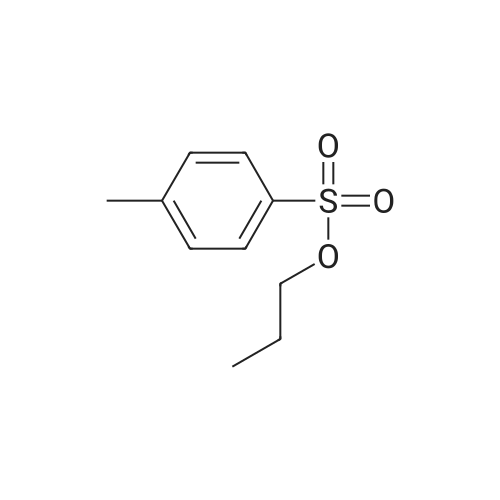
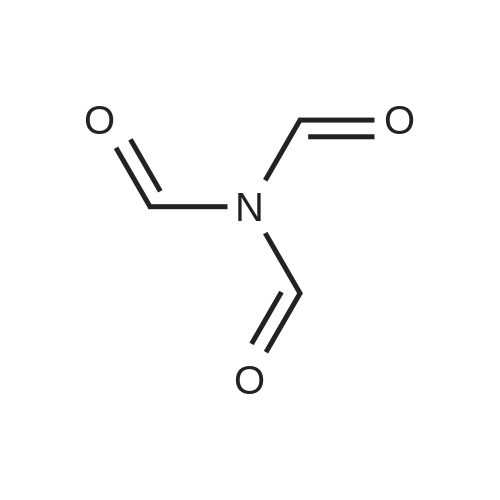
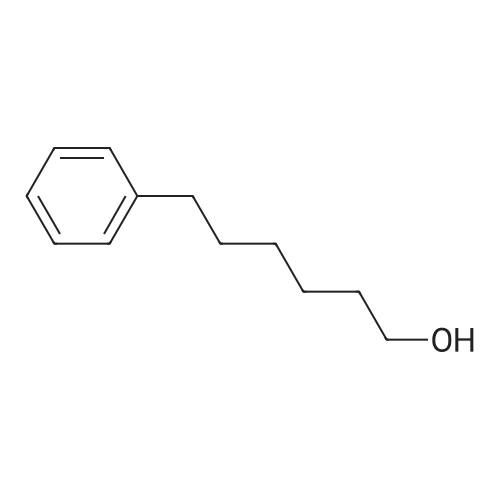
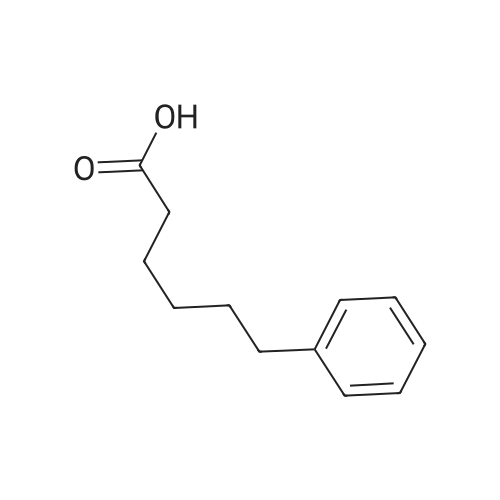
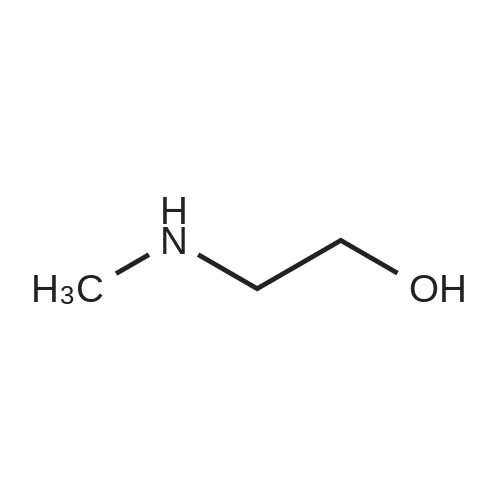
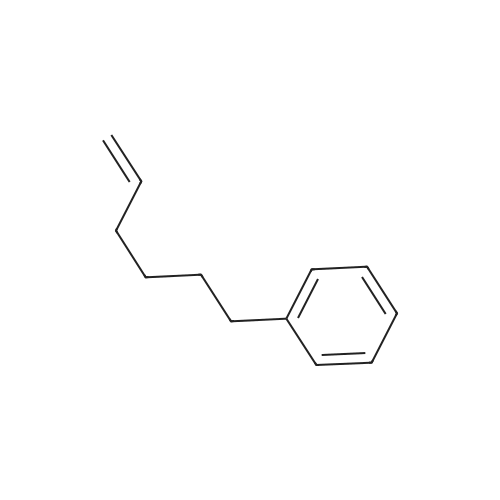
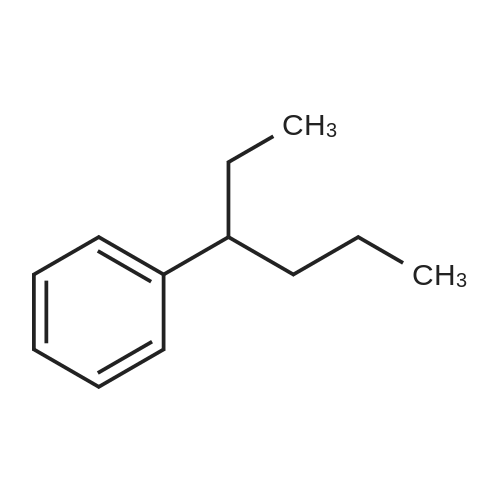
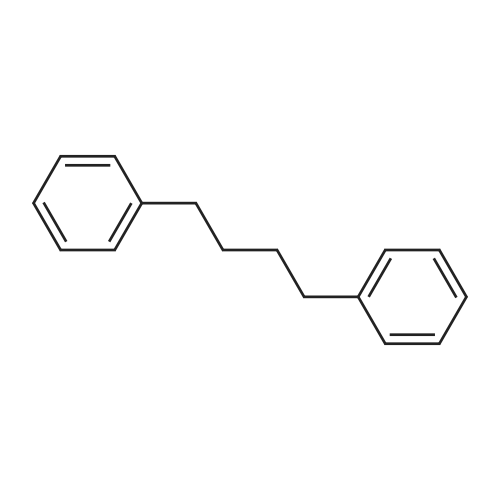
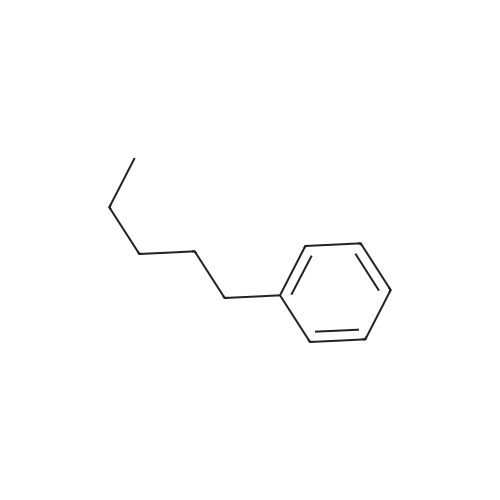
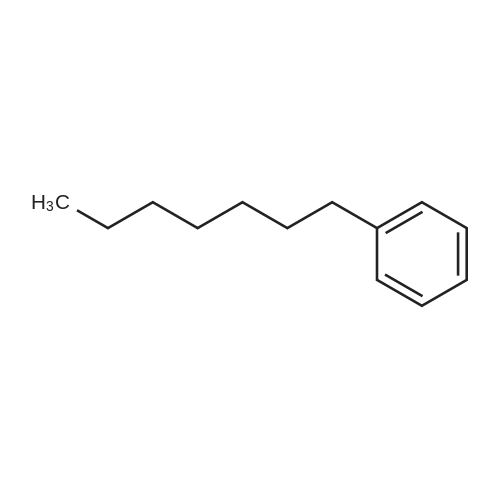
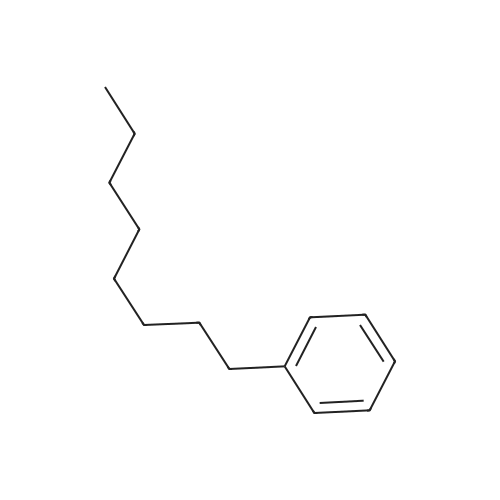
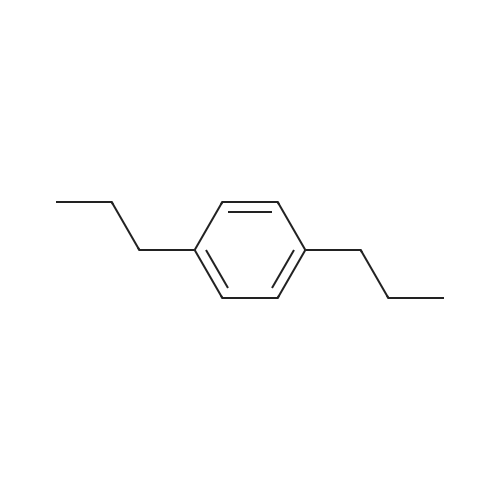
| 75% |
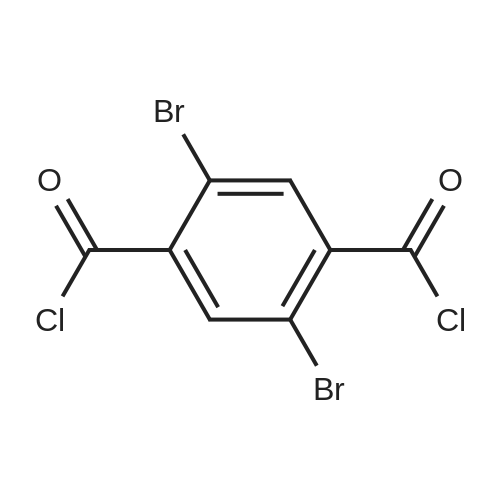
Stage #1: 3,6-dibromo-2,5-phenylenedi(carboxylic acid chloride) With aluminum (III) chloride In dichloromethane at 0℃; Inert atmosphere;
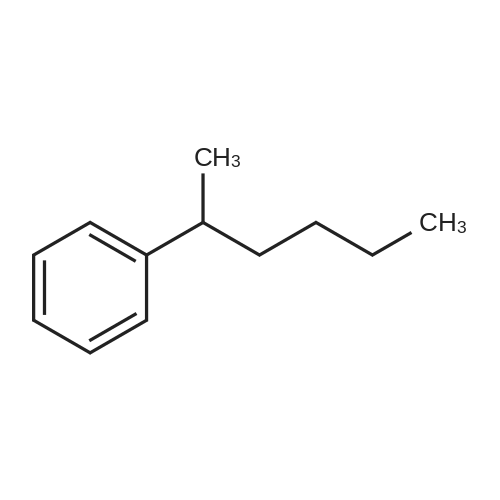
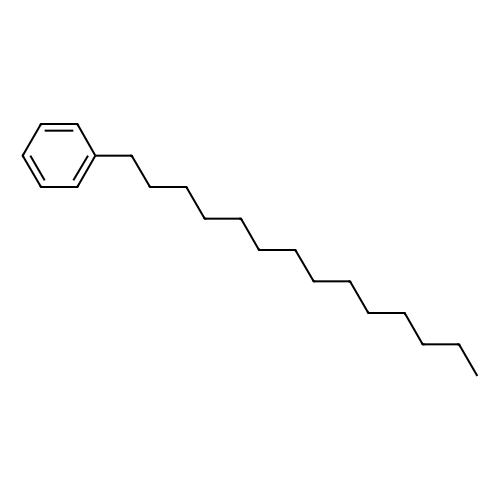
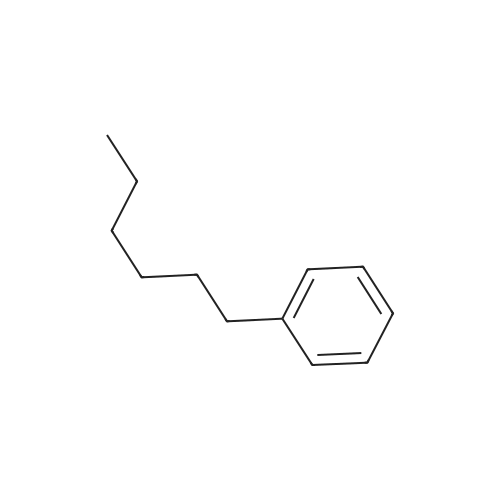
Stage #2: hexylbenzene In dichloromethane at 20℃; Inert atmosphere; |
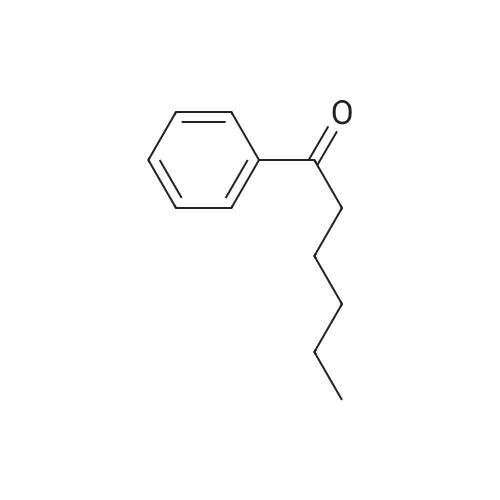
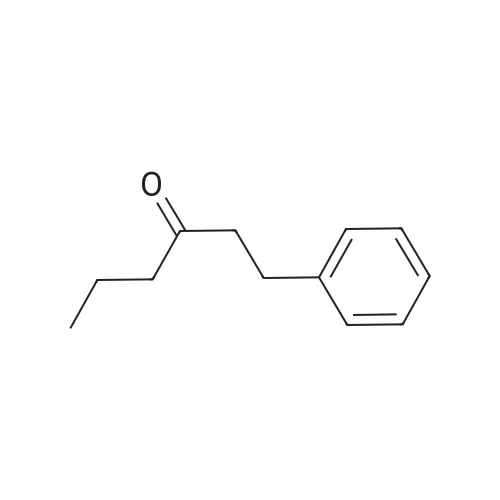
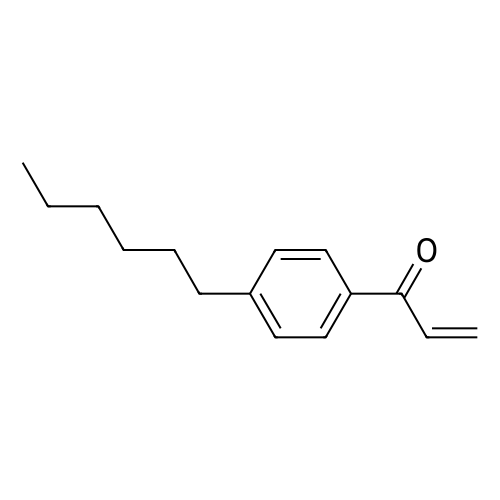
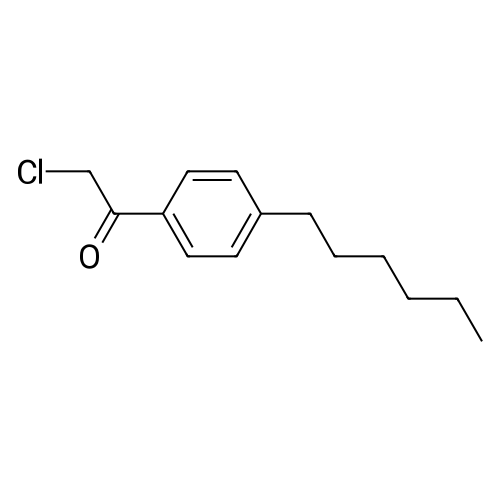
To a solution of 2,5-dibromoterephthaloyl dichloride in 50 mL dryCH2Cl2 was slowly added a solution of anhydrous AlCl3 (7.29 g, 54.7 mmol) in dry CH2Cl2 (70 mL) at 0 °C, the mixture was kept at 0 °Cfor 20 min, then a solution of n-hexylbenzene (13 mL, 69.0 mmol) wasadded slowly. After the addition, the mixture was stirred at roomtemperature overnight. Then ice water and 3M hydrochloric acid solutionwas added to the mixture until it became clear. The mixture waswashed with saturated sodium bicarbonate solution and extracted withCH2Cl2. The organic layer was dried over anhydrous Na2SO4. Afterremoval of the solvent, the crude product was washed with hexane togive the product as a white solid (9.95 g, 75%).1H NMR (400 MHz,CDCl3, δ): 7.79-7.73 (m, 4H), 7.58 (s, 2H), 7.32 (d, J=8.4 Hz, 4H),2.75-2.64 (m, 4H), 1.65 (dt, J=15.4, 7.6 Hz, 4H), 1.40-1.24 (m, 12H),0.93-0.84 (m, 6H); 13C NMR (100 MHz, CDCl3, δ): 193.48, 150.68,143.35, 132.86, 130.54, 129.04, 118.45, 36.23, 31.67, 31.04, 29.00,22.60, 14.13; HRMS (ESI) m/z: [M+Na]+, calcd for C32H36Br2O2,633.0974; found, 633.0973. |
| 141.8 g |
With aluminum (III) chloride In dichloromethane at 0 - 25℃; |
1 Compound 1-3 Synthesis
Into a 3 L reaction flask, add compound 1-5 (100 g, 308.7 mmol, 1eq), add dichloromethane (1 L), add oxalyl chloride (62.6 mL, 740.9 mmol, 2.4 eq), and stir.Add N, N-dimethylformamide (DMF, 0.22 mL, 3.1 mmol, 0.01 eq) to the reaction and stir for 10 minutes.The oil-bath is used to raise the temperature of the bath to 60 ° C. and stir at reflux overnight. After cooling the reaction temperature to room temperature (25 ), the solvent (Dichloromethane (MC)) is concentrated using a reduced pressure pump.Dichloromethane (1 L) was added to the concentrate, followed by aluminum chloride (90.6 g, 679.2 mmol, 2.2 eq).Cool the reactant to 0 ° C using an ice-bath and dilute n-hexylbenzene (174.5 mL, 926.2 mmol, 3 eq) in dichloromethane (200 mL) and slowly add dropwise to the reaction.The reaction is stirred overnight at room temperature (25 ° C.).After completion of the reaction, the reaction solution is slowly poured into cooling water (purified water / ice = 1/1, 1.5 L) and stirred vigorously.The organic layer was separated and then 1N NaOH (in purified water, 1.5 L),Wash each with purified water (1 L).The separated organic layer was dried over anhydrous MgSO 4, filtered, and the organic layer was removed using an evaporatior.The concentrate is recrystallized with Dichloromethane / MeOH and filtered, and the filtered solid is washed with MeOH.The washed solid was dried overnight in a vacuum oven (temp. = 90 ° C.) to afford intermediate compound 1-3 (141.8 g, 2 steps overall 75%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping