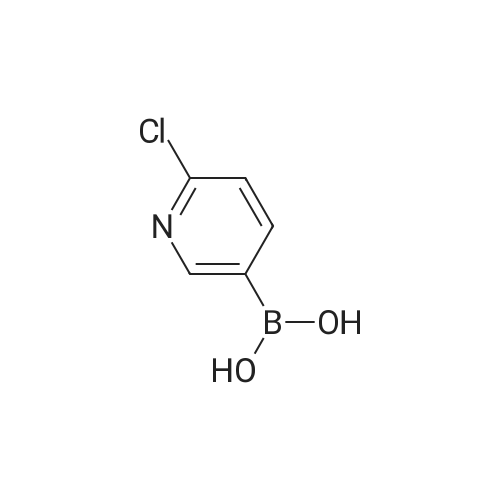
| 81% |
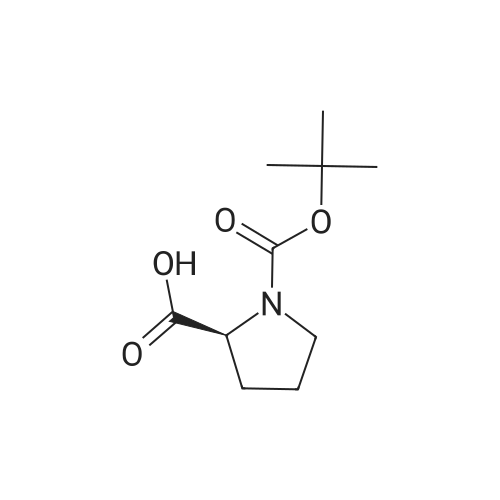
Stage #1: 1-(tert-butoxycarbonyl)-L-proline With N-ethyl-N,N-diisopropylamine; HATU In tetrahydrofuran at 0℃; for 0.5h;
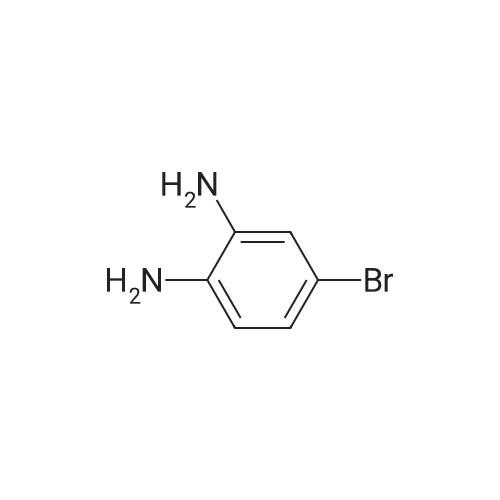
Stage #2: 4-Bromo-benzene-1,2-diamine In tetrahydrofuran at 20℃; for 4h;
Stage #3: With acetic acid at 40℃; |
1.9 10709] Step 9) the Preparation of Compound 1-11
10710] To a solution of compound 1-10 (20 g, 107 mmol) and compound HATU (48.82 g, 128.4 mmol) in THF (250 mE) was added DIPEA (19.5 mE, 118 mmol) at 0° C. After stirring at 0° C. for 0.5 hr, to the solution was added the compound 1-10-2 (25.6 g, 119 mmol) in a portionwise manner, then the reaction mixture was stirred at it for 4 hrs. After the reaction was completed, the reaction was quenched with water (100 mE), the solvent THF was removed, and the resulting mixture was extracted with EtOAc (200 mEx3). The combined organic layers were dried over anhydrous Na2504 and concentrated in vacuo. The residue was dissolved in glacial acetic acid (100 mE). The solution was stirred at 40° C. overnight, and HOAc was removed. The resulting mixture was dissolved in EtOAc (400 mE), washed with Na2CO3 aq (150 mEx3) and dried over anhydrous Na2504 and concentrated in vacuo. The residue was purified by a silica gel colunm chromatography (PE/EtOAc (v/v)=1/2) to give the title compound (35 g, 81%). The compound was characterized by the following spectroscopic data:10711] MS (ESI, pos.ion) mlz: 367.3 [M+H]10712] ‘H NMR (400 MHz, CDC13) ö (ppm): 7.68 (s, 1H),7.42-7.40 (m, 1H), 7.30-7.28 (m, 1H), 5.11-5.09 (m, 1H),3.45-3.43 (m, 2H), 2.94-2.93 (m, 1H), 2.21-2.18 (m, 2H),2.01-1.91 (m, 1H), 1.49 (s, 9H). |
| 71% |
Stage #1: 1-(tert-butoxycarbonyl)-L-proline With N-ethyl-N,N-diisopropylamine; HATU In tetrahydrofuran at 20℃; for 0.166667h;
Stage #2: 4-Bromo-benzene-1,2-diamine In tetrahydrofuran at 20℃;
Stage #3: With acetic acid at 40℃; for 12h; |
1a; 2
To a solution of N-Boc-L-Pro-OH (29 g, 135 mmol) and DIPEA (29 g, 225 mmol) in THF (500 mL) was added HATU (51 g, 135 mmol) at rt. After stirring at rt for 10 min, 4-bromobenzene-l,2-diamine (1-5) (25 g, 135 mmol) was added and the resulting solution was stirred at rt for another several hours. Subsequently, the reaction mixture was concentrated and the residue was diluted with EtOAc (500 mL). The resulting mixture was washed with water for several times (100 mL x 3) and dried with anhydrous Na2S04. The solvent was removed and the residue was dried in vacuo to give a mixture of acylated products, which were used for the next step without further purification.A mixture of acylated products from above in AcOH (1000 mL) was stirred at 40 °C for 12 hrs. After cooling, the reaction mixture was carefully neutralized by adding saturated aqueous sodium bicarbonate solution to adjust the pH value to 8. The resulting mixture was extracted with EtOAc (250 mL x 3). The combined extract was washed with water, and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel chromatography (Petroleum ether/EtOAc = 4/1 (v/v)) to give (S)-tert- butyl 2-(6-bromo-lH-benzo[d]imidazol-2-yl)pyrrolidine-l-carboxylate (la-6) (35 g, 71% yield in 2 steps) as a yellow solid. LC-MS (ESI): m/z 366.1 [M+H]+. |
| 71% |
Stage #1: 1-(tert-butoxycarbonyl)-L-proline With N-ethyl-N,N-diisopropylamine; HATU In tetrahydrofuran at 20℃; for 0.166667h;
Stage #2: 4-Bromo-benzene-1,2-diamine In tetrahydrofuran at 20℃;
Stage #3: With acetic acid at 40℃; for 12h; |
11-1; 11.e; 11.f
To a solution of N-Boc-L-Pro-OH (29 g, 135 mmol) and DIPEA (29 g, 225 mmol) in THF (500 mL) was added HATU (51 g, 135 mmol) at rt. After stirring at rt for 10 min, compound 42 (25 g, 135 mmol) was added and the resulting solution was stirred at rt for another several hours. Subsequently, the reaction mixture was concentrated and the residue was diluted with EtOAc (500 mL). The resulting mixture was washed with ¾0 several times (100 mL x 3) and dried with anhydrous Na2S04. The solvent was removed and the residue was dried in vacuo to give a mixture of crude compounds 43 and 43', which were used for the next step without further purification. LC-MS (ESI): m/z 384.1 (M+H)+. A mixture of crude compounds 43 and 43' obtained from the reaction above in AcOH (1000 mL) was stirred at 40 °C for 12 h. Subsequently, the reaction mixture was carefully neutralized by adding saturated aqueous sodium bicarbonate solution to adjust the pH value to 8. The resulting mixture was extracted with EtOAc several times (250 mL x 3). The extracts were combined, washed with water, and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel chromatography (Petroleum ether/EtOAc = 4/1 (v/v)) to give compound 44 (35 g, 71% yield, two steps from compound 42) as a yellow solid. LC-MS (ESI): m/z 366.1 (M+H)+. |
| 69% |
Stage #1: 1-(tert-butoxycarbonyl)-L-proline With 1,1'-carbonyldiimidazole In pyridine; N,N-dimethyl-formamide at 45℃; for 2h;
Stage #2: 4-Bromo-benzene-1,2-diamine In pyridine; N,N-dimethyl-formamide at 20℃;
Stage #3: With acetic acid at 100℃; for 0.5h; |
1.1
To a solution of Boc-L-Proline (2669 mg, 12.4 mmol) in pyridine/DMF (30 mL, 1/1) was added di(lH-imidazol-l-yl)ketone (2205 mg, 13.6 mmol). The mixture was stirred at 45°C for 2 hours. 4-bromobenzene-l,2-diamine (2319 mg, 12.4 mmol) was added and the mixture was stirred at ambient temperature overnight. The solvent was removed and the residue heated in acetic acid (15 mL) at 100°C for 30 minutes. After concentration of the residue, the mixture was partitioned between ethyl acetate and a saturated sodium bicarbonate solution. The organic phase was separated and washed with water. After drying over Na2SC"4, the mixture was filtered and the filtrate was concentrated in vacuo. The obtained residue was purified by flash chromatography using DCM/EtOAc 90/10 to 50/50, resulting in compound II (3.146 g, 69 %). |
| 69% |
Stage #1: 1-(tert-butoxycarbonyl)-L-proline With pyridine; 1,1'-carbonyldiimidazole In N,N-dimethyl-formamide at 45℃; for 2h;
Stage #2: 4-Bromo-benzene-1,2-diamine In N,N-dimethyl-formamide at 20℃; |
1.1
1.1 preparation of intermediate Ila (PG= Boc; X= Br)Ma; To a solution of Boc-Z-Proline (2669 mg, 12.4 mmol) in pyridine/DMF (30 mL, 1/1) was added di(lH-imidazol-l-yl)ketone (2205 mg, 13.6 mmol). The mixture was stirred at 45°C for 2 hours. 4-bromobenzene-l,2-diamine (2319 mg, 12.4 mmol) was added and the mixture was stirred at ambient temperature overnight. The solvent was removed and the residue heated in acetic acid (15 mL) at 100°C for 30 minutes. Afterconcentration of the residue, the mixture was partitioned between ethyl acetate and a saturated sodium bicarbonate solution. The organic phase was separated and washed with water, after drying over Na2SC"4, the mixture was filtrated and the filtrate was concentrated in vacuum. The obtained residue was purified by flash chromatography using CH2Cl2/EtOAc 90/10 to 50/50, resulting in compound Ila (3.146 g, 69 %). |
| 63% |
Stage #1: 1-(tert-butoxycarbonyl)-L-proline With N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 20 - 25℃; for 1h; Inert atmosphere; Large scale;
Stage #2: With HATU In tetrahydrofuran at 20 - 25℃; Inert atmosphere; Large scale;
Stage #3: 4-Bromo-benzene-1,2-diamine Large scale; Further stages; |
2.1
Step 1. Referring to Scheme 2, N-Boc-L-Proline (4.02 kg, 1.0 eq.) and THF (52.5 L, 15.0 volume) were charged into a 200 L reactor under nitrogen atmosphere. The mixture was cooled to 20 - 25 °C and N, N-diisopropylethylamine (4.8 L, 1.5 eq.) was added over a period of 60 min. Next, HATU (7.11 kg, 1.0 eq.) was slowly added by portion wise over a period of 90 - 120 min at 20 - 25 °C under an atmosphere of nitrogen. After stirring at the same temperature for 15 min, 4-bromo-l, 2-diaminobenzene (3.50 kg, 1.0 eq.) was added into the reactor portion- wise over a period of 90 - 120 min. The resulting reaction mass was stirred at the same temperature. After stirring for 4 - 5 hrs, HPLC analysis indicated that > 97% of 4-bromo-l, 2-diaminobenzene was consumed. The reaction mass was concentrated under vacuum (600 mmHg) to remove THF at < 40 °C and the residue was diluted with ethyl acetate (40.0 L, 10.0 volume) and purified water (25.0 L, 7.0 volume). The resulting mixture was well stirred and the organic layer was separated. Subsequently, the organic layer was washed with purified water (25 L x 3, 7.0 volume) and with saturated brine solution (25 L x1, 7.0 volume) and dried over anhydrous Na2S04. The solvent was removed under high vacuum at 97% of the intermediate was consumed. AcOH was completely distilled off under high vacuum at 40 - 45 °C. The resulting syrup mass was diluted with EtOAc (50.0 L, 14.0 volume) and was purified by washing with water (25.0 L, 7.0 volume) with stirring. The organic layer was separated, washed twice with 5.0 % (w/w) aqueous NaHC03 solution (25.0 L x 2, 7.0 volume), twice with purified water (25.0 L x 2) and once with saturated brine (25 L x 1, 7.0 volume), and dried over anhydrous Na2S04. The solution was treated with active carbon before it was filtered and concentrated under vacuum (600 mrnHg) at 40 - 45 °C to give crude product as a foamy solid (5.20 kg). The residue was suspended with stirring in MTBE (5.2 L, 1.5 volume), the solid was collected by filtration, washed with MTBE (1.75 L, 0.5 volume) and dried in a vacuum tray drier at 40 - 45 °C for 12 hrs to give compound 2-2a (4.20 kg, 63% yield) as pale brown solid with a purity of > 98.0% determined by HPLC analysis. LC-MS (ESI): m/z 366.1 [M + H]+. 1H NMR (400 MHz, -DMSO): δ 12.40 (m, 1H), 7.58 - 7.70 (m, 1H), 7.37 - 7.46 (m, 1H), 7.24 (m, 1H), 4.85 - 4.94 (m, 1H), 3.54 (, 1H), 3.35 - 3.53 (m, 1H), 2.20 -2.32 (m, 1H), 1.88 - 1.96 (m, 3H), 1.38 and 0.98 (s, s, 9H) ppm. |
|
Stage #1: 1-(tert-butoxycarbonyl)-L-proline; 4-Bromo-benzene-1,2-diamine With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane at 20℃; for 19h;
Stage #2: With acetic acid at 65℃; for 1.5h; |
M1.a
EDCI-HCl (2.348 g, 12.25 mmol) was added to a mixture of 4- bromobenzene-l52-diamine (2.078 gs 11.11 mmol), N-Boc-L-proline (2.311 g, 10.74 mmol) and l-hydroxybenzotriazole (1.742 g, 12.89 mmol) in CH2Cl2 (40 mL), and stirred at ambient conditions for 19 h. The mixture was then diluted with CH2Cl2J washed with water (2x), dried (brine; MgSO4), filtered, and concentrated in vacuo to provide a brown foam. Acetic acid (40 mL) was added to the foam, and the mixture was heated at 65 0C for 90 min. The volatile component was removed in vacuo, and the residue was dissolved in EtOAc and washed carefully with saturated NaHCO3 solution (2x), and the organic phase was dried (brine; MgSO4), filtered, and concentrated in vacuo. The resultant crude material was submitted to flash chromatography (silica gel; EtOAc) to provide benzimidazole MIa as a tan foam (2.5 g). 1H NMR (DMSO-de, 6 = 2.5 ppm, 400 MHz): 12.49-12.33 (four br s, IH), 7.71 (d, J = 2, 0.54H), 7.60 (app br s, 0.46H), 7.50 (d, J = 8.6, 0.45H), 7.40 (d, J = 8.4, 0.55H), 7.26 (m, IH), 4.96-4.87 (m, IH), 3.64-3.51 (m, IH), 3.44-3.38 (m, IH), 2.38- 2.21 (m, IH), 1.99-1.85 (m, 3H), 1.39 (s, 3.7H), 1.06 (s, 5.3H). LC/MS: Anal Calcd. for [M+H]+ Ci6H2181BrN3O2: 368.03; found: 368.18. |
|
Multi-step reaction with 2 steps
1.1: HATU; N-ethyl-N,N-diisopropylamine / N,N-dimethyl-formamide / 0.5 h / 20 °C
1.2: 16 h / 20 °C
2.1: acetic acid / 2 h / 85 °C |
|
|
Stage #1: 1-(tert-butoxycarbonyl)-L-proline; 4-Bromo-benzene-1,2-diamine With 4-methyl-morpholine; HATU In N,N-dimethyl-formamide for 16h;
Stage #2: In ethanol at 110 - 130℃; for 48h; Sealed tube; |
AK1
To a mixture of 4-Bromo-benzene-1,2-diamine (5.0 g), Boc-L-proline (6.0 g), and 4- methylmorpholine (5.88 mL) in DMF (100 mL) was added HATU (10.7 g). Reaction mixture was stirred for 16 hours and then concentrated. Residue was dissolved in ethyl acetate and washed with 5% lithium chloride solution (2x), brine and dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 30 to 60% ethyl acetate/hexanes) to a dark brown foam. Brown foam was dissolved in ethanol (100 mL) and heated in a sealed tube at 110- 130°C for 2 days. Reaction mixture was cooled and concentrated. Residue was dissolved in ethyl acetate and extracted with IN HCl (3x). Aqueous layer was basified with 50% NaOH solution to pH 10 and extracted with ethyl acetate (2x). The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 0 to 10% isopropanol/hexanes) to give 2-(6-Bromo-1H-benzoimidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (6.5 g) as an off-white foam: LCMS-ESI+: calc'd for C16H2IBrN3O2: 367.26 (M+H+); Found: 365.8, 367.8 (M+H+). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping