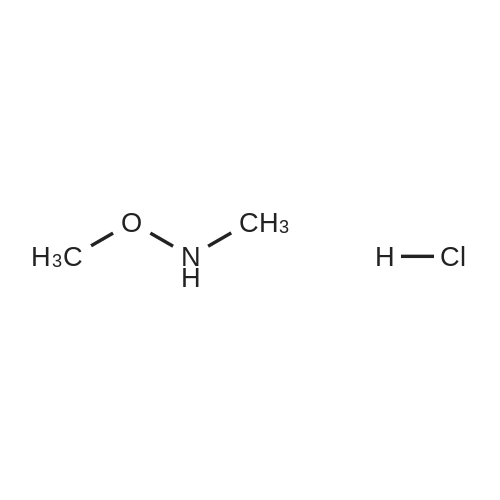
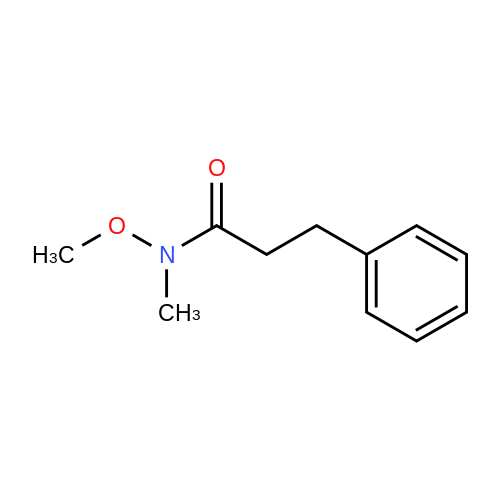
| 99% |
With sulfuric acid for 5h; Reflux; |
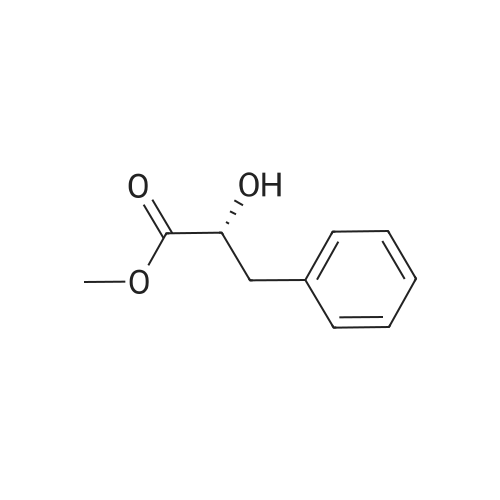
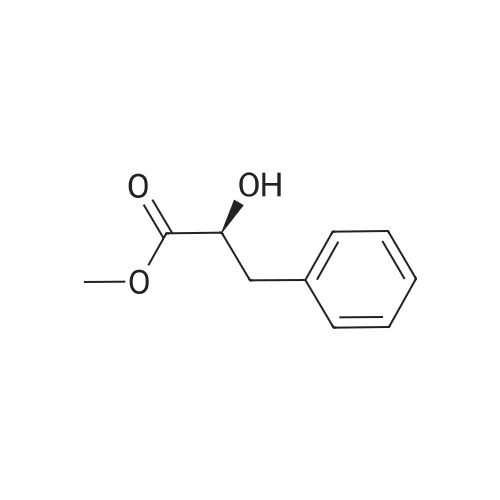
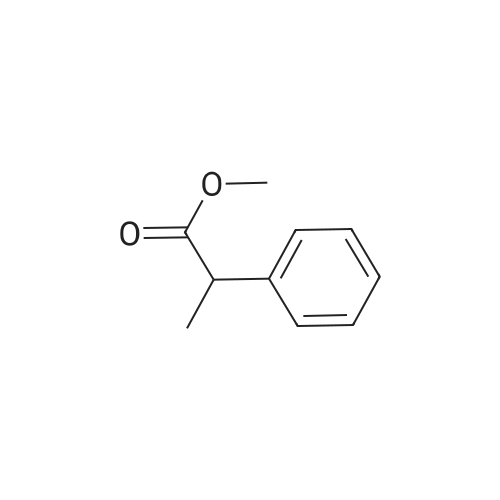
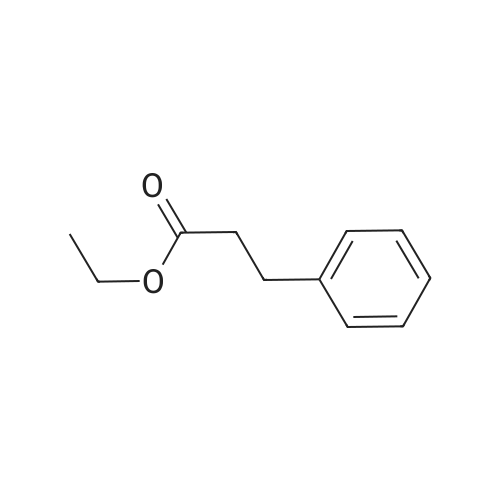
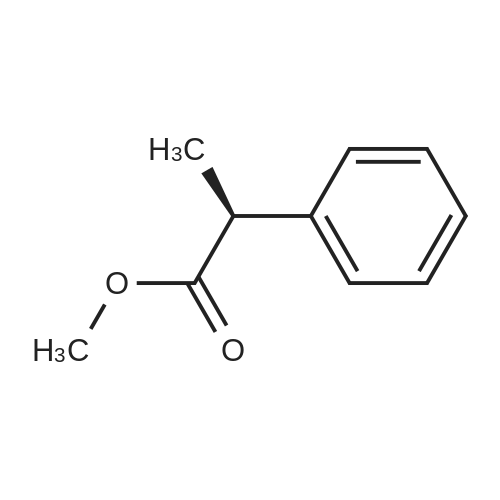
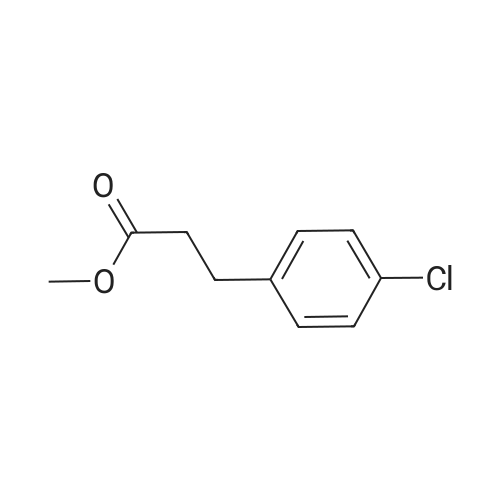
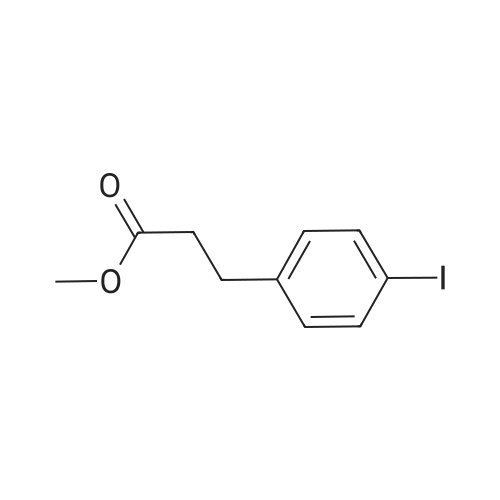
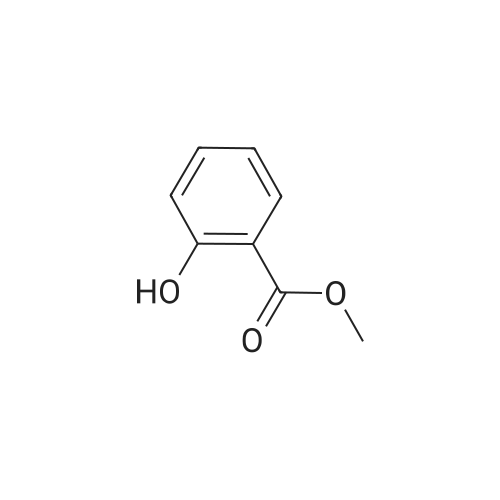
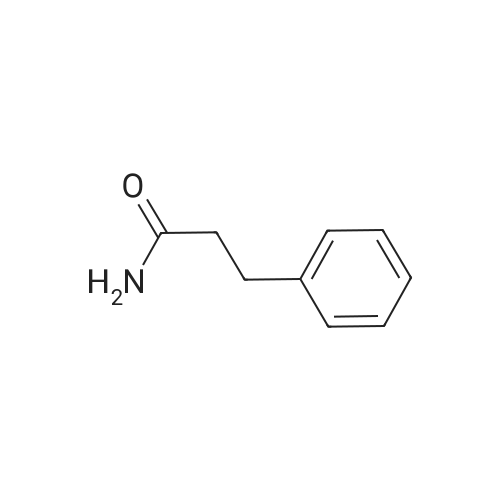
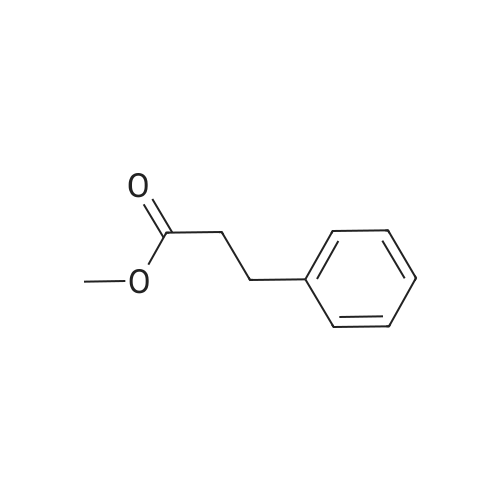
7B 7B. Methyl 3-phenylpropanoate, 82
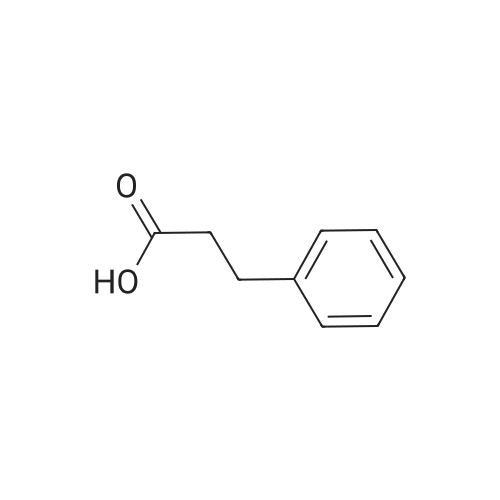
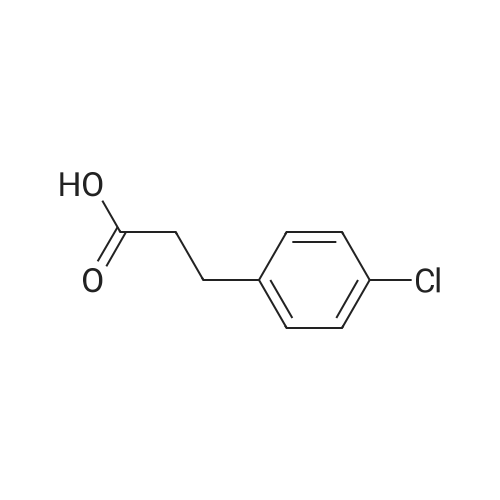
Hydrocinnamic acid (10.0 g, 66.6 mmol, 1 eq.) was dissolved in methanol (90 ml), cone. H2S04 (1 ml) added dropwise with stirring and the reaction mixture was stirred under reflux for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in water (100 ml) and extracted with EtOAc (3 x 50 ml). The combined organic phases were washed with 10 % NaHC03 aq. (2 x 50 ml), brine (50 ml) before being dried (MgS04), filtered and concentrated to give the title compound 82 (10.8 g, 99 %) as a clear, colourless oil. Analytical data consistent with the literature (Black, P. J. et al., Eur. J. Org. Chem. 4367-4378 (2006)). vmax (filmVcm-1 3028, 2952, 1734, 1436, 1194, 1160, 749, 697 *H NMR (400 MHz; CDCI3) δΗ = 2.68 (t, J = 7.8 Hz, 2H, CH2), 3.00 (t, J = 7.8 Hz, 2H, CH2), 3.71 (s, 3H, CH3), 7.25 (m, 3H, ArCH's), 7.33 (m, 2H, ArCH's) 13C NMR (100 MHz; CDCI3) 5C = 31.1 (CH2), 35.8 (CH2), 51.7 (CH3), 126.4 (2 x ArCH), 128.4 (ArCH), 128.6 (2 x ArCH), 140.6 (ArC), 173.5 (C=0) m/z (CI+) 165.1 [MH]+ (20%), 133.1 (100%), 105.1 (55%), 93.1 (51%), 85.0 (57%) |
| 99% |
With sulfuric acid for 5h; Reflux; |
7.7B 7B.
Methyl 3-phenylpropanoate, 82
7B. Methyl 3-phenylpropanoate, 82 Hydrocinnamic acid (10.0 g, 66.6 mmol, 1 eq.) was dissolved in methanol (90 ml), conc. H2SO4 (1 ml) added dropwise with stirring and the reaction mixture was stirred under reflux for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in water (100 ml) and extracted with EtOAc (3*50 ml). The combined organic phases were washed with 10% NaHCO3 aq. (2*50 ml), brine (50 ml) before being dried (MgSO4), filtered and concentrated to give the title compound 82 (10.8 g, 99%) as a clear, colourless oil. Analytical data consistent with the literature (Black, P. J. et al., Eur. J. Org. Chem. 4367-4378 (2006)). νmax (film)/cm-1 3028, 2952, 1734, 1436, 1194, 1160, 749, 697 1H NMR (400 MHz; CDCl3) δH=2.68 (t, J=7.8 Hz, 2H, CH2), 3.00 (t, J=7.8 Hz, 2H, CH2), 3.71 (s, 3H, CH3), 7.25 (m, 3H, ArCH's), 7.33 (m, 2H, ArCH's) 13C NMR (100 MHz; CDCl3) δC=31.1 (CH2), 35.8 (CH2), 51.7 (CH3), 126.4 (2*ArCH), 128.4 (ArCH), 128.6 (2*ArCH), 140.6 (ArC), 173.5 (C=O) m/z (CI+) 165.1 [MH]+ (20%), 133.1 (100%), 105.1 (55%), 93.1 (51%), 85.0 (57%) |
| 98% |
With magnesium chloride; dimethyl dicarbonate at 20℃; for 18h; |
|
| 97% |
With bis(trichloromethyl) carbonate In dichloromethane at 40℃; for 2h; |
|
| 96% |
With sulfuric acid for 2h; Reflux; |
1.a Step a
3 - Phenylpropionic acid 1-1 (3.0g, 20 mmol) was dissolved in methanol (30 ml), a catalytic amount of concentrated sulfuric acid (1.5 ml) was added, and the reaction 2h was refluxed TLC. The organic layer was washed with water and saturated brine and dried under reduced pressure to obtain a colorless oily liquid 1-2 (3.2g, yield 96%). |
| 96% |
With sulfuric acid for 2h; Reflux; |
|
| 95% |
With sodium hydrogen sulfate; silica gel at 20℃; for 5h; |
|
| 95% |
With hydrogen bromide; hydrogen at 40℃; for 3h; Autoclave; |
2.4 Isolated yield
3-phenylpropionic acid (751.0 mg, 5.0 mmol), bromobenzene (78.5 mg, 0.5 mmol), Pd-pol (53.0 mg, Pd: 0.5 mol%), and methanol (10 mL) were placed in autoclave vessel. Autoclave was pressurized with 2 bars of hydrogen gas. The reaction mixture was then warmed to 40 °C temperature and stirred for 3 h. After reaction, the catalyst was filtered through celite bed. Filtrate was added of water (30 mL). The reaction mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with 5% aqueous sodium bicarbonate solution (3 × 15 mL), dried over anhydrous Na2SO4 and filtered. The filtrate was evaporated under vacuum to give methyl 3-phenylpropionate as a pale yellow oil (763.5 mg, yield = 93%). Comparison of its MS and NMR features with those reported in literature [37] confirmed the purity of the obtained product. |
| 95% |
Stage #1: 3-Phenylpropionic acid With Dimethylphenylsilane; pyrographite; palladium dichloride In 1,2-dimethoxyethane at 25 - 40℃; for 4h;
Stage #2: methanol In 1,2-dimethoxyethane at 40℃; for 16h; |
|
| 94% |
With polystyrene-bound tetrafluorophenylbis(triflyl)methane at 27℃; for 29h; |
|
| 93% |
With 4-methyl-morpholine; 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride at 20℃; for 1.5h; |
|
| 93% |
With 4-methyl-morpholine; 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride at 20℃; for 1.5h; |
|
| 93% |
With 4-methyl-morpholine; 2-chloro-4-(diethoxyphosphoryl)-6-methoxy-1,3,5-triazine In tetrahydrofuran at 20℃; for 0.75h; |
3
(Experimental Procedure Example 2) (Ester Formation Reaction Using THF as a Solvent) THF (solvent) (2 mL), 3-phenylpropionic acid (60.1 mg, 0.40 mmol), N-methylmorpholine Compound (III)) (53 μL, 0.48 mmol) was added. Compound (II) (0.44 mmol) was added to the solution, and the mixture was reacted at room temperature. After disappearance of the raw material was confirmed, 4 mL of 1N potassium hydrogen sulfate aqueous solution was added, diluted with dichloromethane, and washed with 1 M hydrochloric acid, saturated sodium bicarbonate aqueous solution and saturated brine. The obtained organic layer was dried over sodium sulfate, filtered, and concentrated. For the obtained mixture, the objective ester compound was quantified using 1 H-q NMR. |
| 92% |
With thionyl chloride; triethylamine at 0 - 40℃; for 12.3333h; Inert atmosphere; |
|
| 91% |
With 4-methyl-morpholine; 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride In water at 20℃; for 2 - 5h; |
|
| 91% |
With sulfuric acid for 4h; Reflux; |
|
| 89% |
With nickel dichloride for 10h; Heating; |
|
| 89.6% |
With thionyl chloride at 20℃; for 3h; |
7 3.2.6. General procedure for preparation of 3-(substituted-phenyl)-propionic acid methyl ester
General procedure: A mixture of substituted dihydrocinnamic acid (2a-e, 0.133 mol) inmethanol (350 mL) was treated slowly with thionyl chloride (20 mL,0.275 mol) drop wise during 1 h. After completion of addition, the reactionmixture was stirred for another 2 h at rt. After completion ofreaction, which was monitored by TLC hexane/ethyl acetate (8:2), thereaction mixture was poured into water (200 mL), extract with ethylacetate (2×400 mL). The extract was washed with water, brine solution,dried over Na2SO4 and concentrated. The crude compound wassubjected to column chromatography on silica gel using hexane/ethylacetate mixtures. The fractions were monitored and the fractions elutedwith ethyl acetate in hexane, 3%/97% (v/v) were combined and concentratedto obtained 3-(substituted phenyl)-propionic acid methyl esters(3a-e) as oily compounds with yields in the range of 87-90%. 3.2.7 3-(Phenyl)-propionic acid methyl ester (3a) Weight: 15.6 g; % yield: 89.6%; 1H NMR (CDCl3, 400 MHz): δ 7.34 (2H, t, J = 8.0 Hz, H-3', 5'), 7.21 (2H, d, J = 7.6 Hz, H-2', 6'), 7.11 (1H, t, J = 7.2 Hz, H-4'), 3.68 (3H, s, OCH3), 2.98 (2H, t, J = 7.6 Hz, H-3), 2.62 (2H, t, J = 7.6 Hz, H-2); 13C NMR (CDCl3, 100 MHz): δ 176.2 (C=O), 146.2 (C-1'), 132.6 (C-3', 5'), 130.8 (C-2', 6'), 128.3 (C-4'), 50.6 (OCH3), 36.8 (C-2), 30.9 (C-3); LC-MS: m/z 165.2 (M + H)+. |
| 88% |
With sulfuric acid for 5h; Reflux; |
Methyl 3-phenylpropanoate (4b).
General procdure: A mixture of 3b (3 g, 20 mmol) and conc H2SO4 (0.5 mL) in methanol (30 mL) was refluxed for 5 h, and most of the solvent was evaporated. The mixture was poured into H2O (30 mL), and extracted with EtOAc (30 mL x 3). The combined organic layer was washed with water, dried, filtered, and condensed to afford 4b (2.9 g) as a white oil, yield 88%. EI-MS m/z 164 (M+); 1H-NMR (CDCl3, 400 MHz): δ 2.59 (t, J=7.6 Hz, 2H), 2.92 (t, J=7.6 Hz, 2H), 3.67 (s, 3 H), 7.18-7.22 (m, 3H), 7.26-7.30 (m, 2H). |
| 83% |
With 4-methyl-morpholine; 2-chloro-4-methoxy-6-(1-octyn-1-yl)-1,3,5-triazine In tetrahydrofuran at 20℃; for 3h; |
3 A method for producing an ester compound of the present invention using a dehydrating condensation agent produced in a reaction system by mixing Compound (II) and NMM (Compound (III))
(Experimental procedure example) THF (solvent) (2 mL) was added to a 10 mL eggplant flask,3-phenylpropionic acid (60.1 mg, 0.40 mmol),N-methylmorpholine (compound (III))(53 μL, 0.48 mmol) was added.Compound (II) (0.44 mmol) was added to the solution,And allowed to react at room temperature.After disappearance of the raw material was confirmed, 4 mL of 1N potassium hydrogen sulfate aqueous solution was added,It was diluted with dichloromethane,Washed with 1 M hydrochloric acid, saturated aqueous sodium hydrogen carbonate solution and saturated brine.The obtained organic layer was dried over sodium sulfate, filtered, and concentrated.For the obtained mixture, the objective ester compound was quantified using 1 H-q NMR. |
| 80% |
With sulfuric acid for 3h; Heating; |
|
| 79% |
With potassium carbonate; dibromotriphenylphosphorane at 20℃; for 24h; |
|
| 77% |
With sulfuric acid at 20℃; for 144h; |
|
| 70% |
With sulfuric acid |
|
| 56% |
With sulfuric acid |
|
| 53% |
With Celite; polystyrene-bound super Broensted acid at 20℃; flow system; |
|
|
With hydrogenchloride |
|
|
With sulfuric acid |
|
| 90 % Chromat. |
at 70℃; for 12h; |
|
|
With sulfuric acid Heating; |
|
|
Heating; |
|
|
With toluene-4-sulfonic acid for 18h; Inert atmosphere; Reflux; |
|
|
Acidic conditions; |
|
|
With thionyl chloride at 20℃; |
37.37a.1
To a solution of 3-phenylpropanoic acid (10 g) in methanol (100 mL) was added thionyl chloride (0.05 mL) dropwise. The resulting mixture was stirred at room temperature overnight. Solvent was removed in vacuo to afford methyl 3-phenylpropanoate (10.896 g) as a light yellow liquid. MS(ES+) m/z 165 (MH+). |
|
With sulfuric acid for 12h; Reflux; |
|
|
With thionyl chloride at 0 - 20℃; for 16h; |
|
|
Reflux; |
|
|
With sulfuric acid for 4h; Reflux; |
|
|
With toluene-4-sulfonic acid at 20℃; for 0.5h; |
|
|
With sulfuric acid for 2h; Inert atmosphere; Reflux; |
|
|
With hydrogenchloride In water Reflux; |
|
|
With hydrogenchloride Reflux; |
General procedure: To a suspension of the appropriate acid 17a-c (1 eq,) in MeOH (1.9 ml*mmol/eq) was added 1, 25 M HCl in MeOH solution (1.9 ml*mmol/eq). The solution was refluxed overnight, then cooled to room temperature and concentrated under vacuum. The residue was partitioned between sat. aq. NaHCO3 solution (30 ml) and EtOAc (3x35 ml). The combined organic extracts were washed with sat. aq. NaHCO3 solution (25 ml) and water (30 ml), dried over MgSO4 and concentrated under vacuum to give the pure title compounds. Intermediate methyl esters were synthesised as described for 18a-c |
|
With sulfuric acid for 3h; Reflux; |
|
|
With sulfuric acid for 24h; Heating; |
|
|
With sulfuric acid |
|
|
With sulfuric acid at 20℃; for 3.08333h; Reflux; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping