Reference:
[1] Free Radical Biology and Medicine, 2011, vol. 50, # 10, p. 1447 - 1457
77
[ 110-15-6 ]
[ 123-11-5 ]
[ 43212-67-5 ]
Reference:
[1] Bulletin de la Societe Chimique de France, 1948, p. 567,569
[2] Journal of the American Chemical Society, 1949, vol. 71, p. 633,637
78
[ 123-11-5 ]
[ 43212-67-5 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1889, vol. 255, p. 293
[2] Free Radical Biology and Medicine, 2011, vol. 50, # 10, p. 1447 - 1457
79
[ 150-90-3 ]
[ 123-11-5 ]
[ 43212-67-5 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1889, vol. 255, p. 293
[2] Bulletin des Societes Chimiques Belges, 1960, vol. 69, p. 356 - 361
80
[ 877-99-6 ]
[ 6647-76-3 ]
[ 119-26-6 ]
[ 120893-20-1 ]
[ 32454-14-1 ]
[ 701-34-8 ]
[ 123-11-5 ]
Reference:
[1] Journal of Organic Chemistry USSR (English Translation), 1988, vol. 24, p. 195 - 198[2] Zhurnal Organicheskoi Khimii, 1988, vol. 24, # 1, p. 217 - 220
81
[ 123-11-5 ]
[ 72035-46-2 ]
Reference:
[1] Patent: WO2018/49094, 2018, A1,
82
[ 141-82-2 ]
[ 123-11-5 ]
[ 5678-45-5 ]
Yield
Reaction Conditions
Operation in experiment
82.3%
With ammonium acetate In ethanol at 75 - 80℃; for 8 h;
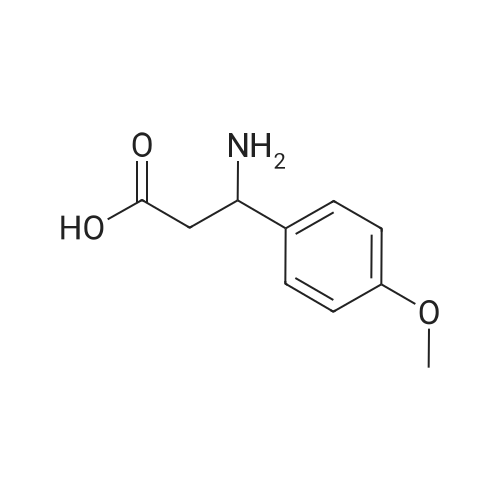
To 150 mL of ethanol were added 28.5 g (0.21 mol) of 4-methoxybenzaldehyde, 21.8 g (0.21 mol) of malonic acid and 32.3 g (0.42 mol) of ammonium acetate, and the mixture was reacted while stirring under reflux (75 to 80°C) for 8 hours. After completion of the reaction, the obtained reaction mixture was stirred at room temperature for 20 hours, and filtered at room temperature to give 23.6 g of 3-amino-3-(4-methoxyphenyl)propionic acid (racemic mixtures) (isolation yield based on 4-methoxybenzaldehyde: 82.3percent) as white powder. Incidentally, physical properties of the 3-amino-3-(4-methoxyphenyl)propionic acid (racemic mixtures) were as follows. 1H-NMR (δ (ppm), D2O) : 2.50 (dd, 1H, J=17.1, 6.8Hz), 2.65 (dd, 1H, J=17.1, 7.8Hz), 3.24 (s, 3H), 6.46 (s, 1H), 6.48 (s, 1H), 6.86 (s, 1H), 6.88 (s, 1H) 13C-NMR (δ (ppm), D2O) : 37.1, 50.7, 55.1, 114.3, 126.9, 128.2, 159.1, 172.7 MS (EI) m/z: 195 (M+) MS (CI, i-C4H10) m/z: 196 (MH+)
70%
With ammonium acetate In butan-1-olReflux
General procedure: A mixture of appropriate aldehyde 2.40 g (1-15), 2.44 g ofmalonic acid and 3.54 g of ammonium acetate (1:1.1:2.3), in 200mLof the 1-butanol was refluxed for 1.5-2 h until the evolution of CO2ceased. The precipitate formed was filtered and washed withboiling 1-butanol (2 x 50 mL), boiling ethanol (2 x 50 mL) and100mL of water. Precipitates were dried at 80-100 °C for 8-10 h.Purity of product was checked by TLC, and yield obtained about65-80percent in each reaction. 4.1.1.1. 3-Amino-3-(4-methoxyphenyl)propanoic acid (A-1).Yield: 70percent, mp 236-8 °C, FT IR ( cm1): 3250-2900 (NH3), 3050 (CHsp2), 1613asym (CO), 1255 (C-O), 1156 (C-N). 1H NMR (500 MHz,DMSO‑d6): d 7.11 (d, 2H, C6H4), 6.77 (d, 2H, C6H4), 4.12 (q, 1H, CHN),3.61 (s, 3H, OCH3), 2.32-2.41 (d, 2H, CHCH2). 13C NMR (126 MHz,DMSO‑d6): d 179.3, 161.8, 137.5, 129.7, 118.4, 59.8, 53.3, 49.1.
Reference:
[1] Patent: EP1621529, 2006, A1, . Location in patent: Page/Page column 40
[2] Chemical Communications, 2011, vol. 47, # 20, p. 5894 - 5896
[3] Journal of Medicinal Chemistry, 2001, vol. 44, # 12, p. 1938 - 1950
[4] Advanced Synthesis and Catalysis, 2010, vol. 352, # 2-3, p. 395 - 406
[5] European Journal of Medicinal Chemistry, 2018, vol. 156, p. 252 - 268
[6] Preparative Biochemistry and Biotechnology, 2013, vol. 43, # 2, p. 207 - 216
[7] Journal of the Chemical Society. Perkin Transactions 1, 2001, # 14, p. 1673 - 1695
[8] Journal of Organic Chemistry, 1985, vol. 50, # 13, p. 2259 - 2263
[9] Advanced Synthesis and Catalysis, 2017, vol. 359, # 9, p. 1570 - 1576
[10] Heterocycles, 1989, vol. 28, # 2, p. 1015 - 1035
[11] Journal fuer Praktische Chemie (Leipzig), 1965, vol. 30, p. 18 - 38
[12] Bulletin de la Societe Chimique de France, 1987, # 6, p. 1079 - 1083
[13] Journal of Medicinal Chemistry, 1996, vol. 39, # 17, p. 3238 - 3240
[14] Synthesis, 2002, # 14, p. 2023 - 2036
[15] Angewandte Chemie - International Edition, 2005, vol. 44, # 45, p. 7466 - 7469
[16] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 6, p. 1356 - 1365
83
[ 123-11-5 ]
[ 5678-45-5 ]
Yield
Reaction Conditions
Operation in experiment
53%
With ammonium acetate; malonic acid In water ethanol
EXAMPLE 18 Preparation of N-(4-Cyanophenyl)-N--[3-(3-(4--methoxyphenyl)propionic acid)]urea To a solution of p-anisaldehyde (40.8 g, 300 mmol) in 100 mL of 95:5 ethanol-water was added ammonium acetate (46.2 g, 600 mmol). The reaction mixture was warmed to 45 °C, and then treated with malonic acid (31.2 g, 300 mmol) in one portion. The resulting suspension was heated at reflux for 18 h, allowed to cool to room temperature, and filtered. The precipitate was recrystallized from 3:1 ethanol-water to give 30.9 g (53percent) of a white solid 3-amino-3-(4--methoxyphenyl)propionic acid: mp 234-235 °C; 1H NMR (300 MHz; HOAc-d4) δ 7.45-6.95 (AB, 4 H, JAB=8.6 Hz), 4.76 (dd, 1 H, J=9.1, 5.2 Hz), 3.79 (s, 3 H), 3.24 (dd, 1 H, J=17.3, 9.1 Hz), and 2.97 (dd, 1 H, J=17.3, 5.2 Hz); 13C NMR(75.5 MHz; HOAc-d4) δ 176.2, 161.2, 129.7, 128.4, 115.1, 55.1, 52.8, and 38.9; IR (KBr): 3424, 2937, 2616, 1613, 1535, 1518, 1407, 1251, 1184, 1027, and 838 cmmin1. Analysis Calculated for C10H13NO3: C, 61.53, H, 6.71; N, 7.18. Found: C, 61.86; H, 6.56; N, 7.10.
Reference:
[1] Patent: EP355819, 1990, A1,
84
[ 141-82-2 ]
[ 123-11-5 ]
[ 943-89-5 ]
[ 5678-45-5 ]
Reference:
[1] Tetrahedron, 2002, vol. 58, # 37, p. 7449 - 7461
[2] Russian Journal of General Chemistry, 2005, vol. 75, # 7, p. 1113 - 1124
85
[ 141-82-2 ]
[ 123-11-5 ]
[ 5678-45-5 ]
[ 830-09-1 ]
Reference:
[1] Archiv der Pharmazie (Weinheim, Germany), 1928, p. 119
Reference:
[1] Synthetic Communications, 2004, vol. 34, # 6, p. 1117 - 1123
[2] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 24, p. 3685 - 3689
88
[ 123-11-5 ]
[ 56-40-6 ]
[ 20839-78-5 ]
Yield
Reaction Conditions
Operation in experiment
33%
Stage #1: for 2 h; Stage #2: at 10 - 20℃;
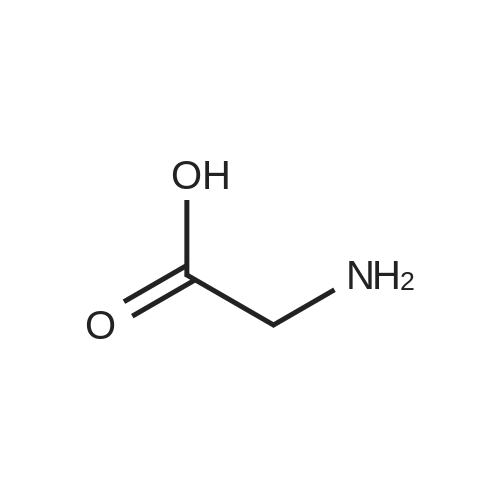
Caution Hydrogen is evolved as a by-product. All flames in thevicinity must be extinguished during the experiment.Glycine (15.0 g, 0.2 mol) was dissolved in aqueous NaOHsolution (8.0 g, 0.2 mol in 200 mL), after which p-methoxybenzaldehyde(27.5 g, 0.2 mol) and ethanol (75 mL) were added[Note 1]. From this point to the end of the experiment, the reaction mixture was stirred using a hot-plate magnetic stirrer.The clear yellow solution, which formed after a few minutes,was heated to 60°C and then stirred without heating for an additional 2 h. After cooling to 10°C in an ice bath, the solution was maintained at that temperature and treated with NaBH4(2.50 g, 0.06 mol), which was added in small portions over a 30-min period. The stirring was continued for the next 30 min without cooling, after which a second portion of p-methoxybenzaldehyde(13.6 g, 0.1 mol) was added. After cooling to108C, the mixture was treated portionwise with NaBH4(1.25 g, 0.03 mol) within 15 min, and stirring was continued for 30 min. The reaction mixture was again cooled to 10°C and subsequently treated with a third portion of p-methoxybenzaldehyde(13.6 g, 0.1 mol) and NaBH4 (1.50 g, 0.04 mol) in the earlier described manner. Then, the reaction mixture was left to stand overnight in a refrigerator (~5°C). Then, the volume of thesolution was reduced to 200mL in a fume hood to remove ethanol and, after cooling, extracted with chloroform (first with100 mL, then twice with 50 mL). The (upper) aqueous layer wasacidified to pH E 6.5 with diluted hydrochloric acid (60 mL;1 : 1, v/v) and left to stand for 2 h. The white powder [Note 2] thathad separated was filtered off, and the volume of the filtrate wasreduced to 100 mL, after which the product started to crystallize.After cooling in an ice bath, the product was filtered off bysuction and quickly washed with ice water (40 mL). Yield:13.2 g (33 percent).
Reference:
[1] Australian Journal of Chemistry, 2016, vol. 69, # 11, p. 1285 - 1291
89
[ 123-11-5 ]
[ 75-04-7 ]
[ 121-44-8 ]
[ 22993-76-6 ]
Yield
Reaction Conditions
Operation in experiment
59%
With sodium hydroxide; NaBH(OAc)3 In tetrahydrofuran; methanol; dichloromethane; water; acetic acid; 1,2-dichloro-ethane
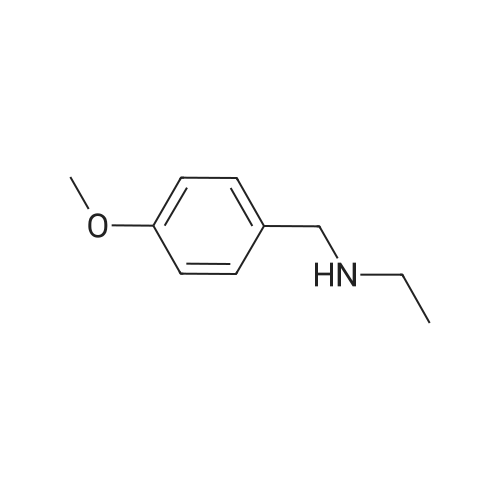
N-Ethyl-4-methoxybenzenemethanamine: To a mixture of anisaldehyde (15.6 g, 115 mmol) and ethylamine (2.0M in THF, 87 mL, 174 mmol) in 1,2-dichloroethane (450 mL) was added glacial acetic acid (10.0 mL, 174 mmol) under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 30 min and then cooled to 0° C. with ice bath. NaBH(OAc)3 (36.9 g, 174 mmol) was added portionwise and the reaction mixture was stirred at room temperature overnight. The mixture was concentrated and the residue was diluted with a basic solution (10 g NaOH in 100 mL of water) to make the solution slightly basic. This aqueous layer was extracted with ether. The combined extracts were washed with water, brine, dried and concentrated. The residue was chromatographed on silica gel (elution with 5percent MeOH in CH2 Cl2 and then 50percent MeOH in CH2 Cl2 containing 4percent Et3 N) to afford the product (11.2 g, 59percent yield) as an oil.
Reference:
[1] Patent: US5677346, 1997, A,
90
[ 123-11-5 ]
[ 22993-76-6 ]
Reference:
[1] Archiv der Pharmazie (Weinheim, Germany), 1942, vol. 280, p. 213,224
91
[ 123-11-5 ]
[ 51307-59-6 ]
Reference:
[1] Canadian Journal of Chemistry, 2013, vol. 91, # 1, p. 43 - 50
[2] RSC Advances, 2014, vol. 4, # 60, p. 31892 - 31903
[3] Journal of the American Chemical Society, 2017, vol. 139, # 25, p. 8428 - 8431
92
[ 123-11-5 ]
[ 43192-31-0 ]
Yield
Reaction Conditions
Operation in experiment
25%
Stage #1: With n-butyllithium; N,N',N'-trimethylenediamine In tetrahydrofuran at -20 - 0℃; for 20.5 h; Stage #2: With carbon tetrabromide In tetrahydrofuran at -78 - 20℃;
Example 4: Scheme Cl; n-BuLi (1 equiv., 147 mmol, 59 ml 2.5 M) was added dropwise to a stirred solution of trimethylethylenediamine (TMEDA) (1.1 equiv., 162 mmol, 17.0 g) in dry THF (80 ml) at -20 0C. After 15 min, /?-anisaldehyde (Cl.1) (1 equiv., 147 mmol, 20.0 g) was added, the mixture was stirred for 15 min and n-butyllithium (n-BuL) (3 equiv.,441 mmol, 176 ml 2.5 M) was added dropwise. The reaction mixture was stirred at 00C for 20 h. The solution was cooled to -78 0C, carbon tetrabromide (2.7 equiv., 397 mmol, 131.6 g) was added and the solution was allowed to warm to room temperature. An aqueous 10percent HCl solution was added and extraction was carried out with dichloromethane. The combined organic extracts were washed with a saturated aqueous sodium thiosulfate solution, water and brine. The organic phase was dried with MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (heptane / ethyl acetate 9:1) to give compound C 1.2 as a white solid (8 g, yield = 25percent). 1H-NMR (δ, DMSO-D6): 3.89 (3H, s), 7.13 (IH, dd, J = 8.7, 2.4 Hz), 7.35 (IH, d, J = 2.4 Hz), 7.83 (IH, d, J = 8.7 Hz), 10.10 (IH, s) ppm
Reference:
[1] Tetrahedron, 2012, vol. 68, # 40, p. 8463 - 8471
[2] Chemical Communications, 2005, # 46, p. 5793 - 5795
[3] Organic and Biomolecular Chemistry, 2007, vol. 5, # 1, p. 143 - 150
[4] Organic Letters, 2011, vol. 13, # 14, p. 3686 - 3689
[5] Patent: WO2008/37784, 2008, A1, . Location in patent: Page/Page column 37
[6] Tetrahedron Letters, 1993, vol. 34, # 50, p. 8097 - 8100
[7] Heterocycles, 1996, vol. 43, # 4, p. 737 - 743
[8] Canadian Journal of Chemistry, 1997, vol. 75, # 6, p. 817 - 824
93
[ 123-11-5 ]
[ 54439-75-7 ]
[ 43192-31-0 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2007, vol. 5, # 1, p. 143 - 150
94
[ 123-11-5 ]
[ 6298-96-0 ]
Reference:
[1] Journal of Organic Chemistry, 1991, vol. 56, # 3, p. 1340 - 1344
Reference:
[1] Advanced Synthesis and Catalysis, 2016, vol. 358, # 20, p. 3298 - 3306
99
[ 123-11-5 ]
[ 84-16-2 ]
Reference:
[1] Journal of the Chinese Chemical Society (Peking), 1946, vol. 13, p. 111,117
100
[ 557-66-4 ]
[ 123-11-5 ]
[ 79865-89-7 ]
Reference:
[1] Journal of Organic Chemistry USSR (English Translation), 1982, vol. 18, # 2, p. 406[2] Zhurnal Organicheskoi Khimii, 1982, vol. 18, # 2, p. 462
101
[ 123-11-5 ]
[ 75-04-7 ]
[ 79865-89-7 ]
Reference:
[1] Journal of Organic Chemistry USSR (English Translation), 1983, vol. 19, # 11, p. 2064 - 2068[2] Zhurnal Organicheskoi Khimii, 1983, vol. 19, # 11, p. 2361 - 2366
102
[ 123-11-5 ]
[ 2393-23-9 ]
[ 854391-95-0 ]
Yield
Reaction Conditions
Operation in experiment
91%
Stage #1: for 4 h; Reflux Stage #2: at 0℃; for 18 h; Stage #3: With hydrogenchloride In 1,4-dioxane; diethyl ether at 0℃; for 2 h;
Pre aration of Intermediate 1-16.A mixture of p-anisaldehyde (1.8 mL, 14.8 mmol) and 4-methoxybenzylamine (1.9 mL, 14.8 mmol) in EtOH (50 mL) was refluxed for 4 h. On cooling to 0 °C, NaBH4 (562 mg, 14.8 mmol) was added and the reaction mixture was stirred for 18 h at rt. On cooling to 0 °C, water (50 mL) and DCM (250 mL) were added. The organic phase was separated and the aqueous layer was extracted with DCM (2 x 250 mL). The combined organic layers were dried (MgS04), filtered and evaporated. The residue was dissolved in Et20 (100 mL) and the mixture was cooled to 0 °C. 4N HCI in dioxane (-10 mL) was added dropwise and the mixture was stirred for 2 h at 0 °C. The resulting white solid was filtered off, washed with Et20/EtOAc to give the desired product (3.99 g, 91 percent) as a white solid.1H NMR (300 MHz, DMSO) δ 9.40 (s, 2H), 7.45 (d, J = 8.7 Hz, 4H), 6.98 (d, J = 8.7 Hz, 4H), 4.03 (s, 4H), 3.77 (s, 6H).
91%
With sodium tetrahydroborate In ethanol at 0 - 20℃; for 22 h; Reflux
Preparation of Intermediate I-16 [0278] [0279] A mixture of p-anisaldehyde (1.8 mL, 14.8 mmol) and 4-methoxybenzylamine (1.9 mL, 14.8 mmol) in EtOH (50 mL) was refluxed for 4 h. On cooling to 0° C., NaBH4 (562 mg, 14.8 mmol) was added and the reaction mixture was stirred for 18 h at rt. On cooling to 0° C., water (50 mL) and DCM (250 mL) were added. The organic phase was separated and the aqueous layer was extracted with DCM (2×250 mL). The combined organic layers were dried (MgSO4), filtered and evaporated. The residue was dissolved in Et2O (100 mL) and the mixture was cooled to 0° C. 4N HCl in dioxane (−10 mL) was added dropwise and the mixture was stirred for 2 h at 0° C. The resulting white solid was filtered off, washed with Et2O/EtOAc to give the desired product (3.99 g, 91percent) as a white solid. [0280] 1H NMR (300 MHz, DMSO) δ 9.40 (s, 2H), 7.45 (d, J=8.7 Hz, 4H), 6.98 (d, J=8.7 Hz, 4H), 4.03 (s, 4H), 3.77 (s, 6H).
Reference:
[1] Chemical Communications, 2014, vol. 50, # 69, p. 9917 - 9920
104
[ 623-33-6 ]
[ 123-11-5 ]
[ 60857-16-1 ]
Yield
Reaction Conditions
Operation in experiment
49%
With sodium cyanoborohydride In methanol at 20℃;
A suspension of glycine ethyl ester hydrochloride (10. 0 G, 71.6 mmol) and NaBH3CN (5.00 g, 79.6 mmol) in MeOH (60 mL) was treated dropwise over 15 min with p-anisaldehyde (11.0 mL, 90.4 mmol). After stirring at room temperature overnight, the solvent was removed in vacuo. The residue was partitioned between CH2CL2 (200 mL) and saturated aqueous NAHC03 (300 mL). The aqueous layer was extracted with CH2C12 (2 x 200 mL) and the combined organic layers were washed with brine, dried over NA2SO4, filtered and the solvent was removed in vacuo. Purification by flash column chromatography (silica gel, hexanes/EtOAc, 90: 10 to 50: 50) gave the title compound (7.77 g, 49percent) as a colorless liquid: MS (ESI) mule 224 (M + H) +.
Stage #1: With potassium hydroxide In ethanol at 20℃; for 0.166667 h; Stage #2: With ammonium hydroxide In ethanol at 37℃; for 24 h;
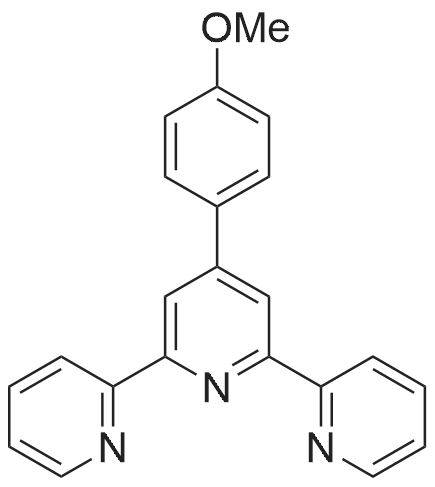
General procedure: Synthesis of 4′-(3-methoxyphenyl)-2,2′:6′,2″-terpyridine (3-MeO-Phtpy), 4′-(2-methoxyphenyl)-2,2′:6′,2″-terpyridine (2-MeO-Phtpy) and 4′-(4-methoxyphenyl)-2,2′:6′,2″-terpyridine (4-MeO-Phtpy) were performed using the methods described previously [27–33]. 2-Acetylpyridine (2.813g, 23.2mmol, 2 eq.) was added to 2-methoxybenzaldehyde, 3-methoxybenzaldehyde or 4-methoxybenzaldehyde (11.6mmol, 1 eq.) dissolved in 50mL ethanol. KOH pellets (46.5mmol, 4 eq.) were added to this solution. The reaction mixture was stirred at room temperature for 10min. NH3 (40mL, 25percent aq.) was slowly added to the reaction mixture. After a 24-h incubation at 37°C, 5mL of 25percent aq. NH3 was added to the reaction mixture again. The flask containing the reaction mixture was cooled to−20°C. The obtained white precipitate in the flask was isolated through filtration and washed with cold ethanol. We further purified the each product using recrystallization in ethanol-H2O. After recrystallization, each product was recovered by filtration, washed with cold ethanol and petroleum ether, and dried under high vacuum for 24h (Scheme 1).
37%
With ammonium hydroxide; potassium hydroxide In ethanol at 20℃; for 24 h;
General procedure: Ketone (20 mmol) (2-acetylpyridine, 2-acetylthiazole or 2-acetylpyrazine)was added to a solution of aldehyde (10 mmol) (4-methoxybenzaldehyde,4-methoxy-1-naphthaldehyde or 6-methoxy-2-naphthaldehyde) in EtOH (75 mL). KOH (1.54 g, 27.5 mmol) and NH3(aq)(35 mL) were then added. The solution was stirred at room temp. for 24 h. The solid was collected by filtration and washed with H2O. Recrystallization from ethanol (L1,L4, L7) or toluene (L2, L3, L5, L6, L8,L9) afforded a crystalline solid. 4′-(4-methoxy-1-phenyl)-2,2′:6′,2″-terpyridine (L1): Yield: 37percent. IR(KBr, cm−1): 1588(s), 1467(m), 1389(m), 1204(m), 1157(m),1069(m), 790(m), 739(m) and 655(m). NMR: 1H NMR (400 MHz,CDCl3) δ 8.73 (d, J=4.1 Hz, 2H, HA1), 8.71 (s, 2H, HB2), 8.67 (d,J=7.9 Hz, 2H, HA4), 7.90–7.85 (m, 4H, HC2+A3), 7.37–7.33 (m, 2H,HA2), 7.03 (d, J=8.7 Hz, 2H, HC3), 3.88 (s, 3H, HC5). 13C NMR(100 MHz, CDCl3) δ 160.66, 156.55, 155.99, 149.90, 149.24, 136.96,130.92, 128.67, 123.87, 121.49, 118.42, 114.46, 55.52. C22H17N3O(339.39 g mol−1): calcd C, 77.86; H, 5.05; N, 12.38percent; found: C, 77.46;H, 5.35; N, 12.25percent. DSC: (I run) Tm=164 °C; (II run) Tg=30, Tc=82and Tm=163 °C.
Reference:
[1] Applied Organometallic Chemistry, 2017, vol. 31, # 10,
[2] Applied Organometallic Chemistry, 2017, vol. 31, # 6,
[3] Chemistry - An Asian Journal, 2018, vol. 13, # 21, p. 3169 - 3172
[4] Chemistry Letters, 2005, vol. 34, # 5, p. 732 - 733
[5] Synthesis, 2005, # 18, p. 3045 - 3050
[6] European Journal of Medicinal Chemistry, 2018, vol. 143, p. 1387 - 1395
[7] MedChemComm, 2018, vol. 9, # 3, p. 525 - 533
[8] Dyes and Pigments, 2019, vol. 163, p. 86 - 101
[9] Medicinal Chemistry Research, 2012, vol. 21, # 9, p. 2723 - 2733,11
[10] Green Chemistry, 2013, vol. 15, # 8, p. 2075 - 2080
[11] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1992, vol. 31, # 8, p. 555 - 556
[12] Synlett, 2005, # 8, p. 1251 - 1254
[13] Journal of the Chemical Society. Perkin Transactions 2, 2001, # 7, p. 1045 - 1050
[14] Inorganic Chemistry, 2017, vol. 56, # 14, p. 8212 - 8222
[15] Journal of the Chemical Society, Dalton Transactions, 2002, # 21, p. 3931 - 3932
[16] Journal of Organic Chemistry, 2011, vol. 76, # 6, p. 1910 - 1913
[17] New Journal of Chemistry, 2011, vol. 35, # 10, p. 2130 - 2135
[18] Polyhedron, 2012, vol. 40, # 1, p. 159 - 167
[19] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2013, vol. 104, p. 48 - 55
[20] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2014, vol. 117, p. 662 - 668
[21] Journal of Inorganic Biochemistry, 2014, vol. 141, p. 17 - 27
[22] Synthesis and Reactivity in Inorganic, Metal-Organic and Nano-Metal Chemistry, 2015, vol. 45, # 8, p. 1097 - 1101
[23] Journal of Coordination Chemistry, 2017, vol. 70, # 3, p. 451 - 462
[24] Monatshefte fur Chemie, 2017, vol. 148, # 5, p. 893 - 900
[25] Chemistry - A European Journal, 2017, vol. 23, # 17, p. 4055 - 4059
110
[ 123-11-5 ]
[ 13104-56-8 ]
Reference:
[1] Synthesis, 2006, # 17, p. 2873 - 2878
With ammonium persulfate; N-chloro-succinimide; oxygen; methylene green; In acetonitrile; at 20℃; for 24h;Irradiation;
General procedure: To an oven-dried flask was added a magnetic stir bar, methylene green (9.1 mg, 0.05 equiv, 0.025 mmol), ammonium peroxodisulfate (11.4 mg, 0.1 equiv, 0.05 mmol), arene/heteroarene (1 equiv, 0.5 mmol), acetonitrile (2.5 mL), and then N-chlorosuccinimide (73.4 mg, 1.1 equiv, 0.55 mmol). The reaction mixture was stirred open to air at room temperature (20 C) in a white LED chamber for 24 h. For substrates that produced a mixture of mono- and dibrominated products upon full conversion, 2.2 equivalents (1.1 mmol) of N-chlorosuccinimide was employed. Upon completion of the reaction, the crude mixture was evaporated under pressure and the chlorinated product was isolated via column chromatography on silica gel.
N-{3-[hydroxy(4-methoxyphenyl)methyl]pyridin-4-yl}pivalamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69%
To a solution of 2.67 g (15.0 mmol, 1 eq.) of pivalamide VII and 6.8 mL (45.0 mmol, 3 eq.) of TMEDA in 120 mL of diethylether, cooled to -78°C, were added 18.0 mL (45.0 mmol, 3 eq., 2.5 M in hexane) of BuLi. After 15 minutes, the solution was brought back to -24°C and stirred 2 hours. Then, the mixture was cooled down to - 78°C before addition of 4.0 mL (33.0 mmol, 2.2 eq) of p-anisaldehyde in 60 mL of THF. The reaction was brought back to 0°C and stirred 2 hours before being stirred at room temperature over night. Then, 120 mL of water were added. The aqueous layer was extracted with 3 x 80 mL of dichloromethane. The organic phase was dried, filtered and concentrated. Precipitation in 30 mL of PE/DCM 1 :1 afforded 3.26 g (10.4 mmol, 69percent) of XIV as a white powder. Rf=0.16 (PE/EtOAc 1 :2). 1H NMR (300 MHz): delta = 9.57 (s, 1 H), 8.36 (d, J = 5.7 Hz, 1 H), 8.23 (d, J = 5.7 Hz, 1 H), 7.87 (s, 1 H), 7.18 (d, J = 8.5 Hz, 2 H), 6.89 (broad, 1 H), 6.83 (d, J = 8.3 Hz, 2 H), 5.78 (s, 1 H), 3.77 (s, 3 H), 1 .08 (s, 9 H) ppm. 13C NMR (75 MHz): delta = 177.9, 159.4, 149.8, 148.4, 145.6, 133.0, 127.5, 126.4, 115.3, 114.0, 73.2, 55.5, 40.2, 27.2 ppm
69%
To a solution of 2.67 g (15.0 mmol, 1 eq.) of pivalamide VII and 6.8 mL (45.0 mmol, 3 eq.) of TMEDA in 120 mL of diethylether, cooled to -78°C, were added 18.0 mL (45.0 mmol, 3 eq., 2.5 M in hexane) of BuLi. After 15 minutes, the solution was brought back to -24°C and stirred 2 hours. Then, the mixture was cooled down to - 78°C before addition of 4.0 mL (33.0 mmol, 2.2 eq) of p-anisaldehyde in 60 mL of THF. The reaction was brought back to 0°C and stirred 2 hours before being stirred at room temperature over night. Then, 120 mL of water were added. The aqueous layer was extracted with 3 x 80 mL of dichloromethane. The organic phase was dried, filtered and concentrated. Precipitation in 30 mL of PE/DCM 1 :1 afforded 3.26 g (10.4 mmol, 69percent) of XIV as a white powder. Rf=0.16 (PE/EtOAc 1 :2). 1H NMR (300 MHz): delta = 9.57 (s, 1 H), 8.36 (d, J = 5.7 Hz, 1 H), 8.23 (d, J = 5.7 Hz, 1 H), 7.87 (s, 1 H), 7.18 (d, J = 8.5 Hz, 2 H), 6.89 (broad, 1 H), 6.83 (d, J = 8.3 Hz, 2 H), 5.78 (s, 1 H), 3.77 (s, 3 H), 1 .08 (s, 9 H) ppm. 13C NMR (75 MHz): delta = 177.9, 159.4, 149.8, 148.4, 145.6, 133.0, 127.5, 126.4, 115.3, 114.0, 73.2, 55.5, 40.2, 27.2 ppm
Step 2: 2- [2- (4-Methoxy-phenyl)-vinyl]-nicotinic acid A mixture of 5.0 G (30.26 MMOL) of the previous compound and 7.74 G (56.8 MMOL) of ANISALDEHYDE is heated at 120°C. 3.9 G (28.6 MMOL) of anhydrous zinc chloride are added- and the whole is heated at 180°C allowing the ethanol formed to be expelled. After 2 hr a solid crystallises and a solution of 4.9 G of sodium hydroxide in 41 ml of water is added. After stirring to disgregation, the inorganic salts are filtered and the filtrate is washed with ethyl ether and neutralised with acetic acid. The solid precipitated is filtered, washed with water and recrystallised from ethanol. Yield : 5.2 G (67 percent).
42%
A mixture of NaH (60percent dispersion in mineral oil, 7.4 g, 0.185 mole), tert-butanol (11.12 g, 0.15 mole), and anhydrous DMF (100 mL) was carefully warmed at 80° C. until gas evolution ceased, then was cooled to 0° C. under argon. A solution of <strong>[1721-26-2]ethyl 2-methylnicotinate</strong> (7.7 mL, 0.050 mole) in anhydrous DMF (17 mL) was added dropwise over 3 min, and the reddish-orange mixture was stirred at 0° C. for 1 hr. A solution of para-anisaldehyde (7.3 mL, 0.060 mL) in anhydrous DMF (17 mL) was added dropwise over 3 min, and the reaction was allowed to warm to RT. The mixture was stirred overnight, then was poured into ice water (200 mL), and the solution was acidified to pH 6 with glacial acetic acid. The volume was adjusted to 1 L with H2O, the flask was scratched with a glass rod, and the mixture was allowed to stand at RT for several hr, then in the refrigerator overnight. The solid was collected by suction filtration and washed with H2O. Recrystallization from boiling absolute EtOH gave the title compound (5.31 g, 42percent, two crops) as yellow needles: 1H NMR (250 MHz, DMSO-d6) delta 8.70 (dd, J=4.6, 1.7 Hz, 1H), 8.18 (dd, J=7.9, 1.7 Hz, 1H), 7.95 (d, J=15.8 Hz, 1H), 7.83 (d, J=15.8 Hz, 1H), 7.57 (d, J=8.7 Hz, 2H), 7.34 (dd, J=7.9, 4.6 Hz, 1H), 6.99 (d, J=8.7 Hz, 2H), 3.79 (s, 3H); MS (ES) m/e 256 (M+H)+.
With diisobutylaluminium hydride; In methanol; toluene; benzene;
(1)2-(4-Methylbenzyloxy)methyl-2-propenol A mixture of p-anisaldehyde (17.31 g, 0.127 mol), <strong>[3513-81-3]2-methylene-1,3-propanediol</strong> (14.00 g, 0.159 mol), and p-TsOH H2 O (121 mg, 0.64 mmol) in dry PhH (100 ml) was stirred and heated at reflux, and the water generated was azeotropically removed in a theoretical amount using a Dean-Stark apparatus. After 70 ml of PhH was distilled off, the resultant red PhH solution containing 2-(4-methoxyphenyl)-5-methylene-1,3-dioxane was cooled with salt-ice, and a solution of diisobutylaluminum hydride (DIBAL-H) in PhMe (1.02M, 100 ml and 1.50M, 67.0 ml; total 0.203 mol) was added dropwise under an atmosphere of N2; at the point when 20 ml of the PhMe solution of DIBAL-H was added, the reaction mixture changed from deep red to light yellow. After the addition was complete, the reaction mixture was allowed to stirr at room temperature overnight. The reaction mixture was ice-cooled, and MeOH (23 ml) was added carefully. H2 O (12 ml) was, then, added dropwise, wherein the mixture became thick and gelatinous, and finally granular. After the stirring was continued at room temperature for an hour, the mixture was filtered through a pad of Celite. The filter cake was washed with PhMe thoroughly. The combined filtrate and washings were dried (MgSO4), and concentrated in vacuo at a bath temperature of 65 C. to give a pale yellow oil (24.88 g). This was distilled in vacuo to give title compound (18.52 g, 70.1percent) as a colorless oil. bp 123-147 C./0.80-0.90 mm; IRmax (film): 3420(s), 1615(s), 1515(s), 1250(s), 1070(s), 1035(s), 918(m), 820(s) cm-1; 1 H-NMR delta: 7.26(d, J=8.8 Hz, 2H), 6.88(d, J=8.8 Hz, 2H), 5.19(br.s, 1H), 5.14(br.s, 1H), 4.45(s, 2H), 4.19(s, 2H), 4.07(s, 2H), 3.81(s, 3H), 1.98(br.s., 1H) ppm.
To a solution of 2-cyanoethylhydrazine (0.81 ml, 10.0 mmol, 1 eq.) in EtOH (5 mL), anisaldehyde (1.21 ml, 10.0 mmol, 1 eq.) was added. The orange solution was stirred at 70 C. for 2 hours. The orange solution was allowed to cool to r.t. and concentrated in vacuo. The residue was dissolved in i-PrOH (8 mL). NaOtBu (991 mg, 10.0 mmol, 1 eq.) was added and the mixture was stirred at 110 C. for 4 hours. The mixture was allowed to cool to r.t. and water (50 mL) was added. The mixture was extracted with Et2O (3*50 mL). The comb. org. phases were extracted with 1N aq. HCl (2*30 mL). The comb. aq. layers were basified to pH 14 with 50% aq. NaOH solution, then extracted with Et2O (3*50 mL). The comb. org. phases were dried over MgSO4 and concentrated in vacuo to give the desired pyrazole as an orange solid. The product was used without further purification. LC-MS (A): tR=1.26 min; [M+H]+=204.20.
General procedure: To a solution of the corresponding aromatic aldehyde (0.27 mmol) in EtOH (0.5 mL) was added urea (0.54 mmol) and CuSO4·5H2O (0.054 mmol). The mixture was stirred at 80 °C for 15 minutes before tetrahydrocurcumin or tetrahydrodemethoxycurcumin (0.27 mmol) was added. The reaction mixture was continued stirring for 24 hours and quenched with water (2 mL). The solution was washed with water (10 mL) and extracted with EtOAc (415 mL). The combined organic phases were washed with brine, dried over MgSO4 and concentrated under reduced pressure to afford crude product as a yellow brown oil. Purification was accomplished by column chromatography eluting with 60percent-75percent EtOAc/hexane to furnish compounds 8-17.
6-chloro-2-[(E)-2-(4-methoxyphenyl)ethenyl]quinazolin-4(3H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With sulfuric acid; at 190℃; for 2h;Microwave irradiation;
6-chloro-2-[(E)-2-(4-methoxyphenyl)vinyl]quinazolin-4(3H)-one 3 g (15 mmol) <strong>[7142-09-8]6-chloro-2-methylquinazolin-4(3H)-one</strong> was mixed with 3.06 g (22.5 mmol) 4- methoxybenzaldehyde and 1 drop concentrated sulphuric acid was given to this mixture. The reaction was carried out in microwave set at 190 C. Reaction time was 2 hour. The crude product was washed with 5% sodium hydrogen carbonate and filtered. The product was crystallized from dimethyl formamide and dried under vacuum. Yield: 4.58 g (95 %) 1H NMR (300 MHz, DMSO-d6) delta ppm 8.02(d, 1H); 7.92(d,1H); 7.81(d,1H); 7.66(d,1H); 7.66(d,1H); 7.61(d,2H); 7.03(d,2H); 6.85(d,1H); 3.81(s,3H). LC-MS (ESI): m/z (M+H)+ 313, Rt: 4.07 min.
6-fluoro-2-(2-(4-methoxybenzylidene)hydrazino)benzothiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With acetic acid; In ethanol; at 80℃; for 0.166667h;Microwave irradiation;
General procedure: 2-(2-Arylidenehydrazino)-6-fluorobenzothiazoles 6a-r. General Procedure D. A mixture of compound 2 (0.0549 g, 0.0003 mol), the appropriate aromatic aldehyde (0.00033 mol) and glacial acetic acid (0.1 mL) in ethanol (5 mL) was heated under microwave (20 W) at 80 °C for 10 min. On cooling, the precipitated solid was collected by filtration, washed with water, dried and crystallized from the appropriate solvent to give the desired compounds 6a-r.
In ethanol; at 70 - 80℃; for 3h;
General procedure: The mixture of <strong>[78364-55-3]6-fluoro-2-hydrazinylbenzo[d]thiazole</strong> (2) (0.01 mol) and benzalde-hyde/substituted benzaldehyde (0.01 mol) was reuxed in ethanol (15 ml) at 70?80 °C for 3 h. The separated product obtained was ltered off, washed withdistilled water and recrystallized from methanol to give the correspondinghydrazone. The product obtained was further dissolved in acetic acid (20 ml) atroom temperature followed by the addition of sodium acetate (0.5 g). Bromine(2 mmol) in acetic acid (10 ml) was added dropwise to the reuxing reactionmixture. After 1 h, the mixture was poured onto crushed ice (100 g). The precipitateobtained was ltered off and crystallized from ethanol-dimethylformamide (1:1) togive crystals of (3a?3t).
(4Z)-2-(4-methoxyphenyl)-4-[(4-methoxyphenyl)methylidene]-1,3-oxazol-5(4H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
With potassium acetate; acetic anhydride; at 100℃;Inert atmosphere;
General procedure: Aromatic aldehyde (1 equiv.) was added to the solution of hippuric acid (1 equiv.) and potassium acetate (1 equiv.) in acetic anhydride under stirring. The mixture was refluxed under argon for 2?4 h at 100 °C and then cooled for the product precipitation. The precipitate was filtered, washed by distilled water and ether and dried on air.
With ZnBr2/SiO2; In neat (no solvent); for 0.1h;Microwave irradiation; Green chemistry;
General procedure: The mixture of 4-(4-chlorophenoxy)aniline (2) (220 mg,1.0 mmol), 3-nitrobenzaldehyde (3e) (155mg, 1.03 mmol),diethyl phosphite (4) (0.13 mL, 1.0 mmol) and SiO2-ZnBr2 (17.1 mg, 6 molpercent) were taken into a flask. Thereaction mass was irradiated with microwave radiationsfor 4 min using Catalyst systems (CATA-4R,), percentpower is 65percent and 465Watts. Ethyl acetate (10 mL) wasadded to the reaction mixture and stirred for 10 min,after completion of the reaction as checked by TLC.The catalyst, SiO2-ZnBr2 was separated by filtrationas residue, washed with ethyl acetate (2×5mL) andcollected residue was dried under vacuum at 100°C toutilize in further reactions. The combined organic layerwas washed with water (10 mL), dried over anhydrousNa2SO4 and concentrated under vacuum at 50°C toobtain crude mass. The pure compound, diethyl (4-(4-chlorophenoxy)phenylamino)(3-nitrophenyl)methylphosphonate (5e) was isolated by column chromatographyusing 20?60percent ethyl acetate:n-hexane as a mobilephase.
With sodium tris(acetoxy)borohydride; In acetic acid; 1,2-dichloro-ethane;
To a mixture of 7-amino-3,4-dihydroisoquinolin-l(2H)-one (122mg, 0.752 mmol) and p-methoxybenzaldehyde (0.2 mL, 1.65 mmol) in DCE (2mL) was added AcOH (4drops) and NaBH(OAc)3 (349 mg, 1.65mmol). After stirring overnight, additional NaBH(OAc)3 (100 mg) was added and stirred for 4 h. Saturated NaHC03 solution and ethyl acetate was added. The organic layer was washed with water and dried over MgSC and concentrated under reduced pressure. The product was purified by silica gel column chromatography (0 to 100% EtOAc in hexane) ESI-MS: 403.9 (MH+). The dihydrosioquinolinone (39mg, 0.096 mmol), methyl (S)-2- (2,6-dichlorobenzamido)-3-(4-iodophenyl)propanoate (46mg, 0.096 mmol), xantphos (3.3 mg, 0.0058 mmol), Pd2dba3 (2mg, 0.002 mmol), and CS2CO3 (44mg, 0.13 mmol) were dissolved in dioxane (ImL). The reaction vessel was degassed and heated at 65 oC for lh and kept stirring at 100 oC for 4 h. The reaction mixture was cooled and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried over MgSC and concentrated. The product was purified by preparative TLC. ESI-MS: 752.9 (MH+). The bis PMB amine (14mg, 0.018mmol) was stirred in TFA (ImL) at 65 oC for 3h and concentrated. ESI-MS: 512.8 (MH+). To a mixture of crude aniline (18mg) in DMF (ImL) was added trimethylamine (0.03 mL), bis-Boc thiourea (16mg, 0.06 mmol), and then CuC^ (8mg, 0.06 mmol). After 30min, water and EtOAc were added and the organic layer was separated, concentrated and purified by preparative HPLC. ESI-MS: 754.9 (MH+). The bis-guanidine was stirred in TFA (50% in DCM, ImL) for 1.5h and concentrated to yield the guanidine (4mg). ESI-MS: 554.8 (MH+). To the crude methyl ester in THF (0.5 mL) was added aq. LiOH (1M, 0.06 mL). After 1.5h, the mixture was acidified with aq. HC1 (1M) and purified by RP-HPLC. condition: 19 100 mm Altantis T3 OBD, solvent A, solvent B', flow rate: 10 ml/min, gradient: 5% solvent B' for 5 min, 5% to 100% solvent B' for 25min, detection: 254nm) 19.5 min, ESI-MS: 540.8 (MH+)
2,3-dihydro-3-hydroxy-5-methoxy-2-(4-methoxyphenyl)-4H-1-benzopyran-4-one[ No CAS ]
[ 123-11-5 ]
Yield
Reaction Conditions
Operation in experiment
2'-Hydroxy-4,6'-dimethoxychalcone (5.0 mg, 0.0176 mmol) was dissolved in 400 muL MeOH and 200 muL H2O , and sodium carbonate (9.3 mg, 0.0880 mmol) was added at room temperature (25°C-28°C) for 30 min in nuclear magnetic tube. Then H2O2 (4.5 muL, 0.0440 mmol) was added in the mixture. After adding H2O2, samples were analysed by LCMS depending on different time (Figure S1).
2,3-dihydro-3-hydroxy-5-methoxy-2-(4-methoxyphenyl)-4H-1-benzopyran-4-one[ No CAS ]
[ 123-11-5 ]
Yield
Reaction Conditions
Operation in experiment
2'-Hydroxy-4,6'-dimethoxychalcone (5.0 mg, 0.0176 mmol) was dissolved in 400 muL MeOH and 200 muL H2O , and sodium carbonate (9.3 mg, 0.0880 mmol) was added at room temperature (25°C-28°C) for 30 min in nuclear magnetic tube. Then H2O2 (4.5 muL, 0.0440 mmol) was added in the mixture. After adding H2O2, samples were analysed by LCMS depending on different time (Figure S1).
dimethyl ((4-methoxyphenyl)((2-methyl-3-(trifluoromethyl)phenyl)amino)methyl)phosphonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92%
With chitosan; In neat (no solvent); at 75℃; for 0.0333333h;Microwave irradiation; Green chemistry;
General procedure: An equimolar mixture of <strong>[54396-44-0]2-methyl-3-(trifluoromethyl)aniline</strong> (0.351 g, 0.002 mol), corresponding aldehyde (0.002 mol), dimethyl phosphite (0.18 ml, 0.002 mol) and chitosan catalyst (10 molpercent) were taken in a reaction glass tube, degassed for 10 min and microwave irradiated at 180 W for 2 min at 60 °C. The progress of the reaction was monitored by TLC using petroleum ether and ethyl acetate (3:7) as solvent. After completion of the reaction, the mixture was diluted with ethyl acetate, washed with water (2 x 15 ml) followed by brine (1 x 10 ml), dried over Na2SO4 and evaporated to dryness. The crude mass was purified by column chromatography on silicagel (100-200 mesh) by using a 7:3 mixture of ethyl acetate in hexane to afford the pure alpha-aminophosphonates.
N'-4-methoxybenzylfluorenylmethylcarbazate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
N4-Methoxybenzyl-fluorenylmethylcarbazate 2 was prepared according to a modification of the reductive amination procedure for carbazate synthesis (Boeglin, D.; Lubell,W. D. ?Aza-Amino Acid Scanning of Secondary Structure Suited for Solid-Phase PeptideSynthesis with Fmoc Chemistry and Aza-Amino Acids with Heteroatomic Side-Chains? J. Comb.Chem. 2005, 7(6), 864-878). A suspension of fluorenylmethylcarbazate (1) in EtOH (0.2 M) was treated with 100 mol percent of 4-methoxybenzaldehyde, heated at reflux for 2 h, and concentrated in vacuo. The resulting methylidene carbazate was dissolved in THE (0.2 M), treated successively with 110 mol percent AcOH and 110 mol percent NaBH3CN, stirred for 1 h, and treated with additionalNaBH3CN if necessary until completion of the reaction was observed by TLC. The mixture was concentrated in vacuo. The residue was dissolved in EtOAc, washed with aqueous KHSO4 (1M) and brine, dried over Na2SO4, and concentrated under reduced pressure to yield a white solid that was dissolved in EtCH and heated at reflux for 1 h. The mixture was concentrated under reduced pressure to yield a residue that was purified by flash chromatography to yieldcarbazate 2 (76 percent) as white solid: Rt 0.53 (30percent EtOAc); 1H NMR (300 MHz, CDCI3) 6 7.78 (d,J= 7.5 Hz, 2H), 7.58 (d, J= 7.6 Hz, 2H), 7.45?7.27 (m, 6H), 6.87 (d, J= 7.2, 2H), 6.35 (bs, 1H),4.48 (bs, 2H), 4.24 (t, J= 2.3 Hz, 1H), 3.94 (bs, 2H), 3.81 (s, 3H); ?3C NMR (75 MHz, CDCI3) 6159.2, 157.2, 143.7, 141.4, 130.3, 129.4, 127.8, 127.1, 125.0, 120.1, 113.9, 66.9, 55.4, 47.2;HRMS mfz calculated for C23H22N203 [M÷HJ 375.1703; found 375.1697.
(+)-diethyl ((4-methoxyphenyl)-2-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)-methyl)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With 3-((3,5-bis(trifluoromethyl)phenyl)amino)-4-(((S)-(6-methoxyquinoline-4-yl))((1S,2S,4S,5R-5-vinylquinuclidine-2-yl)methyl)amino)cyclobutan-3-ene-1,2-dione; In neat liquid; at 50℃; for 48h;
General procedure: Reactions were carried out with <strong>[66521-66-2]4-(pyridin-3-yl)pyrimidin-2-amine</strong> 1 (0.50 mmol), aldehyde 2 (0.50 mmol) and malonate 3 (5 mmol) in the presence of catalyst III or IV (10 molpercent) at 50 °C and stirred for 48h. After completion of the reaction (as observed by TLC), the crude product was purified by preparative TLC (GF254 silica gel: hexane/EtOAc = 7/1), giving the target chiral product
(-)-diethyl ((4-methoxyphenyl)-2-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)-methyl)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92%
With quinine; In neat liquid; at 50℃; for 48h;
General procedure: Reactions were carried out with <strong>[66521-66-2]4-(pyridin-3-yl)pyrimidin-2-amine</strong> 1 (0.50 mmol), aldehyde 2 (0.50 mmol) and malonate 3 (5 mmol) in the presence of catalyst III or IV (10 molpercent) at 50 °C and stirred for 48h. After completion of the reaction (as observed by TLC), the crude product was purified by preparative TLC (GF254 silica gel: hexane/EtOAc = 7/1), giving the target chiral product
With triphenylphosphine; In 1-methyl-pyrrolidin-2-one; at 100℃;Sealed tube; Inert atmosphere;
General procedure: An oven-dried 3-neck round-bottomed ask equipped with amagnetic stir bar was charged with aryl aldehyde (1.0 equiv.) andtriphenylphosphine (1.2 or 1.5 equiv.). The system was sealed withthree PFTE septa, and subsequently evacuated and backlled withN2 three times. Dry NMP was added via syringe transfer (PTFE sy-ringe with oven-dried stainless-steel needle), and the system wasimmersed in a preheated 100C oil bath. Once no solid reagentsremained (approximately 2 min of heating), potassium bromodi-uoroacetate (1.5 or 1.8 equiv.) was added portionwise over 0.5 h,with the rate of addition controlling the evolution of CO2 gas. Onceall of the potassium bromodiuoroacetate was added, the solutionwas allowed to stir for 0.5e1 h. Upon completion, the reaction wascooled to room temperature and then quenched with H2O. Subse-quently, Et2O was added to the reaction, and the mixture waswashed with H2O (ve times), and the aqueous layer was back-extracted with Et2O (two times). The combined organic layerswere dried over Na2SO4 and concentrated. The crude material wasdry-packed onto silica gel and then eluted through a plug of silicagel with EtOAc:hexanes (1:1) to remove triphenylphosphine oxide.Subsequently, H2O2 (30% in H2O) was added to the mother liquorand allowed to react for 30 min to oxidize the residual triphenyl-phosphine. The organic layer was washed with H2O (three times),dried over Na2SO4, concentrated, and subjected to normal phaseash chromatography using EtOAc and hexanes.
2-[(E)-2-(thiophen-2-yl)ethenyl]quinazolin-4(3H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
41%
With sulfuric acid; at 190℃; under 750.075 - 5250.53 Torr; for 1.5h;Microwave irradiation;
General procedure: The 2-methylquinazolin-4(3H)-ones (1 mmol) were mixed with benzaldehydes (1.5 mmol) and 1 drop concentrated sulphuric acid was added to the mixtures. The reaction was carried out in microwave set (Personal Chemistry, Emrys Creator, heating power: 150 W, measured pressure 1-7 bar, reaction time: 1.5 hours) at 190 C. The crude product was washed with 5% sodium hydrogen carbonate and filtered out. The products were crystallized from dimethyl formamide and dried under vacuum.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






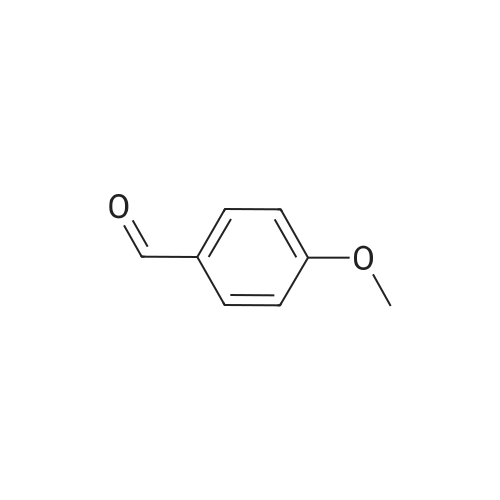

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping