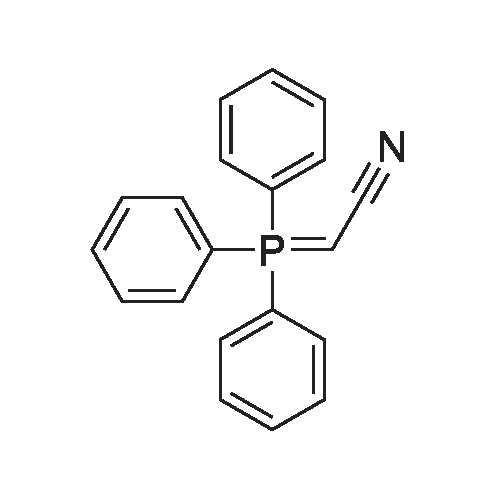
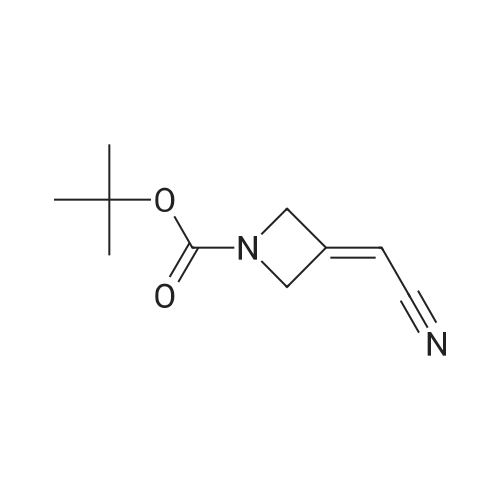
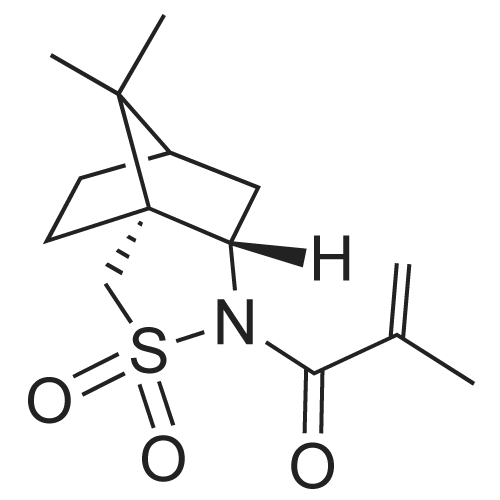
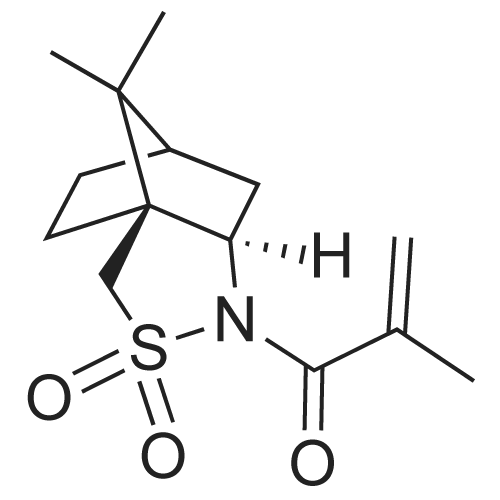
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran at -10 - 0℃; Inert atmosphere Stage #2: at -10 - 20℃; Inert atmosphere
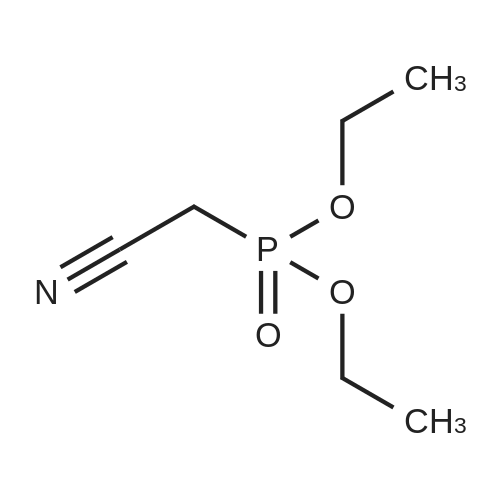
Under a nitrogen atmosphere, in a 3L three-necked bottle, diethyl cyanomethylphosphonate (212.5 g, 1.2 mol) and anhydrous THF (300 ml) were added and dissolved by stirring, and then the temperature of the system was lowered to _10 ~0°C,Under this condition, a solution of t_Bu0K (123.2 g, 1.12 mol) in THF (200 ml) was slowly added dropwise, and the temperature of the control system was maintained at -10 to 0°C. At this temperature, the reaction was effected for 3-4 hours.Then, a solution of Intermediate B (163.0 g, 1.0 mol) in THF (300 ml) was slowly added dropwise during the dropwise addition of the THF solution containing Intermediate B.The temperature of the reaction system was controlled to be -10 ~ 0°C. After the dropwise addition, the mixture was stirred at this temperature for 2-3 hours, then slowly raised to room temperature and allowed to react for 15-18 hours at room temperature.Then the reaction solution was adjusted to pH 3-4 with water (500 ml), saturated NaCl solution (500 ml), 0.5 HC HCl, and extracted with ethyl acetate (500 ml X 2).The organic layers were combined and dried to afford an off-white solid 127,48 in 68.5percent yield.
Reference:
[1] Patent: CN107739328, 2018, A, . Location in patent: Paragraph 0050; 0051; 0058
[2] Patent: WO2016/205487, 2016, A1, . Location in patent: Page/Page column 26
2
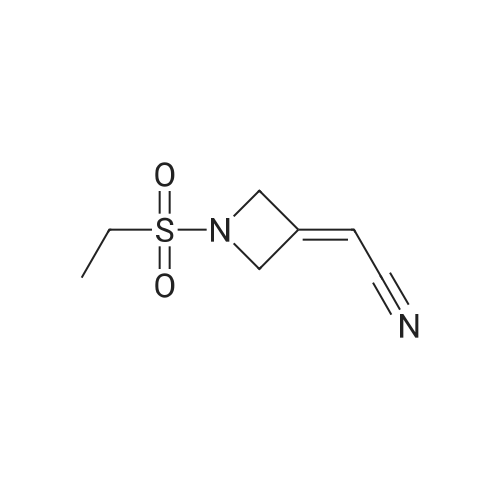
[ 50586-62-4 ]
[ 1187595-85-2 ]
Yield
Reaction Conditions
Operation in experiment
96.4%
Stage #1: With sodium methylate In tetrahydrofuran at 10 - 20℃; for 0.75 h; Inert atmosphere Stage #2: at 30℃; for 11 h;
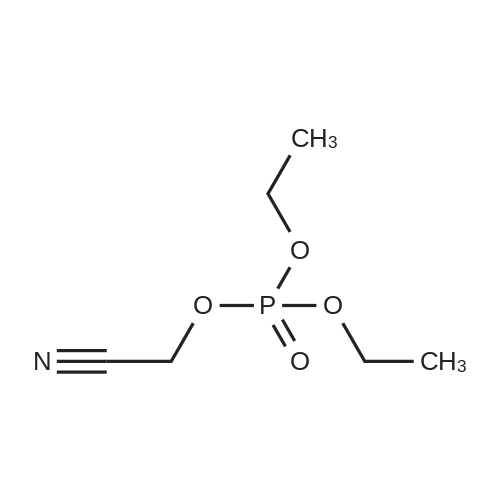
450 g of tetrahydrofuran was added to the 2 L reaction flask. 21.3 g (0.12 mol) of diethyl cyanomethyl phosphate, The system was cooled to 10 ° C, and 7.0 g (0.13 mol) of sodium methoxide was added under nitrogen atmosphere. After the addition, the system gradually warmed to 20 °C. Stir for 45 min. A solution of 16.3 g (0.1 mol) of compound 6 and 80 g of tetrahydrofuran was continuously added dropwise to the system. After 1 hour, the reaction was stirred at 30 ° C for 10 h, and the reaction of the central control material was almost complete. After the system was cooled to 10 ° C, 500 g of ethyl acetate and 300 g of saturated brine were added. After stirring for 5 min, the layers were separated. The aqueous layer was extracted with 300 g of EtOAc EtOAc. The organic layer was washed with 500 g of water. The organic layer is dry, Filtration and concentration of the filtrate gave the crude product of compound 7, The crude product was washed with a small amount of n-hexane and dried to give 16.6 g of Compound 7. Yield: 96.4percent, HPLC ≥ 98.0percent.
Reference:
[1] Patent: CN108129482, 2018, A, . Location in patent: Paragraph 0054; 0062; 0063
3
[ 1314910-43-4 ]
[ 594-44-5 ]
[ 1187595-85-2 ]
Yield
Reaction Conditions
Operation in experiment
98.59%
Stage #1: With N-ethyl-N,N-diisopropylamine In acetonitrile at 0 - 10℃; for 0.166667 h; Stage #2: at 0 - 25℃; for 16 h;
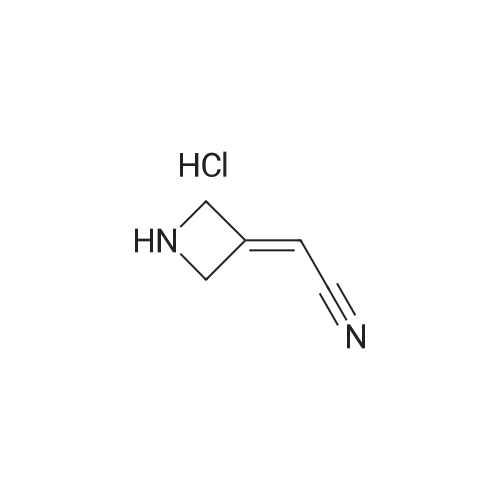
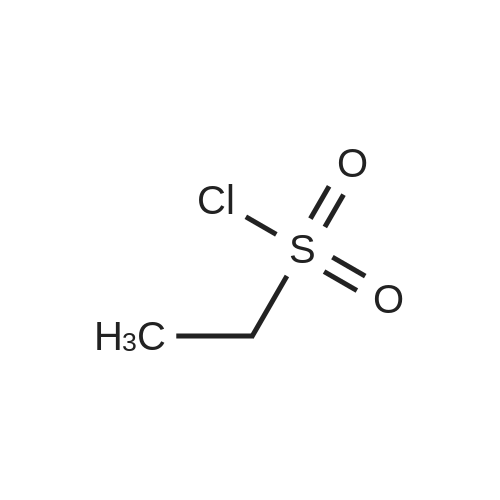
N,N-Diisopropylethylamine (4.5 mL) was added into a reaction vessel containing acetonitrile (50 mL) and 3-(cyanomethylene)azetidine hydrochloride (1.5 g; Formula VII) at about 0°C to about 10°C. The reaction mixture was stirred for about 10 minutes. Ethanesulfonyl chloride (2.22 g) was added into the reaction mixture at about 0°C to about 5°C over about 5 minutes. The temperature of the reaction mixture was raised to about 20°C to about 25 °C, and then the reaction mixture was stirred for about 16 hours. On completion of the reaction, acetonitrile was recovered from the reaction mixture under reduced pressure at about 40°C to about 45°C to obtain an oily residue. Dichloromethane (50 mL) was added into the residue. The contents were washed with a saturated sodium chloride solution (30 mL), followed by complete recovery of dichloromethane under reduced pressure at about 40°C to obtain 2-(l-(ethylsulfonyl)azetidin-3- ylidene)acetonitrile . Yield: 98.59percent
To a solution of 2-(azetine-3-yl) methyl cyanide(2.8 g, 21.4 mmol) and DIPEA(8.3 g, 64.3 mmol) in dichloromethane(30 mL) was added dropwise ethanesulfonyl chloride(4.1 g, 32.1 mmol) at 0°C under the protection of nitrogen, and the temperature was kept below 2°C when dropping. The reaction mixture was stirred at 25°C for reacting for 16 hours. TLC showed that the reaction was completed (petroleum ether/ethyl acetate =1:1). After the reaction mixture was quenched with water, extracted with dichloromethane (30 mL*2). The combined organic phase was washed with saturated salt water(20 mL*2), dried with anhydrous sodium sulfate, filtered, and spun dry. The residue was purified through column chromatography (dichloromethane/ethyl acetate =3/1) to give 2-(1-(ethylsulfonyl)azetine-3-yl)methyl cyanide(1.4 g, 33.0percent yield) as a pale yellow solid. 1H NMR (400MHz, CDCl3) δ = 5.50 - 5.41 (m, 1H), 4.79 (d, J=3.0 Hz, 2H), 4.71 (d, J=2.5 Hz, 2H), 3.06 (q, J=7.4 Hz, 2H), 1.40 (t, J=7.4 Hz, 3H). MS (ESI) Calcd. for C7H10N2O2S [M + H]+ 187, Found 187.
192 mg
With N-ethyl-N,N-diisopropylamine In acetonitrile at 0 - 20℃;
Trifluoroacetic acid (2 mL) was added to a solution of tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (2) (250 mg, 1.29mmol) in dichloromethane (20 mL). The solution was stirred at room temperature for 5 h. Then the reaction mixture was concentrated under reduced pressure to dryness. The residue, which contains the crude desired deprotection product 3, was then suspended ina cetonitrile (20 mL) and cooled to 0 °C. N,N-Diisopropylethylamine (DIPEA) (1.12 mL, 6.44 mmol, 5 equiv.) was then slowly added while keeping the internal temperature below 5 °C. Then ethanesulfonyl chloride (EtSO2Cl) (0.184 mL, 1.94 mmol, 1.5 equiv.) was added over 1 h while keeping the internal temperature below 5 °C. The resulting reaction mixture was stirred overnight at room temperature and then concentrated under reduced pressure. The concentrated residue was then diluted with dichloromethane and was washed with aqueous sodium chloride solution. The aqueous phase was back-extracted with dichloromethane. The combined organic layers were dried over Na2SO4 and the residue was purified using a silica gel column to afford compound 4: Yield 192 mg (81percent); off-white solid; m.p. 58–60 °C; IR: 3294, 3066, 2978, 2222, 1702, 1321, 1142 cm–1. Anal. calcd for C7H10N2O2S: C, 45.15; H, 5.41; N, 15.04; found: C, 45.32; H, 5.35; N, 15.21percent. MS (m/z): 209 [M + Na]+; 1H NMR (300 MHz, CDCl3): δ 1.37 (m, J = 6.7 Hz, 3H), 3.04 (m, J = 7.4 Hz, 2H), 4.69 (t, J = 2.5 Hz, 2H), 4.77 (t, J = 2.8 Hz, 2H), 5.43 (t, J = 2.4 Hz, 1H); 13C NMR (75 MHz, DMSO-d6): δ 7.2, 42.6, 58.5, 58.9, 93.9, 114.9, 156.2, 168.6.
0.192 g
With N-ethyl-N,N-diisopropylamine In acetonitrile at 0 - 5℃;
Compound 2 (0.250g, 1.29mmol) dissolved in acetonitrile was slowly added dropwise trifluoroacetic acid (3ml).Stirred at room temperature for 4 hours gussets tracking.After completion of the reaction, the solvent spin dry, obtained was dissolved in acetonitrile and cooled to 0 deg.] C, was slowly added N, N- diisopropylethylamine (DIEA), maintaining the temperature not higher than 5 .Was slowly added compound 8 (0.184ml, 1.94mmol, 1.5equiv), at T Completion of the reaction followed by TLC, concentrated under reduced pressure, the crude product was diluted with dichloromethane, washed with brine, the aqueous phase was extracted with dichloromethane, the organic phase was concentrated, dried over anhydrous sodium sulfate, and purified by column chromatography to give 0.192g white solid with a yield of 81percent.
Reference:
[1] Patent: EP3290418, 2018, A1, . Location in patent: Paragraph 0083
[2] Journal of Chemical Research, 2016, vol. 40, # 4, p. 205 - 208
[3] Patent: CN105541891, 2016, A, . Location in patent: Paragraph 0084; 0085; 0086; 0087; 0088; 0089; 0090; 0091
5
[ 372-09-8 ]
[ 1187595-85-2 ]
Yield
Reaction Conditions
Operation in experiment
81%
With piperidine In tolueneReflux
A three-necked flask was charged with compound 2 (16.33 g, 100 mmol) Cyanoacetic acid (17.01 g, 200 mmol) And toluene (82 mL), After stirring, piperidine (17.03 g, 200 mmol) was added, Plus after warming to reflux reaction 16-24 hours, The reaction was quenched by cooling to room temperature and adding 0.5 mol / L dilute hydrochloric acid (326 mL). The reaction mixture was separated and extracted with ethyl acetate (163 mL) The organic phase was washed with saturated brine twice (163 mL) Dried over sodium sulfate and concentrated to give Compound 3 (15.08 g, 81percent) by column chromatography on petroleum ether ethyl acetate mixed solvent column chromatography.
Reference:
[1] Patent: CN106496195, 2017, A, . Location in patent: Paragraph 0056-0058
6
[ 16640-68-9 ]
[ 1187595-85-2 ]
Yield
Reaction Conditions
Operation in experiment
67%
at 20℃;
Three-necked flask was charged with triphenylphosphine acetonitrile (33.14 g, 110 mmol) and dichloromethane (96 mL)And then 1- (ethylsulfonyl) azepin-3-one (prepared from Example 3) was added and reacted at room temperature for 3 to 4 hours. The reaction was quenched by the addition of 240 mL of water. The aqueous phase was extracted three times with ethyl acetate (120 mL). The organic phase was washed with saturated brine (120 mL) and concentrated to recrystallize from isopropyl ether and petroleum ether to give 2- [1-(ethylsulfonyl)-3-azetidinylidene] acetonitrile(16.76 g, 4 steps yield 67percent).
Reference:
[1] Patent: CN106946917, 2017, A, . Location in patent: Paragraph 0064-0066
Reference:
[1] Patent: WO2013/40863, 2013, A1,
[2] Journal of Chemical Research, 2016, vol. 40, # 4, p. 205 - 208
[3] Patent: US2016/333015, 2016, A1,
[4] Patent: CN105541891, 2016, A,
[5] Patent: EP3290418, 2018, A1,
9
[ 1187595-85-2 ]
[ 1187594-09-7 ]
Yield
Reaction Conditions
Operation in experiment
91.2%
Stage #1: With potassium carbonate In acetonitrile at 20℃; for 0.5 h; Stage #2: With 1,8-diazabicyclo[5.4.0]undec-7-ene In acetonitrile at 40℃; for 8 h;
200 g of acetonitrile and 20.4 g (0.05 mol) of compound 4 were added to a 1 L reaction flask. 6.9 g (0.05 mol) was added with stirring. Potassium carbonate was stirred at room temperature for 30 min, then 9.3 g (0.05 mol) of compound 7 and 8 g (0.05 mol) of DBU were added. The temperature of the system is raised to 40 ° C After the reaction for 8 hours, the reaction was completed, and the reaction of the starting materials was completed, and the reaction was stopped. Evaporate the solvent under reduced pressure and add to the system. After quenching the reaction with 100 g of water, 200 g of ethyl acetate was added. Stir for 30 min, filter, and rinse the cake with 100 g of fresh ethyl acetate. The filter cake was dried to give a white solid of 17.0 g, HPLC ≥ 99.0percent, yield: 91.2percent.
Reference:
[1] Patent: CN108129482, 2018, A, . Location in patent: Paragraph 0054; 0064; 0065
10
[ 1187595-85-2 ]
[ 1187594-09-7 ]
Reference:
[1] Patent: CN105541891, 2016, A,
[2] Patent: WO2016/205487, 2016, A1,
[3] Patent: WO2016/205487, 2016, A1,
[4] Patent: CN106496195, 2017, A,
[5] Patent: CN106496195, 2017, A,
[6] Patent: CN106496195, 2017, A,
[7] Patent: CN106496195, 2017, A,
[8] Patent: CN106496195, 2017, A,
[9] Patent: CN106946917, 2017, A,
[10] Patent: CN106946917, 2017, A,
[11] Patent: CN106946917, 2017, A,
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
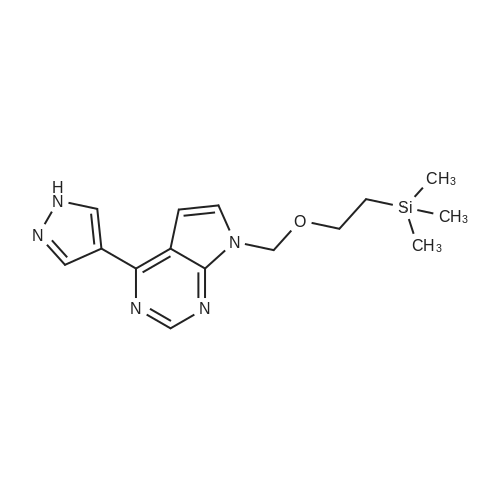
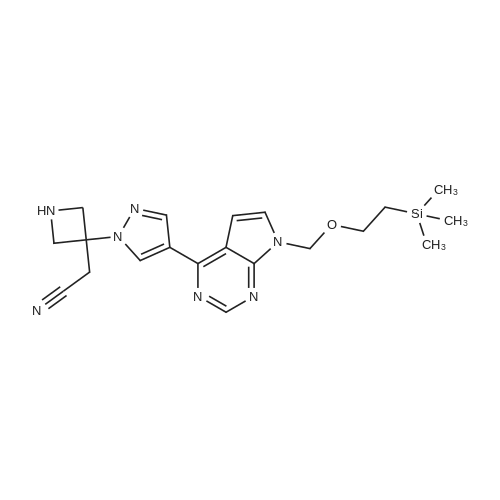
At room temperature, acetonitrile (10 mL) was added to a mixture of 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (9a) (214 mg) and <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (126 mg) to obtain a reaction solution. DBU (120 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified on a preparative silica gel plate, to afford 2-(1-(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (9b) (327 mg, solid), yield: 77%. MS m/z: 502 [M+1]+.
1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 15 - 25℃;Product distribution / selectivity;
To a suspension of 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 440 g, 1.395 mol) and <strong>[1187595-85-2]2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile</strong> (11, 312.4 g, 1.68 mol, 1.2 equiv) in acetonitrile (4.4 L) was added DBU (249.8 mL, 1.67 mol, 1.2 equiv) drop wise to keep the reaction temperature between 15-25 C. After adding DBU, the reaction mixture became homogeneous, but a precipitate appeared in 30 min. The reaction mixture was stirred for 3 h at room temperature. When HPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (11 L). The resulting mixture was stirred at room temperature for additional 30 min and then filtered. The solid cake was washed with water (4 L), MTBE (2 L) and dried in vacuum oven at 35 C. for 24 h to afford crude 2-(1-(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (12, 681 g, 699.8 g theoretical, 97.3% yield) as white solids, which was found to be sufficiently pure for the subsequent reaction without further purification. For 12: 1HNMR (CDCl3, 300 MHz), 6 8.86 (s, 1H), 8.45 (s, 1H), 8.35 (s, 1H), 7.43 (d, 1H), 6.80 (d, 1H), 5.68 (s, 2H), 4.65 (d, 2H), 4.27 (d, 2H), 3.55 (s, 2H), 3.4 (t, 2H), 3.07 (m, 2H), 1.42 (m, 3H), 0.92 (m, 2H), -0.05 (s, 9H) ppm; C22H31N7O3SSi (MW, 501.68), LCMS (EI) m/e 502 (M++H).
1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; at 15 - 25℃;
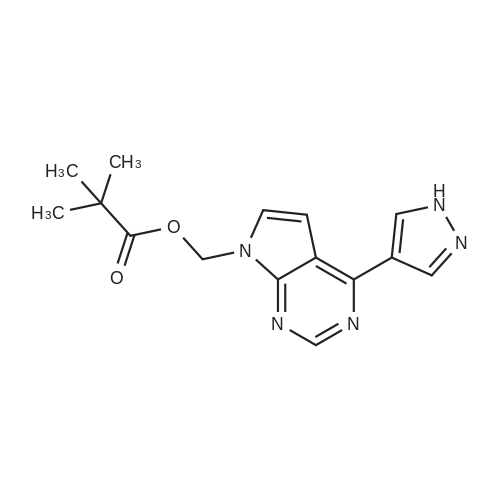
To a suspension of [4-(1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate (19, 10.0 g, 33.4 mmol) and <strong>[1187595-85-2]2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile</strong> (11, 6.22 g, 33.4 mmol, 1.0 equiv) in N,N-dimethylformamide (DMF, 20 mL) was added DBU (254 mg, 1.67 mmol, 0.05 equiv) drop wise to keep the reaction temperature between 15-25 C. After adding DBU, the reaction mixture became homogeneous within 90 min. The reaction mixture was stirred for 3 h at room temperature. When HPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (120 mL) and acetonitrile (80 mL). The resulting mixture was stirred at room temperature for an additional 30 min. The solids were collected by filtration, washed with a mixture of acetonitrile and water (2/3 by volume, 2×20 mL), and dried in vacuum oven at 40-45 C. for 24 h to afford crude (4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (20, 14.5 g, 16.2 g theoretical, 89.5% yield) as white solids, which was found to be sufficiently pure (>98.0% by HPLC) for the subsequent reaction without further purification. For 20: 1HNMR (CDCl3, 300 MHz), delta 8.87 (s, 1H), 8.43 (s, 1H), 8.37 (s, 1H), 7.51 (d, 1H, J=3.6 Hz), 6.76 (d, 1H, J=3.6 Hz), 6.26 (s, 2H), 4.64 (d, 2H, J=9.6 Hz), 4.25 (d, 2H, J=9.6 Hz), 3.41 (s, 2H), 3.09 (q, 2H, J=7.6 Hz), 1.42 (t, 3H, J=7.6 Hz), 1.17 (s, 9H) ppm; C22H27N7O4S (MW, 485.56), LCMS (EI) m/e 486 (M++H).
2.248 g
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; at 5 - 10℃; for 3h;
A suspension of (4-(1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1.5 g, 5.01 mmol) and <strong>[1187595-85-2]2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile</strong> (EAYA) (0.95 g, 5.10 mmol) in DMF (4.5 mL) was cooled to 5 C.-10 C., then 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) (0.045 g, 0.06 mmol) was added dropwise into the solution to keep the temperature between 5 C.-10 C. After adding DBU, the mixture was stirred at 5 C.-10 C. to conduct reaction for 3 hours. When the reaction was deemed to complete, the reaction mixture was quenched with acetonitrile (12 mL) and water (18 mL). The resulting suspension was stirred at room temperature for 1 hour. The solids were collected by filtration, washed with a mixture of acetonitrile and water (2/3 by volume, 3 mL*2), and dried under vacuum to give 4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (2.248 g) as an off-white solid.
N,N-Diisopropylethylamine (4.5 mL) was added into a reaction vessel containing acetonitrile (50 mL) and <strong>[1314910-43-4]3-(cyanomethylene)azetidine hydrochloride</strong> (1.5 g; Formula VII) at about 0C to about 10C. The reaction mixture was stirred for about 10 minutes. Ethanesulfonyl chloride (2.22 g) was added into the reaction mixture at about 0C to about 5C over about 5 minutes. The temperature of the reaction mixture was raised to about 20C to about 25 C, and then the reaction mixture was stirred for about 16 hours. On completion of the reaction, acetonitrile was recovered from the reaction mixture under reduced pressure at about 40C to about 45C to obtain an oily residue. Dichloromethane (50 mL) was added into the residue. The contents were washed with a saturated sodium chloride solution (30 mL), followed by complete recovery of dichloromethane under reduced pressure at about 40C to obtain 2-(l-(ethylsulfonyl)azetidin-3- ylidene)acetonitrile . Yield: 98.59%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 0 - 20℃;Product distribution / selectivity;
A solution of tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (9, 1000g, 5.2 mol) in acetonitrile (7 L) and a 3 N aqueous HCl solution (7 L) was stirred at room temperature for 18 h. When HPLC showed that all the starting material (9) was consumed, the reaction mixture was concentrated under reduced pressure to dryness. The residue, which contains the crude desired deprotection product (10), was then suspended in acetonitrile (12 L) and the resulting suspension was cooled to 0-5 C. Diisopropyethylamine (DIEA, 3.14 L, 18.03 mol, 3.5 equiv) was then slowly added while keeping the internal temperature below 5 C. The resulting homogeneous solution was allowed to cool down to 0 C. and ethane sulfonyl chloride (EtSO2Cl, 730 mL, 7.73 mol, 1.5 equiv) was added over 1 h while keeping the internal temperature below 5 C. The resulting reaction mixture was allowed to gradually warm to room temperature and stirred at room temperature for overnight. When HPLC showed that the reaction was complete, the reaction mixture was concentrated under reduced pressure to a volume of approximately 2 L. The bath temperature of the rotary evaporator is set to not exceed 45 C. The concentrated residue was then diluted with dichloromethane (CH2Cl2, 10 L) and the resulting dichloromethane solution was washed with aqueous sodium chloride solution (10 L). The aqueous phase was back extracted with dichloromethane (CH2Cl2, 5 L). The combined organic layers were dried over anhydrous sodium sulfate (Na2SO4) and the residue was absorbed onto silica gel (SiO2, 1 Kg) under reduced pressure. The bath temperature of the rotary evaporator was set to not exceed 45 C. The material was then loaded onto a silica gel column (SiO2, 2.5 Kg) and eluted with 20-60 % ethyl acetate in heptane to afford 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 882 g, 968.4 g theoretical, 91% yield) as off-white solids. For 11: 1H NMR (CDCl3, 300 MHz) delta 5.46 (m, 1H), 4.77 (m, 2H), 4.70 (m, 2H), 3.05 (q, 2H), 1.39 (t, 3H) ppm; C7H10N2O2S (MW, 186.23), LCMS (EI) m/e 187 (M++H).
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at -10℃; for 7h;
Intermediate No.33Step A-B. ll-(Ethylsulfonv0azetidin-3-ylidenelacetonitriletert-Butyl 3-(cyanomethylidene)azetidine-l-carboxylate (5.0 g, 26 mmol) was dissolved in 4M HC1 in dioxane (25.7 mL) and allowed to stir at ambient temperature for 16 hours. The mixture was concentrated to dryness in vacuo, then dissolved in DCM (30.0 mL) and cooled to -10 C. DIPEA (11.6 g, 90.0 mmol) was added followed by ethanesulfonyl chloride (5.0 g, 39 mmol). The resulting mixture was allowed to stir for 7 hours before the mixture was diluted with water and extracted with DCM. The organic layer was dried over anhydrousNa2S04, filtered and concentrated in vacuo. The residue was purified by MPLC on silica gel (using a gradient elution of 5-70% EtO Ac/heptane). Desired fractions were identified, combined, and concentrated in vacuo to afford the title compound. LRMS (ESI) calc'd for C7H10N2O2S [M+H]+: 187, Found: 187.
Dissolving raw material CF0726Y (0.4 g, 0.0021 mol) in dioxane solution (10 mE) of saturated hydrogen chloride, clearly dissolving it and separating white solid out, spinning solvent dry after raw material disappears, adding THF (10 mE) to dissolve it, then dropwise adding DIPEA (0.8 g, 6.18 mmol, 3 eq), stirring for 10 mi dropwise adding ethylsulfonyl chloride (0.32 g, 0.0025 mol, 1.2 eq), performing room-temperature stirring and ECMS tracking, spinning it dry after the reaction, using EAH20 for extraction, washing it once with NaHCO3 and washing with saturated saline solution, spuming it dry to obtain 0.3 g of yellow oily crude product.
With N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; at 0 - 20℃;
tert-Butyl 3-(cyanomethylene)azetidine-1-carboxylate (2, 82 g, 0.42 mol) was added to THF (850 mL) and the resulting solution was cooled to 0 C. before a 4 M HCl solution in 1,4-dioxane (850 mL, 3.38 mol, 8.0 equiv) was added over 1 h while keeping the temperature<5 C. The resulting reaction mixture was slowly warmed to room temperature and stirred at room temperature for 18 h. When the reaction was deemed complete, the reaction mixture was concentrated under reduced pressure and the residue was placed under high vacuum for an additional 3 h before being treated with THF (900 mL) and diisopropylethyl amine (183 mL, 1.06 mol, 2.5 equiv) at room temperature. The resulting solution was then cooled to 0 C. and ethanesulfonyl chloride (56 mL, 0.59 mol, 1.4 equiv) was added while keeping the reaction temperature<5 C. The ice bath was removed and the reaction was stirred at room temperature for 18 h. When TLC indicated the reaction was complete, the reaction mixture was diluted with ethyl acetate (1 L) and washed with saturated brine (1 L). The aqueous layer was extracted with ethyl acetate (2×500 mL). The combined organic layers were dried over sodium sulphate and concentrated under reduced pressure. The residue was diluted with dichloromethane and absorbed onto silica gel (150 g). This mixture was purified by column chromatography (1.5 Kg silica gel) eluting with heptane (4 L), 10% EtOAc in heptane (4 L), 20% EtOAc in heptane (8L), 30% EtOAc in heptane (12 L), and finally with 40% EtOAc in heptane (12 L) to afford 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (4) and 2-(3-chloro-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile (4B) as a off-white solid (58.1 g, 68% yield), which was found to be an approximately one to one mixture of compound 4 and 4B. For 4: 1H NMR (300 MHz, CDCl3) delta 1.38 (t, 3H), 3.05 (q, 2H), 4.72 (m, 2H), 4.79 (m, 2H), 5.41 (m, 1H); MS: m/z calcd. 187.05; found: 187.1. For 4B: 1H NMR (300 MHz, CDCl3) delta 1.38 (t, 3H), 3.05 (q, 2H), 3.1 (s, 2H), 4.15 (d, 2H), 4.37 (d, 2H); MS: m/z calcd. 222.9; found: 222.9.
1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;Product distribution / selectivity;
To a solution of <strong>[1187595-85-2]2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile</strong> (4) and 2-(3-chloro-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile (4B) obtained from the previous reaction as an approximate one to one mixture (4 and 4B, 184 g, 919 mmol, 1.2 equiv) and 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 241 g, 766 mmol) in acetonitrile (6 L) was added DBU (137 mL, 919 mmol, 1.2 equiv) dropwise over 30 min at room temperature. The resulting reaction mixture was stirred overnight at room temperature. When the reaction was deemed complete, the solvent was removed under reduced pressure. The resulting solid was dissolved in 6 L of ethyl acetate and 2 L of acetonitrile at 40 C. and the solution was washed with a mixture of brine (3 L) and water (1 L). The aqueous layer was extracted with ethyl acetate (3×1.6 L). The combined organic layers were washed with brine (1.6 L) and the solvent was removed under reduced pressure. Toluene (2 L) was added to the residue and the azeotropic distillation was repeated under reduced pressure. The residue was triturated with MTBE (1.5 L, methyl t-butyl ether) and the solids were collected by filtration. The brown solid was dissolved completely in ethyl acetate (3 L) at 50 C. before the solution was treated with charcoal (30 g) and silica gel (30 g). The resulting mixture was stirred at 45 C. for 1 h before being filtered hot through celite. The solvent was removed under reduced pressure and the residue was triturated with MTBE (3 L). The solids were collected by filtration and washed with MTBE (1 L). The solids were then completely dissolved in isopropanol (8.8 L) at 70 C., and the resulting solution was gradually cooled down to room temperature with stirring for overnight. The solids were collected by filtration, washed with isopropanol (1.3 L) and heptane (2×490 mL), and dried in an oven overnight to afford 2-(1-(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (6, 327 g, 384.3 g theoretical, 85% yield) as an off-white solid. For 6: 1H NMR (300 MHz, CDCl3) delta0.00 (s, 9H), 0.99 (m, 2H), 1.49 (t, 3H), 3.15 (q, 2H), 3.49 (s, 2H), 3.60 (m, 2H), 4.30 (d, 2H), 4.70 (d, 2H), 5.76 (s, 2 H), 6.83 (s, 1H), 7.50 (s, 1H), 8.40 (s, 1H), 8.50 (s, 1H), 8.90 (s, 1H); MS: m/z calcd. 502.20; found: 502.3.
1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; for 4h;
A solution of 4-(1H-pyrrol-3-yl)-7-[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine (0.100 g, 0.318 mmol) and <strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong> (0.118 g, 0.636 mmol, prepared as in Example 68) in acetonitrile (1 mL) was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (24 muL, 0.16 mmol). The mixture was stirred for 4 hours. The solvent was evaporated. The residue was partitioned between ethyl acetate and saturated sodium bicarbonate solution, then extracted with two further portions of ethyl acetate. The combined extracts were dried over sodium sulfate, decanted and concentrated. The crude product was stirred with 25% TFA/DCM (8 mL) overnight. The solvents were evaporated. The product was then stirred with ethylenediamine (0.3 mL) in methanol (5 mL). Preparative HPLC-MS (eluting with a gradient of methanol and water containing 0.15% NH4OH) was used to purify the product.1H NMR (300 MHz, CDCl3): delta 8.59 (s, 1H), 7.54 (dd, 1H), 7.27 (d, 1H), 6.95 (dd, 1H), 6.82 (dd, 1H), 6.74 (d, 1H), 4.49 (d, 2H), 4.15 (d, 2H), 3.24 (s, 2H), 3.02 (q, 2H), 1.33 (t, 3H); LCMS (M+H)+: 371.1.
{1-(ethylsulfonyl)-3-[4-(hydroxymethyl)piperidin-1-yl]azetidin-3-yl}acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 3h;
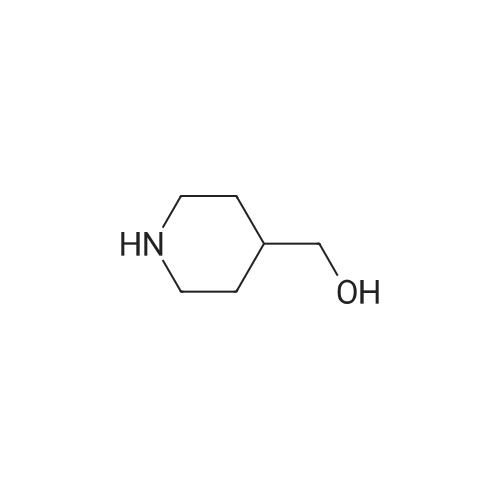
Step 1: {1-(Ethylsulfonyl)-3-[4-(hydroxymethyl)piperidin-1-yl]azetidin-3-yl}acetonitrile To a solution of 4-piperidinemethanol (60 mg, 0.5 mmol) and <strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong> (110 mg, 0.60 mmol, prepared using similar conditions as disclosed in the literature such as WO 2009/114512) in acetonitrile (1.0 mL) was added DBU (20 muL, 0.1 mmol). The resulting mixture was stirred at room temperature for 3 h then diluted with DCM, washed with water and brine. The organic layer was dried over Na2SO4, filtered and then concentrated. The residue was purified by flash chromatography on a silica gel column eluting with MeOH in DCM (0-8%) to give the desired product. LC-MS calculated for C13H24N3O3S (M+H)+: m/z=302.2; found 302.1.
In dichloromethane; at 0 - 25℃; for 16h;Inert atmosphere;
To a solution of 2-(azetine-3-yl) methyl cyanide(2.8 g, 21.4 mmol) and DIPEA(8.3 g, 64.3 mmol) in dichloromethane(30 mL) was added dropwise ethanesulfonyl chloride(4.1 g, 32.1 mmol) at 0C under the protection of nitrogen, and the temperature was kept below 2C when dropping. The reaction mixture was stirred at 25C for reacting for 16 hours. TLC showed that the reaction was completed (petroleum ether/ethyl acetate =1:1). After the reaction mixture was quenched with water, extracted with dichloromethane (30 mL*2). The combined organic phase was washed with saturated salt water(20 mL*2), dried with anhydrous sodium sulfate, filtered, and spun dry. The residue was purified through column chromatography (dichloromethane/ethyl acetate =3/1) to give 2-(1-(ethylsulfonyl)azetine-3-yl)methyl cyanide(1.4 g, 33.0% yield) as a pale yellow solid. 1H NMR (400MHz, CDCl3) delta = 5.50 - 5.41 (m, 1H), 4.79 (d, J=3.0 Hz, 2H), 4.71 (d, J=2.5 Hz, 2H), 3.06 (q, J=7.4 Hz, 2H), 1.40 (t, J=7.4 Hz, 3H). MS (ESI) Calcd. for C7H10N2O2S [M + H]+ 187, Found 187.
192 mg
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 0 - 20℃;
Trifluoroacetic acid (2 mL) was added to a solution of tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (2) (250 mg, 1.29mmol) in dichloromethane (20 mL). The solution was stirred at room temperature for 5 h. Then the reaction mixture was concentrated under reduced pressure to dryness. The residue, which contains the crude desired deprotection product 3, was then suspended ina cetonitrile (20 mL) and cooled to 0 C. N,N-Diisopropylethylamine (DIPEA) (1.12 mL, 6.44 mmol, 5 equiv.) was then slowly added while keeping the internal temperature below 5 C. Then ethanesulfonyl chloride (EtSO2Cl) (0.184 mL, 1.94 mmol, 1.5 equiv.) was added over 1 h while keeping the internal temperature below 5 C. The resulting reaction mixture was stirred overnight at room temperature and then concentrated under reduced pressure. The concentrated residue was then diluted with dichloromethane and was washed with aqueous sodium chloride solution. The aqueous phase was back-extracted with dichloromethane. The combined organic layers were dried over Na2SO4 and the residue was purified using a silica gel column to afford compound 4: Yield 192 mg (81%); off-white solid; m.p. 58-60 C; IR: 3294, 3066, 2978, 2222, 1702, 1321, 1142 cm-1. Anal. calcd for C7H10N2O2S: C, 45.15; H, 5.41; N, 15.04; found: C, 45.32; H, 5.35; N, 15.21%. MS (m/z): 209 [M + Na]+; 1H NMR (300 MHz, CDCl3): delta 1.37 (m, J = 6.7 Hz, 3H), 3.04 (m, J = 7.4 Hz, 2H), 4.69 (t, J = 2.5 Hz, 2H), 4.77 (t, J = 2.8 Hz, 2H), 5.43 (t, J = 2.4 Hz, 1H); 13C NMR (75 MHz, DMSO-d6): delta 7.2, 42.6, 58.5, 58.9, 93.9, 114.9, 156.2, 168.6.
0.192 g
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 0 - 5℃;
Compound 2 (0.250g, 1.29mmol) dissolved in acetonitrile was slowly added dropwise trifluoroacetic acid (3ml).Stirred at room temperature for 4 hours gussets tracking.After completion of the reaction, the solvent spin dry, obtained was dissolved in acetonitrile and cooled to 0 deg.] C, was slowly added N, N- diisopropylethylamine (DIEA), maintaining the temperature not higher than 5 .Was slowly added compound 8 (0.184ml, 1.94mmol, 1.5equiv), at T Completion of the reaction followed by TLC, concentrated under reduced pressure, the crude product was diluted with dichloromethane, washed with brine, the aqueous phase was extracted with dichloromethane, the organic phase was concentrated, dried over anhydrous sodium sulfate, and purified by column chromatography to give 0.192g white solid with a yield of 81%.
2-(1-(ethanesulfonyl)-3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)azetidine-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89.8%
With N,N,N?,N?-tetramethyl-N?-tert-butylguanidine; In tetrahydrofuran; at 65℃; for 3h;
Preparation of 4-(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole solution (107,0 mg/g as a solution in CPME) is conducted using the previously described procedure denoted in Alternate Preparation 5b.The solution of 4-(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (69.41 g, 79.8 mL, 38.27 mmol) is combined with 2-terf-butyl-l, 1,3,3- tetramethylguanidine (0.54 g, 3.15 mmol) followed by a CPME rinse (6 mL) then heated to about 65 C. 2-(l-(Ethylsulfonylazetidin-3-ylidene)acetonitrile (6.00 g, 31.90 mmol) is dissolved in THF (14 mL) and added over 3 hours followed by a THF rinse (2 mL). The reaction is stirred at about 65 C and monitored for completion (typical completion time is two hours post-addition). The solution is cooled to 25 C and concentrated to a wet residue. 1-Propanol (60 mL) is added and the suspension is again concentrated to a wet solid. The solid is suspended in 1 -propanol (90 mL) and heated to 67 C to form a solution. The solution is cooled to 57 C and seed crystals (0.6 g) of the title compound are added. The resulting slurry is held at a temperature of 57 C for 2 hours then cooled to -3 C over 9 hours and held at that temperature for at least 2 hours. The solids are collected by filtration, washed with cold 1 -propanol (2 chi 12 mL) and dried to give the title compound (10.89 g, 89.8%).
85.78%
With potassium carbonate; In 1,4-dioxane; water; at 20 - 25℃;
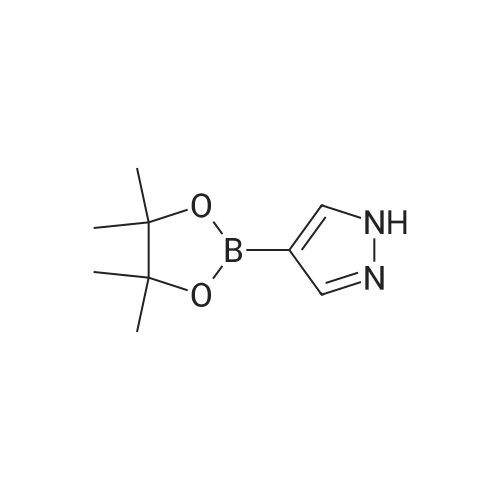
1,4-Dioxane (20 mL) was added into a reaction vessel containing a solution of potassium carbonate (4.5 g) in water (30 mL) at about 20C to about 25 C. 2-(l- (Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (2 g; Formula II) and 4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.30 g; Formula VIII) were added into the reaction mixture at about 20C to about 25 C. The reaction mixture was stirred at about 20C to about 25 C for about 16 hours to about 18 hours. On completion of the reaction, 1,4- dioxane was recovered from the reaction mixture under reduced pressure at about 45 C to obtain a residue. Ethyl acetate (20 mL) was added into the residue, and the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45 C to obtain {1-(ethylsulfonyl)-3-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile. Yield: 85.78%. Mass: 381.4 [M + H]+
85%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 2h;
pyrazole-4-boronic acid pinacol ester (14 g, 72.1 mmol) was added to 100 mL of acetonitrile at room temperature, Was added <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (18 g, 96.7 mmol) and DBU (20 mL) After stirring at room temperature for 2 hours, Ethyl acetate (500 mL) and water (500 mL) were added, Extraction, The aqueous layer was extracted once with 50 mL of ethyl acetate, Combined organic layer, And then washed with 50mL saturated sodium chloride once, Dried over anhydrous sodium sulfate, filter, Concentrated under reduced pressure to give 23 g of a yellow solid. Yield 85%
84%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In isopropyl alcohol; acetonitrile; at 60℃; for 4h;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (11) (156 mg, 0.80 mmol, 1.01 equiv.), compound 4 (149 mg, 0.80 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (0.061 mL, 0.408 mmol) in acetonitrile (20 mL) was heated at 60 C for 4 h. After cooling, the solvent was removed under reduced pressure. The residue was purified by flash chromatography on a silica gel column eluting with methanol in dichloromethane (0-60%) to afford the desired compound 12: Yield 255 mg (84%); m.p. 120-122 C; IR: 3280, 2978, 1639, 1554, 1369 cm-1. Anal. calcd for C16H25BN4O4S: C, 50.54; H,6.63; N, 14.73; found: C, 50.67; H, 6.74; N, 14.62%. MS (m/z): 403 [M+ Na]+; 1H NMR (300 MHz, DMSO-d6): delta 1.22 (t, J = 7.3 Hz, 3H), 1.27(s, 12H), 3.19 (m, J = 7.3 Hz, 2H), 3.59 (s, 2H), 4.14 (d, J = 9.0 Hz, 2H), 4.44 (d, J = 9.0 Hz, 2H), 7.77 (s, 1H), 8.35 (s, 1H); 13C NMR (75 MHz, DMSO-d6): delta 4.6, 24.9, 39.5, 43.2, 55.5, 58.2, 83.2, 116.6, 135.8, 145.6.
84%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In isopropyl alcohol; at 20 - 60℃; for 3.5h;
In isopropanol solvent, was added Compound 10: 1,8-diazabicyclo [5,4,0] undec-7-ene (0.061 mL), Compound 9 (0.156g, 0.80mmol, 1.01equiv) , compound 4 (0.149g, 0.80mmol), at room temperature the formation of a suspension, was refluxed for half an hour, to form a uniform solution.The reaction was refluxed for 3 hours at 60 deg.] C, TLC trace.Cooling to room temperature, vacuum filtration, and purified by column chromatography to give a white solid 0.255 g, 84% yield.
7-((2-(trimethylsilyl)ethoxy)methyl)-7H,7'H-4,5'-bipyrrolo[2,3-d]pyrimidine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H,7'H-[4,5'-bipyrrolo[2,3-d]pyrimidin]7'-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
32%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 32 - 36℃; for 40h;
7 - ((2- (trimethylsilyl) ethoxy) methyl) -7H, 7'H-4,5'- associated pyrrolo [2,3-d] pyrimidine (Compound C, 224mg, 0.61 mmol, 1.0eq) was dissolved in acetonitrile (5mL) was added2- (l- (ethylsulfonyl) azetidin-3-ylidene) acetonitrile(136mg, 0.73mmol, 1.2eq) After dropping DBU (111mg, 0.73mmol, 1.2eq), 36 degrees C reaction 20h, TLC did not detect completion of the reaction, the addition of 2- (1- (ethylsulfonyl) azetidin butane-3-ylidene) acetonitrile (136mg, 0.73mmol, 1.2eq), DBU (111mg, 0.73mmol, 1.2eq), the addition was complete the reaction 32 degrees C 20h. The reaction was monitored by TLC (Rf = 0.2, PE: EA = 1: 1) After the unreacted starting material, spin dry solvent, column chromatography (PE: EA = 1: 1) gave a yellow oil compound D107mg, yield: 32%.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
Dissolving raw material CF0726F (0.1 g, 0.00027 mol) in acetonitrile (5 mE), adding SM2 (0.1 g, 0.00054 mol, 2 eq) and DI3U (0.1 g, 0.00066 mol, 2.4 eq), performing room-temperature stirring overnight, spinning it dry after the reaction, using H20/EA for extraction, spinning the organic phase dry and making it pass the colunm to obtain 0.19 g of product (PE/EA=3:1-1: 1).
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
Dissolving raw material CF0726F (0.2 g, 0.00052 mol) in acetonitrile (5 mE), adding 5M2 (0.2 g, 0.00108 mol, 2.1 eq) and DI3U (0.2 g, 0.00132 mol, 2.5 eq), performing room-temperature stirring overnight, spinning it dry afier the reaction, using H20/EA for extraction, spinning the organic phase dry and making it pass the column to obtain 0.1 g of product (PE/EA=3:1-1:1).
Under a nitrogen atmosphere, in a 3L three-necked bottle, diethyl cyanomethylphosphonate (212.5 g, 1.2 mol) and anhydrous THF (300 ml) were added and dissolved by stirring, and then the temperature of the system was lowered to _10 ~0C,Under this condition, a solution of t_Bu0K (123.2 g, 1.12 mol) in THF (200 ml) was slowly added dropwise, and the temperature of the control system was maintained at -10 to 0C. At this temperature, the reaction was effected for 3-4 hours.Then, a solution of Intermediate B (163.0 g, 1.0 mol) in THF (300 ml) was slowly added dropwise during the dropwise addition of the THF solution containing Intermediate B.The temperature of the reaction system was controlled to be -10 ~ 0C. After the dropwise addition, the mixture was stirred at this temperature for 2-3 hours, then slowly raised to room temperature and allowed to react for 15-18 hours at room temperature.Then the reaction solution was adjusted to pH 3-4 with water (500 ml), saturated NaCl solution (500 ml), 0.5 HC HCl, and extracted with ethyl acetate (500 ml X 2).The organic layers were combined and dried to afford an off-white solid 127,48 in 68.5% yield.
35%
To a suspension of potassium tert-butoxide (0.15 g, 1.366 mmol) in dry tetrahydrofuran (4 mL) was added diethyl (cyanomethyl)phosphonate (0.23 mL, 1.47 mmol) at 0 C. and the mixture stirred for 3.5 hours. Then a solution of 1-(ethylsulfonyl)azetidin-3-one (0.2 g, 1.22 mmol) in tetrahydrofuran was added 0 C. and the mixture stirred for another 3 hours. The reaction mixture temperature was raised to ambient temperature and stirred for an additional 15 hours. The reaction mixture was poured onto ice water and the pH of the reaction mixture was adjusted to 4.0 with 0.5N hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with water, brine, dried over anhydrous sodium sulphate, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (5% methanol/dichloromethane) to provide 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile the as a yellow oil (0.48 g, 35% yield): 1H NMR (400 MHz, DMSO-d6) delta 5.88 (d, J=2.4 Hz, 1H), 4.74-4.76 (m, 2H), 4.66-4.67 (m, 2H), 3.14-3.21 (m, 2H), 1.21-1.29 (m, 3H).
With N-ethyl-N,N-diisopropylamine; In isopropyl alcohol; at 0 - 10℃; for 20h;
Diethyl cyanomethylphosphonate (48.6 g, 274 mmol) is added to an IPA solution (225 mL) of l-(ethylsulfonyl)azetidin-3-one (41 g, 251 mmol). The resulting solution is cooled to 0 C and DIPEA (44.2 g, 348 mmol) is added at a rate such that the temperature is maintained at 5 C. The mixture is stirred for 1 hour and seeded with [1-(ethylsuifony2)azetidin-3-y2idene]acetonitrile. The mixture is stirred an additional 3 hours mamtaimng the temperature at 0-5 C and then warmed to 10 C and stirred an additional 16 hours. The suspension is cooled to 0 C then heptane (225 mL) is added over at least an hour, and the mixture is stirred for an additional hour at 0 C. The reaction mixture is filtered and the resulting precipitate is rinsed with 1 ;2 IP A/ heptane (120 mL) and dried at 30 C for at least 12 hours to give the title compound (41 g, 83.4%) as a white powder. The material (30 g, 159 mmol) is purified by a seeded recrystalltzation in an IPA (120 mL) / water (12 mL) mixture to give the title compound (28.7 g, 90.7%). Melting poim=68 C, ES/MS m/z 187.0527 [M+H]+: NMR (400 MHz, de-DMSO) delta 5.89 (quintet, J= 2,5 Hz, III), 4.76 (q, J= 3.1 Hz, 2H), 4.67 (dd, J= 2.6, 5.7 Hz, 2H), 3.21 (q, J= 7.3 Hz, 2H), 1.21 (t, J- 7.3 Hz, 3H), 13C NMR (500 MHz, d6-DMSO) delta 7.3, 42.5, 58.7, 59.1, 94.0, 115.0, 156.3.
A three-necked flask was charged with compound 2 (16.33 g, 100 mmol) Cyanoacetic acid (17.01 g, 200 mmol) And toluene (82 mL), After stirring, piperidine (17.03 g, 200 mmol) was added, Plus after warming to reflux reaction 16-24 hours, The reaction was quenched by cooling to room temperature and adding 0.5 mol / L dilute hydrochloric acid (326 mL). The reaction mixture was separated and extracted with ethyl acetate (163 mL) The organic phase was washed with saturated brine twice (163 mL) Dried over sodium sulfate and concentrated to give Compound 3 (15.08 g, 81%) by column chromatography on petroleum ether ethyl acetate mixed solvent column chromatography.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 35 - 40℃;
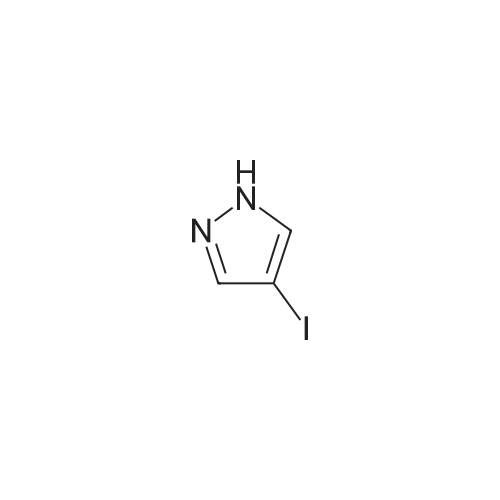
A three-necked flask was charged with 3 (18.62 g, 100 mmol) 4 - iodo - pyrazole4a (19.40g, 100mmol) And acetonitrile (93ml), After stirring, DBU (15.20 g, 100 mmol) was added, Add the temperature after warming to 35 ~ 40 C overnight. The reaction was quenched by removing the acetonitrile, adding 0.5 mol / L dilute hydrochloric acid (186 mL), the aqueous phase was extracted three times with ethyl acetate (93 mL), washed twice with organic saturated brine (93 mL) Sodium sulfate, After concentration, compound 5a (29.66 g, 78%) was isolated by column chromatography using ethyl acetate petroleum ether mixed solvent column chromatography.
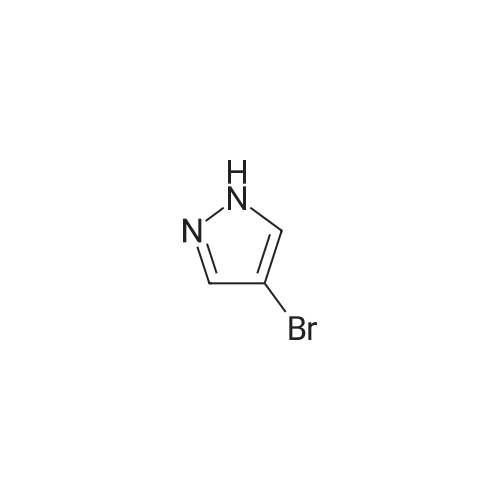
2-(3-(4-bromo-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 35 - 40℃;
A three-necked flask was charged with 3 (18.62 g, 100 mmol) 4-bromopyrazole 4b (14 · 70g, 100mmol) And acetonitrile (93 ml), After mixing, add DBU (15.20 g, 100 mmol) and add the temperature to 35 ~ 40 C overnight. The reaction was quenched by the addition of partially acetonitrile and quenched with 0.5 mol / L dilute hydrochloric acid (186 mL). The aqueous phase was extracted three times with ethyl acetate (93 mL). The organic phase was washed with saturated brine (93 mL), dried over sodium sulfate, After concentration, compound 5b (27.66 g, 83%) was isolated by column chromatography on a mixture of ethyl acetate and petroleum ether
53%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 0 - 20℃; for 16h;
To a stirred solution of <strong>[1187595-85-2]2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile</strong> (0.48 g, 2.58 mmol) in dry acetonitrile (15 mL) was added 4-bromo-1H-pyrazole (0.37 g, 2.58 mmol) at 0 C. After 10 minutes, 1,8-diaza-bicyclo(5.4.0)undec-7-ene (0.39 g, 2.57 mmol) was added and the solution was stirred at ambient temperature for 16 hours. The reaction mixture was diluted with acetonitrile and the pH was adjusted to 6.0 with 0.5N hydrochloric acid and concentrated in vacuo. The residue was extracted with ethyl acetate, washed with water, brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (50% ethyl acetate/hexane) to provide 2-(3-(4-bromo-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile as an off-white solid (0.45 g, 53% yield): MS (ES) m/z 335.1 (M+2H).
Three-necked flask was charged with triphenylphosphine acetonitrile (33.14 g, 110 mmol) and dichloromethane (96 mL)And then 1- (ethylsulfonyl) azepin-3-one (prepared from Example 3) was added and reacted at room temperature for 3 to 4 hours. The reaction was quenched by the addition of 240 mL of water. The aqueous phase was extracted three times with ethyl acetate (120 mL). The organic phase was washed with saturated brine (120 mL) and concentrated to recrystallize from isopropyl ether and petroleum ether to give 2- [1-(ethylsulfonyl)-3-azetidinylidene] acetonitrile(16.76 g, 4 steps yield 67%).
4-(5,5-dimethyl-1,3,2-dioxaborate-2-yl)-1H-pyrazole[ No CAS ]
2-(3-(4-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-1H-pyrazole-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 35 - 40℃;
A mixture of <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (18.62 g, 100 mmol), 4-(5,5-dimethyl-1,3,2-dioxane (18.00g, 100 mmol) and acetonitrile (93 mL). After stirring, DBU (3.04 g, 20 mmol) was added and the temperature was raised to 35 to 40 C overnight. The reaction was quenched by partial acetonitrile and saturated ammonium chloride (93 mL). The aqueous phase was extracted three times with ethyl acetate (93 mL). The organic phase was washed with saturated brine twice (93 mL), dried over sodium sulfate, (3-(4-(5,5-dimethyl-1,3,2-dioxaborolan-2-yl)-1H-(3-methylphenyl)yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile 8 (31.50 g, 86%).
3-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrole[2,3-b]pyridin-4-yl)[3,2-c]pyridine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(3-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrole[2,3-d]pyridine Yl)-1H-pyrrole [3,2-c] pyridin-1-yl)azetidin-3-ylidene)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 15 - 20℃; for 0.5h;Inert atmosphere;
A solution of 3- (1 - ((2- (trimethylsilyl) ethoxy) methyl) -1H-pyrrole [2,3-b] pyridin-4-yl)[3,2-c] pyridine (Compound D, 1.05 g, 2.88 mmol)Was dissolved in acetonitrile (20 mL)DBA (657 mg, 4.32 mmol) was added dropwise after addition of 2- (1- (ethylsulfonyl) azetidin-3-ylidene) acetonitrile (536 mg, 2.88 mmol)Nitrogen protection,15 ~ 20 reaction 0.5h, TLC detection reaction finished, spin dry solvent, column chromatography (PE: EA = 1: 1) to obtain a yellow solid (compound E) 1.20g, Yield: 76%.
8-(1H-pyrazole-4-yl)-[1,2,4]triazolo[1,5-a]pyridin-2-amine[ No CAS ]
2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-a]pyridine-8-yl)pyrazole-1-yl]-1-ethylsulfonylazetidine-3-yl]acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
49.22%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 26℃; for 12h;
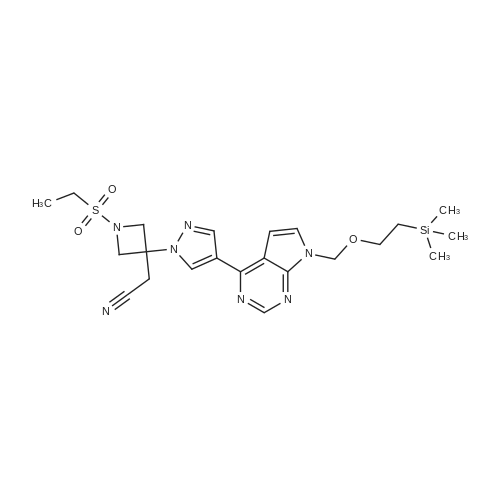
To 8-(1H-pyrazole-4-yl)-[1,2,4]triazolo[1,5-a]pyridin-2-amine(50 mg, 249.7 umol) and 2-(1-ethylsulfonyl-azetidine-3-ylene) acetonitrile(56 mg, 299.7 umol), dissolved in acetonitrile(8 mL), was added dropwise DBU(46 mg, 299.7 umol). The reaction mixture was stirred at 26C for reacting for 12 hours. After LC-MS showed that the reaction was completed, the reaction mixture was distilled under reduced pressure to remove acetonitrile. To the residue was added water(10 mL), and then extracted with EA(10 mL*3). The organic phase was combined, washed with saturated salt water(10 mL), dried with anhydrous sodium sulfate, filtered, and distilled under reduced pressure to remove the filtrate. The residue was purified through silica gel column chromatography(DCM/MeOH=20/1) to give 2-[3-[4-(2-amino-[1,2,4]triazolo [1,5-a]pyridine-8-yl)pyrazole-1-yl]-1-ethylsulfonyl- azetidine-3-yl]acetonitrile(50 mg, 49.22% yield) as a white solid. MS (ESI) Calcd. for C16H18N8SO2[M+H]+ 387, Found 387.
5-(1H-pyrazole-4-yl)-[1,2,4]triazolo[1,5-c]pyrimidin-2-amine[ No CAS ]
2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-c]pyrimidin-5-yl)pyrazole-1-yl]-1-ethylsulfonylazetidine-3-yl] acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69.2%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; acetonitrile; at 26℃; for 16h;
To a suspension of Intermediate 1(150 mg, 745.6 umol) slightly dissolved in acetonitrile(4 mL) and DMF(2 mL) was added Intermediate 9 (208 mg, 1.1mmol) and DBU(227mg, 1.5 mmol). The reaction mixture was stirred at 26C for 16 hours. LC-MS showed that the reaction was completed. The precipitated solid was filtered, collected, washed with cold acetonitrile(5 mL), and dried under reduced pressure to give 2-[3-[4-(2-amino-[1,2,4]triazolo[1,5-c]pyrimidin-5-yl) pyrazole-yl]-1-ethylsulfonylazetidine-3-yl] acetonitrile(200 mg, 69.2% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) delta = 9.16 (s, 1H), 8.69 (s, 1H), 8.16 (d, J=6.3 Hz, 1H), 7.31 (d, J=6.0 Hz, 1H), 6.56 (s, 2H), 4.53 (d, J=9.0 Hz, 2H), 4.28 (d, J=9.0 Hz, 2H), 3.70 (s, 2H), 3.25 (q, J=7.4 Hz, 2H), 1.25 (t, J=7.4 Hz, 3H). MS (ESI) Calcd. for C15H17N9O2S [M + H]+ 388, Found 388.
N-[8-(1H-pyrazole-4-yl)-[1,2,4]triazolo[1,5-a]pyrazine-2-yl]cyclopropanecarboxamide[ No CAS ]
N-[8-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl]pyrazole-4-yl]-[1,2,4]triazolo[1,5-a]pyrazine-2-yl]cyclopropanecarboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
40 mg
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In N,N-dimethyl-formamide; at 40℃; for 16h;
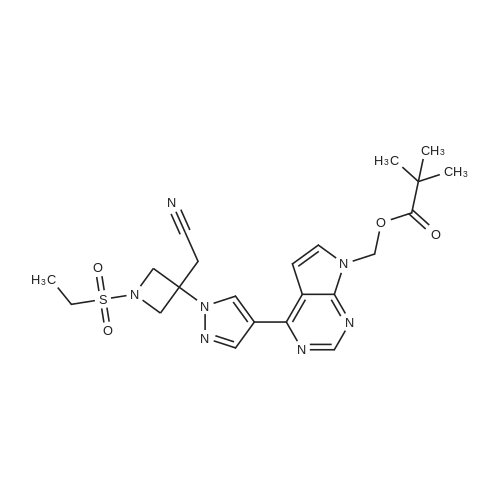
To N-[8-(1H-pyrazole-4-yl)-[1,2,4]triazolo[1,5-a]pyrazine-2-yl] cyclopropanecarboxamide (120 mg, 445.7 umol) and 2-(1-ethylsulfonylazetidin-3-ylidene) acetonitrile (125 mg, 668.5 umol) dissolved in DMF(10 mL), was added DBU(136 mg, 891 umol). The resulting mixture was stirred at 40C for reacting for 16 hours. After LC-MS showed that the reaction was completed, the solvent was spun dry under reduced pressure, and the residue was dissolved in EtOAc(50 mL). The resulting solution was washed with 1N of HCl(10 mL) and saline water(20 mL), the organic phase was dried with anhydrous Na2SO4 and the solvent was spun dry. The residue was purified through preparative thin layer chromatography (PE/EA=1:4) to give a crude compound, which was purified and freeze-dried through preparative HPLC(HCl) to give N-[8-[1-[3-(cyanomethyl)-1-ethylsulfonylazetidin-3-yl] pyrazole-4-yl)-[1,2,4]triazolo[1,5-a]pyrazine-2-yl] cyclopropanecarboxamide (40 mg) as a white solid. 1H-NMR (400 MHz, MeOD-d4) delta = 8.07 (d, J = 8 Hz, 2H), 7.86 (dd, J = 7.2, 13.2 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.44-7.42 (m, 1H), 4.19 (s, 2H), 3.44 (d, J = 4.8 Hz, 4H), 3.26 (d, J = 4.8 Hz, 4H), 2.95 (m, 1H), 0.87 (m, 2H), 0.74 (m, 2H). MS (ESI) Calcd. for C19H21N9O3S [M+H]+456, Found 456.
N-(5-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidine-3-yl)-1H-pyrazole-4-yl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropyl carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 25℃; for 16h;Inert atmosphere;
To a solution of Intermediate 6(1.5 g, 5.6 mmol) and Intermediate 9(1.5 g, 7.8 mmol) in acetonitrile(15 mL) was added dropwise DBU(1.7 g, 11.2 mmol) under the protection of nitrogen. The reaction mixture was stirred at 25C for 16 hours. TLC(petroleum ether/ethyl acetate=0:1) detected that the reaction was completed. The reaction mixture was poured into methanol(200 mL) at 0C, a great number of solids were precipitated immediately, stirred for 10 min, and then filtered. The resulting solid was washed with methanol(5 mL) and dried under vacuum to give N-(5-(1-(3-(cyanomethyl)-1-(ethylsulfony)azetidine-3-yl)-1H-pyrazole-4-yl]-[1,2,4] triazolo[1,5-a]pyridin-2-yl)cyclopropyl carboxamide(1.60 g, 60% yield) as a product. 1H NMR (400MHz, DMSO-d6) delta = 9.25 (s, 1H), 8.79 (s, 1H), 7.76 - 7.70 (m, 1H), 7.64 (d, J=7.0 Hz, 1H), 7.60 (d, J=8.5 Hz, 1H), 4.49 (d, J=9.0 Hz, 2H), 4.32 (d, J=9.0 Hz, 2H), 3.68 (s, 2H), 1.26 (t, J=7.3 Hz, 3H), 0.91 - 0.85 (m, 4H). MS (ESI) Calcd. for C20H22N8O3S [M+H]+ 455, Found 455.
450 g of tetrahydrofuran was added to the 2 L reaction flask. 21.3 g (0.12 mol) of diethyl cyanomethyl phosphate, The system was cooled to 10 C, and 7.0 g (0.13 mol) of sodium methoxide was added under nitrogen atmosphere. After the addition, the system gradually warmed to 20 C. Stir for 45 min. A solution of 16.3 g (0.1 mol) of compound 6 and 80 g of tetrahydrofuran was continuously added dropwise to the system. After 1 hour, the reaction was stirred at 30 C for 10 h, and the reaction of the central control material was almost complete. After the system was cooled to 10 C, 500 g of ethyl acetate and 300 g of saturated brine were added. After stirring for 5 min, the layers were separated. The aqueous layer was extracted with 300 g of EtOAc EtOAc. The organic layer was washed with 500 g of water. The organic layer is dry, Filtration and concentration of the filtrate gave the crude product of compound 7, The crude product was washed with a small amount of n-hexane and dried to give 16.6 g of Compound 7. Yield: 96.4%, HPLC ? 98.0%.
200 g of acetonitrile and 20.4 g (0.05 mol) of compound 4 were added to a 1 L reaction flask. 6.9 g (0.05 mol) was added with stirring. Potassium carbonate was stirred at room temperature for 30 min, then 9.3 g (0.05 mol) of compound 7 and 8 g (0.05 mol) of DBU were added. The temperature of the system is raised to 40 C After the reaction for 8 hours, the reaction was completed, and the reaction of the starting materials was completed, and the reaction was stopped. Evaporate the solvent under reduced pressure and add to the system. After quenching the reaction with 100 g of water, 200 g of ethyl acetate was added. Stir for 30 min, filter, and rinse the cake with 100 g of fresh ethyl acetate. The filter cake was dried to give a white solid of 17.0 g, HPLC ? 99.0%, yield: 91.2%.
6-(1H-pyrazol-4-yl)-9-((2-(trimethylsilyl)ethoxy)methyl)-9H-purine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(4-(9-((2-(trimethylsilyl)ethoxy)methyl)-9H-purin-6-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (10 mL) was added to a mixture of 6-(1H-pyrazol-4-yl)-9-((2-(trimethylsilyl)ethoxy)methyl)-9H-purine (6c) (200 mg) and <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (118 mg) to obtain a reaction solution. DBU (120 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified on a preparative silica gel plate (DCM:MeOH=10:1), to afford 2-(1-(ethylsulfonyl)-3-(4-(9-((2-(trimethylsilyl)ethoxy)methyl)-9H-purin-6-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (6d) (289 mg, yellow oil), yield: 91%. MS m/z: 503 [M+1]+.
4-(1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carboxamide[ No CAS ]
4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 2h;
At room temperature, <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (71.8 mg, 0.3 mmoL) and DBU (60 mg, 0.39 mmol) were sequentially added to a solution of 4-(1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carboxamide (7c) (130 mg, 0.36 mmol) in acetonitrile (3 mL), and the reaction was performed at room temperature for 2 h. TLC indicated the reaction was complete, the reaction was added with water (10 mL), extracted with ethyl acetate (10 mL*3), and the ethyl acetate layer was combined, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated to afford 4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carboxamide (7d) (130 mg, yield: 78%) as a colorless oil. MS (ESI, m/z): 544 [M+H]+.
2-(1-(ethylsulfonyl)-3-(4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89.1%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 1h;
Compound 4-(1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (8c) (200 mg, 0.64 mmol), <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (118 mg, 0.64 mmol) and acetonitrile (15 mL) were added to a 50 mL reaction flask, DBU (116 mg, 0.76 mmol) was added after the reaction solution was stirred until homogeneous. The reaction was performed at room temperature for 1 h, and monitored by thin layer chromatography. After the reaction was complete, the reaction solution was quenched with water, concentrated under reduced pressure, and purified by column chromatography on silica gel, to afford compound 2-(1-(ethylsulfonyl)-3-(4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (8d) (285 mg, yield: 89.1%, yellow solid). MS (ESI, m/z): 500.2 [M+H]+.
4-(1H-imidazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-imidazol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
13 mg
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (2 mL) was added to a mixture of 4-(1H-imidazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (10d) (30 mg) and <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (20 mg) to obtain a cloudy reaction solution. DBU (20 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified on a preparative silica gel plate, to afford 2-(1-(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-imidazol-1-yl)azetidin-3-yl)acetonitrile (10e) (13 mg, brown solid). MS (ESI, m/z): 502 [M+H]+.
6-chloro-4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine[ No CAS ]
2-(3-(4-(6-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
177 mg
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (10 mL) was added to a mixture of 6-chloro-4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (11c) (160 mg) and <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (85 mg) to obtain a reaction solution. DBU (100 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified by preparative flash chromatography, to afford 2-(3-(4-(6-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile (11d) (177 mg, solid product). MS m/z: 536 [M+1]+.
4-(1H-pyrrol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carbonitrile[ No CAS ]
4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrrol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
4-(1H-pyrrol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carbonitrile (17c) (142 mg, 0.42 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (17d) (85 mg, 0.46 mmol) were dissolved in acetonitrile (5 mL), DBU (0.12 mL) was then added, and the reaction was stirred at room temperature. After TLC indicated the reaction was complete, the reaction was rotary evaporated to dryness, and purified by column chromatography, to afford 4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrrol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carbonitrile (17e) (200 mg, milk white oil, yield: 91%). MS (ESI, m/z): 525 [M+H]+.
4-(1H-pyrrol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(3-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrrol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
4-(1H-pyrrol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (28c) (283 mg, 0.9 mmol) and <strong>[1187595-85-2]2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile</strong> (28d) (183 mg, 0.99 mmol) were dissolved in acetonitrile (10 mL), the reaction system was added with DBU (0.3 mL), and then stirred at room temperature overnight. The reaction solution was concentrated, and purified by column chromatography, to afford 2-(1-(ethylsulfonyl)-3-(3-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrrol-1-yl)azetidin-3-yl)acetonitrile (28e) (240 mg, milk white oil, yield: 53%). MS (ESI, m/z): 500 [M+H]+.
4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrrole-2-carbonitrile[ No CAS ]
1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrrole-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
51%
With potassium tert-butylate; In acetonitrile; at 20℃; for 24h;
Compound (39e) (1.65 g, 4.86 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (1.36 g, 7.30 mmol) were added to acetonitrile (50 mL), the reactants were dispersed in the solvent, potassium tert-butoxide (1.37 g, 12.17 mmol) was added, and the reaction was stirred at room temperature for 24 h. After TLC (PE:EA=1:1) indicated the reaction was complete, the reaction was quenched with water, extracted with EA, the organic phase was dried over anhydrous sodium sulfate, concentrated, and purified by preparative flash chromatography (PE:EA=1:1), to afford compound (39f) (1.30 g, yellow oil), yield: 51%. MS m/z: 526 [M+1]+.
4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrrole-3-carbonitrile[ No CAS ]
1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrrole-3-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (10 mL) was added to a system of compound (60b) (200 mg, 0.59 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (164 mg), and a cloudy reaction solution was obtained. DBU (269 mg) was added, the reaction solution became clear, and was stirred at room temperature overnight. The reaction solution was concentrated, the residue was dissolved in DCM, and purified on a preparative silica gel plate (PE:EA=1:2), to afford compound (60c) (232 mg, light yellow solid), yield: 75%. MS m/z: 526 [M+1]+.
4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrrole-2-carbonitrile[ No CAS ]
1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrrole-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃;
DMF (20 mL) was added to compound (61e) (260 mg, 0.77 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (215 mg, 1.15 mmol), the reactants were dispersed in the solvent, potassium carbonate (319 mg, 2.31 mmol) was then added, and the reaction was stirred overnight at room temperature. After LC-MS indicated the substrate disappeared, the reaction solution was poured into water, extracted with EA, the organic phase was dried over anhydrous sodium sulfate, concentrated, and purified on a preparative silica gel plate (PE:EA=1:1), to afford compound (61f) (159 mg, oil), yield: 39%. MS m/z: 525 [M+1]+.
4-(3-chloro-1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine[ No CAS ]
2-(3-(3-chloro-4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 3h;
At room temperature, <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (169 mg, 0.91 mmoL) and DBU (105 mg, 0.69 mmol) were sequentially added to a solution of compound (48g) (159 mg, 0.46 mmol) in acetonitrile (5 mL), and the reaction was performed for 3 h. After thin layer chromatography indicated the reaction was complete, the reaction solution was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography on silica gel, to afford compound (48h) (177 mg, white solid), yield: 73%. MS (ESI, m/z): 535 [M+H]+.
4-(3-methyl-1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(3-methyl-4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (15 mL) was added to compound (49b) (200 mg, 0.59 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (112 mg) to obtain a cloudy reaction solution, DBU (249 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified on a preparative silica gel plate (DCM:EA=2:1), to afford compound (49c) (220 mg, white solid). Yield: 73%. MS m/z: 516 [M+1]+.
5-chloro-4-(1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine[ No CAS ]
2-(3-(4-(5-chloro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (3 mL) was added to compound (50c) (130 mg, 0.37 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (76 mg, 0.41 mmol) to obtain a cloudy reaction solution. DBU(170 mg) was then added, the system became clear, and was stirred at room temperature overnight. The reaction solution was concentrated under reduced pressure, and purified on a preparative silica gel plate (PE:EA=2:1), to afford compound (50d) (141 mg, oil). Yield: 71%. MS m/z: 535 [M+1]+.
4-(4-methyl-1H-pyrrol-3-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(3-methyl-4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrrol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (5 mL) was added to compound (56c) (108 mg, 0.33 mmol) and <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (92 mg), to obtain a cloudy reaction solution. DBU (150 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction solution was concentrated, the residue was dissolved in DCM, and purified on a preparative silica gel plate (PE:EA=1:1), to afford compound (56d) (88 mg, yellow solid), yield: 53%. MS m/z: 515 [M+1]+.
4-(1H-pyrazol-3-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(3-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (2 mL) was added to a mixture of 4-(1H-pyrazol-3-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin (3c) (45 mg) and <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (35 mg) to obtain a reaction solution. DBU (25 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified on a preparative silica gel plate, to afford 2-(1-(ethylsulfonyl)-3-(3-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (3d) (75 mg, brown solid), yield: 105% (containing some solvents). MS (ESI, m/z): 502 [M+H]+.
7-(1H-pyrazol-4-yl)-3-((2-(trimethylsilyl)ethoxy)methyl)-3H-imidazo[4,5-b]pyridine[ No CAS ]
2-(1-(ethylsulfonyl)-3-(4-(3-((2-(trimethylsilyl)ethoxy)methyl)-3H-imidazo[4,5-b]pyridin-7-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, acetonitrile (6 mL) was added to a mixture of 7-(1H-pyrazol-4-yl)-3-((2-(trimethylsilyl)ethoxy)methyl)-3H-imidazo[4,5-b]pyridine (4c) (175 mg) and <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (103 mg) to obtain a reaction solution. DBU (100 mg) was then added, and the reaction was stirred at room temperature overnight. The reaction was concentrated, and purified on a preparative silica gel plate (DCM:MeOH=20:1), to afford 2-(1-(ethylsulfonyl)-3-(4-(3-((2-(trimethylsilyl)ethoxy)methyl)-3H-imidazo[4,5-b]pyridin-7-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (4d) (214 mg, oil), yield: 77%. MS (ESI, m/z): 502 [M+H]+.
2-cyano-4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine[ No CAS ]
4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 2h;
2-cyano-4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (20d) (219 mg, 0.64 mmol), <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (131 mg, 0.71 mmol) and acetonitrile (10 mL) were sequentially added to a 50 mL reaction flask, DBU (108 mg, 0.76 mmol) was added, and the reaction was performed at room temperature for 2 h. The reaction was monitored by thin layer chromatography. After the reaction was complete, the reaction solution was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography on silica gel, to afford 4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine-2-carbonitrile (20e) (280 mg, yield: 83%, white solid). MS (ESI, m/z): 527 [M+H]+.
4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 2h;
4-(1H-pyrazol-4-yl)-5-cyano-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine (5c) (259 mg, 0.76 mmol), <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (155 mg, 0.84 mmol) and acetonitrile (10 mL) were sequentially added to a 50 mL reaction flask, and DBU (119 mg, 0.84 mmol) was then added. The reaction was performed at room temperature for 2 h, and monitored by thin layer chromatography. After the reaction was complete, the reaction solution was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography on silica gel, to afford 4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-pyrazol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine-5-carbonitrile (5d) (305 mg, yield: 76%, white solid). MS (ESI, m/z): 526 [M+H]+.
4-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1H-pyrazole-3-carbonitrile[ No CAS ]
4-(1-(3-(cyanomethyl)-1-(ethylsulfonyl)azetidin-3-yl)-1H-(3-cynaopyrazole)-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
48%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;
At room temperature, <strong>[1187595-85-2]2-<strong>[1187595-85-2][1-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile</strong></strong> (521 mg, 2.80 mmoL) and DBU (127 mg, 0.84 mmol) were sequentially added to a solution of a compound (36e) (189 mg, 0.56 mmol) in acetonitrile (2 mL), and the reaction was allowed to proceed overnight. After thin layer chromatography indicated the reaction was complete, the reaction solution was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography on silica gel, to afford compound (36f) (140 mg, light yellow solid), yield: 48%. MS (ESI, m/z): 526 [M+H]+.
2-{1-(ethanesulfonyl)-3-[4-(1H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]azetidin-3-yl}acetonitrile[ No CAS ]
2-[3-(4-{1-[3-(cyanomethyl)-1-(ethanesulfonyl)azetidin-3-yl]-1H-pyrazol-4-yl}-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-1-(ethanesulfonyl)azetidin-3-yl]acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 0 - 5℃; for 3h;Inert atmosphere;
1,8-Diazabicyclo[5.4.0]undec-7ene (DBU, 0.01 g, 0.68 mmol) was added with stirring at 0-5C to a suspension of 8 (10.0 g, 0.027 mol) and <strong>[1187595-85-2]2-[1-(ethanesulfonyl)azetidin-3-ylidene]acetonitrile</strong> (5.01 g, 0.027 mol) in acetonitrile (80 mL). The mixture was stirred for ~3 h (TLC) and diluted with diisopropyl ether (100 mL), the precipitate was filtered off and taken in additional diisopropyl ether (50 mL), the mixture was stirred, and the precipitate was filtered off and dried. Yield 12.0 g (80%), white solid. 1 H NMR spectrum (300 MHz, DMSO-d6), delta, ppm: 1.21-1.29 m (6H), 3.18-3.28 m (4H), 3.62 s (2H), 3.70 s (2H), 4.28 d (4H, J = 9.0 Hz), 4.65 d (4H, J = 9.0 Hz), 7.29 d (1H, J = 3.6 Hz), 7.91 d (1H, J = 3.6 Hz), 8.52 s (1H), 8.78 s (1H), 8.99 s (1H). 13C NMR spectrum (300 MHz, DMSO-d6), deltaC, ppm: 150.98, 150.47, 150.41, 140.06, 130.01, 127.56, 121.64, 116.67, 116.63, 113.96, 100.84, 56.16, 52.32, 43.35, 43.31, 26.91, 26.86, 7.43. Mass spectrum: m/z 558.09 [M + H]+ .
2-[3-(4-{1-[3-(cyanomethyl)-1-(ethanesulfonyl)azetidin-3-yl]-1H-pyrazol-4-yl}-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-1-(ethanesulfonyl)azetidin-3-yl]acetonitrile[ No CAS ]






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping