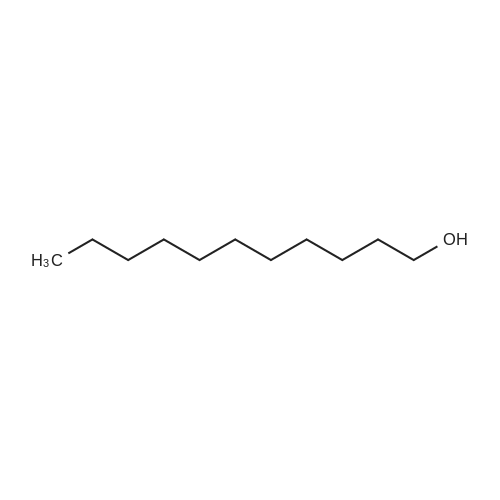
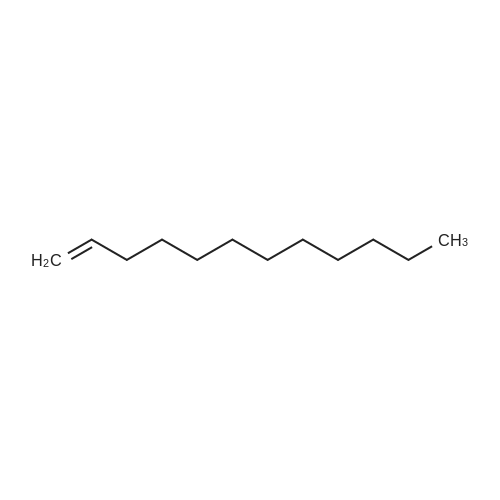
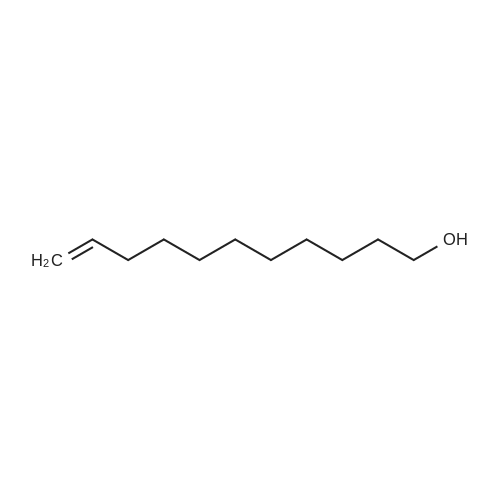
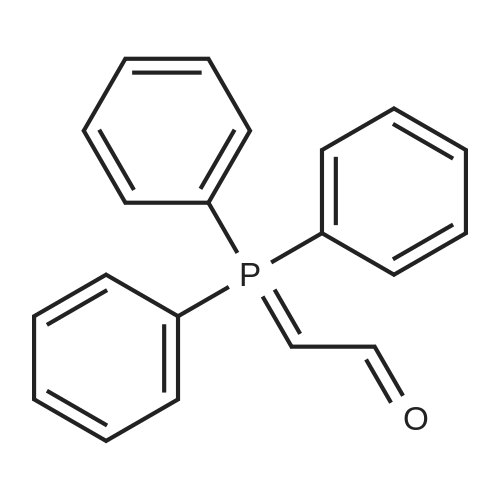
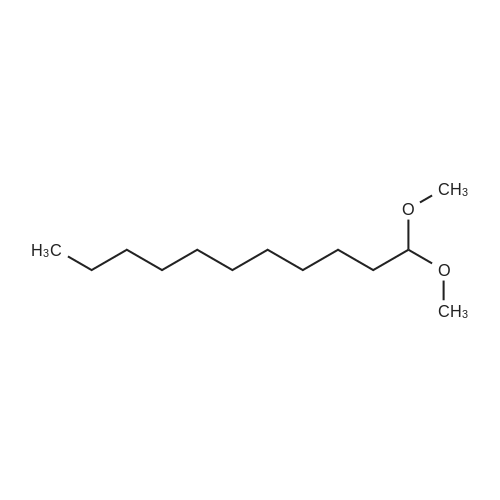
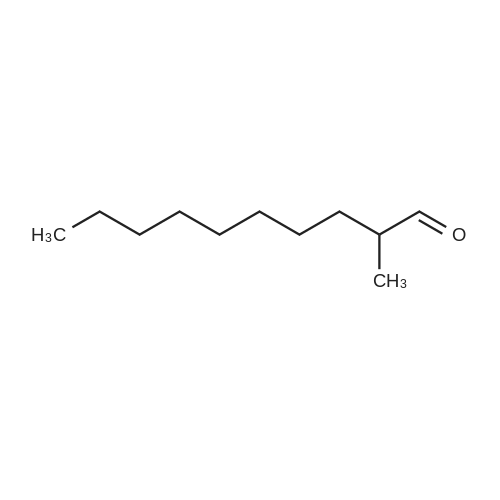
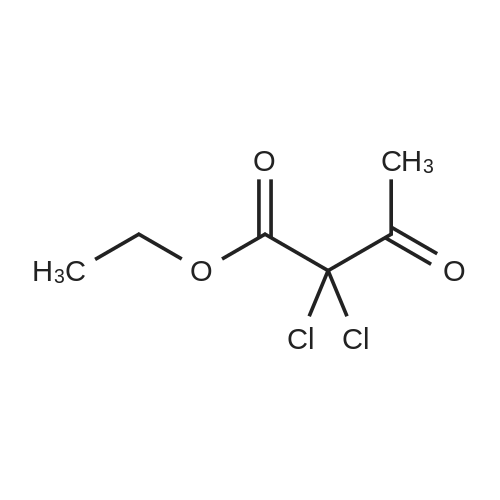
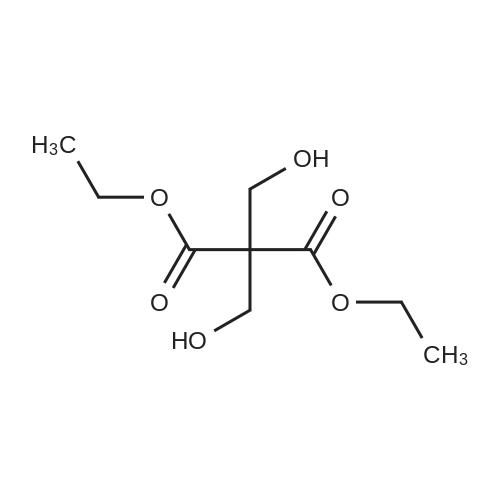
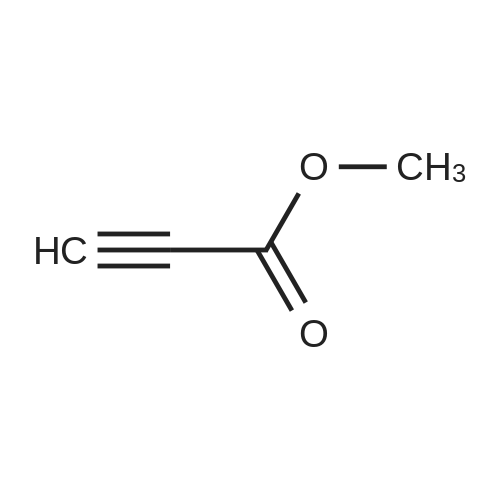
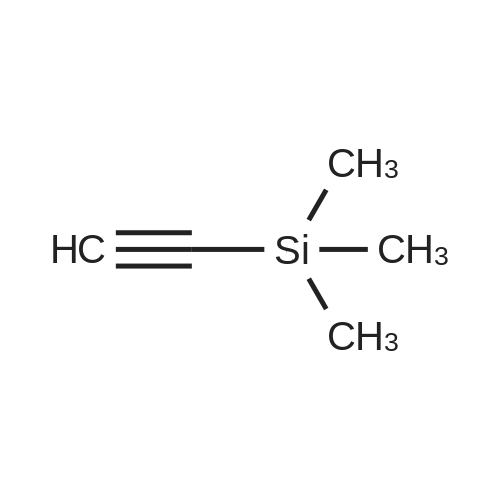
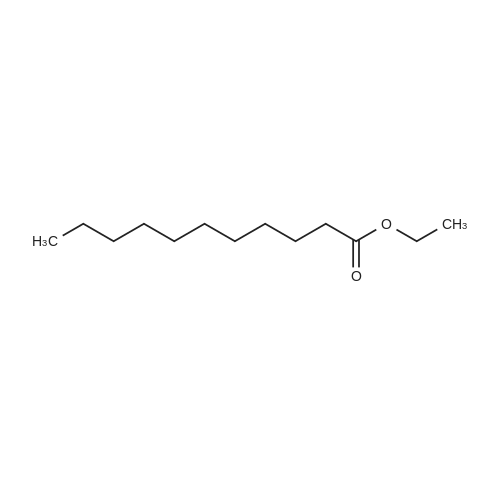
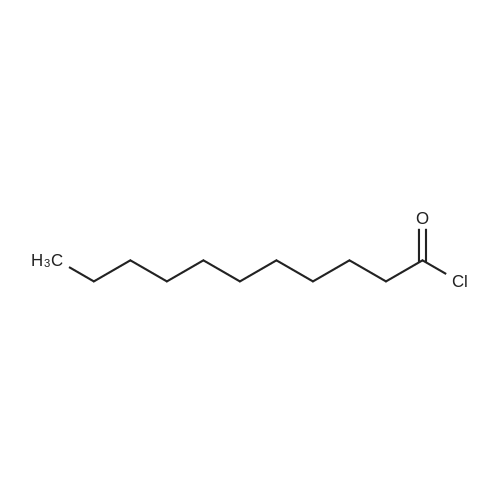
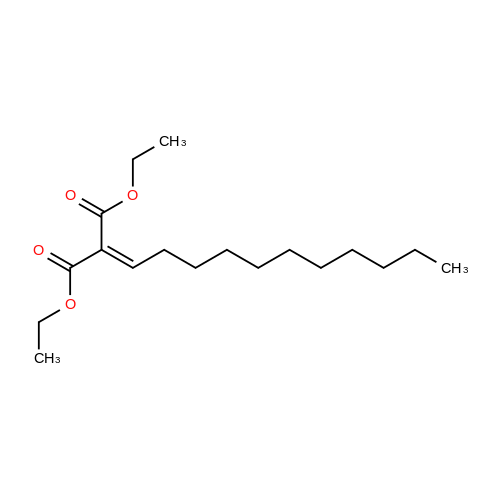
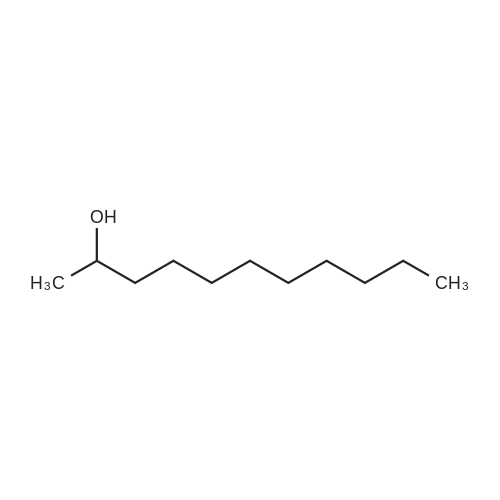
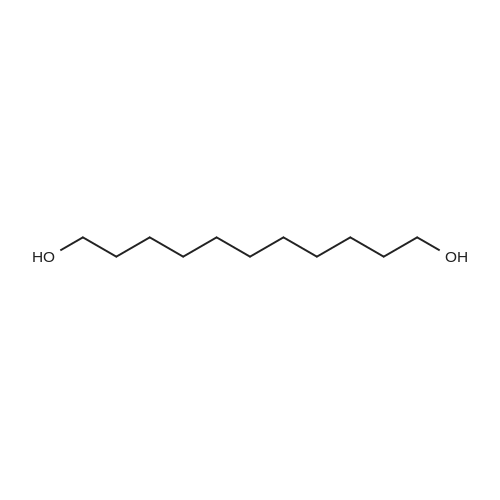
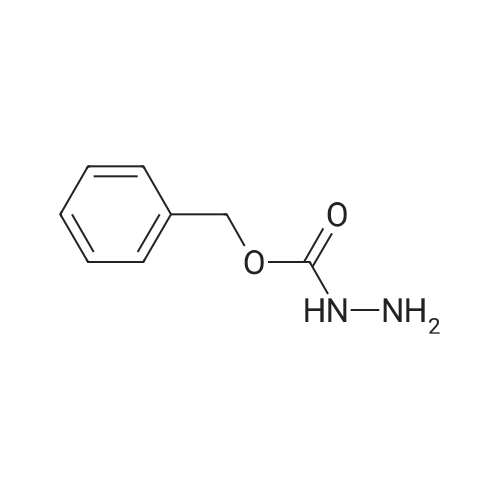
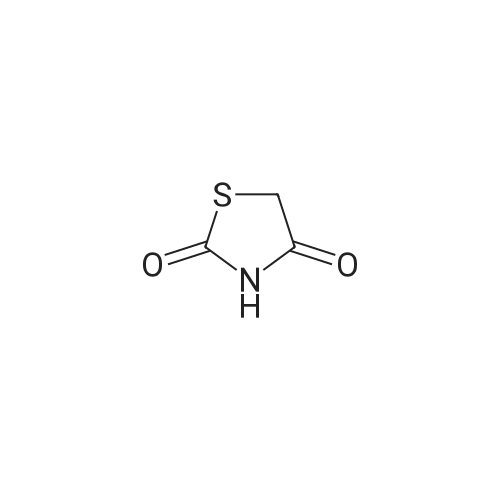
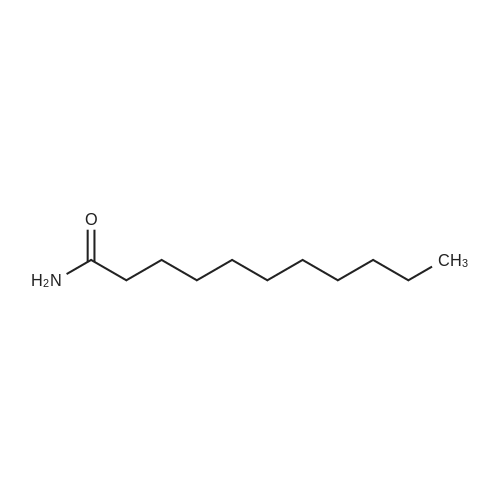
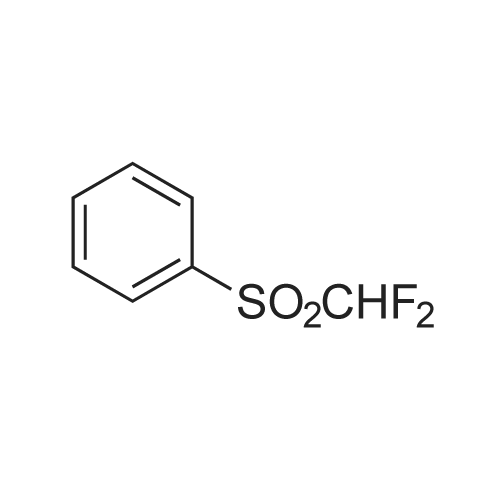
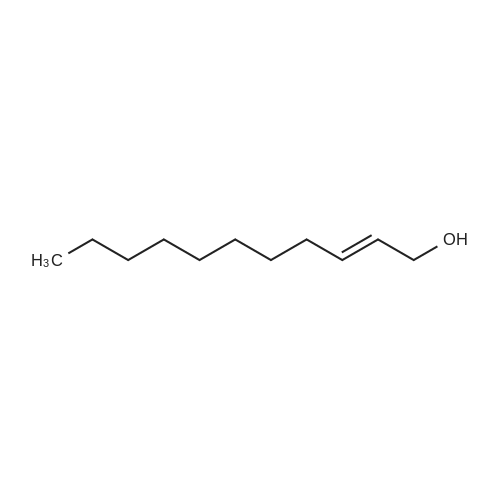
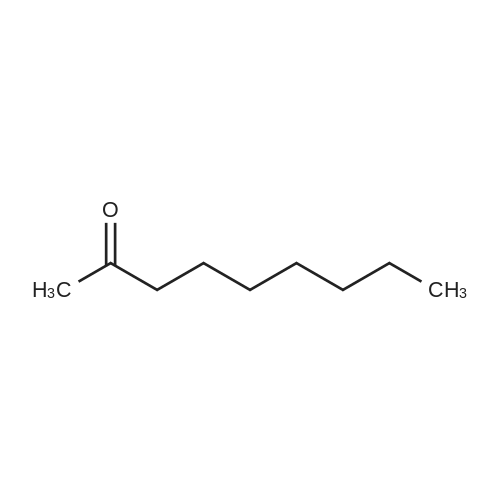
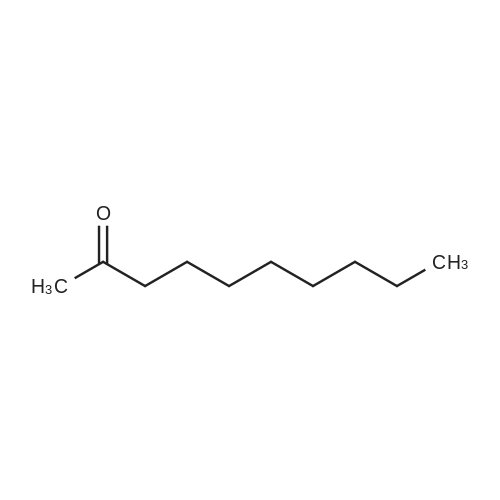
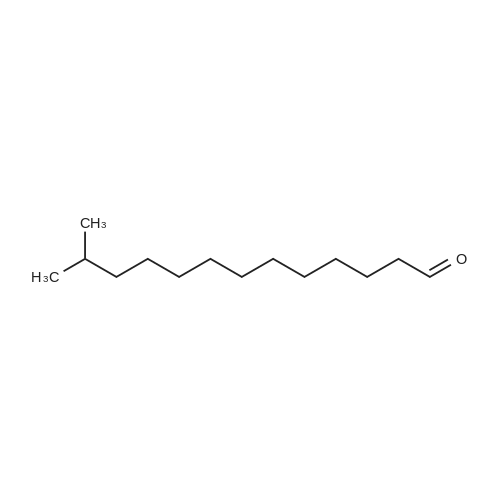
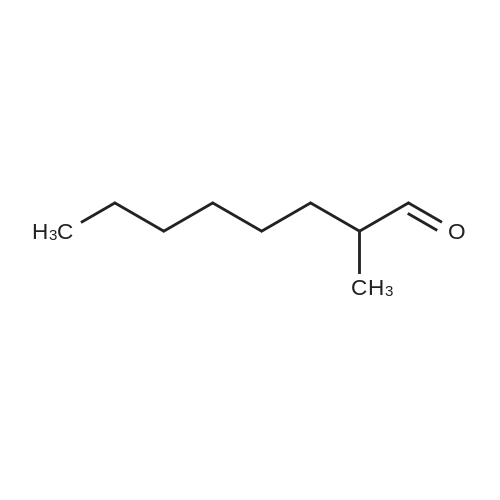
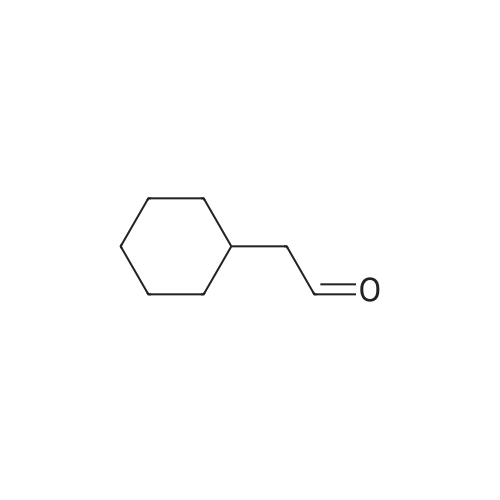
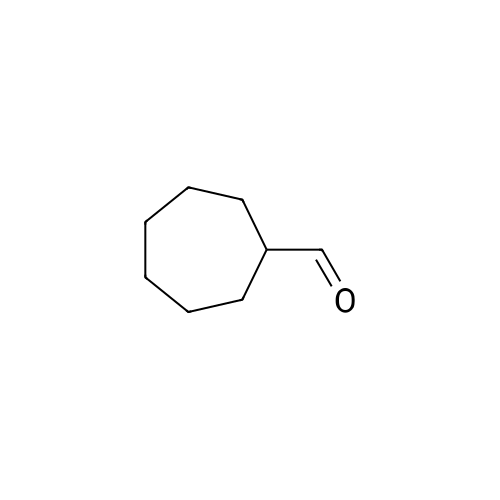
|
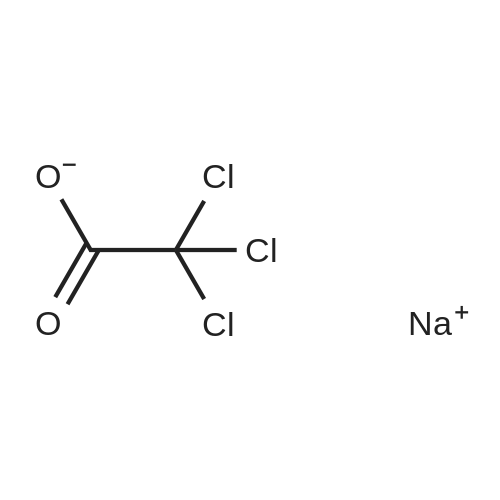
With trisodium tris(3-sulfophenyl)phosphine; hydrogen at 80℃; for 8h; Yield given. Yields of byproduct given. Title compound not separated from byproducts; |
|
|
With triisooctylamine-tristriphenylphosphine trisulfonate salt; triisooctyl amine; hydrogen In acetone at 130℃; for 6h; Yield given. Yields of byproduct given; |
|
|
With hydrogen at 100℃; for 12h; Title compound not separated from byproducts; |
|
|
With alkylated poly(propylene imine) dendrimer; 4-diphenylphosphanobenzoic acid; hydrogen In tetrahydrofuran at 60℃; |
|
|
With MeO-PEG-PPh2; hydrogen at 70℃; |
|
|
With Calix[4]arenesulfonic acid; hydrogen; triphenylphosphine In water; toluene at 50℃; Title compound not separated from byproducts; |
|
|
With polyether phosphite; hydrogen In n-heptane at 100℃; for 4h; |
|
|
With partly methylated β-cyclodextrin; trisodium tris(3-sulfophenyl)phosphine; hydrogen at 80℃; for 8h; other chemically modified and intact cyclodextrins; |
|
|
With hydrogen In n-heptane; water at 100℃; for 4h; |
|
|
With carbon dioxide; hydrogen at 80℃; for 1h; |
|
|
With hydrogen; fipronilβ-cyclodextrin; 5,10,15,20-tetrakis(4-sulfonatophenyl)-21H,23H-porphyrin In water at 80℃; for 18h; |
|
|
With mono-2-hydroxy-3-trimethylammoniopropyl-α-cyclodextrin; trisodium tris(3-sulfophenyl)phosphine; hydrogen In water for 6h; |
|
|
With hydrogen; randomly methylated β-cyclodextrin; trilithium salt of 3,3',3''-phosphanotriylbenzenecarboxylic acid In water at 80℃; for 3h; |
|
|
With randomly methylated-β-cyclodextrin; sodium salt of trisulfonated triphenylphosphine; hydrogen In water at 80℃; for 6h; |
|
|
With tetrasulfonated 1,3-bis(diphenylphosphino)propane; hydrogen; randomly methylated β-cyclodextrin In water at 80℃; for 24h; |
|
|
With hydrogen; tris(4-(1H,1H,2H,2H-perfluorodecyl)phenyl)phosphite In toluene at 80℃; for 1h; |
|
|
With heptakis(2,3-di-O-methyl-6-O-sulfopropyl)-β-cyclodextrin; trisodium tris(3-sulfophenyl)phosphine; hydrogen In water at 80℃; for 2h; |
|
|
With hydrogen at 120℃; for 2h; Title compound not separated from byproducts.; |
|
|
With hydrogen at 50℃; for 2h; |
|
|
With n-Undecane; hydrogen at 80℃; for 6h; Title compound not separated from byproducts.; |
|
|
With hydrogen In water at 125℃; for 4h; |
|
|
With hydrogen In water at 125℃; for 1h; |
|
|
With hydrogen In water at 125℃; for 35h; |
|
|
With [κ2-{3-iPr2P-2-O-indene}Rh(COD)]; hydrogen In tetrahydrofuran at 45℃; for 12h; regioselective reaction; |
|
|
With dicarbonylacetylacetonato rhodium (I); trisodium tris(3-sulfophenyl)phosphine; hydrogen In water at 80℃; for 24h; |
|
|
With dicarbonylacetylacetonato rhodium (I); C24H18O3PS(1-)*C43H73N3O34*ClH*Na(1+); hydrogen In water at 80℃; for 6h; Autoclave; regioselective reaction; |
|
|
With (acetylacetonato)dicarbonylrhodium (l); trisodium tris(3-sulfophenyl)phosphine; hydrogen In water at 80℃; for 2h; Inert atmosphere; |
|
|
With dicarbonylacetylacetonato rhodium (I); hydrogen; trisodium triphenylphosphine‐3,3′,3″‐trisulfonate at 80℃; for 3h; |
|
|
With acetylacetonatodicarbonylrhodium(l); C98H148N2O70; C24H16O9PS3(3-)*3Na(1+); hydrogen In water at 80℃; for 9h; Schlenk technique; Autoclave; regioselective reaction; |
|
|
With {Rh(COD)Cl}2(Ph2POC6H4OPPh2); hydrogen In tetrahydrofuran at 60℃; for 6h; Autoclave; Inert atmosphere; regioselective reaction; |
General procedure for catalytic hydroformylation reaction:
in a typical experiment, to a high pressure reactor (autoclave) of 100 mL capacity, Rh phosphinite complex (0.002 mmol, 2 mg), olefin (5 mmol), and THF (15 mL) were added. The reactor was then flushed with nitrogen, followed by syngas(1:1 mixture of CO and H2 gas) at room temperature; next, the reaction was pressurized to 30 bar syngas and heated to 60 C at a stirring speed of 600 rpmfor 6 h. After completion of reaction, the reactor was cooled to room temperature, and the remaining CO/H2 gas was carefully vented, and the reactor was opened. The reaction mixture was analyzed by gas chromatography (GC). |
|
With hydrogen In toluene at 100℃; Autoclave; Inert atmosphere; |
|
|
With acetylacetonatodicarbonylrhodium(l); N,N'-Dimethylurea; trisodium tris(3-sulfophenyl)phosphine; hydrogen at 90℃; for 1h; Autoclave; Inert atmosphere; regioselective reaction; |
|
|
Stage #1: carbon monoxide With acetylacetonatodicarbonylrhodium(l); trisodium tris(3-sulfophenyl)phosphine; hydrogen at 90℃; for 1h; Autoclave;
Stage #2: 1-Decene at 90℃; for 1h; Autoclave; |
2. Experimental
In a typical hydroformylation experiment, [Rh(acac)(CO)2] (5.5mg;21 μmol; 1 eq.), TPPTS (60mg; 105 μmol; 5 eq.) and solvent constituents at solid state (4.2 g of N,N′-dimethylurea and 1.8 g of RAME-β-CD (w/w (%)=70/30) for example) were charged into a 25 mL autoclave. Air was replaced by 20 bar of CO/H2 (1/1) and after heating at 90 °C, the mixture was stirred using a multipaddle unit (1500 rpm) for 1 h for an incubation period. The stirring was then stopped and after cooling and depressurization, 1-Decene (6 g; 42 mmol; 2000 eq.) was introduced under nitrogen in the autoclave. The medium was heated at 90 °C and then stirred (1500 rpm) for 1 h at this temperature under 50 bar of CO/H2(1/1). |
|
With (acetylacetonato)dicarbonylrhodium (l); hydrogen In water at 80℃; for 6h; Autoclave; Inert atmosphere; |
2.4. General procedure for hydroformylation reaction
In all experiments, the stirring speed and the CO/H2 pressure were fixed to 1500 rpm and 50 bar, respectively. In a typical experiment, Rh(acac)(CO)2 (0.021 mmol), phosphane(0.107mmol) and RAME-β-CD (0.21mmol) were dissolved in 6 mL of water. The resulting aqueous phase and an organic phase composed of 1-decene (10.5mmol) were charged under an atmosphere of N2 into the 25mL reactor which was heated at 80 °C. The autoclave was then pressurized with 50 bar of CO/H2 (1/1) and mechanical stirring equipped with a multipaddle unit was started. The pressure was kept constant over the reaction time of 6h by feeding syngasvia a pressure controller. At the reaction’s end, the autoclave was cooled down to room temperature and depressurized. The medium is then decanted and the organic phase analyzed by GC. For kinetic measurements, the time corresponding to the addition of CO/H2 was considered as the beginning of the reaction. |
|
With dicarbonylacetylacetonato rhodium (I); hydrogen; triphenylphosphine In toluene at 90℃; for 2h; Inert atmosphere; |
1.1c Example 1 - Hvdroformylation of Biorefinery Olefins and Unsaturated Esters
All olefin feedstock materials were pre-treated by heating to 200 °C for 2 hours to reduce peroxide value to less than 0.5 meq/kg. Methyl 9-decenoate was 98.8% pure. It contained 1 .0% methyl 8-decenoate and 0.2% methyl decanoate. 9,12-Tridecadienoate was 95.8% pure. It contained 0.25% saturated C13 FAME and 0.62% of mono-unsaturated C13 FAME. The C10 olefin consisted mainly of 1 - decene (91 .8%), internal C10 olefins (2.5%). The balance consisted of other olefins and FAMES. (0375) [0181] Hydroformylation experiments were conducted in a 3-ounce Fisher- Porter tube equipped with a 20 mm cross-shaped stir bar, a digital pressure gauge capable of measuring pressure to a tenth of a PSI, a sealable port for the (0376) introduction of liquid reagents, a vent line to de-pressure the reactor through the headspace, and a gas manifold capable of delivering nitrogen, hydrogen, or syngas at a pressure of at least 100 psig. Gas was delivered to the Fisher-Porter tube through a dip-tube. The Fischer-Porter tube was submerged in a silicone oil bath. Heating and stirring was provided by a Magnetic Stirrer/Hotplate. [0182] Except where otherwise noted, manipulation of chemicals was performed with standard air-free lab techniques. The reactor was loaded, in a dry- box, with Rh(acac)(CO)2 (ca 5 to 6 mg), triphenylphosphine, and olefinic substrate (ca 27 mmole), and undecane (ca 0.5 g as an internal standard). The sealed reactor was brought out of the dry box and then attached to the gas manifold. It was pressured to 95 psig with nitrogen followed by venting to 0 psig (2 times). Toluene was added through sealable port to bring total volume to 27 mL. The stir rate was set to 1500 rpm creating a deep vortex. The reactor was pressured to 95 psig and vented to 0 psig three times with nitrogen then 3 times with hydrogen. The reactor was then pressured to 95 psig with syngas, vented to 0 psig. Finally, the reactor was pressurized and heated to specified reaction conditions. Pressure was maintained by continuous syngas feed set at the specified pressure. The reaction mixture was analyzed by GC after the indicated time. (0377) [0183] Table 3 shows the conditions for hydroformylation experiments performed on four different substrates, denominated as Examples 1 a to 1 c. (0378) Table 3 (0379) (0380) [0184] Table 4 shows the results from the hydroformylation of the four substrate materials shown in Table 3. The "linear" addition of the formyl group refers to addition at the terminal carbon atom, while "branched" addition refers to addition of the formyl group at the non-terminal carbon. Table 4 |
|
With dicobalt octacarbonyl; C20H39P; hydrogen; sodium acetate In toluene at 110℃; for 22h; Autoclave; regioselective reaction; |
III-General Procedure for hydroformylation-reduction for Table 4 and 5
General procedure: Lim-10 (5-10 mol%), 2-ethylhexanoic acid (10 mol%) and KOH/EtOH (1.0 M soln.; 2.5 mol%) were loaded in to a glass liner containing a magnetic stirring bar and flushed with a continuous stream of argon. Solid Co2(CO)8 (5 mol%) was weighed promptly and added to the liner. Then, under argon protection, alkene (2.0 mmol) and solvent (5 mL) were added. The glass liner was then inserted into a stainless steel autoclave, capped and purged three times with CO and then pressurized at room temperature to 200 psi of CO and 400 psi of H2. The autoclave was heated at 110 C for 22 h and then cooled to room temperature before releasing the excess gases. The reaction mixture was then transferred to a flask using 5-10 mL of MeOH to which was added NaBH4 (1.2 equiv.) at 0 C and the consumption of the RCHO monitored by TLC. Upon completion of the reaction, the resulting mixture was acidified to PH~6 and extracted with ether (3x 50 mL). The combined organic layers were dried with anhydrous MgSO4 and the solvent was removed in vacuo to give an oil residue which was purified by flash chromatography on silica gel with hexane / EtOAc (v/v, 5:1 to 2:1) as the eluant to obtain inseparable mixture of alcohols in 53-99 % yields. |
|
With dicarbonylacetylacetonato rhodium (I); C56H82NO40PS2(2-)*2Na(1+); hydrogen In water at 80℃; for 3h; Inert atmosphere; Autoclave; chemoselective reaction; |
|
|
With acetylacetonatodicarbonylrhodium(l); C18H12O9PS3(3-)*3C44H84O16P(1+); hydrogen at 95℃; for 0.5h; Autoclave; |
10 Two-phase hydroformylation of 1-octene with Rh(acac)(CO)2/[CH3(OCH2CH2)16Ρ(C2H5)3]3[(S03-)3-1}
General procedure: In an inert atmosphere, A stainless steel autoclave was charged with Rh (acac) (C0) 2 [CH3 (OCH2CH2) 16P (C2H5) 3] 3 [(S03-) 3-1] (Acac) (CO) 2 = 10: 1 (molar ratio), 1-octene / Rh (acac) 2 = 10000: 1 (molar ratio), Then pressurized with synthetic gas (H2 / C0 = 1: 1) to 5.0 MPa, the reaction temperature was 95 ° C, the reaction time 0.5 hours, and then rapidly cooled to room temperature, venting the synthesis gas after the autoclave, the system naturally separated into two phases, the lower layer is an ionic liquid phase containing a rhodium catalyst, the upper layer is an organic phase, and n-heptane can also be added for extraction,and the organic phase containing the product aldehyde is obtained through simple two-phase separation. The gas chromatographic analysis results are as follows: The conversion of 1-octene was 7.1%, the selectivity of aldehyde was 83.4%, the molar ratio of normal aldehyde to isomeric aldehyde was 2.6: 1, and the T0F value was 1184h-1. |
|
With hydrogen at 110℃; for 12h; Autoclave; |
|
|
With HRh(CO)(TPPTS)<SUB>3</SUB>; hydrogen at 120℃; for 0.25h; Autoclave; Inert atmosphere; Green chemistry; regioselective reaction; |
2.2 Hydroformylation andCatalyst Recycling
General procedure: All hydroformylation was carried out in a 60mL stainlesssteel autoclave equipped with a magnetic stirrer. HRh(CO)(TPPTS)3, TPPTS or BISBIS, PEG and long chain alkenewere introduced to the autoclave under a nitrogen atmosphere.The autoclave was closed, fushed with syngas threetimes, and then pressurized with syngas (H2:CO = 1:1) todesired pressure and heated to needed temperature for a specifedtime. After the reaction was fnished, the autoclave wascooled to room temperature and carefully depressurized. Theproduct phase was separated from the PEG phase containingwater soluble rhodium catalyst by simple phase separation.The products were analyzed by GC (PANNA A91, KB-1,30m × 0.25mm × 0.50μm, FID). In catalyst recycling experiments,the PEG phase and new portion of long chain alkenewere introduced to the autoclave under a nitrogen atmosphere.The other operations were the same as the formerhydroformylation. |
|
With bis(triphenylphosphine)iminium tetracarbonylhydridoferrate(0); hydrogen; triphenylphosphine In methanol at 100℃; for 24h; Autoclave; regioselective reaction; |
|
|
With dicarbonyl(acetylacotonato)rhodium(I); trisodium tris(3-sulfophenyl)phosphine; hydrogen In water at 80℃; for 22h; chemoselective reaction; |
|
|
With acetylacetonatodicarbonylrhodium(l); 3C43H81N3O16*C18H15O9PS3; hydrogen at 95℃; for 0.5h; Autoclave; |
8 Rh(acac)(CO)2/[Ph(EO)16N+H=C(N(CH3)2)2]3[(SO3-)3-1]/1-decene system under two-phase hydroformyl Chemical reaction
The olefin was changed to 1-decene, and the remaining reaction conditions and steps were the same as in Example 3. The gas chromatographic analysis showed that: 1-deceneThe conversion rate was 8.8%, the selectivity of aldehyde was 62.7%, the molar ratio of normal aldehyde to isomeric aldehyde was 2.5:1, |
|
With hydrogen In toluene at 79.84℃; for 6h; Autoclave; |
|
|
With HRh(CO)(TPPTS)<SUB>3</SUB>; trisodium tris(3-sulfophenyl)phosphine; hydrogen In methanol at 80℃; for 1h; Autoclave; |
|
|
With hydrogen In toluene at 70℃; for 7h; Autoclave; chemoselective reaction; |
|
|
With (acetylacetonato)dicarbonylrhodium (l); trisodium tris(3-sulfophenyl)phosphine; hydrogen In water at 80℃; for 6h; Autoclave; chemoselective reaction; |
|
|
With rhodium(III) chloride trihydrate; trisodium tris(3-sulfophenyl)phosphine; hydrogen at 85℃; for 5h; Autoclave; Inert atmosphere; Ionic liquid; chemoselective reaction; |
|
|
With hydrogen; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene; palladium(II) iodide In 1,4-dioxane at 100℃; for 16h; Overall yield = 99 percentChromat.; |
|
|
With (acetylacetonato)dicarbonylrhodium (l); hydrogen; C40H36N4O4P2 In toluene at 100℃; for 1h; Autoclave; |
9-10 Example 9:
In a 50ml autoclave, add 0.25mmol bidentate phosphine ligand 1, 0.025mmol Rh(acac)(CO)2, 50mmol1-decene, 5ml toluene, and then fill with synthesis gas (CO:H2=1:1) Replace the reactor three times, fill it with synthesis gas again, keep the total pressure in the reactor at 3MPa, quickly raise the temperature to 100°C and start stirring, stop stirring after 1 hour of reaction, and quickly cool to room temperature, and take out the reaction solution for analysis. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping