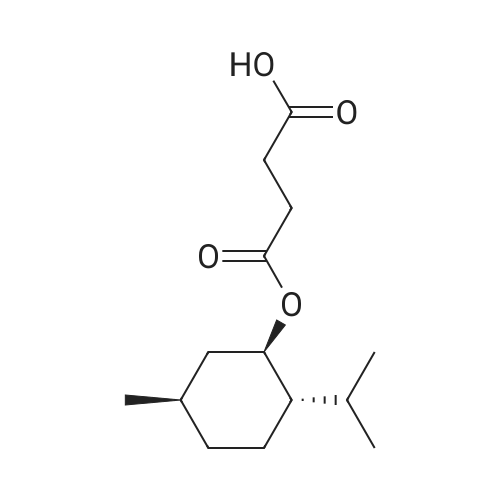
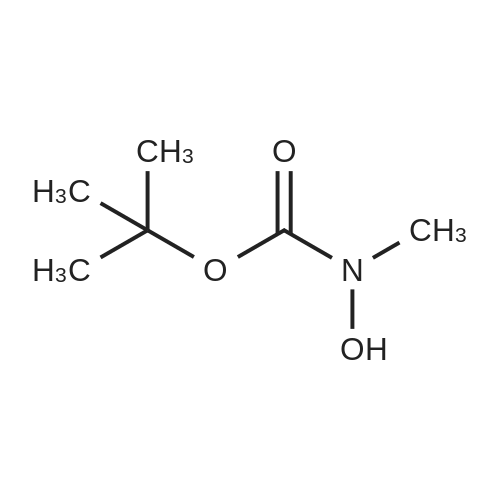
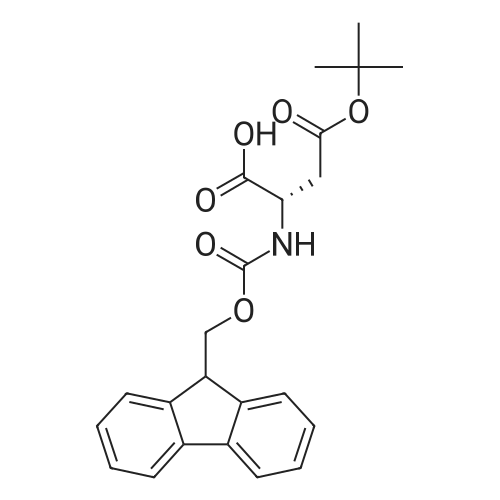
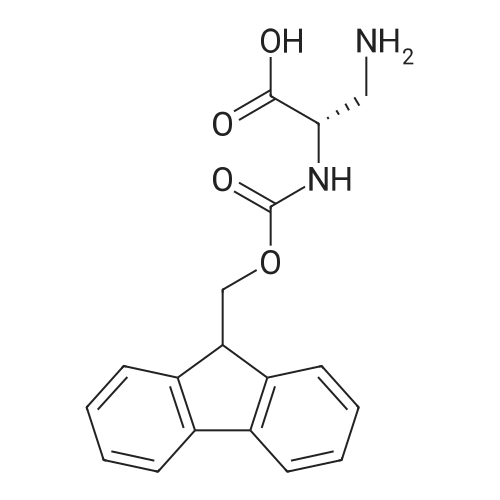
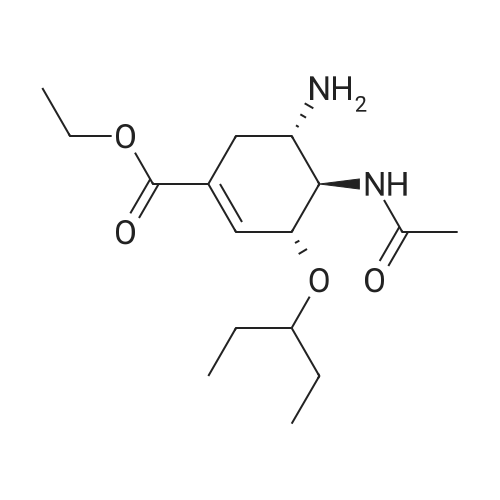
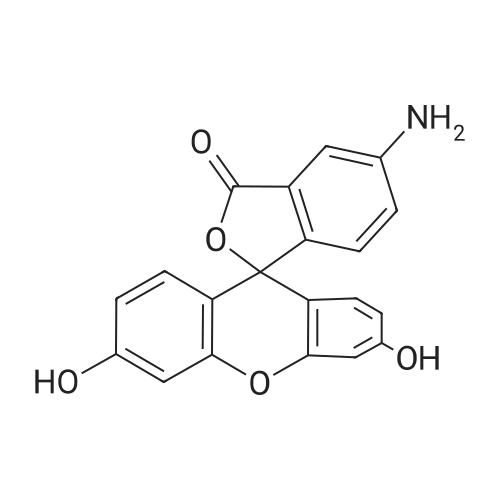
There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
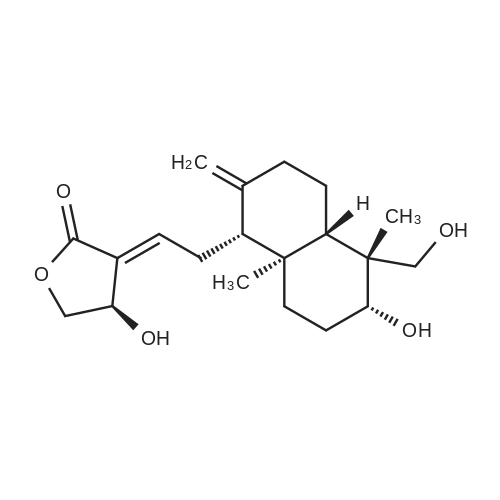
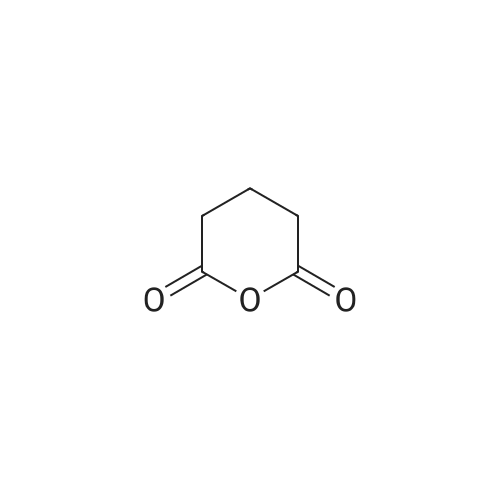
Structure of 108-30-5 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Tetrahedron, 2008, vol. 64, # 19, p. 4347 - 4353
[2] Journal of the Chemical Society, 1930, p. 1733,1739
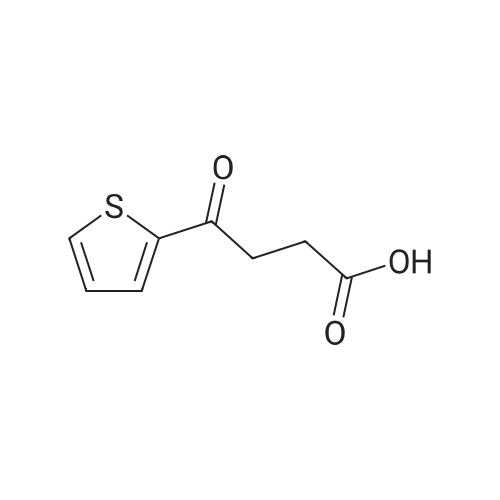
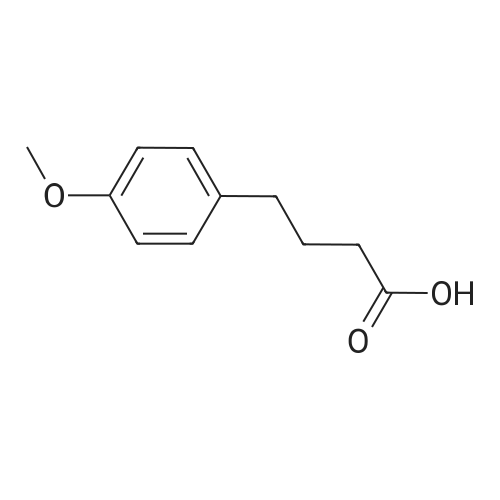
3
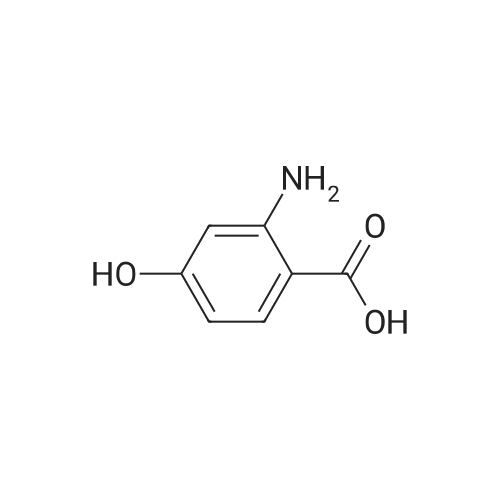
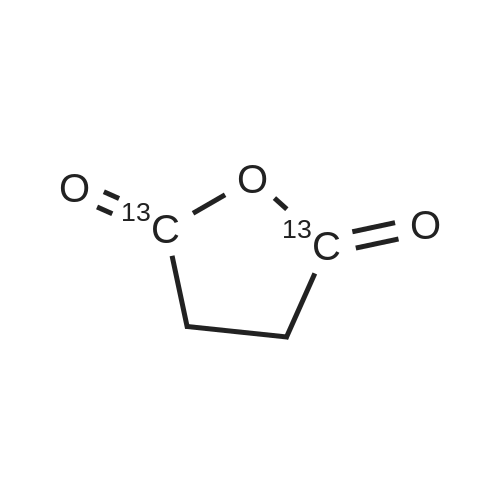
[ 108-30-5 ]
[ 7726-95-6 ]
[ 5470-44-0 ]
[ 1122-12-9 ]
Reference:
[1] Journal of the Chemical Society, 1930, p. 1733,1739
4
[ 188290-36-0 ]
[ 108-30-5 ]
[ 4653-08-1 ]
Yield
Reaction Conditions
Operation in experiment
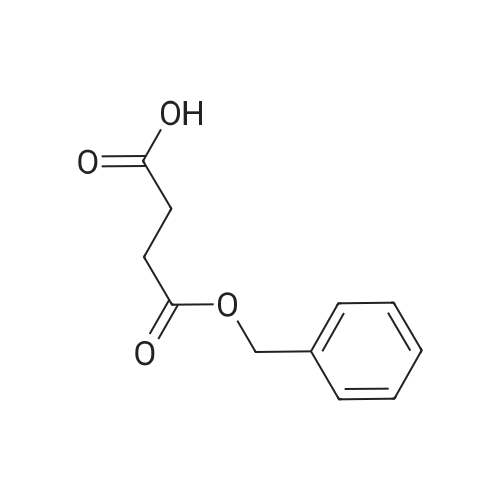
89%
Stage #1: With aluminum (III) chloride In 1,1-dichloroethane at 20℃; for 1 h; Heating / reflux Stage #2: With hydrogenchloride In 1,1-dichloroethane; water at 20℃; for 0.166667 h;
Thiophene [(5.] [0GM,] 0.059moles) and succinic anhydride (7.13gm, 0.0713 moles) were taken up in dichloroethane (50 ml). Aluminium chloride (17.43gm, 0.131 moles) was added at room temperature and the resulting mixture heated under reflux for lhr with stirring. The reaction mixture was cooled to room temperature, diluted with 30 ml of 1 : [1] mixture of water and [CONC.] hydrochloric acid. After stirring for 10 min the separated solid was filtered under suction, washed with water and dilute hydrochloric acid (50 ml). The solid was dried at room temp to give 9.8 gm (89percent 0 of the title compound, mp [103-107°] C. [LH NMR] (CDC13, [8)] : 7.69 (dd, [1H),] 7.58 (dd, 1H), 7.07 (dd, lH), 3.9 (bs, 1H), 3.23 (t, 2H), 2.74 (t, 2H).
87%
With aluminum (III) chloride In dichloromethane at 20 - 35℃; for 19 h; Inert atmosphere
Dihydrofuran-2,5-dione (30 g, 300 mmol) and thiophene (24 ml, 300 mmol) were added to a 250 ml RBF and DCM (120 ml) was added. The mixture was stirred at room temperature under nitrogen until the succinic anhydride was dissolved (colorless solution) following this aluminum trichloride (48 g, 360 mmol) was added to the mixture portion wise over 30 min (the solution became dark red/brown). The reaction mixture was stirred at room temperature under nitrogen for 17 h and then heated to 35°C under nitrogen for 2 h.The reaction mixture was allowed to cool down to room temperature and was poured into a mixture of ice, concentrated HCl (20 ml) was added. The mixture was stirred for 30 minand was then partitioned between DCM (200 ml) and water (100 ml). A solid crashed out in the aqeuous phase and was filtered. The organic phase was washed with water (100 ml), dried over magnesium sulfate, filtered and concentrated in vacuo and combined with the solid obtained previously. The combined material was filtered and dried to afford 4-oxo-4-(thiophen-2-yl)butanoic acid (48g, 262 mmol) 87percent yield as a white solid. 1H NMR (METHANOL-d4) d: 7.88 - 8.00 (m, 1 H), 7.79 - 7.88 (m, 1 H), 7.22 (t, J = 4.3 Hz, 1 H), 3.69 (s, 1 H), 3.29 (t, J = 6.4 Hz, 2 H), 2.69 - 2.76 (m, 2 H) ES- 183.
81%
With aluminum (III) chloride In dichloromethane at 0 - 20℃; for 17 h;
Add 24 ml of thiophene and 30 g of succinic anhydride and 200 ml of dichloromethane in a 500 ml three-necked flask, stir and cool to 0-5 °C, add 36 g of anhydrous aluminum trichloride in batches, stir at room temperature for 17 hours after the addition, and then The reaction solution was poured into 500 ml of crushed ice, stirred for half an hour, and the organic layer was separated. The organic layer was washed with 500 ml of water, then dried over magnesium sulfate and evaporated to dryness. The mixture was dried by blowing at 50-60 °C for 12 hours to obtain a solid 44.7 g, and the yield was 81percent.
77%
at 20℃; for 5 h;
Example 5.; Preparation of dichloror6,6'-bis(dimethylsilanediyl)di(η5-(7-phenyl-5,6-dihvdro-4/-/- indeno[5,4-.pound.>1thiophene-6-yl)1zirconium.; The preparation is described in the following scheme. KOH 6,7-dihydro-1-benzothiophen-4(5/-/)-one was obtained with an overall yield of 43percent via the acylation of thiophene by succinic anhydride followed by reduction of the ketone formed and by the cyclisation of 4-(2-thienyl)butanoic acid in the presence of the laton's reagent. Next, 4,5,5a,6-tetrahydro-7/-/-indeno[5,4-ιb]thiophen-7-one was synthesised from 6,7-dihydro-1 -benzothiophen-4(5H)-one with an overall yield of 30percent. The latter product was further reacted with one equivalent of PhLi in ether followed by acidification of the reaction medium to give a mixture of 7-phenyl-5,5a- dihydro-4H-indeno[5,4-ib]thiophene and 7-phenyl-5,8-dihydro-4H-indeno[5,4- .pound.>]thiophene as shown in the scheme belowThe lithium salt of this ligand was treated with 0.5 eqv of Me2SiCl2 in THF to form the respective ιb/s(cyclopentadienyl)dimethylsilane. This b/s-cyclopentadienyl ligand was isolated as a mixture of the Me2Si-bhdging ligands involving ca. 70percent of the desired isomers with a yield of 17percent by repeating flash chromatography on Silica Gel 60. It was metallated with 2 eqv of "BuLi in toluene-hexanes, and then with metallic salt ZrCI4(THF)2. This mixture was stirred overnight at room temperature and then filtered through glass frit. Crystals precipitated from the filtrate at a temperature of -3O0C were collected and dried in vacuum. On the evidence of 1H NMR spectroscopy, this product isolated with a yield of 13percent is a mixture of me- and meso-complexes in a ratio of 1 to 5. This 1H NMR spectrum is shown in figure 9. The meso-isomer was characterised by X-ray crystal structure analysis. Figure 10 is the ORTEP representation of the molecular structure of this meso-complex with thermal ellipsoids drawn at the 50percent probability level.The key geometric paramerers of this structure are:- bond lengths Zr-Cp(c) and Zr-Cp(c)' are 2.239(1 ) Angstroms;- angle between two cyclopentadienyl planes is 59.2°.
63%
Stage #1: With aluminum (III) chloride In dichloromethane at 0℃; for 2 h; Stage #2: at 0 - 20℃; for 5 h;
General procedure: General procedure 6 (GP6) for Friedel Crafts reaction: Representative experimental procedure for the Friedel Crafts using heterocycles with the anhydride. To solution of succinic anhydride (1 eq.) in anhydrous DCM was added anhydrous A1C13 (1.2 eq) at 0 C and the reaction mixture was stirred for 2 h at 0 °C. Then the aromatic heterocycle was added dropwise over a period of 1 h. The reaction mixture was stirred for 4 h at room temperature. 50 ml of ice-cold H20 was added at 0 °C, followed by acidification with 2N HC1 to pH 2. The aqueous phase was extracted with DCM (3 times). The combined organic extracts were dried over Na2S04, filtered and evaporated in vacuo. The crude mixture was purified by recrystallization or flash column chromatography to afford the compounds 9, 10.
38%
Stage #1: With aluminum (III) chloride In dichloromethane at 0 - 20℃; for 8 h; Stage #2: With hydrogenchloride In dichloromethane; water at 0℃;
EXAMPLE 13; 4-Oxo-4-(thiophen-2-yl)butanoic acidAnhydrous aluminium chloride (15.8 g, 0.12 mol) was added to a solution of succinic anhydride (1 1.9 g, 0.12 mol) in dry CH2CI2 (50 Ml) and the reaction mixture was cooled to 0 °C. A solution of thiophene (10.0 g, 0.12 mol) in CH2CI2 (50 Ml) was then added dropwise maintaining the same temperature. The reaction mixture was allowed to warm up to room temperature and stirred for 8 h. The mixture was then cooled to 0 °C and the Ph was adjusted to ~3 using 6N HCI. The organic product was extracted with CH2CI2. The combined extracts were washed with water and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by column chromatography (silica 60-120 mesh, eluant 5-10percent MeOH in CH2CI2), to get 4- oxo-4-(thiophen-2-yl)butanoic acid (8.3 g, yield 38percent). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1 H), 8.00 - 7.98 (m, 2H), 7.26 - 7.23 (m, 1 H), 3.21 - 3.18 (m, 2H), 2.58 - 2.55 (m, 2H). MS (ESI) m/z: Calculated for C8H803S: 184.02; found: 184.9 (M+H)+
Reference:
[1] Patent: WO2004/26848, 2004, A1, . Location in patent: Page 33
[2] European Journal of Pharmacology, 2014, vol. 727, # 1, p. 1 - 7
[3] Patent: CN108341797, 2018, A, . Location in patent: Paragraph 0008; 0017
[4] Patent: WO2010/76188, 2010, A1, . Location in patent: Page/Page column 17-19
[5] Archiv der Pharmazie, 1988, vol. 321, # 10, p. 735 - 738
[6] Journal of Medicinal Chemistry, 2016, vol. 59, # 5, p. 2222 - 2243
[7] Patent: WO2017/63910, 2017, A1, . Location in patent: Page/Page column 19
[8] European Journal of Medicinal Chemistry, 1998, vol. 33, # 11, p. 867 - 877
[9] Patent: WO2011/88187, 2011, A1, . Location in patent: Page/Page column 60
[10] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2000, vol. 39, # 8, p. 614 - 619
[11] Journal of the Chemical Society, 1954, p. 4162,4165
[12] Journal of the American Chemical Society, 1935, vol. 57, p. 1611,1614
[13] Bulletin des Societes Chimiques Belges, 1956, vol. 65, p. 874,891
[14] Tetrahedron Letters, 1984, vol. 25, # 47, p. 5439 - 5440
[15] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2000, vol. 39, # 5, p. 334 - 338
[16] European Journal of Medicinal Chemistry, 2009, vol. 44, # 8, p. 3147 - 3157
[17] European Journal of Medicinal Chemistry, 2013, vol. 69, p. 490 - 497
[18] Patent: WO2015/95261, 2015, A1, . Location in patent: Page/Page column 100
[19] Patent: WO2015/89842, 2015, A1, . Location in patent: Page/Page column 98-99
[20] Archiv der Pharmazie, 2017, vol. 350, # 10,
[21] Patent: US2423709, 1945, ,
5
[ 108-30-5 ]
[ 4653-08-1 ]
Reference:
[1] Patent: US4299769, 1981, A,
6
[ 96-43-5 ]
[ 108-30-5 ]
[ 80885-91-2 ]
[ 70685-06-2 ]
Reference:
[1] Patent: US4299769, 1981, A,
7
[ 96-43-5 ]
[ 108-30-5 ]
[ 70685-06-2 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1950, vol. 69, p. 1083,1103
8
[ 1003-09-4 ]
[ 108-30-5 ]
[ 52240-28-5 ]
Reference:
[1] Journal of Organic Chemistry, 2001, vol. 66, # 22, p. 7283 - 7286
[2] Organic Syntheses, 2002, vol. 79, p. 204 - 204
[3] Journal of the Chemical Society, 1954, p. 4162,4165
[4] Recueil des Travaux Chimiques des Pays-Bas, 1950, vol. 69, p. 1083,1103
[5] Nippon Kagaku Zasshi, 1957, vol. 78, p. 779,783[6] Chem.Abstr., 1960, p. 22559
9
[ 1003-09-4 ]
[ 108-30-5 ]
[ 80885-88-7 ]
[ 52240-28-5 ]
Reference:
[1] Patent: US4299769, 1981, A,
10
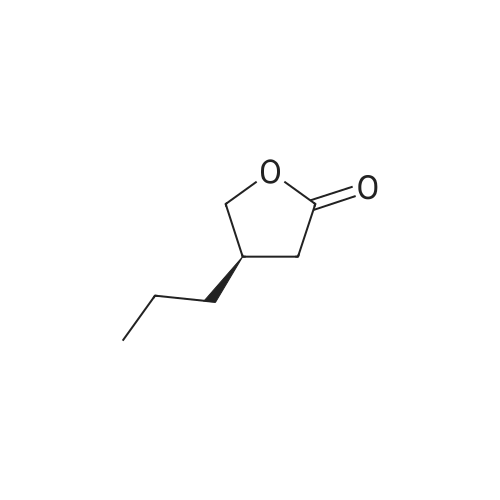
[ 108-30-5 ]
[ 1165952-91-9 ]
[ 6708-37-8 ]
Reference:
[1] Australian Journal of Chemistry, 1981, vol. 34, # 8, p. 1719 - 1727
11
[ 108-30-5 ]
[ 72-14-0 ]
[ 116-43-8 ]
Yield
Reaction Conditions
Operation in experiment
67%
at 20℃; for 6 h;
Compound 27j was synthesized by the method outlined in FIG. 1. Briefly, Compound 27j was synthesized by first complexing succinic anhydride with sulfathiazole by reacting these compounds at room temperature for 6 hours in tetrahydrofuran (THF) in the presence of triethylamine (TEA) to produce succinylsulfathiazole as a white solid (67percent yield). The succinylsulfathiazole was then reacted for 2 hours at room temperature with O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TSTU) in dichloromethane in the presence of TEA to produce the corresponding succinimidyl ester, which was reacted with 1-(4-aminobutyl)-2-{1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl}acetamide in dichloromethane in the presence of TEA at room temperature for 16 hours to produce Compound 27j as a yellow gummy mass (45percent yield). Compound 27j was characterized by NMR, two-dimensional NMR, and mass spectroscopy.
Reference:
[1] Pharmaceutical Chemistry Journal, 1997, vol. 31, # 9, p. 471 - 473
[2] Patent: US2007/292352, 2007, A1, . Location in patent: Page/Page column 45
[3] Patent: US2324013, 1941, ,
[4] Journal of the American Chemical Society, 1942, vol. 64, p. 1572,1573, 1574
[5] Patent: US2324014, 1941, ,
[6] Rum. Med. Rev., 1957, vol. 2, p. 96
[7] Patent: US2391853, 1942, ,
[8] Rum. Med. Rev., 1957, vol. 2, p. 96
[9] Patent: CH242247, 1944, ,
[10] Patent: CH242247, 1944, ,
Stage #1: at -5 - 20℃; for 18 h; Stage #2: With hydrogenchloride In water at 0 - 20℃; for 0.5 h;
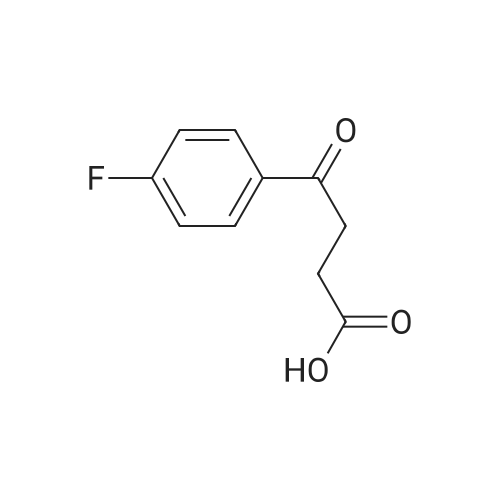
4-(4-FIuorophenyI)-4-oxobutanoic acid 142. To a stirred mixture of succinic anhydride (1.0Og5 10 mmol) in fluorobenzene (5.6 mL, 60 mmol), cooled to -5°C, was added aluminium chloride (2.66 g, 20 mmol). The reaction temperature was maintained at -5°C for Ih before being allowed to warm slowly to r.t. over 17h. The mixture was poured into a stirred solution of aqueous HCl (30 mL of 18percent) at 0°C and stirring was continued for a further 30 min while the mixture was allowed to warm to r.t. The resulting white powder was collected by filtration and washed with water. Yield 1.67g, 85percent: 1H NMR .pound. (400 MHz, CD3OD) 2.70-2.74 (2H, m), 3.32-3.35 (2H5 m), 6.64 (IH, s), 7.20-7.26 (2H5 m), 8.06-8.10 (2H, m); LC/MS (APCI) m/z 195.17 (M-H)'; HPLC tτ = 1.53 min (>99percent) 80percent MeCN in H2O.
83%
With aluminum (III) chloride In dichloromethane for 2 h; Cooling with ice; Reflux
Dichloromethane (50 ml) was added to a 250 ml reaction flask, 10.6 g (0.11 mol) of fluorobenzene and 14.7 g (0.11 mol) of anhydrous aluminum trichloride were added. The reaction mixture was stirred evenly in an ice-water bath, and succinic anhydride was slowly added dropwise 10.1. After 100percent of a solution of g (0.1 mol) in methylene chloride was added dropwise, the temperature was gradually raised to reflux and the reaction was carried out for 2 hours. After the reaction is completed, add 150ml of water, cool to 0°C, add 100ml of concentrated hydrochloric acid, stir at room temperature for 1 hour, stand for stratification, wash the organic layer with saturated aqueous sodium bicarbonate solution and saturated brine 150ml, and concentrate to dryness under reduced pressure. HPLC The purity was 98.0percent, and it was purified by recrystallization from toluene to obtain 16.3 g of 4-(4-fluorophenyl)-4-oxobutanoic acid in a yield of 83percent. The purity by HPLC was 99.8percent, and the content of ortho-substitution by-product was 0.03percent.
81.5%
at 100℃; for 2 h;
In a 500 ml three-necked reaction flask equipped with a mechanical stirrer, a condenser, and a thermometer,17.1 g (0.171 mol) of succinic anhydride and 105 g (1.09 mol) of fluorobenzene were added and dissolved with stirring. 60 g (0.306 mol) of anhydrous aluminum chloride was added in one portion,The mixture was stirred at 100 ° C for 2 hours, and decomposed by adding 165 ml of hydrochloric acid at a concentration of 10percent for 30 minutes.The other product was obtained in the same manner as in Example 1, m.p. 105-107 ° C, the yield of this step was 81.5percent, and the overall yield was 46.7percent
71%
Stage #1: at -9 - 20℃; for 4.5 h; Stage #2: at 0℃;
4-(4-Fluoro-phenyl)-4-oxo-butyric acid. Succinic anhydride (1.0 g, 0.01 mmol) was dissolved in 4-fluorobenzene (3.75 mL; 0.04 mmol) and cooled to -9 CC under nitrogen. AICI3 (2.67 g, 0.02 mmol) was added, and the temperature was kept between -9 and 0 °C for 4.5 hours. The reaction was allowed to warm to room temperature and left over night. The reaction mixture was poured into aqueous 4 M HCI (10 mL) at 0 0C, filtered and washed with water. The solid was recrystallized from toluene to give 1.40 g of a colorless solid (71 percent). LC/MS (an10n8): Rt 0.26 min, m/z 195 [M-H], 413 [2M-2H+Na]; 1H NMR (CDCI3, 300 MHz) δ 2.84 (t, J = 6.5 Hz, 2H), 3.31 (t, J = 6.5 Hz, 2H), 7.16 (m, 3H), 8.03 (m, 2H).
62%
With aluminum (III) chloride In dichloromethane at 20℃; Inert atmosphere
General procedure: AlCl3 (1.0 g, 7.5 mmol)was added portion-wise to a solution of succinic anhydride (0.5 g, 5 mmol) and anaromatic compound (6 mmol) in CH2Cl2 (5 mL) at room temperature. The mixture wasstirred at room temperature for overnight, and the mixture was poured into 1N HClsolution (10 mL), extracted with ethyl acetate (15 mL x 3). The organic layer was driedover MgSO4. The solvent was removed under reduced pressure, and the crude productwas purified by recrystallized from ethyl acetate and hexane.
59.6%
Stage #1: With aluminum (III) chloride In dichloromethane at 20℃; for 2 h; Stage #2: With hydrogenchloride; water In dichloromethane
Fluorobenzene (50 mL, 530 mmol) and aluminum trichloride (156 g, 1.17 mol) were added to 500 mL of methylene chloride, and the reaction mixture was stirred. Succinic anhydride (50 g, 500 mmol) was added to the stirrring reaction mixture all at once, and the reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched by cautious addition of 10percent HCl, and the reaction mixture was added to 500 mL of water. The aqueous mixture was extracted twice with 250 mL of methylene chloride, and the combined organic layers were dried (MgSO4), and evaporated under reduced pressure to give 62 g (316 mmol, 59.6percent) of 4-(4-fluoro-phenyl)-4-oxo-butyτic acid as a crude solid. MS: 197 (M+H)+.
59.6%
Stage #1: With aluminum (III) chloride In dichloromethane at 20℃; for 2 h; Stage #2: With hydrogenchloride; water In dichloromethane
Fluorobenzene (50 mL, 530 mmol) and aluminum trichloride (156 g, 1.17 mol) were added to 500 mL of methylene chloride, and the reaction mixture was stirred. Succinic anhydride (50 g, 500 mmol) was added to the stirrring reaction mixture all at once, and the reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched by cautious addition of 10percent HCl, and the reaction mixture was added to 500 mL of water. The aqueous mixture was extracted twice with 250 mL of methylene chloride, and the combined organic layers were dried (MgSO4), and evaporated under reduced pressure to give 62 g (316 mmol, 59.6percent) of 4-(4-fluoro-phenyl)-4-oxo-butyric acid as a crude solid. MS: 197 (M+H)+.
59.6%
Stage #1: With aluminum (III) chloride In dichloromethane at 20℃; for 2 h; Stage #2: With hydrogenchloride In dichloromethane; water
Step 1: 4-(4-Fluoro-phenyl)-4-oxo-butyric acid Fluor obenzene (50 mL, 530 mmol) and aluminum trichloride (156 g, 1.17 mol) were added to 500 mL of methylene chloride, and the reaction mixture was stirred. Succinic anhydride (50 g, 500 mmol) was added to the stirrring reaction mixture all at once, and the reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched by cautious addition of 10percent HCl, and the reaction mixture was added to 500 mL of water. The aqueous mixture was extracted twice with 250 mL of methylene chloride, and the combined organic layers were dried (MgSO4), and evaporated under reduced pressure to give 62 g (316 mmol, 59.6percent) of 4-(4-fluoro-phenyl)-4-oxo-butyric acid as a crude solid. MS: 197 (M+H)+. Step 2: 4-Oxo-4-(4-phenylsulfanyl-ρhenyl') -butyric acid
59.6%
With aluminum (III) chloride In dichloromethane at 20℃; for 2 h;
Step 1: 4-(4-Fluoro-phenyl)-4-oxo-butyric acid Fluorobenzene (50 mL, 530 mmol) and aluminum trichloride (156 g, 1.17 mol) were added to 500 mL of methylene chloride, and the reaction mixture was stirred. Succinic anhydride (50 g, 500 mmol) was added to the stirring reaction mixture all at once, and the reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched by cautious addition of 10percent HCl, and the reaction mixture was added to 500 mL of water. The aqueous mixture was extracted twice with 250 mL of methylene chloride, and the combined organic layers were dried (MgSO4), and evaporated under reduced pressure to give 62 g (316 mmol, 59.6percent) of 4-(4-fluoro-phenyl)-4-oxo-butyric acid as a crude solid. MS: 197 (M+H)+.
59.6%
With aluminum (III) chloride In dichloromethane at 20℃; for 2 h;
Preparation 27-Benzenesulfonγl-3,4-dihγdro-2H-naphthalen-l-oneThe synthetic procedure described in this Preparation was carried out according to the process shown in Scheme E. <n="45"/>SCHEME E4-(4-Fluoro-phenγl)-4-oxo-butγric acid Fluorobenzene (50 niL, 530 nimol) and aluminum trichloride (156 g, 1.17 mol) were added to 500 mL of methylene chloride, and the reaction mixture was stirred. Succinic anhydride (50 g, 500 mmol) was added to the stirrring reaction mixture all at once, and the reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched by cautious addition of 10percent HCl, and the reaction mixture was added to 500 mL of water. The aqueous mixture was extracted twice with 250 mL of methylene chloride, and the combined organic layers were dried (MgSCU), and evaporated under reduced pressure to give 62 g (316 mmol, 59.6percent) of 4-(4-fluoro-phenyl)-4-oxo-butyric acid as a crude solid. MS: 197 (M+H)+.
Reference:
[1] Helvetica Chimica Acta, 1995, vol. 78, # 3, p. 647 - 662
[2] Patent: WO2007/96647, 2007, A2, . Location in patent: Page/Page column 148-149
[3] Patent: CN107556287, 2018, A, . Location in patent: Paragraph 0036; 0037; 0038; 0039; 0040; 0052; 0053
[4] Patent: CN106187863, 2016, A, . Location in patent: Paragraph 0153; 0154; 0155; 0156; 0157; 0104-0108
[5] Tetrahedron Letters, 2016, vol. 57, # 24, p. 2583 - 2586
[6] Chemistry - A European Journal, 2017, vol. 23, # 40, p. 9501 - 9504
[7] Patent: WO2007/62797, 2007, A1, . Location in patent: Page/Page column 16-17
[8] Asian Journal of Chemistry, 2012, vol. 24, # 8, p. 3553 - 3556
[9] Chemistry Letters, 2015, vol. 44, # 11, p. 1503 - 1505
[10] Organic Process Research and Development, 2004, vol. 8, # 2, p. 291 - 292
[11] Patent: WO2006/66748, 2006, A1, . Location in patent: Page/Page column 26
[12] Patent: WO2006/66790, 2006, A1, . Location in patent: Page/Page column 74; 75
[13] Patent: WO2006/66745, 2006, A1, . Location in patent: Page/Page column 24
[14] Patent: US2007/293526, 2007, A1, . Location in patent: Page/Page column 30
[15] Patent: WO2007/147771, 2007, A2, . Location in patent: Page/Page column 43-44
[16] Chemistry - An Asian Journal, 2017, vol. 12, # 3, p. 355 - 360
[17] Journal of the American Chemical Society, 1948, vol. 70, p. 3177
[18] Bulletin de la Societe Chimique de France, 1965, p. 1308 - 1315
[19] Journal of the American Chemical Society, 1967, vol. 89, p. 386 - 390
[20] Journal of Organic Chemistry, 1961, vol. 26, p. 2667 - 2669
[21] Journal of Medicinal Chemistry, 1984, vol. 27, # 9, p. 1099 - 1101
[22] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 3, p. 1177 - 1180
[23] European Journal of Medicinal Chemistry, 2013, vol. 67, p. 352 - 358
[24] Medicinal Chemistry Research, 2015, vol. 24, # 11, p. 3775 - 3784
[25] Chemical Biology and Drug Design, 2015, vol. 86, # 4, p. 619 - 625
[26] Tetrahedron Letters, 2016, vol. 57, # 30, p. 3334 - 3338
[27] Archiv der Pharmazie, 2017, vol. 350, # 10,
[28] Organic Letters, 2018, vol. 20, # 9, p. 2787 - 2791
14
[ 108-30-5 ]
[ 79-03-8 ]
[ 765-69-5 ]
Reference:
[1] Australian Journal of Chemistry, 1982, vol. 35, # 4, p. 799 - 808
15
[ 108-30-5 ]
[ 60-32-2 ]
[ 55750-53-3 ]
Yield
Reaction Conditions
Operation in experiment
77.4%
at 20℃; for 10 h; Reflux
Compound 1 (150 g, 1.53 mol) was added to a stirred solution of Compound 2 (201 g, 1.53 mol) in HOAc (1000 mL). After the mixture was stirred at r.t. for 2 h, it was heated at reflux for 8 h. The organic solvents were removed under reduced pressure and the residue was extracted with EtOAc (500 mL x 3), washed with H20. The combined organic layers was dried over Na2S04 and concentrated to give the crude product. It was washed with petroleum ether to give compound 3 as white solid (250 g, 77.4 percent).
Reference:
[1] Gazzetta Chimica Italiana, 1895, vol. 25 II, p. 26
17
[ 108-30-5 ]
[ 64-17-5 ]
[ 14794-31-1 ]
Reference:
[1] Tetrahedron, 2010, vol. 66, # 13, p. 2340 - 2350
[2] Journal of Heterocyclic Chemistry, 1987, vol. 24, # 6, p. 1573 - 1579
18
[ 108-30-5 ]
[ 75-65-0 ]
[ 15026-17-2 ]
Yield
Reaction Conditions
Operation in experiment
100%
With dmap; 1-hydroxy-pyrrolidine-2,5-dione In toluene for 48 h; Reflux
The monoester of succinate was prepared as described by Srinivasan,Uttamchandani and Yao [42]: Tert-butanol (10 mL) was added to asolution of succinic anhydrate (6.04 g, 60.40 mmol), Nhydroxysuccinimid (2.53g, 22.01 mmol) and DMAP (0.88 g, 7.23 mmol) in toluene (100 mL)and the solution was heated for 48 h under reflux conditions. After cooling down to rt, twolayers formed in the reaction vessel (brown oil and clear, colourless solution). The crudesolution was diluted with EtOAc (50 mL) and washed with citric acid (10 percent, 2 × 50 mL) andbrine. The organic layer was dried over Na2SO4, the solvent evaporated and the crude productwas recrystallized from Et2O/PE (1:3, 25 mL) to yield the title compound in quantitativeyield.
77%
With dmap; 1-hydroxy-pyrrolidine-2,5-dione; triethylamine In toluene for 24 h; Inert atmosphere; Reflux
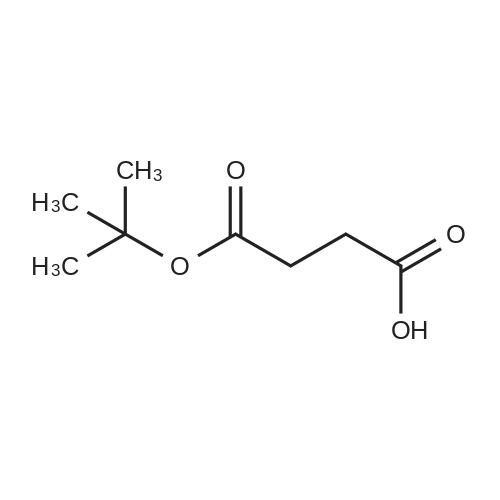
Succinic anhydride 7 (10 g, 100 mmol), N-hydroxysuccinimide (3.34 g, 0.30 mmol), and DMAP (1.17 g, 1.00 mmol) were dissolved in toluene (50 mL). tert-Butanol (11.7 mL, 124 mmol) and Et3N (3.03 g, 4.17 mL, 30.0 mmol) were added sequentially. The suspension was heated under reflux for 24 h. The solution was cooled and diluted with EtOAc (50 mL) and was washed with citric acid (10percent w/v, 100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to give a brown solid. The solid was recrystallised with ether and petroleum ether (10:90) at -20 °C to give succinic acid mono-tert-butyl ester 8 (13.3 g, 76.4 mmol, 77percent) as fawn crystals
77%
With dmap; 1-hydroxy-pyrrolidine-2,5-dione; triethylamine In toluene
Succinic anhydride (10 g, 100 mmol) was reacted with anhydrous t-butyl alcohol (17 mL) in anhydrous toluene (150 mL) in the presence of dimethylaminopyridine (1.8 g, 15 mmol), N-hydroxysuccinamide (3.45 g, 30 mmol), and trimethylamine (4.2 mL, 30 mmol) to yield the ring opened mono-tert-butylsuccinate product (4). The product was purified by ethyl acetate and water extractions followed by recrystallization of the isolated products of the organic phase in 1:3 ether:hexanes (77percent). The pure compound (4) (5.57 g, 31.9 mmol) was then reacted with tert-Butyldimethylchlorosilane (TBDMS) protected diethanolamine (24.6 mmol) in a 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (6.60 g, 34.4 mmol) mediated coupling reaction in anhydrous N,N-dimethylformamide (DMF) (30 mL) overnight. The resulting product (5) was purified by removing the DMF via rotovap, performing ethyl acetate and water extractions, and further purifying the product by performing silica gel liquid column chromatography using ethyl acetate and hexanes as the mobile phase (69.8percent). Deprotection of the TBDMS groups was performed by reacting the purified product from the previous step with iodine in methanol (2percent w/v) at reflux for three hours. After reaction, Na2S2O3 was added until the reaction mixture became clear and was concentrated on rotary evaporator. To purify the t-butyl protected carboxylate diol monomer (5), methylene chloride and water extractions were performed followed by silica gel liquid column chromatography using methylene chloride and methanol as the mobile phase to purify the products extracted into the organic phase. The product was dried under reduced pressure to yield pure monomer (6) (61percent). Synthesis of the monomer was confirmed by 1HNMR.
64%
With dmap; 1-hydroxy-pyrrolidine-2,5-dione; triethylamine In toluene for 24 h; Inert atmosphere; Reflux
Succinic anhydride, 3 (5.0g, 49.96mmol) was suspended in 30mL of toluene under nitrogen atmosphere. N-Hydroxysuccinimide (1.72g, 14.98mmol), 4-(dimethylamino)pyridine (0.610g, 4.9mmol), dry tert-butyl alcohol (15mL, 150mmol), and Et3N (2.10mL, 14.98mmol) were added to the solution. Dissolution occurred upon refluxing it for one day. After cooling, ethyl acetate (20mL) was added, and the organic layer was washed three times with 10percent citric acid and once with brine, and dried over Na2SO4, filtered, and evaporated to afford 5.40g (64percent yield) of brown solid. Recrystallization with ether gave white crystals of 4. The structure was confirmed by 1H NMR. Mp: 49–50°C (lit.22 49–51°C) 1H NMR (500MHz, CDCl3) δ 2.61 (t, J=6.7Hz, 2H), 2.56–2.49 (m, 2H), 1.43 (s, 9H); 13C NMR (125MHz, CDCl3) 178.7, 171.6, 81.2, 30.2, 29.3, 28.2.
Reference:
[1] Beilstein Journal of Organic Chemistry, 2017, vol. 13, p. 2428 - 2441
[2] Journal of Medicinal Chemistry, 2012, vol. 55, # 23, p. 10460 - 10474
[3] Journal of Organic Chemistry, 2003, vol. 68, # 17, p. 6679 - 6684
[4] Organic Letters, 2006, vol. 8, # 4, p. 713 - 716
[5] Angewandte Chemie - International Edition, 2017, vol. 56, # 6, p. 1643 - 1647[6] Angew. Chem., 2017, vol. 129, # 6, p. 1665 - 1669,5
[7] Synthesis, 2000, # 10, p. 1369 - 1371
[8] Tetrahedron, 2014, vol. 70, # 44, p. 8343 - 8347
[9] Patent: US2017/172147, 2017, A1, . Location in patent: Paragraph 0081
[10] Journal of Organic Chemistry, 1994, vol. 59, # 17, p. 4862 - 4867
[11] Helvetica Chimica Acta, 2004, vol. 87, # 6, p. 1545 - 1560
[12] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 20, p. 5073 - 5077
[13] Biomacromolecules, 2017, vol. 18, # 1, p. 113 - 126
[14] Tetrahedron Letters, 1984, vol. 25, # 41, p. 4623 - 4626
[15] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 18, p. 4004 - 4009
[16] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 7, p. 3501 - 3518
[17] Patent: WO2010/124046, 2010, A1, . Location in patent: Page/Page column 28
[18] ChemMedChem, 2010, vol. 5, # 4, p. 567 - 574
19
[ 108-30-5 ]
[ 57-14-7 ]
[ 1596-84-5 ]
Reference:
[1] Journal of Organic Chemistry, 1973, vol. 38, p. 2058 - 2061
20
[ 108-30-5 ]
[ 100-51-6 ]
[ 2304-94-1 ]
Reference:
[1] Angewandte Chemie, 1977, vol. 89, p. 570 - 572
[2] Synthesis, 1976, p. 329 - 330
21
[ 108-30-5 ]
[ 578-58-5 ]
[ 61495-09-8 ]
[ 1685-84-3 ]
Reference:
[1] Patent: US3978116, 1976, A,
22
[ 108-30-5 ]
[ 108-90-7 ]
[ 26673-32-5 ]
Reference:
[1] Journal of the Chemical Society, 1952, p. 279
23
[ 108-30-5 ]
[ 108-86-1 ]
[ 35656-89-4 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2005, vol. 3, # 14, p. 2503 - 2504
24
[ 108-30-5 ]
[ 108-88-3 ]
[ 4619-20-9 ]
Yield
Reaction Conditions
Operation in experiment
99.3%
at 50 - 60℃; for 2 h;
Dissolve 75.7 g of succinic anhydride in 457.5 ml of toluene.220.5 g of anhydrous AlCl3 are added batchwise. The reaction mixture is controlled at a temperature of 50-60°C.After stirring for 2 hours,The end of the reaction was confirmed by TLC. With vigorous stirring,The reaction solution was slowly added dropwise to 1250 ml of 1 mol/L ice hydrochloric acid.Gradually precipitated milky solids,After the addition was complete, the mixture was stirred for 0.5 hours and filtered.After the filter cake was washed with toluene and ice water,Withdrawing the filter cake with 50 ml of ethyl acetate;Slowly add the solution and add 500ml of petroleum ether to crystallize.After stirring for another 0.5 hours,Filter and dry at 40°C for 2 hours to obtain Compound II144g.Yield 99.3percent.[Compound II intermediate] white solid,
95%
With aluminum (III) chloride In dichloromethane at 20℃;
General procedure: The general procedure (Scheme 1) for the preparation of compounds (2a, 2b and 2c): succinicanhydride (1 equiv, 10 mmol) was reacted with an appropriate aromatic compound (substitutedbenzene, 1 equiv, 10 mmol) in DCM (20 mL) in the presence of anhydrous aluminium chloride(1.5 equiv, 15 mmol). The reaction mixture was stirred under anhydrous conditions overnight at roomtemperature and then ice-cold diluted hydrochloric acid solution was added dropwise. A solid massseparated out which was filtered and purified by recrystallization to give 2a, 2b and 2c [44].
80%
With aluminum (III) chloride In dichloromethane at 20℃; Inert atmosphere
General procedure: AlCl3 (1.0 g, 7.5 mmol)was added portion-wise to a solution of succinic anhydride (0.5 g, 5 mmol) and anaromatic compound (6 mmol) in CH2Cl2 (5 mL) at room temperature. The mixture wasstirred at room temperature for overnight, and the mixture was poured into 1N HClsolution (10 mL), extracted with ethyl acetate (15 mL x 3). The organic layer was driedover MgSO4. The solvent was removed under reduced pressure, and the crude productwas purified by recrystallized from ethyl acetate and hexane.
69%
at 0 - 20℃; for 3 h;
To a suspension of succinic anhydride (1 g, 10.2 mmol, 1 eq)in toluene (4.6 mL, 44.8 mmol, 4.4 eq) kept at 0 C, aluminumchloride (2.7 g, 20.4 mmol, 2 eq) was added in small portions.The resulting suspension was stirred at room temperature for3 h, then poured onto ice and acidified with HCl conc. The aqueoussolution was extracted with EtOAc and the combined organiclayers were dried (Na2SO4) and concentrated under reducedpressure. The residue was triturated with petroleum ether to furnish4 as a white solid (1.35 g, 69percent). Rf = 0.36 (petroleum ether/EtOAc 4:6), mp 125–128 C; 1H NMR (400 MHz, Chloroform-d) d7.88 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 3.29 (t, J = 6.6 Hz,2H), 3.00 (br s, 1H, OH, exch. with D2O), 2.80 (t, J = 6.0 Hz, 2H),2.41 (s, 3H).
Reference:
[1] Organic Letters, 2016, vol. 18, # 3, p. 504 - 507
[2] Patent: CN107628947, 2018, A, . Location in patent: Paragraph 0053; 0054; 0055; 0056; 0057
[3] Russian Journal of General Chemistry, 2014, vol. 84, # 9, p. 1825 - 1829[4] Zhurnal Obshchei Khimii, 2014, vol. 84, # 9, p. 1825 - 1829,5
[5] Molecules, 2018, vol. 23, # 3,
[6] Tetrahedron Letters, 1994, vol. 35, # 19, p. 3017 - 3020
[7] Chemical and pharmaceutical bulletin, 2002, vol. 50, # 10, p. 1407 - 1412
[8] Synthetic Communications, 2003, vol. 33, # 18, p. 3109 - 3113
[9] Journal of Chemical Research - Part S, 2003, # 10, p. 650 - 651
[10] Organic Syntheses, 2002, vol. 79, p. 204 - 204
[11] Synthetic Communications, 2001, vol. 31, # 10, p. 1467 - 1475
[12] Chemistry Letters, 2015, vol. 44, # 11, p. 1503 - 1505
[13] Journal of Organic Chemistry, 1990, vol. 55, # 11, p. 3546 - 3552
[14] Journal of the Chinese Chemical Society, 2016, vol. 63, # 9, p. 739 - 750
[15] Journal of Organic Chemistry USSR (English Translation), 1984, p. 1604 - 1608[16] Zhurnal Organicheskoi Khimii, 1984, vol. 20, # 8, p. 1760 - 1764
[17] Bioorganic and Medicinal Chemistry, 2018, vol. 26, # 1, p. 295 - 307
[18] Journal of the Chilean Chemical Society, 2010, vol. 55, # 1, p. 74 - 77
[19] Organic Process Research and Development, 2004, vol. 8, # 2, p. 291 - 292
[20] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2000, vol. 39, # 8, p. 614 - 619
[21] Organic Letters, 2014, vol. 16, # 6, p. 1740 - 1743
[22] Chemistry - An Asian Journal, 2017, vol. 12, # 3, p. 355 - 360
[23] Australian Journal of Chemistry, 1990, vol. 43, p. 1547 - 1557
[24] Tetrahedron, 1985, vol. 41, # 8, p. 1509 - 1516
[25] Medicinal Chemistry Research, 2014, vol. 23, # 1, p. 146 - 157
[26] Journal of the Indian Chemical Society, 1948, vol. 25, p. 323,327
[27] Journal of the Chinese Chemical Society (Peking), 1943, vol. 10, p. 116[28] Chem.Abstr., 1944, p. 2953
[29] Journal of the Chemical Society, 1933, p. 434,436
[30] Annales de Chimie (Cachan, France), 1882, vol. <5> 26, p. 435
[31] Chemische Berichte, 1895, vol. 28, p. 3215
[32] Bulletin de la Societe Chimique de France, 1888, vol. <2> 49, p. 448
[33] Chemische Berichte, 1901, vol. 34, p. 3828
[34] Journal of the American Chemical Society, 1967, vol. 89, # 14, p. 3487 - 3494
[35] Bulletin of the Chemical Society of Japan, 1979, vol. 52, p. 3033 - 3042
[36] Journal of Organic Chemistry, 1962, vol. 27, p. 857 - 863
[37] Indian Journal of Chemistry - Section A Inorganic, Physical, Theoretical and Analytical Chemistry, 2005, vol. 44, # 4, p. 715 - 718
[38] Journal of Organic Chemistry, 1973, vol. 38, # 7, p. 1388 - 1395
[39] European Journal of Medicinal Chemistry, 1996, vol. 31, # 9, p. 683 - 692
[40] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2002, vol. 41, # 10, p. 2160 - 2171
[41] Journal of Solution Chemistry, 2007, vol. 36, # 3, p. 345 - 356
[42] Chemistry of Heterocyclic Compounds, 2007, vol. 43, # 3, p. 292 - 297
[43] Acta Poloniae Pharmaceutica - Drug Research, 2008, vol. 65, # 2, p. 223 - 228
[44] Acta Poloniae Pharmaceutica - Drug Research, 2008, vol. 65, # 4, p. 441 - 447
[45] Acta Poloniae Pharmaceutica - Drug Research, 2008, vol. 65, # 3, p. 353 - 362
[46] European Journal of Medicinal Chemistry, 2009, vol. 44, # 6, p. 2372 - 2378
[47] European Journal of Medicinal Chemistry, 2010, vol. 45, # 6, p. 2283 - 2290
[48] Arkivoc, 2010, vol. 2010, # 9, p. 40 - 53
[49] Journal of Molecular Structure, 2010, vol. 980, # 1-3, p. 59 - 65
[50] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 3, p. 1023 - 1026
[51] Acta Poloniae Pharmaceutica - Drug Research, 2010, vol. 67, # 5, p. 555 - 562
[52] Journal of Heterocyclic Chemistry, 2011, vol. 48, # 4, p. 864 - 867
[53] Acta Poloniae Pharmaceutica - Drug Research, 2011, vol. 68, # 2, p. 231 - 236
[54] Journal of the Chilean Chemical Society, 2011, vol. 56, # 3, p. 778 - 780
[55] Journal of Enzyme Inhibition and Medicinal Chemistry, 2011, vol. 26, # 5, p. 742 - 748
[56] Journal of the Chilean Chemical Society, 2011, vol. 56, # 4, p. 856 - 859
[57] Journal of the Serbian Chemical Society, 2011, vol. 76, # 12, p. 1617 - 1626
[58] European Journal of Medicinal Chemistry, 2013, vol. 67, p. 352 - 358
[59] Organic Process Research and Development, 2003, vol. 7, # 6, p. 1079 - 1082
[60] Organic Letters, 2014, vol. 16, # 7, p. 2026 - 2029
[61] Journal of Enzyme Inhibition and Medicinal Chemistry, 2014, vol. 28, # 3, p. 552 - 559
[62] Letters in Drug Design and Discovery, 2015, vol. 12, # 6, p. 500 - 504
[63] Medicinal Chemistry Research, 2015, vol. 24, # 11, p. 3775 - 3784
[64] Tetrahedron Letters, 2016, vol. 57, # 30, p. 3334 - 3338
[65] Patent: CN106349104, 2017, A, . Location in patent: Paragraph 0066
[66] Archiv der Pharmazie, 2017, vol. 350, # 10,
[67] Organic Letters, 2018, vol. 20, # 9, p. 2787 - 2791
[68] Advanced Synthesis and Catalysis, 2018, vol. 360, # 15, p. 2894 - 2899
25
[ 108-30-5 ]
[ 4294-57-9 ]
[ 4619-20-9 ]
[ 76609-13-7 ]
[ 13145-56-7 ]
Reference:
[1] Journal of the Indian Chemical Society, 1994, vol. 71, # 6-8, p. 469 - 474
26
[ 108-30-5 ]
[ 4619-20-9 ]
Reference:
[1] Chemische Berichte, 1901, vol. 34, p. 3828
[2] Chemische Berichte, 1914, vol. 47, p. 1113
27
[ 108-30-5 ]
[ 7446-70-0 ]
[ 108-88-3 ]
[ 4619-20-9 ]
Reference:
[1] Chemische Berichte, 1901, vol. 34, p. 3828
[2] Chemische Berichte, 1895, vol. 28, p. 3215
[3] Bulletin de la Societe Chimique de France, 1888, vol. <2> 49, p. 448
28
[ 108-30-5 ]
[ 108-86-1 ]
[ 6340-79-0 ]
Yield
Reaction Conditions
Operation in experiment
93.4%
Stage #1: at 0 - 20℃; for 100 h; Inert atmosphere Stage #2: at 0℃; for 1 h; Inert atmosphere
Succinic anhydride 3 (5.0 g, 50 mmol) and bromobenzene 2 (48 g, 300 mmol) were cooled to 0°C. Aluminium chloride (13.3 g, 100 mmol) was added and the mixture was stirred for 4 h at 0°C under nitrogen atmosphere. The reaction was allowed to warm to room temperature and stirred for 96 h under nitrogen atmosphere. The reaction was cooled to 0°C and concentrated HCI (125 mL) was added and reaction stirred under nitrogen for a further 1 h. The reaction was filtered and washed with water (1 L) to obtain a pale yellow solid which was recrystallised from toluene to yield 4-(4- bromophenyl)-4-oxobutanoic acid 4 (12 g, 46.68 mmol, 93.4percent). MS (ESI-QUADRUPOLE) m/z: calc. for Ci0H9BrO3: 255.97, 257.97; Found : 258.0 (1 1 ), 257.0 (98), 256.0 (12), 255.0 (100), 213.0 (18), 21 1 .0 (17) (negative ions). HPLC: tr = 24.4 min 1 H NMR (500 MHz, ofe-DMSO) δ: 2.59 (2 H, t, J = 6.5 Hz, CH2), 3.21 (2 H, t, J = 6.5 Hz, CH2), 7.88 (2 H, d, J = 8.8 Hz, ArH), 7.96 (2 H, d, J = 8.8 Hz, ArH), 12.19 (1 H, br s, OH).
82%
With hydrogenchloride; aluminum (III) chloride In dichloromethane at 0 - 20℃; for 4 h;
Step 1: Synthesis of 4-(4-bromophenyl)-4-oxobutanoic acid (37A, R═Br) (0320) Anhydrous aluminum trichloride (29.1 g, 218 mmol) was suspended in dichloromethane (120 mL) and cooled to 0° C. Bromobenzene (35.1 g, 224 mmol) was added carefully. When the addition was complete, succinic anhydride (10.0 g, 100 mmol) was added in ten portions carefully. Then the mixture was warmed to room temperature and stirred for 4 h. TLC showed the reaction was complete, 6N HCl (50 mL) was added dropwise. The solid was filtered, washed with distilled water (10 mL×2) and dried in vacuo to afford 37A, R═Br as a white solid (22 g, yield 82percent).
60%
With aluminum (III) chloride In dichloromethane at 20℃; Inert atmosphere
General procedure: AlCl3 (1.0 g, 7.5 mmol)was added portion-wise to a solution of succinic anhydride (0.5 g, 5 mmol) and anaromatic compound (6 mmol) in CH2Cl2 (5 mL) at room temperature. The mixture wasstirred at room temperature for overnight, and the mixture was poured into 1N HClsolution (10 mL), extracted with ethyl acetate (15 mL x 3). The organic layer was driedover MgSO4. The solvent was removed under reduced pressure, and the crude productwas purified by recrystallized from ethyl acetate and hexane.
55%
With aluminum (III) chloride In cyclohexane at 80℃; for 2 h;
Step A: Bromobenzene (150 ml, 1.42 mol) was added in one portion to a mixture of aluminum chloride (109 g, 0.82 mmol) and succinic anhydride (41 g, 0.41 mol) in cyclohexane (200 ml) at 80° C. and the resulting mixture was stirred at 80° C. for 2 hours. After cooling below 40° C., the mixture was slowly poured into 6 M HCl (200 ml) and ice, and yellow solids precipitated. Methyl tert-butyl ether was added to dissolve the solids and the phases were separated. The organic layer was washed with water three times and was extracted into 2 M NaOH. The aqueous extracts were acidified to pH 1 with 6 M HCl, extracted with methyl tert-butyl ether, dried with magnesium sulfate and concentrated to a residue. This material was recrystallized by dissolving the residue in 50:35:15 cyclohexane/toluene/isopropanol at 70° C. followed by cooling to room temperature, filtering and azeotroping the resulting solids with hexanes to give the desired compound (57 g, 55percent) as a white solid: 1H NMR (300 MHz, DMSO-d6) δ 7.89-7.93 (m, 2H), 7.72-7.77 (m, 2H), 3.23 (t, J=6.0 Hz, 2H), 2.58 (t, J=6.6 Hz, 2H).
Reference:
[1] Molecular Crystals and Liquid Crystals Science and Technology Section A: Molecular Crystals and Liquid Crystals, 2001, vol. 365, p. 181 - 188
[2] Journal of Organic Chemistry, 2001, vol. 66, # 22, p. 7283 - 7286
[3] Organic Syntheses, 2002, vol. 79, p. 204 - 204
[4] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 13, p. 4345 - 4359
[5] Organic Process Research and Development, 2014, vol. 18, # 1, p. 215 - 227
[6] Patent: WO2016/119017, 2016, A1, . Location in patent: Page/Page column 22; 30-31
[7] Canadian Journal of Chemistry, 2006, vol. 84, # 10, p. 1487 - 1503
[8] Journal of the American Chemical Society, 2001, vol. 123, # 17, p. 3940 - 3952
[9] Journal of Materials Chemistry, 2001, vol. 11, # 12, p. 3068 - 3077
[10] Patent: US9138427, 2015, B2, . Location in patent: Page/Page column 268
[11] Organic Letters, 2016, vol. 18, # 3, p. 504 - 507
[12] Synthetic Communications, 2003, vol. 33, # 18, p. 3109 - 3113
[13] Journal of Medicinal Chemistry, 2008, vol. 51, # 24, p. 8088 - 8095
[14] Journal of Chemical Research - Part S, 2003, # 10, p. 650 - 651
[15] Egyptian Journal of Chemistry, 1998, vol. 41, # 1-6, p. 339 - 349
[16] Asian Journal of Chemistry, 2012, vol. 24, # 8, p. 3553 - 3556
[17] Chemistry - A European Journal, 2017, vol. 23, # 40, p. 9501 - 9504
[18] Organic Letters, 2014, vol. 16, # 6, p. 1740 - 1743
[19] Acta Poloniae Pharmaceutica - Drug Research, 2009, vol. 66, # 5, p. 513 - 521
[20] Chemistry Letters, 2015, vol. 44, # 11, p. 1503 - 1505
[21] Patent: US2006/52378, 2006, A1, . Location in patent: Page/Page column 112-113
[22] Tetrahedron, 1985, vol. 41, # 8, p. 1509 - 1516
[23] Organic letters, 1999, vol. 1, # 13, p. 2117 - 2120
[24] Chemistry - An Asian Journal, 2017, vol. 12, # 3, p. 355 - 360
[25] Annales de Chimie (Cachan, France), 1938, vol. <11> 9, p. 447,491[26] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1939, vol. 208, p. 1228
[27] Journal of the American Chemical Society, 1938, vol. 60, p. 170,174
[28] Bulletin of the Chemical Society of Japan, 1979, vol. 52, p. 3033 - 3042
[29] Bulletin de la Societe Chimique de France, 1971, p. 2092 - 2094
[30] Journal of Organic Chemistry, 1962, vol. 27, p. 76 - 78
[31] Journal of Organic Chemistry, 1964, vol. 29, # 8, p. 2109 - 2116
[32] Journal of Organic Chemistry, 1990, vol. 55, # 11, p. 3546 - 3552
[33] Indian Journal of Chemistry - Section A Inorganic, Physical, Theoretical and Analytical Chemistry, 2005, vol. 44, # 4, p. 715 - 718
[34] Journal of Solution Chemistry, 2007, vol. 36, # 3, p. 345 - 356
[35] Patent: WO2007/96647, 2007, A2, . Location in patent: Page/Page column 101
[36] Patent: US2004/9998, 2004, A1, . Location in patent: Page/Page column 35
[37] Acta Poloniae Pharmaceutica - Drug Research, 2008, vol. 65, # 2, p. 223 - 228
[38] Acta Poloniae Pharmaceutica - Drug Research, 2008, vol. 65, # 3, p. 353 - 362
[39] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 3, p. 1177 - 1180
[40] Acta Poloniae Pharmaceutica - Drug Research, 2010, vol. 67, # 5, p. 555 - 562
[41] Acta Poloniae Pharmaceutica - Drug Research, 2011, vol. 68, # 2, p. 231 - 236
[42] Journal of the Serbian Chemical Society, 2011, vol. 76, # 12, p. 1617 - 1626
[43] Journal of Enzyme Inhibition and Medicinal Chemistry, 2012, vol. 27, # 1, p. 92 - 96
[44] European Journal of Medicinal Chemistry, 2013, vol. 67, p. 352 - 358
[45] Journal of Solution Chemistry, 2013, vol. 42, # 1, p. 239 - 250
[46] Medicinal Chemistry Research, 2015, vol. 24, # 11, p. 3775 - 3784
[47] Tetrahedron Letters, 2016, vol. 57, # 30, p. 3334 - 3338
[48] ACS Medicinal Chemistry Letters, 2017, vol. 8, # 4, p. 395 - 400
[49] Archiv der Pharmazie, 2017, vol. 350, # 10,
[50] Advanced Synthesis and Catalysis, 2018, vol. 360, # 15, p. 2894 - 2899
29
[ 108-30-5 ]
[ 108-86-1 ]
[ 6340-79-0 ]
[ 2051-95-8 ]
Reference:
[1] Journal of Organic Chemistry, 1992, vol. 57, # 1, p. 366 - 370
With aluminum (III) chloride In dichloromethane at 20℃; Inert atmosphere
General procedure: AlCl3 (1.0 g, 7.5 mmol)was added portion-wise to a solution of succinic anhydride (0.5 g, 5 mmol) and anaromatic compound (6 mmol) in CH2Cl2 (5 mL) at room temperature. The mixture wasstirred at room temperature for overnight, and the mixture was poured into 1N HClsolution (10 mL), extracted with ethyl acetate (15 mL x 3). The organic layer was driedover MgSO4. The solvent was removed under reduced pressure, and the crude productwas purified by recrystallized from ethyl acetate and hexane.
Reference:
[1] Organic Letters, 2016, vol. 18, # 3, p. 504 - 507
[2] Organic Preparations and Procedures International, 1995, vol. 27, # 5, p. 550 - 552
[3] Chemistry Letters, 2015, vol. 44, # 11, p. 1503 - 1505
[4] Organic Process Research and Development, 2004, vol. 8, # 2, p. 291 - 292
[5] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2000, vol. 39, # 8, p. 614 - 619
[6] Journal of the Chinese Chemical Society, 2016, vol. 63, # 9, p. 739 - 750
[7] Pharmazie, 2002, vol. 57, # 7, p. 448 - 452
[8] Journal of the American Chemical Society, 2002, vol. 124, # 39, p. 11827 - 11832
[9] European Journal of Medicinal Chemistry, 2009, vol. 44, # 9, p. 3798 - 3804
[10] Acta Poloniae Pharmaceutica - Drug Research, 2009, vol. 66, # 5, p. 513 - 521
[11] Journal of Coordination Chemistry, 2012, vol. 65, # 21, p. 3860 - 3868,9
[12] Synthetic Communications, 2001, vol. 31, # 10, p. 1467 - 1475
[13] European Journal of Medicinal Chemistry, 1987, vol. 22, # 1, p. 45 - 57
[14] Collection of Czechoslovak Chemical Communications, 1986, vol. 51, # 11, p. 2617 - 2625
[15] Journal of Medicinal Chemistry, 2012, vol. 55, # 2, p. 824 - 836
[16] Chemistry and Industry (London, United Kingdom), 1940, p. 402
[17] Journal of the Chemical Society, 1940, p. 1030
[18] Patent: US2130989, 1935, ,
[19] DRP/DRBP Org.Chem.,
[20] Justus Liebigs Annalen der Chemie, 1955, vol. 596, p. 1,223
[21] Tetrahedron, 1964, vol. 20, p. 195 - 209
[22] Journal of Pharmaceutical Sciences, 1977, vol. 66, # 4, p. 466 - 476
[23] Journal of Solution Chemistry, 2007, vol. 36, # 3, p. 345 - 356
[24] Acta Poloniae Pharmaceutica - Drug Research, 2008, vol. 65, # 3, p. 353 - 362
[25] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 3, p. 1177 - 1180
[26] Journal of Medicinal Chemistry, 2008, vol. 51, # 24, p. 8088 - 8095
[27] European Journal of Medicinal Chemistry, 2010, vol. 45, # 6, p. 2283 - 2290
[28] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 3, p. 1023 - 1026
[29] Acta Poloniae Pharmaceutica - Drug Research, 2010, vol. 67, # 5, p. 555 - 562
[30] Journal of Enzyme Inhibition and Medicinal Chemistry, 2012, vol. 27, # 1, p. 92 - 96
[31] Letters in Drug Design and Discovery, 2013, vol. 10, # 7, p. 651 - 660
[32] European Journal of Medicinal Chemistry, 2013, vol. 67, p. 352 - 358
[33] Journal of Solution Chemistry, 2013, vol. 42, # 1, p. 239 - 250
[34] Journal of Enzyme Inhibition and Medicinal Chemistry, 2014, vol. 28, # 3, p. 552 - 559
[35] Archiv der Pharmazie, 2017, vol. 350, # 10,
[36] European Journal of Medicinal Chemistry, 2018, vol. 143, p. 1474 - 1488
[37] Patent: US2130989, 1935, ,
[38] DRP/DRBP Org.Chem.,
With dmap; triethylamine In dichloromethane at 20℃;
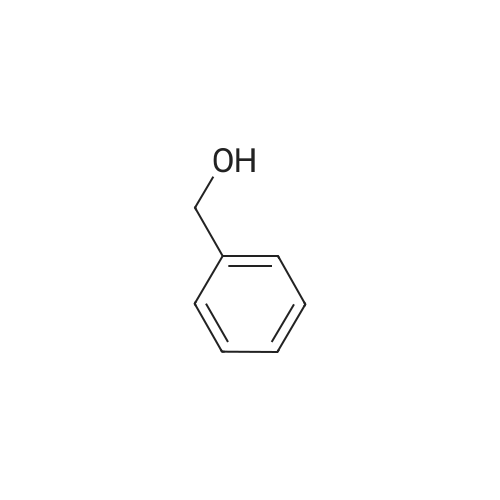
a) Succinic acid monobenzyl ester To the ice-cooled solution of benzyl alcohol (12.96 g, 120 mmol) in methylene chloride (300 mL) was added triethylamine (25 mL, 180 mmol), 4-dimethylaminopyridine (610 mg, 5 mmol) followed by dropwise addition of succinic anhydride (10 g, 100 mmol). The reaction mixture was stirred at room temperature overnight and washed with 1N HCl (2*100 mL) and brine (100 mL). The organic layer was dried (Na2SO4) and concentrated. The residue was dissolved in ethyl acetate (150 mL) and extracted with saturated aqueous NaHCO3 (3*150 mL). The combined aqueous layers were acidified with concentrated HCl to pH=1-2, and extracted with methylene chloride (4*250 mL). The combined organic layers were dried (Na2SO4) and concentrated to give the title acid (19.5 g, 93.8percent). 1H NMR (400 MHz, CDCl3) δ: 11.80-10.50 (br, 1H), 7.35 (m, 5H), 5.15 (s, 2H), 2.70 (m, 4H).
89%
With N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 20℃;
General procedure: To a solution of succinic anhydride (1.0g, 10mmol) in DMF (4mL) was added benzyl alcohol (0.94mL, 9.09mmol) and DIEA (1.93mL, 11mmol) at 0°C. The reaction mixture was stirred at room temperature overnight and was evaporated in Speed-vac. The residue was dissolved in ethyl acetate (50mL) and washed with saturated NaCl (10mL×2). The organic solution was extracted with aqueous NaHCO3 (5M, 5mL×3) and the aqueous extractions were combined, acidified to pH 4 by adding citric acid (5M), extracted with ethyl acetate (30mL×3). The EtOAc extractions were combined, washed with saturated NaCl, and dried over Na2SO4. Solvent was removed by evaporation in vacuo to give a white solid (1.66g, 89percent); 1H NMR (200MHz, CDCl3) δ 7.38 (s, 5H), 5.18 (s, 2H), 2.73–2.71 (m, 4H); LC–MS (ESI−) 207.0 (100percent).
88%
With dmap In tetrahydrofuran for 18 h; Reflux
A solution of succinic anhydride (1.0 g, 10 mmol), benzyl alcohol (1.14 g,11 mmol), and DMAP (54 mg, 0.44 mmol) in THF (5 mL) was refluxed for 18 h. The mixture was bacified with sat. NaHCO3, and washed with AcOEt. The aqueous layer was then acidified, and the precipiated solid was extracted with AcOEt. The solid was recrystalized to afford succinic acid monobenzyl ester as colorless needles, 1.85 g, 88percent. 1H NMR (400 MHz, CDCl3) d. 2.65–2.73 (m, 4H,CH2CH2), 5.15 (s, 2H, PhCH2), and 7.31–7.38 ppm(s, 5H, aromatic).
85%
With dmap; triethylamine In dichloromethane at 20℃; for 18 h;
Method B: Succinic anhydride (5 g, 50.0 mmol) was dissolved in anhydrous DCM (40 mL). Benzyl alcohol (5.69 mL, 55.0 mmol), triethylamine (7.50 mL, 55.0 mmol), and a catalytic amount of DMAP were added to this solution. The resulting clear solution was stirred at room temperature for 18h, after which the, all the volatiles were removed under vacuum. The crude residue was taken up in diethyl ether (200 mL) and was extracted with 2N NaOH (2 χ 75 mL). The aqueous extracts were carefully acidified to pH 2 with concentrated HC1 and then extracted with diethyl ether (2 χ 100 mL). The organic layer was dried (Na2S04), filtered and concentrated to give the title compound as a white solid (8.84 g, 85 percent). M.p. 52-54 °C, lit. 56-57 °C; 1H NMR (300 MHz, acetone-tfc): δ 2.68-2.71 (m, 4H, 2 χ CH2), 5.14 (s, 2H, CHiAr), 7.34-7.36 (m, 5H, ArH).
77%
With pyridine; dmap In dichloromethane
3-Hydroxyflavone-3-hemisuccinate (15) was produced according to the reaction outlined in Figure 3. Reaction of succinic anhydride (11) and benzyl alcohol (12) in the presence of 4-dimethylaminopyridine (DMAP) and pyridine in dichloromethane produced the succinic acid monobenzyl esteT (13) as white crystalline flakes in 77percent yield. This protected succinic acid derivative was coupled to 3-hydroxyflavone (1) in the presence of DCC and DMAP, forming flavone-3-hemisuccinate monobenzyi ester(14) as yellow or brown oil that solidified upon standing, with yields of up to 96percent produced.The deprotection of the monobenzyl ester to form the corresponding hemisuccinate using a Pd(OAc)2 in the THF:EtOH;acetic acid solvent system, a larger scale reaction was undertaken to yield the requiτed 3-hydrosyflavone-3-hernisuccinate (15).
71%
With dmap In dichloromethane at 0 - 25℃; for 96 h;
Example 14; 1-(((4-(benzyloxy)-4-oxobutanoyl)oxy)methyl)-1-methyl-4-(2-methyl-10H-benzo[b]thieno[2,3-e][1,4]diazepin-4-yl)piperazin-1-ium iodide (Compound 14)Synthesis of 4-(benzyloxy)-4-oxobutanoic acidSuccinic anhydride (7 g, 70.0 mmol) and benzyl alcohol (8.7 mL, 83.9 mmol) were combined in dichloromethane (350 mL) at 0° C. and DMAP (0.85 g, 7.0 mmol) was added portion-wise. The reaction was allowed to gradually warm to 25° C. and stirred for 4 days. The reaction mixture was washed with 1M HCl (3.x.200 mL) then water (300 mL). The organic phases were then extracted with aq saturated NaHCO3 (3.x.300 mL). This was then acidified with conc HCl until pH 1 resulting in a solid precipitating which was filtered then dissolved in dichloromethane. The dichloromethane was dried (MgSO4) and concentrated in vacuo to give 4-(benzyloxy)-4-oxobutanoic acid (10.36 g, 71percent).1H-NMR (300 MHz, CDCl3) δ 7.41-7.29 (5H, m), 5.15 (2H, s), 2.74-2.63 (4H, m).
70.47%
at 100℃; for 2 h;
A mixture of 5.0 g (0.05 mol) of succinic acid anhydride, 5.4 g (0.05 mol) of benzyl alcohol, 16.25 g (0.05 mol) of cesium carbonate, and 50 ml of N,N-dimethylforamide was stirred at 100° C. for 2 hours. The reaction mixture was cooled to room temperature, and then poured into 200 ml of ethyl acetate. The mixture was washed with saturated aqueous NaCl containing 0.35 N HCl (3*100 mL). The ethyl acetate phase, which contains the product, was collected and dried over anhydrous MgSO4. The MgSO4 was removed via filtration, and the ethyl acetate was removed under reduced pressure. The crude product was purified by crystallization in diethyl ether and hexane to provide 7.34 g of succinic acid monobenzyl ester, 70.47percent.
63.29%
With dmap In tetrahydrofuran at 50℃; for 5 h;
Succinic anhydride 1 (5.00 g, 49.96 mmol), benzyl alcohol (5.94 g, 54.96 mmol)And 4-dimethylaminopyridine (DMAP, 61 mg, 0.50 mmol) was added to 50 mL of tetrahydrofuran, and the mixture was heated to 50 ° C and stirred under heating for 5 hours.The solvent was removed under reduced pressure, 100 mL of ethyl acetate was added to the residue, washed with saturated sodium bicarbonate (100 mL) with,Discard the organic layer, adjust the water layer to pH=2 with dilute hydrochloric acid (1 mol/L), and filter.The filter cake was dried to obtain a white solid of 6.58 g, and the yield was 63.29percent.
57%
Stage #1: at 100℃; for 0.5 h; Microwave irradiation Stage #2: With water In ethyl acetate at 20℃; for 2 h;
Succinic acid anhydride (0.66 g, 6.6 mmol) and benzyl alcohol (0.65 g, 6.0 mmol) were dissolved in CH2Cl2 (5 mL) and the mixture was kept under stirring in a microwave reactor (Biotage Initiator 8) at 100 °C (pressure: ca 7 bar) for 30 min. The solvent was removed under reduced pressure and the residue was taken up in EtOAc (80 mL) and water (10 mL). The mixture was stirred at rt for 2 h, washed with 5percent KHSO4 (2 * 20 mL) and brine (20 mL), and dried over Na2SO4. The solvent was removed under reduced pressure and the colorless oil was subjected to column chromatography (eluent: CH2Cl2/EtOAc 10:1 to CH2Cl2/EtOAc 2:1). The volatiles were removed from the eluate under reduced pressure, CH2Cl2 (20 mL) was added, the solvent was evaporated, and the process repeated. Product 10 was obtained as colourless oil, which crystallized during drying in vacuo to give a white compact solid (0.71 g, 57percent) mp 55-56 °C (lit.;31 mp 56-57 °C). Rf = 0.3 (CH2Cl2/EtOAc 5:1). 1H NMR (300 MHz, [D4]MeOH): δ (ppm) 2.57-2.68 (m, 4H), 5.13 (s, 2H), 7.26-7.38 (m, 5H). C11H12O4 (208.21).
57.7%
With dmap In tetrahydrofuran at 50℃; for 5 h;
The succinic anhydride (10g, 0.1mol), benzyl alcohol (11.88g, 0.11mol), 4- dimethylaminopyridine (DMAP) (120mg) was added to a 100ml flask, THF (50ml) as solvent, the outside temperature 50 ° C reaction 5h. The reaction solution was concentrated, ethyl acetate was added, the organic layer was successively washed with water, saturated NaHCO3 solution, saturated brine, dried over anhydrous Na2SO4, and concentrated to a solid with isopropyl ether / acetone and recrystallized to give a white solid 12g, yield 57.7percent,
48%
for 4 h; Reflux
Benzyl succinic acid was synthesized by following a known procedure.33 Here, succinic anhydride (30 g, 300 mmol) was dissolved in benzyl alcohol (31.6 mL, 33 g, 300 mmol), and the resulting solutionwas heated at reflux for 4 h. The reaction mixture was dissolvedin ether (100 mL), and the insoluble succinic acid wasremoved by filtration. The filtrate was extracted with saturatedaqueous Na2CO3 (3 100 mL), and the combined aqueous extractswere acidified with 2 M HCl (2.0 L). The precipitate was collectedby filtration and dried under vacuum to give 2 (30 g, 48percent). 1HNMR (CDCl3, 300 MHz) d 7.35 (m, 5H), 5.15 (s, 2H), 2.70 (m, 4H).13C NMR (CDCl3, 75 MHz) d 177.7, 172.1, 135.7, 128.8, 128.5,128.4, 66.9, 29.1, 29.0.
250 g
With toluene-4-sulfonic acid In 5,5-dimethyl-1,3-cyclohexadiene for 5 h; Reflux
A solution of Succinic anhydride (184 grams, 1.84 mol), Benzyl alcohol (200 grams, 1.849 mol) and PTSA (1 gram, 5.25 mmol) in Xylene (1200 ml) was stirred at reflux temperature for 5 hours. The reaction mixture was cooled to room temperature, poured onto 10percent Sodium bicarbonate solution (2500 ml), aqueous layer washed with Ethyl acetate (500 ml), pH made acidic with dil HCl, extracted with Chloroform, dried over Sodium sulphate, distilled under reduced pressure, precipitated the residue by using Hexane to give pure Succinic acid mono benzyl ester (250 grams) as white powder with a melting point of 61-63° C. The product was characterized by IH NMR (CDCl3) δ 2.70 (m, 4H, CH2X2), 5.20 (s, 2H, CH2), 7.35 (m, 5H, Ar), 10.25 (bs, 1H, COOH).
18.5 g
With pyridine In tetrahydrofuran at 20℃; for 120 h;
10.0 g (100 mmol) of succinic anhydride, 10 ml of THF (tetrahydrofuran), 10.8 g (100 mmol) of benzyl alcohol and 4 ml of pyridine were combined and stirred for 5 days at ambient temperature. The solution was diluted with toluene. The mixture was extracted with a saturated solution of sodium hydrogen carbonate. The water phase was separated, and acidified with hydrochloric acid. The product was extracted with toluene to yield 18.5 g of succinic acid monobenzyl ester.
Reference:
[1] Synthetic Communications, 2013, vol. 43, # 10, p. 1397 - 1403
[2] Journal of Organic Chemistry, 2013, vol. 78, # 23, p. 12207 - 12213
[3] Bioorganic and Medicinal Chemistry, 2001, vol. 9, # 6, p. 1589 - 1600
[4] Journal of Organic Chemistry, 2001, vol. 66, # 12, p. 4115 - 4121
[5] Patent: US2005/107355, 2005, A1, . Location in patent: Page/Page column 38
[6] Journal of Medicinal Chemistry, 1989, vol. 32, # 2, p. 357 - 367
[7] Angewandte Chemie - International Edition, 2011, vol. 50, # 15, p. 3450 - 3453
[8] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 23, p. 7507 - 7514
[9] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 14, p. 4004 - 4010
[10] Chemistry - A European Journal, 2008, vol. 14, # 34, p. 10745 - 10761
[11] Tetrahedron, 2012, vol. 68, # 14, p. 2943 - 2949
[12] Patent: WO2014/71457, 2014, A1, . Location in patent: Page/Page column 50; 51
[13] Organic and Biomolecular Chemistry, 2015, vol. 13, # 38, p. 9850 - 9861
[14] Journal of Pharmacy and Pharmacology, 2002, vol. 54, # 3, p. 349 - 364
[15] Patent: WO2006/94357, 2006, A1, . Location in patent: Page/Page column 25; 3/19
[16] Asian Journal of Chemistry, 2013, vol. 25, # 17, p. 9701 - 9703
[17] Patent: US2011/166128, 2011, A1, . Location in patent: Page/Page column 63
[18] Patent: US2007/142331, 2007, A1, . Location in patent: Page/Page column 11
[19] Tetrahedron, 1989, vol. 45, # 24, p. 7783 - 7794
[20] Patent: CN108743953, 2018, A, . Location in patent: Paragraph 0021
[21] Chemistry - A European Journal, 2015, vol. 21, # 41, p. 14376 - 14381
[22] Bioorganic and Medicinal Chemistry, 2002, vol. 10, # 4, p. 1171 - 1179
[23] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 21, p. 6303 - 6322
[24] Patent: CN105732733, 2016, A, . Location in patent: Paragraph 0035
[25] Tetrahedron Letters, 1996, vol. 37, # 31, p. 5427 - 5430
[26] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 12, p. 3597 - 3601
[27] Acta Chemica Scandinavica (1947-1973), 1955, vol. 9, p. 1674,1678
[28] Tetrahedron Letters, 1984, vol. 25, # 41, p. 4623 - 4626
[29] Die Pharmazie, 1995, vol. 50, # 6, p. 382 - 387
[30] Organic Letters, 2004, vol. 6, # 22, p. 4133 - 4136
[31] Patent: US2010/286225, 2010, A1, . Location in patent: Page/Page column 4
[32] Synthetic Communications, 2012, vol. 42, # 23, p. 3441 - 3454
[33] Patent: US2014/142199, 2014, A1, . Location in patent: Paragraph 0218-0220
[34] Bioscience, Biotechnology and Biochemistry, 2016, vol. 80, # 1, p. 128 - 134
[35] Chemical Communications, 2016, vol. 52, # 30, p. 5254 - 5257
[36] Patent: WO2016/193309, 2016, A1, . Location in patent: Page/Page column 26
[37] Patent: WO2017/147220, 2017, A1, . Location in patent: Paragraph 00263-00264
[38] Synlett, 2017, vol. 28, # 20, p. 2881 - 2885
34
[ 108-30-5 ]
[ 121-44-8 ]
[ 103-40-2 ]
Yield
Reaction Conditions
Operation in experiment
83%
With hydrogenchloride; benzyl alcohol In tetrahydrofuran; ethyl acetate
Butanedioic acid monobenzylester 21 To a solution of 4.13 mL (4.32 g, 40 mmol) of benzyl alcohol and 4.40 g (44 mmol) of succinic anhydride in 100 mL of THF at 0° C. was added at once 12.24 mL (8.90 g, 88 mmol) of Et3 N. The mixture was allowed to come to room temperature and stirred for 18 h at which time it was concentrated under vacuum to a viscous oil and partitioned between 100 mL of 1 N HCl and 100 mL of EtOAc. The EtOAc layer was extracted with 84 mL of 5percent NaHCO3 solution. The aqueous layer was acidified with concentrated HCl solution to pH 1 and extracted with two 50 mL portions of EtOAc. The EtOAc layers were combined and washed with brine, dried (Mg SO4), filtered, and concentrated to give 6.9 g (83percent) of 21 as a white chunky solid: 1 H NMR (CDCl3) 2.70 (s, 4H), 5.22 (s, 2H), 7.38 (s, 5H).
83%
With hydrogenchloride; benzyl alcohol In tetrahydrofuran; ethyl acetate
Butanedioic acid monobenzylester 21 To a solution of 4.13 mL (4.32 g, 40 mmol) of benzyl alcohol and 4.40 g (44 mmol) of succinic anhydride in 100 mL of THF at 0° C. was added at once 12.24 mL (8.90 g, 88 mmol) of Et3 N. The mixture was allowed to come to room temperature and stirred for 18 h at which time it was concentrated under vacuum to a viscous oil and partitioned between 100 mL of 1N HCl and 100 mL of EtOAc. The EtOAc layer was extracted with 84 mL of 5percent NaHCO3 solution. The aqueous layer was acidified with concentrated HCl solution to pH 1 and extracted with two 50 mL portions of EtOAc. The EtOAc layers were combined and washed with brine, dried (Mg SO4), filtered, and concentrated to give 6.9 g (83percent) of 21 as a white chunky solid: 1 H NMR (CDCl3) 2.70 (s, 4H), 5.22 (s, 2H), 7.38 (s, 5H).
Reference:
[1] Patent: US5175257, 1992, A,
[2] Patent: US5112953, 1992, A,
35
[ 108-30-5 ]
[ 1927-62-4 ]
[ 103-40-2 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2009, vol. 57, # 10, p. 1167 - 1170
36
[ 108-30-5 ]
[ 100-51-6 ]
[ 103-43-5 ]
[ 103-40-2 ]
Reference:
[1] Chemische Berichte, 1902, vol. 35, p. 4080
37
[ 108-30-5 ]
[ 1575561-08-8 ]
[ 1575561-09-9 ]
[ 4373-41-5 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2014, vol. 74, p. 278 - 301
38
[ 108-30-5 ]
[ 2216-69-5 ]
[ 3562-99-0 ]
Yield
Reaction Conditions
Operation in experiment
86.4%
With aluminum (III) chloride In dichloromethane at 33 - 37℃; for 6 h;
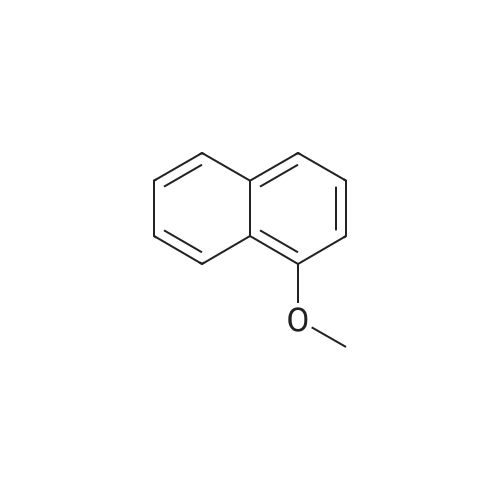
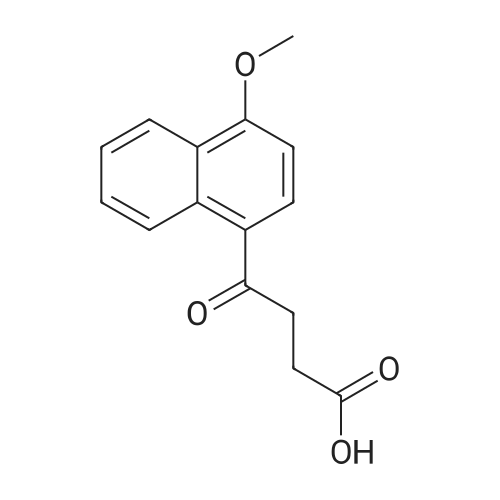
15.8 g of 1-methoxynaphthalene and 10.0 g of succinic anhydride were dissolved in 120 mL of dichloromethane,Stir,Cooling to 1 ~ 3 ° C,Divided into three batches of anhydrous aluminum trichloride 15.0 grams,The addition process takes about 20 minutes,The solution was then heated to 35 ± 2 ° C,Insulation reaction 6 hours (5.5 ~ 6.5 hours range),After the reaction is complete,The reaction solution was poured into an ice-water mixture (200 g of ice and 300 g of water) for 30 minutes,Standing, analysiscrystal,Filter,The filtrate was heated and distilled to recover dichloromethane,The cake is the crude of the ketone ketone;And then the crude ketoprofen water as a solvent by 2-3 times recrystallization,Activated carbon decolorization,Demon ketone boutique.The mass of the present product of the chamballone was 22.3 g,Melting point of 176 ~ 179 ° C,The yield was 86.4percent.
Reference:
[1] Patent: CN104370734, 2016, B, . Location in patent: Paragraph 0016-0018
[2] Helvetica Chimica Acta, 1932, vol. 15, p. 907,914
[3] Journal of the University of Bombay, Science: Physical Sciences, Mathematics, Biological Sciences and Medicine, 1936, vol. 5/2, p. 73,75
[4] Journal of the American Chemical Society, 1940, vol. 62, p. 2750,2752
[5] Journal of the American Chemical Society, 1951, vol. 73, p. 897,899
[6] Journal of the American Chemical Society, 1951, vol. 73, p. 326,327
[7] Kogyo Kagaku Zasshi, 1953, vol. 56, p. 887,889[8] Chem.Abstr., 1955, p. 6904
[9] Journal of the American Chemical Society, 1936, vol. 58, p. 2314,2316
[10] Bulletin de la Societe Chimique de France, 1968, p. 627 - 631
[11] Fortschr. Teerfarbenfabr. Verw. Industriezweige, vol. 14, p. 285
39
[ 108-30-5 ]
[ 75-15-0 ]
[ 7446-70-0 ]
[ 2216-69-5 ]
[ 3562-99-0 ]
Reference:
[1] Journal of the American Chemical Society, 1936, vol. 58, p. 2314,2316
[2] Helvetica Chimica Acta, 1932, vol. 15, p. 907,914
[3] Journal of the University of Bombay, Science: Physical Sciences, Mathematics, Biological Sciences and Medicine, 1936, vol. 5/2, p. 73,75
40
[ 108-30-5 ]
[ 7446-70-0 ]
[ 630-20-6 ]
[ 2216-69-5 ]
[ 3562-99-0 ]
Reference:
[1] Journal of the American Chemical Society, 1936, vol. 58, p. 2314,2316
[2] Helvetica Chimica Acta, 1932, vol. 15, p. 907,914
[3] Journal of the University of Bombay, Science: Physical Sciences, Mathematics, Biological Sciences and Medicine, 1936, vol. 5/2, p. 73,75
41
[ 108-30-5 ]
[ 7446-70-0 ]
[ 98-95-3 ]
[ 2216-69-5 ]
[ 3562-99-0 ]
Reference:
[1] Journal of the American Chemical Society, 1936, vol. 58, p. 2314,2316
[2] Helvetica Chimica Acta, 1932, vol. 15, p. 907,914
[3] Journal of the University of Bombay, Science: Physical Sciences, Mathematics, Biological Sciences and Medicine, 1936, vol. 5/2, p. 73,75
42
[ 108-30-5 ]
[ 57-88-5 ]
[ 1510-21-0 ]
Yield
Reaction Conditions
Operation in experiment
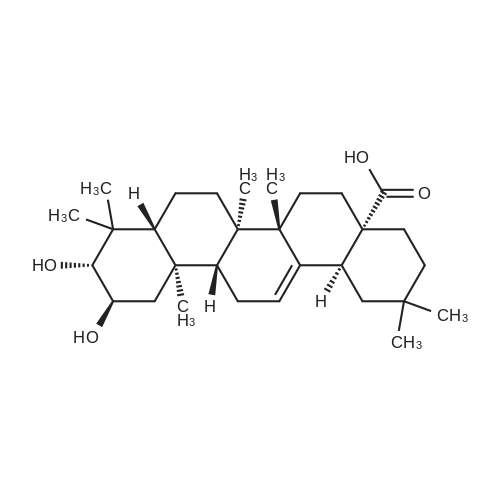
96%
at 20℃; for 168 h;
General procedure: Succinic anhydride (14.7mmol; 1.57equiv) and DMAP (1.584mmol, 0.17equiv) was added to a solution of (3β)-lanosta-8,24-dien-3-ol (1) or (3β)-cholest-5-en-3-ol (11) (9.37mmol) in pyridine (28mL). The reaction mixture was stirred over 7days at r.t. After stopping the reaction, the resulting mixture was poured onto ice, and hydrochloric acid was added to adjust pH=7, extracted with chloroform, and dried over sodium sulfate. Evaporation of the solvent gave a solid which was purified by column chromatography, affording the products (2) or (12) in 79percent or 96percent yield, respectively, and with >99.9percent analytical purity (Schemes 1–2). The analytical data of the products 2 and 12 are presented in the Supplementary material.
95%
Stage #1: at 20℃; for 24 h; Stage #2: With hydrogenchloride In 1,4-dioxane; water
7.6 grams of cholesterol and 2.36 grams of succinic anhydride were dissolved in 100 ml of 1,4-dioxane in a round-bottomed flask. A reaction catalyst, 2.9 grams of 4-(dimethylamino)pyridine (DMAP), was added thereto, and the mixture was stirred at room temperature for 24 hours. The reaction mixture was introduced into an HCl solution in order to precipitate the cholesterol succinate (9.1 g; yield=95percent).
95%
With dmap In dichloromethane at 20℃; for 15 h;
DMAP (1.58g, 12.9mmol) and succinic anhydride (1.29g, 12.9mmol) were added to a solution of cholesterol (5g, 12.9mmol) in CH2Cl2 (50mL). The solution was stirred at room temperature for 15h. Progress of the reaction was monitored by TLC. After completion of the reaction, solvent was removed in vacuo and the resultant residue was dissolved in chloroform (50mL). The organic layer was subsequently washed with aq. HCl (1N) (2×50mL), saturated NaHCO3 (2×50mL), brine solution (50mL) and dried over anhydrous Na2SO4, followed by solvent evaporation in vacuo. The crude product was purified by column chromatography (SiO2, 20percent EtOAc-hexane) to afford CS [29], as a white amorphous powder. Yield=5.98g (95percent); Rf 0.5 (1:9 MeOH/CHCl3); mp: 178.85°C from DSC trace, second cycle; [α]D20=−36.02 (c 1.00, CHCl3); FT-IR (Neat): ν 2930, 1704, 1434, 1314, 1178, 998cm−1; 1H NMR (400MHz, CDCl3): δ 0.5–2.1 (m, 41H, cholesterol), 2.35 (d, J=7.0Hz, 2H, CH2), 2.60 (t, J=6.0Hz, 2H, CH2), 2.65 (t, J=6.0Hz, 2H, CH2), 4.50 (m, 1H, OCH), 5.37 (m, 1H, CH); 13C NMR (100MHz, CDCl3): δ 177.6, 171.6, 139.5, 122.7, 74.2, 56.7, 56.1, 50.0, 42.2, 39.7, 39.5, 37.9, 36.9, 36.5, 36.2, 35.8, 31.9, 31.9, 29.2, 28.21, 28.0, 27.7, 24.2, 23.8, 22.8, 22.5, 21.0, 19.3, 18.7, 11.8; HRMS: m/z C31H50O4Na calcd. 509.3607, found 509.3608.
93%
at 90℃; for 24 h; Inert atmosphere
ChMS was synthesized according to a previously reported protocol [33], which is shown in Scheme 1. The cholesterol (2.5 g) was dissolved in 6 mL of pyridine, and succinic anhydride (1.8 g) was added. The reaction was allowed to proceed at 90 °C for 24 h with stirring under a nitrogen atmosphere. After completion of the reaction, the dark brown colored reaction mixture was cooled to room temperature and poured into distilled water. The resulting precipitate was thoroughly washed with distilled water. The obtained product was dried under reduced pressure and purified by recrystallization from acetone. The recrystallized product was dried under vacuum at room temperature (yield = 2.92 g; 93percent). IR (KBr, cm−1): 3200–2500 (−COOH), 2980–2839 (CH2, CH), 1709 (C=O), 1181 (C−O−C).
92%
at 80℃; for 3 h;
Cholesteryl Hemisuccinate (CHEMS, ): Succinic anhydride (1.55 gm, 15.5 mmol) was added to a solution of cholesterol (5 gm, 12.9 mmol) in pyridine (10 ml). After heating at 80° C. for 3 h, the reaction mixture was diluted with DCM and the organic phase was washed with 10percent HCl (50 ml) followed by a water wash (50 ml). The organic phase was separated, dried over Na2SO4 to obtain cholesterol hemisuccinate as white solid. The solid was recrystallized from DCM and hexane (5.78 gm, 92percent yield, and melting point 176-178° C.). 1H NMR (300 MHz, CDCl3): δ 5.47 (d, vinylic-H, J=5.0), 4.70-4.60 (m, 1H), 2.75-2.65 (m, 4H), 2.38-2.35 (d, 7H), 2.20-1.80 (m, 12H), 1.60-1.40 (m, 12H), 0.78-0.75 (m, 3H).
89%
Stage #1: With triethylamine In ethyl acetate for 12 h; Heating / reflux Stage #2: With hydrogenchloride In methanol; water; ethyl acetate
Cholesterylhemisuccinate (2) [0391] Warm a solution of 38.7 g (100 mmol) cholesterol, 20.0 g (200 mmol) succinic acid anhydride, 0.6 g (5 mmol) DMAP and 51.2 ml (400 mmol) triethylamine in 500 ml ethyl acetate for 12 hours with reflux. Add 100 ml ethyl acetate and 100 ml methanol, then extract with 200 ml 2 N HCl. Add an additional 100 ml ethyl acetate, then extract the organic phase twice with 150 ml each time of a mixture (2:1) of 0.2 N HCl and methanol. Concentrate the organic phase to a small volume and take up the residue in 300 ml methanol. Stir the resultant suspension for 15 minutes at room temperature. Add 300 ml water to completely precipitate the product. Siphon off the raw product, wash twice with 200 ml water each time, vacuum-dry, then recrystallize out of 200 ml diisopropyl ether. The yield is 43.4 g 2 as a colorless powder.
85.3%
at 90℃; for 24 h;
5 g of cholesterol and 1.55 g of succinic anhydride were dissolved in 90 mL of pyridine,90 reflux 24h,Cool to room temperature,Dilute hydrochloric acid (pH 1.0, 100 mL) Produce a white precipitate,filter,After the filter cake was washed with ethyl acetate dissolved in water,Dichloromethane / methanol column chromatography,A white solid (cholesterol succinate, 5.24 g, 85.3percent) was obtained.100 mg of cholesterol succinate monoester was dissolved in 10 mL of chloroform,Stirred at room temperature,65 mg of 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (EDCI) was added,46 mg of 1-hydroxybenzotriazole (HOBt) and 150 μL of triethylamine,After 1 h 62.5 mg of ADIBO,After stirring at room temperature overnight,After completion of the reaction, wash with water (10 mL × 3)The organic layer was concentrated,Dichloromethane / methanol column chromatography,Obtained as a light yellow solid (azabicyclooctynecholesterol, 64 mg, 40.3percent).
80.1%
at 20℃; for 24 h;
The Chol-COOH was synthesized successfully according to the method reported by Yu et al. [30]. Briefly, cholesteroland succinic anhydride (SA) were dissolved in pyridine and kept for 24 h with constant stirring at room temperature.Then, deionized water was added to the above solution. The produced precipitate was obtained by filtration and dried.The crude product was recrystallized in acetone to obtain the white powder, Chol-COOH.
72%
With triethylamine In acetone at 25℃; for 24 h; Inert atmosphere
3-Cholesteryloxycarbonylpropanoic acid was synthesized by the reported method.30 A solution of cholesterol (3.89 g, 10 mmol), succinic anhydride (1.27 g, 13 mmol) and triethylamine (1.5 mL, 15 mmol) in dry acetone (60 mL) was heated to reflux under nitrogen atmosphere for 24 h. The acetone was then evaporated and resulting precipitate was recrystallized twice from glacial acetic acid, white solid 3.5 g, 72 percent, yield. 1H NMR (500 MHz, CDCl3) δ 5.37 (d, 1H, J = 4.0 Hz), 4.63 (m, 1H), 2.68 (t, 2H, J = 6.0 Hz), 2.61 (t, 2H, J = 7.0 Hz), 2.32 (d, 2H, J = 9.0 Hz), 2.02-0.91 (m, 33H), 0.86 (dd, 6H, J = 4.0 Hz), 0.68 (s, 3H).
71%
With dmap In dichloromethane at 20℃;
To a solution of cholesterol (l.Og, 2.5 mmoles) in dry dichloromethane was added succinic anhydride (0.25g, 2.5mmoles) and 4-dimethyaminopyridine (0.305g, 2.5mmoles) under nitrogen atmosphere, which was allowed to stir overnight at room temperature. The reaction mixture was extracted with chloroform (5OmL) and washed with ammonium chloride solution (3x25mL) followed by brine solution (3x20 mL). The chloroform extract was dried over sodium sulfate and evaporated on a rotary evaporator. Silica gel column chromatographic purification of the resulting residue using 230-400 mesh silica gel size and 5-6percent methanol in chloroform (v/v) as the eluent afforded the title compound as a white solid (2.0 g, 71percent yield, Rf = 0.5, 10:90, v/v, methanolxhloroform).1H NMR (400 MHz, CDCl3): δ/ppm= 0.6-2.30 [m, 43H, cholesteryl skeleton]; 2.60 [t, Chol-OCO-C/ir]; 2.70 [t, ChOl-OCO-CH2-CH2-COOH]; 4.60 [m, IH5 H30 (Choi)]; 5.40 [m, IH5 H6 (Choi)].
70%
With pyridine In n-heptane for 21 h; Reflux
A solution of cholesterol (5.80 g, 15 mmol), succinic anhydride (1.50 g, 15 mmol), pyridine (1.00 mL), and dry heptane (150 mL) were heated to reflux for 21 h and cooled to room temperature. The resulting precipitate was recrystallized twice from acetone. Yield: 70percent.
55%
With dmap In dichloromethaneReflux
Example 6: Preparation of 2-((2-(2,4-difluorophenyl)-l,3-di(lH-l,2,4-triazol-l-yl)propan- 2-yloxy)(ethoxy)phosphoryloxy)ethyl (3S,8R,9S,10R,13S,14S,17R)-10,13,17-trimethyl-17- ((R)-6-methylheptan-2-yl)-2,3,4,7,8,9,10,ll,12,13,14,15,16,17-tetradecahydro-lH- cyclopenta [a] phenanthren-3-yl succinateScheme 6cholesterol 55percent 6-163percent 6-268percentAs shown in Scheme 6 above, cholesterol was reacted with succinic anhydride in the presence of 4-dimethylaminopyridine in re fluxing dichloromethane to afford in 55percent yield the carboxylic acid 6.1, which was converted into its acid chloride with thionyl chloride in toluene at 65°C and further reacted with ethylene glycol in the presence of triethylamine in dichloromethane at room temperature to give derivative 6.2 in 63percent yield. The reaction of 6.2 with the freshly prepared phosphorylating reagent ethyl Λ/.iV-diisopropylchlorophosphoramidite, in the presence ofN,N- diisopropylethylamine (Hnig's base) in dry tetrahydrofuran at room temperature, subsequent coupling with fluconazole (FLC) in the presence of tetrazole followed by oxidation with 30percent aqueous hydrogen peroxide afforded in a one-pot process the desired fosfluconazole derivative in 68percent yield after chromatography. The resulting product was characterized by proton nuclear magnetic resonance and mass spectrometry as follows:- 1H NMR (400MHz, CDCl3): 58.39 (d, J = 14.9 Hz, 2H), 7.81 (s, 2H), 7.11 (m, IH), 6.88 (m, IH), 6.77 (m, IH), 5.35 (m, IH), 5.18 (m, 4H), 4.59 (m, IH), 4.24 (m, 2H), 4.21-4.01 (m, 4H), 2.61 (m, 4H), 2.30 (m, 2H), 1.98 (m, 2H), 1.83 (m, 2H), 1.62-1.03 (m, 25H), 1.00(s, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 and 0.84 (dd, J = 1.8 Hz, 6H), 0.67 (s, 3H)- MS (APCI): 928 (M) White powderA melting point DSC characterization of the product is presented in Figure 2.
31%
Reflux
General procedure: A solution of cholesterol (38.6 g, 100 mmol) and dicarboxylic acid anhydride (10 mmol) was refluxed in dry pyridine (100 mL) for 8-10 h., The reaction mixture was concentrated under reduced pressure using rotary evaporator to produce a wet residue, which was dissolved in 50 mL of boiling acetone, and on cooling it gave colorless product. The crude compound was purified by two recrystallizations from acetone and one from absolute ethanol. 4-(Cholesteryloxy)-4-oxobutanoic acid (1). Yield 15.1 g, 31percent, mp 175-176°C as reported [15].
Reference:
[1] Russian Journal of Organic Chemistry, 2002, vol. 38, # 8, p. 1189 - 1191
[2] Steroids, 2013, vol. 78, # 14, p. 1347 - 1352
[3] Patent: US2005/201972, 2005, A1, . Location in patent: Page/Page column 15
[4] Polymer, 2016, vol. 86, p. 98 - 104
[5] Arzneimittel-Forschung/Drug Research, 1994, vol. 44, # 11, p. 1259 - 1264
[6] Reactive and Functional Polymers, 2012, vol. 72, # 11, p. 832 - 838
[7] Chemical Communications, 2010, vol. 46, # 29, p. 5358 - 5360
[8] Patent: US9173839, 2015, B2, . Location in patent: Page/Page column 12
[9] Journal of Medicinal Chemistry, 2010, vol. 53, # 21, p. 7632 - 7638
[10] Monatshefte fur Chemie, 1998, vol. 129, # 3, p. 319 - 327
[11] Patent: US2003/229037, 2003, A1, . Location in patent: Page/Page column 38, 7
[12] Angewandte Chemie - International Edition, 2006, vol. 45, # 41, p. 6889 - 6893
[13] Pharmazie, 2008, vol. 63, # 6, p. 434 - 438
[14] Patent: CN106478718, 2017, A, . Location in patent: Paragraph 0100
[15] Tetrahedron Letters, 2009, vol. 50, # 30, p. 4314 - 4317
[16] RSC Advances, 2015, vol. 5, # 9, p. 6985 - 6992
[17] Pharmaceutical Research, 2017, vol. 34, # 10, p. 2172 - 2184
[18] Journal of Molecular Structure, 2008, vol. 881, # 1-3, p. 83 - 89
[19] Bulletin of the Korean Chemical Society, 2014, vol. 35, # 5, p. 1343 - 1348
[20] Patent: WO2007/22030, 2007, A2, . Location in patent: Page/Page column 55
[21] Journal of Materials Chemistry C, 2015, vol. 3, # 41, p. 10925 - 10933
[22] Dyes and Pigments, 2016, vol. 132, p. 48 - 57
[23] Journal of Materials Chemistry, 2007, vol. 17, # 44, p. 4699 - 4704
[24] Chemistry - A European Journal, 2007, vol. 13, # 29, p. 8231 - 8239
[25] Angewandte Chemie - International Edition, 2014, vol. 53, # 43, p. 11532 - 11537[26] Angew. Chem., 2014, vol. 126, # 43, p. 11716 - 11721,6
[27] RSC Advances, 2017, vol. 7, # 23, p. 14176 - 14185
[28] Chemical and Pharmaceutical Bulletin, 1988, vol. 36, # 8, p. 3060 - 3069
[29] Patent: WO2010/105910, 2010, A1, . Location in patent: Page/Page column 31-33
[30] Molecules, 2014, vol. 19, # 9, p. 13177 - 13187
[31] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 8, p. 787 - 791
[32] Bulletin of the Chemical Society of Japan, 2007, vol. 80, # 10, p. 1975 - 1980
[33] International Journal of Pharmaceutics, 2010, vol. 397, # 1-2, p. 147 - 154
[34] International Journal of Pharmaceutics, 2011, vol. 408, # 1-2, p. 173 - 182
[35] Polymer, 2012, vol. 53, # 16, p. 3566 - 3576
[36] Carbohydrate Polymers, 2013, vol. 94, # 1, p. 17 - 23
[37] Patent: US2014/363490, 2014, A1, . Location in patent: Paragraph 0070; 0071
[38] Patent: CN106432338, 2017, A, . Location in patent: Paragraph 0091
[39] Patent: CN106554368, 2017, A, . Location in patent: Paragraph 0040
[40] Patent: WO2017/137958, 2017, A1, . Location in patent: Paragraph 00195
[41] Patent: CN108424434, 2018, A, . Location in patent: Paragraph 0033; 0035
43
[ 108-30-5 ]
[ 57-88-5 ]
[ 1510-21-0 ]
Reference:
[1] Journal of the American Pharmaceutical Association (1912-1977), 1944, vol. 33, p. 107[2] Chem.Abstr., 1944, p. 2765
[3] Magyar biol. Kutatointezet Munkai, 1941, vol. 13, p. 334[4] Chem.Abstr., 1942, p. 484
44
[ 108-30-5 ]
[ 2216-51-5 ]
[ 77341-67-4 ]
Yield
Reaction Conditions
Operation in experiment
99.9%
at 110℃; for 8 h; Autoclave; Inert atmosphere; Neat (no solvent)
Synthetic Example 1 Synthesis of l-menthyl-(l-menthoxyethyl)succinic ester Compound 1 To a 100 ml autoclave, 10.00 g (63.99 mmol) of l-menthol and 6.40 g (63.96 mmol) of succinic anhydride were added, flushed with nitrogen gas, and then stirred at 110° C. for 8 hours. After cooling to room temperature, 100 ml of hexane was added thereto. The precipitated crystals were filtered and the filtrate, hexane phase was concentrated to obtain 16.61 g of l-menthylsuccinic acid. The yield was >99.9percent.
78.6%
With dmap In tetrahydrofuran for 12 h; Reflux
(-)- Monomenthyl succinate was synthesized using the common method esterification of (-)-methanol and succinic anhydridein THF with the catalyst DMAP. Yield: 78.6percent. M.p. 61–63 °C.FR-IR (KBr, cm−1): 2917–3440, 1727, 1710, 1388, 1285, 1224, 1177, 1037, 1011. 1HNMR (CDCl3, ppm): .4.67–4.74 (m, 1H), 2.67–2.72 9 (t, 2H), 2.58–2.63 (t, 2H),0.72–2.01 (broad, 18H).
With Candida cylindracea lipase In diethyl ether at 20℃; for 24 h; Schlenk technique; Resolution of racemate; Enzymatic reaction
A dry Schlenk tube was charged with rac-alcohol (1 equivalent) and succinic anhydride (1 equiv.) dissolved in 2 mL of diethyl ether. The reaction was initiated by the addition of 100 mg of CCL. The reaction mixture was shaken at room temperature for 24 h. After removal of the lipase by filtration, the filtrate was shaken with 1 M Na2CO3 solution, and the remaining alcohol and the produced monoester succinate were separated by liquid–liquid extraction. The remaining enantiomer was obtained from the organic layer and the aqueous phase was washed with an organic solvent and treated by adding 1 M NaOH solution to obtain the other enantiomer. The enantiomeric excesses values were quantified by chiral GC analyses. Chiral GC: Chiralsil-DEX CB: (Tcolumn = 120 °C. flow: 1,2 mL/min); dl-(±)-menthyl acetate: td-(+) = 9.58 min; tl-(-) =10.85 min. dl-(±)-menthol: td-(+) = 13.25 min; tl-(-) = 13.69 min.
Reference:
[1] Research on Chemical Intermediates, 2018, vol. 44, # 11, p. 6847 - 6860
Reference:
[1] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1924, vol. 179, p. 405[2] Bulletin de la Societe Chimique de France, 1926, vol. <4> 39, p. 672
48
[ 108-30-5 ]
[ 490-99-3 ]
[ 77341-67-4 ]
Reference:
[1] Journal of the Chemical Society, 1912, vol. 101, p. 116
49
[ 108-30-5 ]
[ 2216-51-5 ]
[ 77341-67-4 ]
[ 115936-72-6 ]
Reference:
[1] Annales de Chimie (Cachan, France), 1886, vol. <6> 7, p. 483
Stage #1: for 6 h; Reflux Stage #2: With hydrogenchloride In acetone at 0 - 20℃; for 8 h;
1-Dimethylamino-3-[2-[2-(3-methoxyphenyl)ethyl]phenoxy]-2-propanol (29.8 g, 0.090 mol)And succinic anhydride (11.3 g, 0.113 mol)Soluble in acetone (200ml),Heating and refluxing for 6 hours,The ice bath is cooled to 0~5 °C,Passing dry hydrogen chloride gas,To a pH of 1 to 2, a white solid precipitated,Slowly warm to room temperature and continue to stir for 8 hours.filter,The filter cake is dried and recrystallized from acetone.Obtained white solid sarpogrelate hydrochloride (38.8g, yield 92.5percent), purity 99.8percent
Reference:
[1] Journal of Medicinal Chemistry, 1990, vol. 33, # 6, p. 1818 - 1823
53
[ 108-30-5 ]
[ 125700-67-6 ]
Reference:
[1] Patent: US9290760, 2016, B2,
54
[ 108-30-5 ]
[ 14949-00-9 ]
[ 78851-85-1 ]
Yield
Reaction Conditions
Operation in experiment
96%
at 100℃; for 12 h;
To solution of compound 3 (0.40 g, 2.22 mmol) in N,N-dimethylformamide was added succinic anhydride (0.22 g, 2.22 mmol). The reaction mixture was heated at 100 oC for 12 hr. After reaction completion, solvent was evaporated to obtained 0.60 g of compound 4 in 96 percent yields (compound was used without purification). 1H NMR (400 MHz, DMSO-d6) δ 8.33 (s, 2H), 2.78 (t, J = 6.6 Hz, 2H), 2.62 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 174.08, 172.38, 164.52, 162.15, 30.77, 29.08; HRMS (ESI+): m/z Calcd for C6H9N4O5S2 [M+H]+: 281.0014, Found: 281.0009.
33%
at 50℃;
EC3105 (178 mg, 0.988 mmol) was dissolved in DMF (1.6 mL). To this solution was added succinic anhydride (98.8 mg, 1 eq.) The solution was heated to 50 oC overnight. The reaction was diluted with water and loaded onto a Biotage C18 column (0.1percent TFA/ACN) and purified. After freeze drying, EC3108 (92mg, 33percent yield) was recovered as a white solid. ESI-MS [M+H]+ = 281.1.13C NMR (125 MHz, DMSO-d6): δ 173.5, 171.9, 164.9, 161.2, 30.4, 28.5.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 5, p. 915 - 921
[2] Journal of Medicinal Chemistry, 2016, vol. 59, # 10, p. 5077 - 5088
[3] Patent: WO2017/161144, 2017, A1, . Location in patent: Page/Page column 69
[4] Patent: US2005/143366, 2005, A1, . Location in patent: Page/Page column 18
[5] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 16, p. 4851 - 4856
55
[ 108-30-5 ]
[ 116574-71-1 ]
[ 116574-71-1 ]
[ 140695-84-7 ]
Reference:
[1] Organic Process Research and Development, 2001, vol. 5, # 4, p. 415 - 420
[2] Organic Process Research and Development, 2001, vol. 5, # 4, p. 415 - 420
56
[ 108-30-5 ]
[ 63968-64-9 ]
[ 88495-63-0 ]
Yield
Reaction Conditions
Operation in experiment
96%
Stage #1: With sodium tetrahydroborate; cation exchange resin In tetrahydrofuran at 20 - 35℃; for 0.666667 h; Stage #2: With triethylamine In tetrahydrofuran at 20 - 35℃; for 1 h;
Artemisinin (500MG) and cation exchange resin (LG) are stirred in tetrahydrofuran (10ML) at room temperature for 5 minutes. Sodium borohydride (250mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20- 35 degree C). After completion of the reaction succinic anhydride (250mg) and triethylamine (0. 7ML) are added at room temperature and the reaction mixture is stirred further for 1 hours at room temperature. The resin is filtered. After usual worlcup and column chromatography of the crude product (710MG), 480mg of pure artesunic acid (yield = 96percent W/W) is obtained.
93%
Stage #1: With sodium tetrahydroborate; D-glucose In 1,4-dioxane at 20 - 30℃; for 2 h; Stage #2: at 20 - 30℃; for 2 h;
Artemisinin (500mg) and polyhydroxy compound (dextrose, 2.5g) are stirred in 1,4-dioxan (15ML) at room temperature for 5 minutes. Sodium borohydride (2.5g) is added slowly for 10 minutes and the reaction mixture is stirred for about 2 hours at room temperature (20- 30° C). After completion of the reaction (Checked by TLC), succinic anhydride (250 mg) and anion exchange (basic) resin (1.5g) are added at room temperature and the reaction mixture is stirred further for 2 hours at room temperature. Cold water (50 ml) is added to the reaction mixture and pH is adjusted between 6-7 with dilute acetic acid and extracted with 40percent ethyl acetate in hexane (3 x 25 ml). The combined extract is washed with water (50 ml). The ethyl acetate n-hexane extract is dried over anhydrous sodium sulphate and evaporation of the solvent yield 655 mg of crude artesunic acid which upon purification over silica gel (1: 5 ratio) with 20-30percent ethyl acetate in hexane, furnish pure artesunic acid in 93percent w/w (465 mg) yield (according to CO-TLC). After drying the pure a-artesunic acid, mp 140-142° C is characterized by spectral analysis
93%
Stage #1: With sodium tetrahydroborate; cation exchange resin In tetrahydrofuran at 20 - 35℃; for 0.75 h; Stage #2: With sodium hydrogencarbonate In tetrahydrofuran at 20 - 35℃; for 1.25 h;
Artemisinin (500 mg) and cation exchange resin (LG) are stirred in tetrahydrofuran (15 ml) for 5 minutes. Sodium borohydride (2.4gm) is added slowly and the reaction mixture is stirred for 45 minutes at room temperature (20-35 degree C). After completion of the reduction reaction, succinic anhydride (245 mg) and sodium bicarbonate (3.5g) are added and the reaction mixture is further stirred for 1.25 hours. After usual workup and purification of impure reaction product (650 mg), pure artesunic acid in 93percent w/w yield is obtained.
91.2%
Stage #1: With sodium tetrahydroborate; D-glucose In 1,4-dioxane at 20 - 30℃; for 2 h; Stage #2: With triethylamine In 1,4-dioxane at 20 - 30℃; for 2 h;
Artemisinin (500 mg), polyhydroxy compound (dextrose, 2. 0G) are stirred in 1,4-dixan (10 ml). Sodium borohydride (2.5g) is added slowly for 10 minutes and the reaction mixture is stirred for about 2 hours at room temperature (20-30° C). After completion of the reduction step, succinic anhydride (250 mg) and triethylamine (LML) are added and the reaction mixture is further stirred for 2 hours at room temperature (20-30 degree C). After usual work up and purification of crude product (690mg) through column chromatography (1: 4 ratio) 91.2percent pure artesunic acid is obtained.
89.6%
Stage #1: With sodium tetrahydroborate; D-glucose In 1,4-dioxane at 20 - 30℃; for 2 h; Stage #2: With sodium hydrogencarbonate In 1,4-dioxane at 20 - 30℃; for 2 h;
Artemisinin (500 mg) and polyhydroxy compound (dextrose, 2g) are stirred in dioxan (15 ml) for 5 minutes. Sodium borohydride (2.4gm) is added slowly and the reaction mixture is stirred for 2 hours at room temperature (20-30 degree C). After completion of the reduction step succinic anhydride (250 mg) and sodium bicarbonate (3. 5G) are added and the reaction mixture is further stirred for 2 hours. After usual workup and purification of impure reaction product (650 mg), 89.6percent w/w pure artesunic acid is obtained.
87.4%
Stage #1: With sodium tetrahydroborate; D-glucose In tetrahydrofuran at 20 - 30℃; for 2 h; Stage #2: With triethylamine In tetrahydrofuran at 20 - 30℃; for 2 h;
Artemisinin (500 mg), polyhydroxy compound (dextrose, 2. 0G) are stirred in tetrahydrofuran (10 ml). Sodium borohydride (2. 5G) is added slowly for 10 minutes and the reaction mixture is stirred for about 2 hours at room temperature. After completion of the reduction step succinic anhydride (250 mg) and triethylamine (LML) are added and the reaction mixture is further stirred for 2 hours at room temperature. After usual work up and purification of the crude product (615mg) through column chromatography 87.4percent pure artesunic acid is obtained.
85%
Stage #1: With sodium tetrahydroborate; cation exchange resin In 1,4-dioxane at 20 - 35℃; for 0.75 - 0.916667 h; Stage #2: With triethylamine In 1,4-dioxane at 20 - 35℃; for 1.25 h;
ARTEMISININ (500MG) and cation exchange resin (LG) are stirred in 1,4 dioxan (10ML) at room temperature for 5 minutes. Sodium borohydride (250mg) is added slowly for 10 minutes and the reaction mixture is stirred for about 30 minutes at room temperature (20-35 degree C). After completion of the reaction succinic anhydride (250mg) and triethylamine (0. 7ML) are added slowly at room temperature and the reaction mixture is stirred further for 1.25 hours at room temperature. After usual work up and purification of the crude artesunic acid (680mg) pure product in 91.7percent w/w is obtained.
With dmap In 1,2-dichloro-ethane at 45℃; for 6 h; Industrial scale
Weigh 5 kg of dihydroartemisininIn the jacketed stainless steel reactor R1,Adding 50.0 L dichloroethane,Room temperature stirring dissolved,To prepare a solution; weigh the succinic anhydride 10kg in a jacketed stainless steel reactor R2, add 40.0L dichloroethane, add 50g DMAP, stirring at room temperature, made of solution;The solution in the reactor R1 was added dropwise to the reactor R2 for 4 hours and the mixture was stirred for 2 hours. The pH of the reaction solution was adjusted to 5 with hydrochloric acid and concentrated to a solvent-free solution. 25L dichloroethane and 25L water, stirring extraction, liquid separation, 25L dichloroethane repeated extraction time;The organic phases were combined, dried over anhydrous sodium sulfate, fine filtered, concentrated under reduced pressure,Artesunate 6.56 kg,Yield 97.0percentHPLC detection purity 99.9percent
Reference:
[1] Patent: CN106946904, 2017, A, . Location in patent: Paragraph 0030; 0031
58
[ 108-30-5 ]
[ 102-54-5 ]
[ 1273-94-5 ]
Reference:
[1] Doklady Akademii Nauk SSSR, 1956, vol. 106/111, p. 675 - 677[2] Doklady Akademii Nauk SSSR, 1956, vol. 111, p. 362 - 364
[3] , Gmelin Handbook: Fe: Org.Verb.A1, 2.5.7.6.5, page 156 - 161,
[4] , Gmelin Handbook: Fe: Org.Verb.A1, 2.5.7.6.5, page 156 - 161,
[5] Journal of the American Chemical Society, [6] Journal of the American Chemical Society, 1957, vol. 79, p. 3416 - 3420
59
[ 108-30-5 ]
[ 786593-06-4 ]
Yield
Reaction Conditions
Operation in experiment
93.4%
With triethylamine In ethyl acetate for 8 h; Inert atmosphere; Reflux; Large scale
Add andrographolide 5kg and 50L reactorSuccinic anhydride 10kg,Triethylamine 7.5kg,Ethyl acetate 20L,First sealed the reactor, pumping negative pressure,Fill it with nitrogen again and repeat it three times under nitrogen protection.Warm up to reflux for 8 hours,TLC analysis, no material andrographolide, that is, after the end of the reaction, stop heating, cooling to 40 ~ 50 °C, vacuum distillation at this temperature,Dehydrated andrographolide disuccinate hemi-ester crude oil;Dehydration of Andrographolide Disuccinic Acid Half-ester:Dissolve the crude dehydrated andrographolide disuccinate hemiester oil with dichloromethane.The dehydrated andrographolide disuccinic acid half ester solution in dichloromethane isWeigh 200 kg of 200-300 mesh silica gel and load the column to load the solution of the dehydrated andrographolide disuccinic acid half ester in dichloromethane.Elution was performed with a mixture of dichloromethane:methanol:formic acid=500:15:0.5, and the sample was subjected to TLC analysis. The purified dehydrated andrographolide disuccinic acid half ester eluent was collected and distilled under reduced pressure.Pure dehydroandrographolide disuccinic acid half ester was obtained in 7.1 kg, and the yield was 93.4percent.
Reference:
[1] Patent: CN107857747, 2018, A, . Location in patent: Paragraph 0043-0045; 0048-0050; 0053-0055; 0058-0060
Thiophene [(5.] [0GM,] 0.059moles) and succinic anhydride (7.13gm, 0.0713 moles) were taken up in dichloroethane (50 ml). Aluminium chloride (17.43gm, 0.131 moles) was added at room temperature and the resulting mixture heated under reflux for lhr with stirring. The reaction mixture was cooled to room temperature, diluted with 30 ml of 1 : [1] mixture of water and [CONC.] hydrochloric acid. After stirring for 10 min the separated solid was filtered under suction, washed with water and dilute hydrochloric acid (50 ml). The solid was dried at room temp to give 9.8 gm (89% 0 of the title compound, mp [103-107] C. [LH NMR] (CDC13, [8)] : 7.69 (dd, [1H),] 7.58 (dd, 1H), 7.07 (dd, lH), 3.9 (bs, 1H), 3.23 (t, 2H), 2.74 (t, 2H).
87%
With aluminum (III) chloride; In dichloromethane; at 20 - 35℃; for 19h;Inert atmosphere;
Dihydrofuran-2,5-dione (30 g, 300 mmol) and thiophene (24 ml, 300 mmol) were added to a 250 ml RBF and DCM (120 ml) was added. The mixture was stirred at room temperature under nitrogen until the succinic anhydride was dissolved (colorless solution) following this aluminum trichloride (48 g, 360 mmol) was added to the mixture portion wise over 30 min (the solution became dark red/brown). The reaction mixture was stirred at room temperature under nitrogen for 17 h and then heated to 35C under nitrogen for 2 h.The reaction mixture was allowed to cool down to room temperature and was poured into a mixture of ice, concentrated HCl (20 ml) was added. The mixture was stirred for 30 minand was then partitioned between DCM (200 ml) and water (100 ml). A solid crashed out in the aqeuous phase and was filtered. The organic phase was washed with water (100 ml), dried over magnesium sulfate, filtered and concentrated in vacuo and combined with the solid obtained previously. The combined material was filtered and dried to afford 4-oxo-4-(thiophen-2-yl)butanoic acid (48g, 262 mmol) 87% yield as a white solid. 1H NMR (METHANOL-d4) d: 7.88 - 8.00 (m, 1 H), 7.79 - 7.88 (m, 1 H), 7.22 (t, J = 4.3 Hz, 1 H), 3.69 (s, 1 H), 3.29 (t, J = 6.4 Hz, 2 H), 2.69 - 2.76 (m, 2 H) ES- 183.
81%
With aluminum (III) chloride; In dichloromethane; at 0 - 20℃; for 17h;
Add 24 ml of thiophene and 30 g of succinic anhydride and 200 ml of dichloromethane in a 500 ml three-necked flask, stir and cool to 0-5 C, add 36 g of anhydrous aluminum trichloride in batches, stir at room temperature for 17 hours after the addition, and then The reaction solution was poured into 500 ml of crushed ice, stirred for half an hour, and the organic layer was separated. The organic layer was washed with 500 ml of water, then dried over magnesium sulfate and evaporated to dryness. The mixture was dried by blowing at 50-60 C for 12 hours to obtain a solid 44.7 g, and the yield was 81%.
77%
aluminum (III) chloride; In dichloromethane; at 20℃; for 5h;
Example 5.; Preparation of dichloror6,6'-bis(dimethylsilanediyl)di(eta5-(7-phenyl-5,6-dihvdro-4/-/- indeno[5,4-£>1thiophene-6-yl)1zirconium.; The preparation is described in the following scheme. KOH 6,7-dihydro-1-benzothiophen-4(5/-/)-one was obtained with an overall yield of 43% via the acylation of thiophene by succinic anhydride followed by reduction of the ketone formed and by the cyclisation of 4-(2-thienyl)butanoic acid in the presence of the laton's reagent. Next, 4,5,5a,6-tetrahydro-7/-/-indeno[5,4-iotab]thiophen-7-one was synthesised from 6,7-dihydro-1 -benzothiophen-4(5H)-one with an overall yield of 30%. The latter product was further reacted with one equivalent of PhLi in ether followed by acidification of the reaction medium to give a mixture of 7-phenyl-5,5a- dihydro-4H-indeno[5,4-ib]thiophene and 7-phenyl-5,8-dihydro-4H-indeno[5,4- £>]thiophene as shown in the scheme belowThe lithium salt of this ligand was treated with 0.5 eqv of Me2SiCl2 in THF to form the respective iotab/s(cyclopentadienyl)dimethylsilane. This b/s-cyclopentadienyl ligand was isolated as a mixture of the Me2Si-bhdging ligands involving ca. 70% of the desired isomers with a yield of 17% by repeating flash chromatography on Silica Gel 60. It was metallated with 2 eqv of "BuLi in toluene-hexanes, and then with metallic salt ZrCI4(THF)2. This mixture was stirred overnight at room temperature and then filtered through glass frit. Crystals precipitated from the filtrate at a temperature of -3O0C were collected and dried in vacuum. On the evidence of 1H NMR spectroscopy, this product isolated with a yield of 13% is a mixture of me- and meso-complexes in a ratio of 1 to 5. This 1H NMR spectrum is shown in figure 9. The meso-isomer was characterised by X-ray crystal structure analysis. Figure 10 is the ORTEP representation of the molecular structure of this meso-complex with thermal ellipsoids drawn at the 50% probability level.The key geometric paramerers of this structure are:- bond lengths Zr-Cp(c) and Zr-Cp(c)' are 2.239(1 ) Angstroms;- angle between two cyclopentadienyl planes is 59.2.
63%
General procedure: General procedure 6 (GP6) for Friedel Crafts reaction: Representative experimental procedure for the Friedel Crafts using heterocycles with the anhydride. To solution of succinic anhydride (1 eq.) in anhydrous DCM was added anhydrous A1C13 (1.2 eq) at 0 C and the reaction mixture was stirred for 2 h at 0 C. Then the aromatic heterocycle was added dropwise over a period of 1 h. The reaction mixture was stirred for 4 h at room temperature. 50 ml of ice-cold H20 was added at 0 C, followed by acidification with 2N HC1 to pH 2. The aqueous phase was extracted with DCM (3 times). The combined organic extracts were dried over Na2S04, filtered and evaporated in vacuo. The crude mixture was purified by recrystallization or flash column chromatography to afford the compounds 9, 10.
38%
EXAMPLE 13; 4-Oxo-4-(thiophen-2-yl)butanoic acidAnhydrous aluminium chloride (15.8 g, 0.12 mol) was added to a solution of succinic anhydride (1 1.9 g, 0.12 mol) in dry CH2CI2 (50 Ml) and the reaction mixture was cooled to 0 C. A solution of thiophene (10.0 g, 0.12 mol) in CH2CI2 (50 Ml) was then added dropwise maintaining the same temperature. The reaction mixture was allowed to warm up to room temperature and stirred for 8 h. The mixture was then cooled to 0 C and the Ph was adjusted to ~3 using 6N HCI. The organic product was extracted with CH2CI2. The combined extracts were washed with water and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by column chromatography (silica 60-120 mesh, eluant 5-10% MeOH in CH2CI2), to get 4- oxo-4-(thiophen-2-yl)butanoic acid (8.3 g, yield 38%). 1H NMR (400 MHz, DMSO-d6) delta 12.18 (s, 1 H), 8.00 - 7.98 (m, 2H), 7.26 - 7.23 (m, 1 H), 3.21 - 3.18 (m, 2H), 2.58 - 2.55 (m, 2H). MS (ESI) m/z: Calculated for C8H803S: 184.02; found: 184.9 (M+H)+
Under N2 succinic anhydride (6) (11.9 g, 119 mmol) was dissolved in dry CH2Cl2 (550 mL). Anhydrous AlCl3 (19.0 g, 142 mmol) was added at 0 C and the reaction mixture was stirred for 30 min. Then thiophene (5) (10.0 g, 119 mmol) dissolved in CH2Cl2 (100 mL) was added over a period of 60 min and the reaction mixture was stirred for 4 h at rt. Then water was added and mixture was extracted with CH2Cl2 (3 * 300 mL). The organic layer was dried (Na2SO4) and concentrated in vacuum, (dichloromethane/MeOH = 85/15, Rf = 0.18). Pale yellow solid, mp 72-82 C, yield 14.0 g, (65%). Crude purity: 92%, tR = 10.7 min. FT-IR (neat): nu (cm-1) = 3100 (O-H), 1694 (C=O), 1655 (HOC=O). Exact mass (ESI): m/z = calcd. for (C8H8O3S)H 185.0292, found 185.0298. 1H NMR (DMSO-d6): delta (ppm) = 2.56 (t, J = 6.7 Hz, 2H, COCH2), 3.18 (t, J = 6.7 Hz, 2H, CH2CO2H), 7.23 (t, J = 4.8 Hz, 1H, 4-CH), 7.68 (dd, J = 4.8/0.9 Hz, 1H, 3-CH), 7.72 (dd, J = 4.8/0.9 Hz, 1H, 5-CH), 12.2 (bs, 1H, CO2H). 13C NMR (DMSO-d6): delta (ppm) = 27.7 (1C, COCH2), 32.4 (1C, CH2CO2H), 128.7 (1C, C-4), 133.1 (1C, C-3), 134.5 (1C, C-5), 143.3 (1C, C-2), 173.6 (1C, CO2H), 191.6 (1C, C=O).
With aluminum (III) chloride; In dichloromethane; at 20℃;
Step 1: S23-SM (28.5 mL, 0.36 mol) was dissolved in anhydrous DCM (170 mL) and the solution was added to a suspension of dihydrofuran-2,5-dione (35.7 g, 0.36 mol) and A1C13 (104 g, 0.36 mol) in anhydrous DCM (350 mL) at room temperature. The reaction was stirred over night. The mixture was added to a solution of 250 mL of cold HC1 and 200 m/l of cold water at 0C. The mixture was extracted twice with DCM. The combined organic layers were washed with NaOH (4N aq., 500 mL) solution, and extracted with DCM. After acidification with HC1 (4N aq.), the aqueous phase was extracted with DCM, dried over Na2SO4, concentrated to affordS23-1.
With aluminum (III) chloride; In dichloromethane; at 20℃;
Step 1: S23-SM (28.5 mL, 0.36 mol) was dissolved in anhydrous DCM (170 mL) and the solution was added to a suspension of dihydrofuran-2,5-dione (35.7 g, 0.36 mol) and A1C13 (104 g, 0.36 mol) in anhydrous DCM (350 mL) at room temperature. The reaction was stirred over night. The mixture was added to a solution of 250 mL of cold HCl and 200 m/1 of cold water at 0 C. The mixture was extracted twice with DCM. The combined organic layers were washed with NaOH (4N aq., 500 mL) solution, and extracted with DCM. After acidification with HCl (4N aq.), the aqueous phase was extracted with DCM, dried over Na2S04, concentrated to afford S23-1.
With aluminum (III) chloride; In dichloromethane; at 33 - 37℃; for 6.0h;
15.8 g of 1-methoxynaphthalene and 10.0 g of succinic anhydride were dissolved in 120 mL of dichloromethane,Stir,Cooling to 1 ~ 3 C,Divided into three batches of anhydrous aluminum trichloride 15.0 grams,The addition process takes about 20 minutes,The solution was then heated to 35 ± 2 C,Insulation reaction 6 hours (5.5 ~ 6.5 hours range),After the reaction is complete,The reaction solution was poured into an ice-water mixture (200 g of ice and 300 g of water) for 30 minutes,Standing, analysiscrystal,Filter,The filtrate was heated and distilled to recover dichloromethane,The cake is the crude of the ketone ketone;And then the crude ketoprofen water as a solvent by 2-3 times recrystallization,Activated carbon decolorization,Demon ketone boutique.The mass of the present product of the chamballone was 22.3 g,Melting point of 176 ~ 179 C,The yield was 86.4%.
Compound 4 (1 g; 0.88 mmol; 1 eq.) is dissolved in 20 ml of anhydrous DMF under an inert atmosphere, in a clean and dry 100 ml round-bottomed flask. Succinic anhydride (0.135 g; 1.34 mmol; 1.5 eq.) dissolved in 6 ml of anhydrous DMF is added. The reaction medium is left under an inert atmosphere for 18 hours at ambient temperature. The reaction is stopped with 120 muL of water and the solution is then precipitated from 200 ml of acetone. The solid obtained is filtered off and then dried in a desiccator. The solid is taken up in a minimum of water, the insoluble material is filtered off and the filtrate is lyophilized. Compound 5 is obtained with a 55percent yield. TLC: Rf=0.6 eluent: DMF/BuOH/H2O 1/2/1 (v/v/v) M.p.: 160° C. (decomposition) 1H NMR pyridine-d5 delta (ppm): 8.75 (s, 1H, NH-CD); 7.9-7.6 (14OH); 5.8-5.55 (m, 7H, H1-CD); 4.95-3.95 (m, H3-CD/H6-CD/H6'-CD/H5-CD/H4-CD/H2-CD); 3 (m, 4H, Hb/Hc) ESI-MS+: m/z measured at 1234.5 [M+H]+, calculated at 1234.4 for C46H76NO37
poly(ethylene glycol), α-terminated with 4-vinylbenzyloxy group, ω-terminated with 2-carboxylatoethylcarboxy group, potassium salt, Mn 1.8E3 Da; monomer(s): 4-vinylbenzyl alcohol; ethylene oxide; succinic anhydride[ No CAS ]
Part A; Succinic anhydride (10.0 g, 100 mmol) was added to a suspension of 4-amino-2- ethoxymethyl-alpha,alpha-dimethyl-lH-imidazo[4,5-c]quinoline-l-ethanol (31.4 g, 100 mmol, Example 99 in US Patent No. 5,389,640) in toluene (about 500 mL) and the reaction mixture was heated to reflux. Additional succinic anhydride (10.0 g) was added after 24 hours and again after 72 hours. The reaction was heated for a total of 6 days and then allowed to cool. A white solid was isolated by filtration. The solid was twice triturated with methanol, filtered, and dried. The resulting solid was suspended in methanol (300 mL). The suspension was heated at reflux for 2 hours and then allowed to cool. A solid was isolated by filtration and then dried under high vacuum at 100 0C to provide 29.6 g of l-[2-(ethoxymethyl)-l-(2-hydroxy-2-methylpropyl)-lH-imidazo[4,5-c]quinolin-4- yl]pyrrolidine-2,5-dione.
In methanol; toluene;
Part A Succinic anhydride (10.0 g, 100 mmol) was added to a suspension of 4-amino-2-ethoxymethyl-alpha,alpha-dimethyl-1H-imidazo[4,5-c]quinoline-1-ethanol (31.4 g, 100 mmol, Example 99 in U.S. Pat. No. 5,389,640) in toluene (about 500 mL) and the reaction mixture was heated to reflux. Additional succinic anhydride (10.0 g) was added after 24 hours and again after 72 hours. The reaction was heated for a total of 6 days and then allowed to cool. A white solid was isolated by filtration. The solid was twice triturated with methanol, filtered, and dried. The resulting solid was suspended in methanol (300 mL). The suspension was heated at reflux for 2 hours and then allowed to cool. A solid was isolated by filtration and then dried under high vacuum at 100 C. to provide 29.6 g of 1-[2-(ethoxymethyl)-1-(2-hydroxy-2-methylpropyl)-1H-imidazo[4,5-c]quinolin-4-yl]pyrrolidine-2,5-dione.
In toluene; for 168h;Heating / reflux;
A lL-round bottom flask, equipped with a Dean-Stark trap, was charged with A- amino-alpha,alpha-dimethyl-2-ethoxymethyl-lH-imidazo[4,5-c]quinolin-l-ethanol (i.e. l-[4- amino-2-(ethoxymethyl)-lH-imidazo[4,5-c]quinolin-l-yl]-2-methylpropan-2-ol (31.4 g, 100 mmol), synthesis of which is described, for example, at U.S. Pat. No. 5,389,640, Example 99. Anhydrous toluene (500 mL) was added followed by succinic anhydride (10.0 g, 100 mmol) and the mixture was heated to reflux for 24 hours. Another 10.0 g succinic anhydride was added to the reaction mixture and heating was continued for 2 days. The reaction was still not complete so an additional 10.0 g of succinic anhydride was added and heating was continued for 4 days. The reaction mixture was then cooled and filtered to give a white powder. The white powder was stirred in refluxing methanol (300 mL), cooled and filtered to give a white solid. The treatment with refluxing methanol was repeated two more times to give a white powder that was dried by suction and then under vacuum at 100 0C to give 29.6 g of l-[2-(ethoxymethyl)-l-(2-hydroxy-2-methylpropyl)- lH-imidazo[4,5-c]quinolin-4-yl]pyrrolidine-2,5-dione.
15 g (0.902 mole) of <strong>[18979-61-8]rucinol</strong> was dissolved in pyridine solvent under atmosphere of nitrogen gas and 27.1 g (0.27 mole) of succinic anhydride was added to the solution. After then, the solution was subject to a reaction at room temperature for 24 hours under a state that the light was blocked. After the completion of the reaction, pyridine was adjusted to pH 2 by adding hydrochloric acid and thus converted into a salt form, and chloroform and water were added to extract and remove it. Similarly, an excessive succinic anhydride was also extracted and removed with the water layer. 4.06 g (0.011 mole) of <strong>[18979-61-8]rucinol</strong>-carboxyl acid obtained as above was dissolved in dichloromethane solvent under atmosphere of nitrogen gas and carboxyl group was activated by using 3.09 ml (0.022 mole) of triethylamine and 2.11 g (0. 011 mole) of p- toluenesulfonylchloride. After that, phytosphingosine, sphinganine or sphingadiene 7.75 g (0.024 mole) was slowly added to the solution and then was subject to a reaction for 24 hours while warming. After the completion of the reaction, the reaction was stopped by extracting with distilled water, and refined with an adsorption-chromatography by silica gel, so that compounds of the above formulas 1 to 3 were obtained. The succinate, which was introduced in <strong>[18979-61-8]rucinol</strong> by reacting with succinic anhydride, was confirmed with 1H NMR (6 = 2.6 ppm, t, 4H), and finally methylene of sphingolipid (8 = 1. 2 ppm, m, 48H and 8 = 0.9 ppm, t, 6H) and benzene group of <strong>[18979-61-8]rucinol</strong> (8 = 6.9 ppm, 6.35 ppm, d, t, 5H) were confirmed. Accordingly, it was confirmed that the derivative having <strong>[18979-61-8]rucinol</strong> and sphingolipid coupled was synthesized.
With sodium acetate; In water; acetic anhydride; benzene;
EXAMPLE 47 STR151 2-Bromo-6-(N-succinimido)-naphthalene (29) A solution of <strong>[7499-66-3]2-amino-6-bromonaphthalene</strong> (0.565 g, 2.54 mmol) and succinic anhydride (0.508 g, 5.08 mmol) in 13 mL of benzene was refluxed for 1.5 hours. The resulting precipitate was isolated by filtration, washing with additional benzene. After drying in vacuo, 0.780 g of a solid was obtained which was dissolved in 12 mL of acetic anhydride along with sodium acetate (0.595 g, 7.26 mmol) and refluxed for 3 hours. After cooling to room temperature, the reaction mixture was quenched with 3 mL of water and was evaporated to dryness under high vacuum. Purification by flash chromatography through 150 g of silica gel (1:4 EtOAc/CH2 Cl2) yielded 0.670 g (86percent) of the title compound as a white solid. 1 H-NMR (300 MHz, CDCl3): delta 2.95 (bs, 4H), 7.41 (dd, J=8.8, 1.9 Hz, 1H), 7.59 (dd, J=8.8, 1.7 Hz, 1H), 7.73 (d, J=8.8 Hz, 1H), 7.79 (s, 1H), 7.84 (d, J=8.8 Hz, 1H), 8.04 ppm (s, 1H). FAB-MS: m/e=504,506 (M+H).
With hydrogenchloride; sodium carbonate;aluminium trichloride; In thiophene; water; nitrobenzene;
EXAMPLE 1 1-[3-(Acetylthio)-3-(2-thenoyl)propionyl]-L-proline A solution of 0.3 mole of thiophene in 100 ml. of nitrobenzene is added dropwise with stirring to a cooled mixture of 30 g. (0.3 mole) of succinic anhydride, 87.8 g. (0.66 mole) of aluminum chloride and 300 ml. of nitrobenzene. The mixture is cooled and stirred for 30 minutes and then allowed to stand at room temperature overnight. The mixture is poured onto ice and 50 ml. of concentrated hydrochloric acid. The nitrobenzene is removed by steam distillation. The aqueous layer is decanted and the residue is mixed with 650 ml. of water and 20 g. of sodium carbonate. The mixture is heated, mixed with diatomaceous earth and filtered. The filtrate is acidified with concentrated hydrochloric acid and the solid which separates is filtered and washed with water. Recrystallization from water gives 53% of 4-oxo-4-(2-thienyl)butyric acid, m.p. 121-123 C.
1-[N-(3-carboxy-1-oxopropyl)-L-alanyl]-L-proline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; ethyl acetate;
(a) 1-[N-(3-Carboxy-1-oxopropyl)-L-alanyl]-L-proline, phenylmethyl ester Diisopropylethylamine (0.85 ml., 4.9 mmole) is added to a mixture of succinic anhydride (0.49 g., 4.9 mmole) and <strong>[13485-59-1]L-alanyl-L-proline</strong>, phenylmethyl ester, tosylate salt (2.2 g., 4.9 mmole) in tetrahydrofuran (50 ml.). The resulting mixture is stirred at 25 for 17 hours, after which it is concentrated. The residue is redissolved in ethyl acetate and is washed with water and extracted with 10% aqueous sodium bicarbonate solution. The aqueous extract is acidified with hydrochloric acid and then extracted with ethyl acetate. The ethyl acetate phase is dried and concentrated to give 1-[N-(3-carboxy-1-oxopropyl)-L-alanyl]-L-proline, phenylmethyl ester as a clear oil.
caesium carbonate; In 1,4-dioxane; for 4h;Heating / reflux;
A mixture of 1.50 g (0.01 mol) of <strong>[4170-90-5]2,4,6-trimethylbenzyl alcohol</strong>, 1.00 g (0.01 mol) of succinic acid anhydride, 3.25 g (0.01 mol) of cesium carbonate, and 100 ml of dioxane was stirred at reflux temperature for 4 hours. The reaction mixture was cooled to room temperature and then filtered to remove the solid cesium carbonate. The solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (acetone-hexane) to provide 1.167 g of succinic acid mono-(2,4,6-trimethylbenzyl) ester (46.62%).
To a solution of N-methyl-N-boc hydroxylamine3 (536 mg, 3.65 mmol) in ichloromethane (8 mL) were added DMAP (134 mg, 1.10 mmol) and succinic anhydride (548 mg, 5.48 mmol). The resulting mixture was stirred for 16 h at room temperature. The reaction mixture was poured into a saturated solution of ammonium chloride (10 mL) and extracted with ethyl acetate (2 x 40 mL). The combined organic layer was washed with water (20 mL) and dried over sodium sulfate. The solvent was removed in vacuo and the crude product was purified by silica flash column chromatography using 45percent EtOAc/hexane as eluent to give the corresponding acid as a white solid (693 mg, 2.81 mmol, 77percent). 1H NMR (CDCl3, 400 MHz): delta (ppm) 1.42 (s, 9 H), 2.65-2.70 (m, 4 H), 3.17 (s, 3 H).
With triethylamine; In tetrahydrofuran; at 0℃;Cooling with ice;
Example 5 4-(2-(2-(tert-butoxycarbonylamino)ethoxy)ethylamino)-4-oxobutanoic acid In a one-necked flask (250 mL), 20.155 g (98.7 mmol) of the compound 4 and 50 mL of THF (dried over molecular sieves) were dissolved and the reaction mixture was cooled on an ice-water bath. To the flask, a mixture prepared by dissolving 12.844 g (128.4 mmol) of succinic anhydride in 50 mL of THF was added slowly, and then 42 mL of triethylamine was also added slowly with stirring on an ice-water bath. After reaction completion, the solution was dried to remove the solvent, and the resultant concentrate was dissolved in chloroform, washed with 5% citric acid twice, saturated brine once, dried over anhydrous MgSO4, filtered, and dried to yield the title compound 5.
With triethylamine; In tetrahydrofuran;Cooling with ice;
Example 5 4-(2-(2-(tert-butoxycarbonylamino)ethoxy)ethylamino)-4-oxobutanoic acid In a one-necked flask (250 mL), 20.155 g (98.7 mmol) of the compound 4 and 50 mL of THF (dried over molecular sieves) were dissolved and the reaction mixture was cooled on an ice-water bath. To the flask, a mixture prepared by dissolving 12.844 g (128.4 mmol) of succinic anhydride in 50 mL of THF was added slowly, and then 42 mL of triethylamine was also added slowly with stirring on an ice-water bath. After reaction completion, the solution was dried to remove the solvent, and the resultant concentrate was dissolved in chloroform, washed with 5% citric acid twice, saturated brine once, dried over anhydrous MgSO4, filtered, and dried to yield the title compound 5.
General procedure: One molar equivalent of 2-aminothiazole derivatives in 50 mL THF were mixed with 1 molar equiv of succinic anhydride or glutaric anhydride. The reaction solution was refluxed at 65 C for 7 h, and then evaporated under vacuum to yield crude product. The crude product was dissolved in a cooled aqueous solution of K2CO3/KOH, and then filtered to discard the insoluble solid. The combined aqueous phase was acidified by 1 M HCl to pH 1.0 to obtain milk-white deposits. The deposits were then recrystallized from ethyl acetate/ethanol to obtain a pure milk-white powder.
With dmap; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 48h;
[00869] Specifically, beta-estradiol 17-acetate (1) and a 10-fold excess of succinic anhydride (2) were stirred together in DMF with 5 DIEA and DMAP for 48 hours at room temperature. After 48 hours, the reaction solvent was evaporated in vacuo and the product (3) was re-suspended in ethyl acetate. The reaction product (3) was washed with aqueous HC1 (0.0 IN) two times followed by a single wash with water. The ethyl acetate was evaporated in vacuo and the product (3) was re-suspended in a mixture of MeOH/acetonitrile/water. The estrogen derivative product (3) was purified by reverse-phase HPLC. Lyophilized product (3) and the resin-bound peptide were mixed in HOBt/DIC/NMP for 4 hours, filtered, treated with TFA, and cleaved from the resin by HF treatment. The peptide-estrogen conjugate was purified using reverse-phase HPLC and characterized by ESI mass spectrometry.
With dmap; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 48h;
Specifically, 3-estradiol 17-acetate (1) and a 10-fold excess of succinic anhydride (2) were stirred together in DMF with5percent DIEA and DMAP for 48 hours at room temperature. After48 hours, the reaction solvent was evaporated in vacuo and the product (3) was re-suspended in ethyl acetate. The reaction product (3) was washed with aqueous HC1 (0.0 iN) two times followed by a single wash with water. The ethyl acetate wasevaporated in vacuo and the product (3) was re-suspended ina mixture of MeOH/acetonitrile/watet The estrogen derivative product (3) was purified by reverse-phase HPLC. Lyophilized product (3) and the resin-bound peptide were mixed in H013t/DIC/NMP for 4 hours, filtered, treated with TFA,and cleaved from the resin by HF treatment. The peptideestrogen conjugate was purified using reverse-phase HPLC and characterized by ESI mass spectrometry.
In tetrahydrofuran; at 110℃; for 1.5h;Microwave irradiation;
4-((2-cyanobenzo[d]thiazol-6-yl)amino)-4-oxobutanoic acid (1). 6- aminobenzo[d]thiazole-2-carbonitrile (2.0 g, 11.4 mmol), succinic anhydride (1.3 g, 13 mmol) and THF (15 mL) were placed in a 25 mL vessel and heated in a microwave synthesizer for 90 minutes at 110°C. Upon cooling, the reaction mixture was triturated with Et20 and filtered, dried and evaporated to give 3.1 g of the product as a light yellow solid (99percent). IH-NMR (d6-DMSO, 300 MHz): delta 12.15 (s, 1H), 10.45 (s, 1H), 8.71 (s, 1H), 8.16 (d, 1H, J = 8.2 Hz), 7.70 (d, 1H, J= 8.2 Hz), 2.62 (m, 2H), 2.55 (m, 2H). ESI-MS: Calc. C12H10N3O3S +: m/z 276.3; found m/z 276. tert-butyl (18-((2-cyanobenzo [d] thiazol-6-yl)amino)- 15,18-dioxo-4,7, 10-trioxa-14- azaoctadecyl)carbamate (2). Compound 1 (4.93g, 17.9 mmol), tert-butyl (3-(2-(2-(3- aminopropoxy)ethoxy)ethoxy)propyl)carbamate (7.40 g, 23.1 mmol) and DCM:DMF (10: 1, 100 mL) were stirred together in a 250 mL round bottomed flask at room temperature. ED AC (4.0 g, 20.9 mmol) was added and the reaction was stirred for 20h. The solvent was evaporated and purified by normal phase chromatography with DCM/MeOH as solvent to give 6.62 g of a white solid (64percent). IH-NMR (d3-ACN, 300 MHz): delta 9.21 (s, NH), 8.62 (d, 1H, J = 2.0 Hz), 8.09 (d, 1H, J = 8.4 Hz), 7.63 (d, 1H, J = 8.4 Hz), 6.65 (bs, NH), 5.40 (bs, NH), 3.5 (m, 12 H), 3.28 (m, 2H), 3.06 (m, 2H), 2.65 (m, 2H), 2.51 (m, 2H), 2.70 (m, 4H), 1.40 (s, 9H). ESI-MS: Calc. C27H40N5O7S +: m/z 578.7; found m/z 578.4. Nl-(3-(2-(2-(3-aminopropoxy)ethoxy)ethoxy)propyl)-N4-(2-cyanobenzo[d]thiazol-6- yl)succinamide hydrochloride (3). Compound 2 (6.62 g, 11.5 mmol) was stirred in a 500 mL round bottomed flask with DCM (200 mL) and triisopropylsilane (1 mL). A 4.0 M solution of HCl in dioxane (30 mL, 120 mmol) was added and stirred at room temperature for 3h. The solvent was evaporated to give 6.4 g of a yellow hygroscopic solid (98percent). 1H-NMR (d6-DMSO, 300 MHz): delta 10.62 (s, 1H), 8.74 (d, 1H, J = 2.0 Hz), 8.15 (d, 1H, J = 8.4 Hz), 7.77 (d, 1H, J = 8.4 Hz), 3.4 (m, 16 H), 3.28 (m, 2H), 3.08 (m, 2H), 2.80(m, 2H), 2.61 (m, 2H), 2.41 (m, 2H), 1.80, (m, 2H), 1.60 (m, 2H). ESI-MS: Calc. C22H32N505S +: m/z 478.59; found m/z 478.2. Immobilized cyanobenzothiazole-magnetic cellulose (4). Carboxymethyl magnetic cellulose (7.24g, 30-50 muiotaeta, Iontosorb MG CM) was taken up in a 250 mL round bottomed flask with compound 3 (800 mg, 1.53 mmol) in DMF (100 mL). ED AC (387 mg, 2.01 mmol) was added, and the reaction was stirred for 20h at room temperature. The particles were filtered on a frit and rinsed first with DMF (200 mL) then 25percent EtOH (300 mL) and stored as a 50percent> suspension at 4°C.
General procedure: A mixture of succinic anhydride (1.0 eq) and dry alcohol (1 eq) in toluene (4 mL) was heated at reflux 2.5 h. The toluene was removed under reduced pressure and the residue was taken up in water, and the solution was extracted with dichloromethane, dried (MgSO4), and evaporated to afford monoalkylsuccinic acid. A solution of monoalkyl succinic acid (1.2 eq) and thionyl chloride (1.4 eq) in benzene (5 mL) was refluxed for 3 h. Subsequently, the majority of the SOCl2 and benzene were removed by distillation. The mixture was cooled down to room temperature and dried under a vacuum to give a crude chlorocarbony-alkyl ester. A solution of chlorocarbonyl-alkyl ester in dichloromethane (5 mL) was added to a round flask containing 14 (1 eq) by cannula, and subsequently added pyridine (1.4 eq). The resulting solution was stirred at room temperature overnight, and quenched by adding water. The solution was extracted with ethyl acetate, dried (MgSO4), and evaporated to give a residue which was purified by column chromatography (Al2O3), eluting by ethyl acetate /hexane (1:15) to provide 25?43.
4-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethoxy)-4-oxobutanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In dichloromethane; for 3h;
[00171 ] Synthesis of compound 9. N-(2-hvdroxyethyl)-1 H-pyrrole-2-5-dione (141 mg, 1 .00 mmol) was dissolved in dichloromethane (1 mL), triethylamine (101 mg, 1 .00 mmol) was added, followed by succinic anhydride. After 3 hours, solvent was evaporated.
An oseltamivir-PEG-Succinyl-Diaminopropionyl-Asp-Cys-conjugate (a novel water soluble neuraminidase inhibitor for the detection of influenza viruses) has been synthesized for direct attachment to inorganic surfaces. This conjugate was prepared from Fmoc-Cys(4-methoxytrityl)-Wang resin, swollen with CH2Cl2 followed by DMF. A solution of 20% piperidine in DMF was added to the resin, and argon was bubbled for 5 minutes. The resin was washed with DMF and isopropyl alcohol. Formation of free amine was assessed by the Kaiser test. After swelling the resin in DMF, a solution of Fmoc-Asp-(OtBu)-OH, DAPA-OH, succinic anhydride, oseltamivir-PEG-amine, and HBTU, HOBt plus DIPEA in DMF was added. Argon was bubbled for 2 hours, and resin was washed with DMF and i-PrOH. The coupling efficiency was assessed by the Kaiser Test. The above sequence was repeated for the 4 required sequential coupling steps. The final compound was cleaved from the resin using a TFA/H2O/iPr3SiH/ethanedithiol cocktail (37:1:1:1) and concentrated under vacuum. The concentrated product was precipitated in diethyl ether and dried under vacuum. The crude product was purified by preparative RP-HPLC using aqueous CH3CN and then freeze-dried to yield the desired compound as a white solid.
A oseltamivir-PEG-Succinyl-Diaminopropionyl-Asp-Cys-conjugate (a novel water soluble neuraminidase inhibitor for the detection of influenza viruses) has been synthesized for direct attachment to inorganic surfaces. This conjugate was prepared from Fmoc-Cys(4-methoxytrityl)-Wang resin, swollen with CH2Cl2 followed by DMF. A solution of 20% piperidine in DMF was added to the resin, and argon was bubbled for 5 minutes. The resin was washed with DMF and isopropyl alcohol. Formation of free amine was assessed by the Kaiser test. After swelling the resin in DMF, a solution of Fmoc-Asp-(OtBu)-OH, Diaminopropionic acid, succinic anhydride, oseltamivir-amine, and HBTU, HOBt plus DIPEA in DMF was added. Argon was bubbled for 2 hours, and resin was washed with DMF and i-PrOH. The coupling efficiency was assessed by the Kaiser Test. The above sequence was repeated for the 4 required sequential coupling steps. The final compound was cleaved from the resin using a TFA/H2O/iPr3SiH/ethanedithiol cocktail (37:1:1:1) and concentrated under vacuum. The concentrated product was precipitated in diethyl ether and dried under vacuum. The crude product was purified by preparative RP-HPLC using aqueous CH3CN and then freeze-dried to yield the desired compound as a white solid.
A oseltamivir-3-pentyloxy-PEG-Succinyl-Diaminopropionyl-Asp-Cys-conjugate (a novel water soluble neuraminidase inhibitor for the detection of influenza viruses) has been synthesized for direct attachment to inorganic surfaces. This conjugate was prepared from Fmoc-Cys(4-methoxytrityl)-Wang resin, swollen with CH2Cl2 followed by DMF. A solution of 20% piperidine in DMF was added to the resin, and argon was bubbled for 5 minutes. The resin was washed with DMF and isopropyl alcohol. Formation of free amine was assessed by the Kaiser test. After swelling the resin in DMF, a solution of Fmoc-Asp-(OtBu)-OH, DAPA-OH, succinic anhydride, oseltamivir-3-pentyloxy-Peg amine, and HBTU, HOBt plus DIPEA in DMF was added. Argon was bubbled for 2 hours, and resin was washed with DMF and i-PrOH. The coupling efficiency was assessed by the Kaiser Test. The above sequence was repeated for the 4 required sequential coupling steps. The final compound was cleaved from the resin using a TFA/H2O/iPr3SiH/ethanedithiol cocktail (37:1:1:1) and concentrated under vacuum. The concentrated product was precipitated in diethyl ether and dried under vacuum. The crude product was purified by preparative RP-HPLC using aqueous CH3CN and then freeze-dried to yield the desired compound as a white solid.
4-((3,5-dichlorobenzyl)amino)-4-oxobutanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In 1,4-dioxane; at 80℃; for 0.5h;
(a) Succinic anhydride (1 eq) and (3,5-dieh1oropheny{)memanamine (I eq) in dioxane was heaied to 80 C for 30 min. The solvent was evaporated off and residue was purified via flash column chromatography directly eluting with 20% MeOH in DCM to yield 4-((3,5-dichlorobenzyl)amino)-4-oxobutanoic acid.
[00243] 2-Hydroxyethyl maleimide (120 mg, 0.85 mmol, 2.00 equiv) was suspended in THF (4.0 mL) then A/-methyl morpholine (93 mu, 0.85 mmol, 2.0 equiv) was added followed by succinic anhydride (85 mg, 0.85 mmol, 2.0 equiv). The solution was stirred at room temperature for 16 hours, then N- methyl morpholine (93 mu, 0.85 mmol, 2.0 equiv) was added followed by isobutyl chloroformate (1 10 mu, 0.85 mmol, 2.0 equiv) and the solution was stirred at room temperature for 45 minutes. Water (10 mL) and ethyl acetate (10 mL) were added and the layers were separated.
2-Hydroxyethyl maleimide (120 mg, 0.85 mmol, 2.00 equiv.) was suspended in THF (4.0 mL) then N-methyl morpholine (93 uL, 0.85 mmol, 2.0 equiv.) was added followed by succinic anhydride (85 mg, 0.85 mmol, 2.0 equiv.). The solution was stirred at room temperature for 16 hours, then N-methyl morpholine (93 uL, 0.85 mmol, 2.0 equiv.) was added followed by isobutyl chloroformate (110 uL, 0.85 mmol, 2.0 equiv.) and the solution was stirred at room temperature for 45 minutes. Water (10 mL) and ethyl acetate (10 mL) were added and the layers were separated. Solvent was then evaporated, then the activated ester was dissolved in DMF and hydroxy(2- methylethoxyacetoxy)oxalplatin(IV) (233 mg, 0.43 mmol, 1.0 equiv.) was added and solution was stirred sat 50 °C for 18 hours. Solvent was evaporated from this mixture and the crude product was purified on silica gel (0-8percent MeOH/DCM gradient) to afford the desired product with residual MeOH. The residue was dissolved in MeOH (2 mL) and the resulting solution was added to TBME (50 mL) to obtain a white precipitate (compound 13) that was filtered and dried under vacuum. Compound 13 was obtained as an off- white solid (131 mg, 40percent yield). HPLC/MS (method B): 3.41 minutes, M+H = 771
The <strong>[3681-99-0]puerarin</strong>(4.16 g, 0. Olmol),Succinic anhydride(1.98 g, 0.012 mol)Was added to dichloromethane (80 ml), DMAP was added(100 mg), heated to reflux,Reaction 24h; reaction is completed,Spin drying solvent,Add water 20ml,Preparative column separation and purification,Collect fractions,Concentrated to 20 mL volume,The pH was adjusted to 8 with sodium bicarbonate and the aqueous solution was poured into ethanol(200 ml)A white solid precipitated,filter,Drying in vacuo gave a white solid (3.41 g, 63.3percent).
4-oxo-4-((4-(4-chlorophenoxy)phenyl)amino)butanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
In toluene; for 12h;Reflux;
General procedure: Anhydrides of succinic or glutaric acid (1.1 eq.) were added to the solution of the corresponding phenoxyaniline(1 eq.) in toluene (200 eq.). The mixture was refluxed for 12 h and then cooled to room temperature. The precipitate that formed was filtered off and dissolved in CH2Cl2. The solution was washed with a saturated solution NH4Cl. The organic layer was dried on anhydrous Na2SO4. Finally, the solvent was evaporated to obtain compounds 4a-h.
With pyridine; at 48℃; for 9h;Darkness; Sealed tube;
1, take 50mL conical flask,Followed by adding 1.012 g of <strong>[113507-06-5]moxidectin</strong>,20 mL of anhydrous pyridine and 0.5 g of succinic anhydride,The bottle sealed with sealer placed at 48 ,240rpm, dark conditions, the reaction 9h.2. After completion of step 1, pyridine is removed by rotary evaporation to give the product.3, the product of step 2 was dissolved in 50mL of ethyl acetate,Then add 15mL 1g / 100mL aqueous sodium hydroxide solution, mix well and let stand for stratification, discard the organic phase,The aqueous phase was collected and the pH adjusted to 4-5 with 2M hydrochloric acid solution.4, take the liquid phase obtained in step 3,Extract three times with ethyl acetate (each adding an equal volume of ethyl acetate to the liquid,The organic phase was collected on each extraction) and the organic phases were combined.5, take the organic phase obtained in Step 4, add an excess of anhydrous sodium sulfate, then filtered and the filtrate was collected.6, take the filtrate obtained in step 5, 4500g centrifuged 15min, the supernatant was collected.7, take the supernatant obtained in Step 6, after rotary evaporation, the product (yellow oil).8. Take the product from Step 7 and recrystallize three times to give the product (brown powder).
[3-(methoxycarbonyl)propanoyloxy]methyl(methoxy)(phenyl)silane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With CBV780; at 140℃; for 0.5h;Microwave irradiation;
The same procedure as in Example 1 was repeated with varying reaction conditions (such as catalysts, starting materials, reaction temperature, reaction time and the like), to carry out reactions, and the analyses of the resulting products. The results of the yields of the respective products as measured by gas chromatography or nuclear magnetic resonance spectroscopy are shown in Table 1-ito Table 1-7.
4-[(4'-hydroxy-3,3',5,5'-tetraisopropylbiphenyl)-4-oxy]-4-oxobutyric acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
54.59%
In dimethyl sulfoxide; at 90℃; for 5h;
4,4?-dihydroxy-3,3?,5,5?-tetraisopropylbiphenyl (5.00 g, 14.10 mmol) was dissolved in DMSO (20 mL), and succinic anhydride (1.41 g, 14.09 mmol) was then added thereto and heated at 90° C. for a reaction for 5 h. The reaction solution was cooled to room temperature, into which water was added, extracted with ethyl acetate, dried over anhydrous sodium sulfate, and filtered to remove sodium sulfate. The filtrate was purified with petroleum ether-ethyl acetate eluent to give [4-(4?-hydroxy-3,3?,5,5?-tetraisopropylbiphenyl)oxy]-4-carbonylbutyric acid (3.50 g, 54.59percent) as a white solid. 1H NMR (300 MHz, CDCl3) delta 11.10 (s, 1H), 7.65 (s, 2H), 7.51 (s, 2H), 5.35 (s, 1H), 3.07-3.04 (m, 4H), 2.71 (s, 4H), 1.20-1.18 (d, 24H)
9,10-bis(2-carboxyethyl)carbonyloxyanthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With triethylamine; In N,N-dimethyl acetamide; at 60℃; for 2h;
2.1 g (10 mmol) of 9,10-dihydroxyanthracene, 2.5 g (25 mmol) of succinic anhydride and 15 g of N, N-dimethylacetamide were added to a three-necked flask equipped with a stirrer, To a yellow-green slurry, a solution of 2.0 g (20 mmol) of triethylamine in 4 g of N, N-dimethylacetamide was added dropwise. It became a slightly brown slurry. Subsequently, the mixture was heated at 60 ° C. for 2 hours to obtain a brown solution. After cooling to room temperature, it was poured into a 3percent sulfuric acid aqueous solution, and a large amount of ocher-colored precipitation occurred. The precipitate formed was suction filtered, washed with water and dried to obtain 2.7 g (6.5 mmol) of an ocher powder of 9,10-bis (2-carboxyethyl) carbonyloxyanthracene.Isolated yield to 9,10-dihydroxyanthraceneWas 65 molpercent.
4-(2-carbamoyl-5-methylanilino)-4-oxobutanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
With acetic acid; at 20℃; for 2h;
To a stirred solution of 2-amino-4-methyl-benzamide (190 mg, 1.27 mmol) in AcOH (5 mL),succinic anhydride (151 mg, 1.52 mmol) was added at RT and stirred for 2 h (TLC indicatedcomplete consumption of starting material). The reaction mixture was diluted with water (20mL), solid was precipitated out, filtered and dried under vacuum to get the required 4-(2-carbamoyl-5-methyl-anilino)-4-oxo-butanoic acid (250 mg, 89percent) which was used for nextstep without further purification.1H NMR [300 MHz, DMSO-d6]: J 12.37 (brs, 1H), 11.88 (s, 1H), 8.33 (s, 1H), 8.20 (s, 1H),7.70 (d, J = 8.1 Hz, 1H), 7.64 (s, 1H), 6.91 (d, J = 7.8 Hz, 1H), 2.53-2.50 (m, 4H), 2.31 (s,3H).LCMS: m/z: 273.50 [M+Nat.
4-[(3-carbamoyl-2-thienyl)amino]-4-oxobutanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With acetic acid; at 20℃; for 2h;
To a stirred solution of <strong>[14080-51-4]2-aminothiophene-3-carboxamide</strong> (300 mg, 2.11 mmol) in acetic acid(3 mL) was added succinic anhydride (253 mg, 2.53 mmol) at RT. The the reaction mixturewas stirred at RT for 2 h. (TLC indicated complete consumption of starting material). Thereaction mixture was then diluted with water (20 mL) and precipitation was formed. Themixture was filtered and the obtained solid was dried under vacuum to provide 4-[(3-carbamoyl-2-thienyl)amino]-4-oxo-butanoic acid (410 mg, 69%), which was used for nextstep without further purification.1H NMR [400 MHz, DMSO-d6]: J 12.20 (s, 1H), 12.14 (s, 1H), 7.89 (s, 1H), 7.50 (brs, 1H),7.39 (d, J = 5.6 Hz, 1H), 6.93 (d, J = 6.0 Hz, 1H), 2.67 (t, J = 6.4 Hz, 2H), 2.56 (t, J = 6.8 Hz,2H).LCMS: m/z: 265.40 [M+Nat.
With benzotriazol-1-ol; O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate;
Manual synthesis using FMOC solid phase synthesis strategy. Using FMOC-Gly-Wang resin (glycine bonding amount is0.698mmol/g) as starting material, 20percent hexahydropyridine/DMF as deprotecting reagent, N-methylmorpholine and HBTU as liveChemical reagent.The specific synthesis step of the AP2H polypeptide sequence is as follows: the weighed FMOC-Gly-Wang resin is loaded into the sand core funnel,DMF was swollen for 30 minutes and then washed three times with DMF. Add 20percent hexahydropyridine/DMF solution at a ratio of 30 mL/g resin, magneticAfter stirring for 5 minutes, the mixture was drained and repeated twice to remove the FMOC protecting group. Wash the resin with DMF six times, stirring for 1 minute each time, pumpingDry for 10 seconds. Dissolving a mixture of FMOC-AA-OH with HBTU and HOBt with 0.4 mol/L N-methylmorpholine/DMF solution, will liveThe amino acid solution after the addition is added to the resin, and after the magnetic stirring reaction for 2 hours, the Kaiser test is performed, and the resin is not blue-violet.The color indicates complete reaction, otherwise the reaction time is extended, or the coupling reaction is repeated once, and then the resin is washed six times with DMF. repeatThe above steps, in order to complete the extension of the polypeptide chain, complete the synthesis of the AP2H polypeptide sequence (IHGHHIISVG), and then remove the peptide amino group.End FMOC protecting group.After the AP2H polypeptide sequence is synthesized, the FMOC protecting group is removed, and a 3-fold molar amount of succinic anhydride is added, and the coupling is coupled.After the beam is washed, add 3 times the molar amount of FMOC-NH-NH2, HBTU and HOBt to react overnight, after the reaction is finished, remove the FMOC protection.Base, using freshly prepared lysate (95percent TFA, 2.5percent H2O, 2.5percent TIS, v/v/v) to AP2H-hydrazideThe product is cleaved from the resin, and the obtained crude product is purified by Shimadzu semi-preparative liquid chromatography, pure liquid chromatography and mass spectrometry. The spectrum was characterized. The results are shown in Figures 2 and 3.
In the first step, pure water (50 ml), <strong>[24103-75-1]4-methoxy-2-methylpyridine</strong> compound (1) (10 g) and succinic anhydride (8 g) were placed in a three-necked flask equipped with a stirrer, a thermometer and a condenser. After the water bath is heated to 80 C, potassium permanganate (6 g total) is added in three batches, the temperature is controlled at 84 C, the potassium permanganate to be added is completely reacted, and the solution in the reaction flask is dark brown and purple-free. Then add the next batch of potassium permanganate, and then proceed. After all the potassium permanganate has been reacted, keep stirring at 82 C for at least 45 minutes, remove the manganese dioxide by hot filtration, and wash the water twice with hot distilled water. Manganese oxide until the filtrate is a colorless transparent solution to obtain a solution containing the potassium 1-methoxy-5-hydroxy-benzopyridine-3-carboxylate compound (2) without isolation of 1-methoxy-5 - hydroxy-benzopyridin-3-carboxylic acid potassium compound (2); In the second step, the solution containing the potassium 1-methoxy-5-hydroxy-benzopyridine-3-carboxylate compound (2) in the first step is adjusted to a pH of 3.25 with concentrated hydrochloric acid to stirred at 30 min. In a minute, it was concentrated in a vacuum evaporator at a temperature of 46 C. After evaporation to dryness, ethanol was refluxed at 68 C for 2 h. After the reflux was completed, the mixture was cooled and suction filtered, and the filtrate was taken and dried to give 1-methoxy. -5-hydroxy-benzopyridine-3-carboxylic acid compound (3) (10.36 g), the yield was 86.2% by high performance liquid chromatography






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +
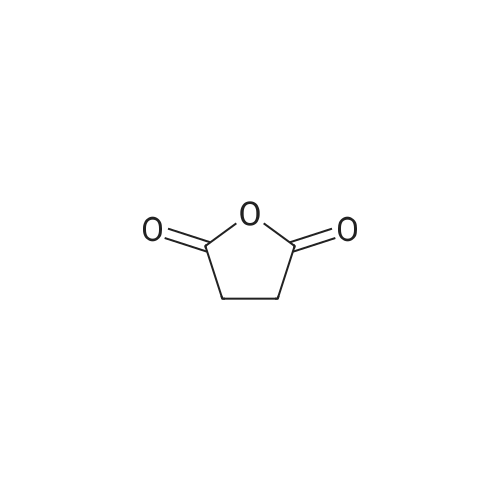

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping