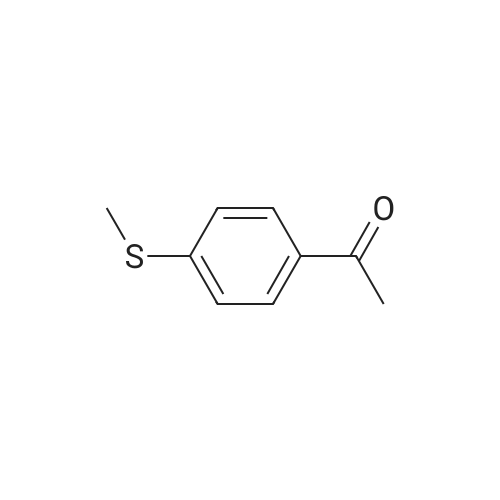
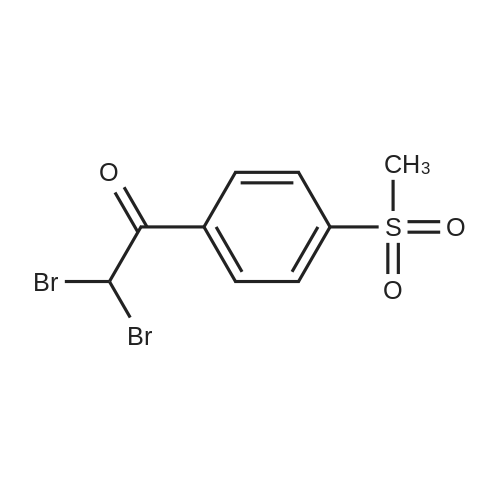
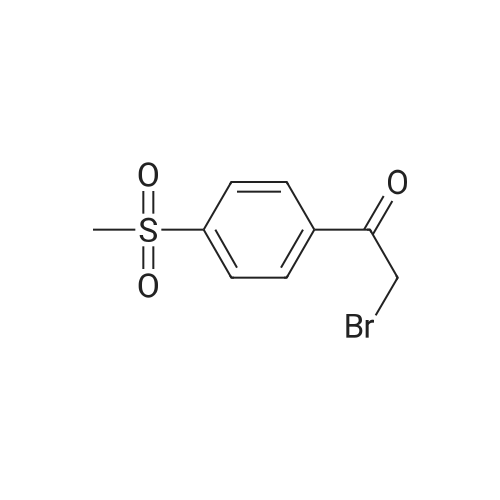
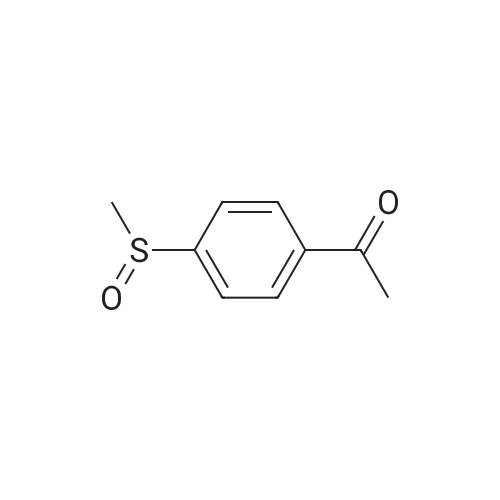
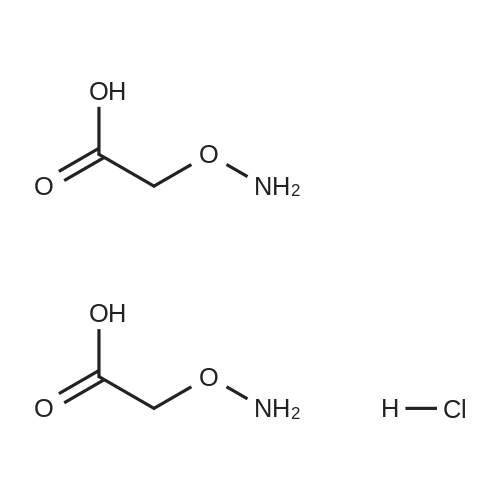
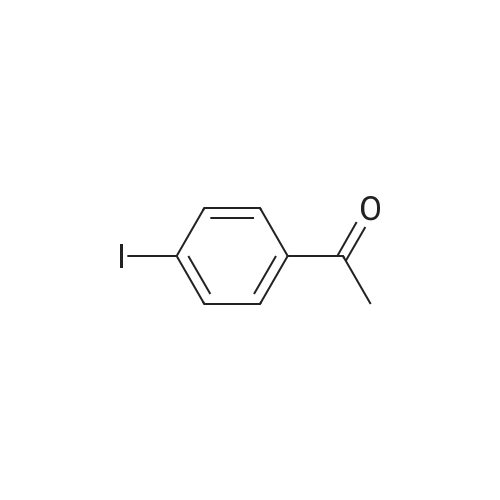
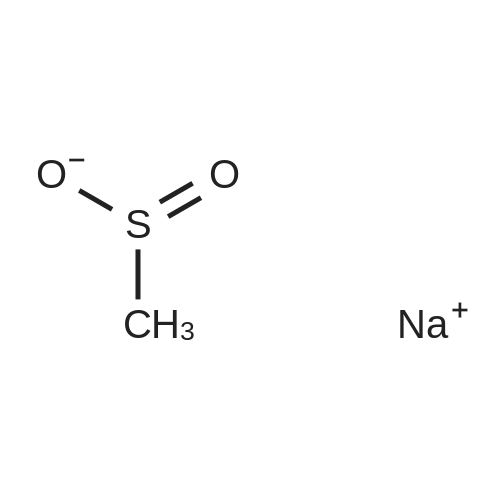
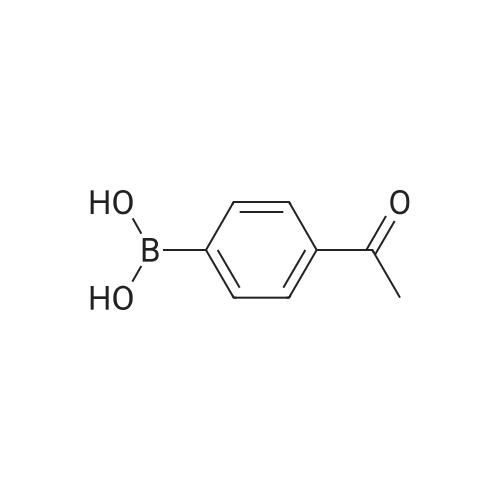
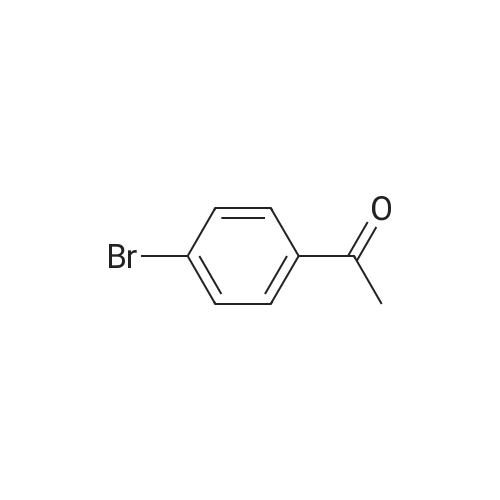
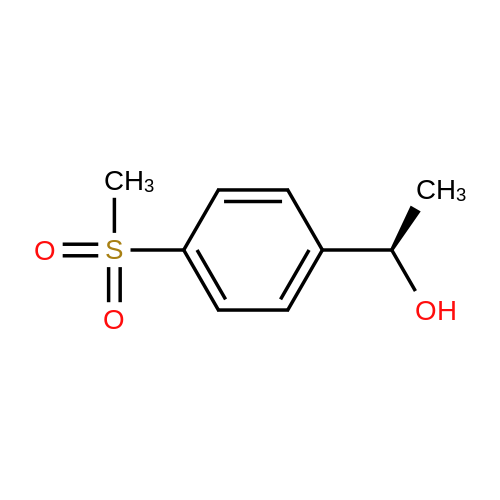
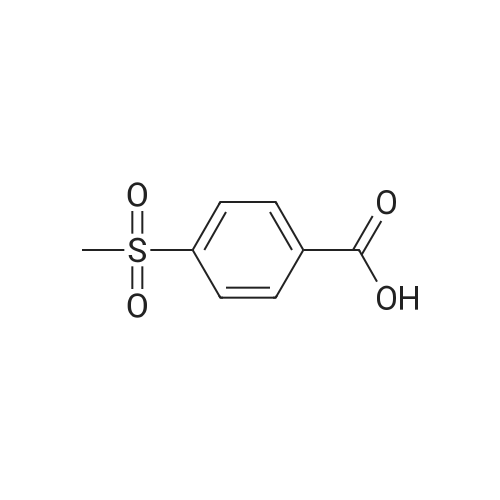
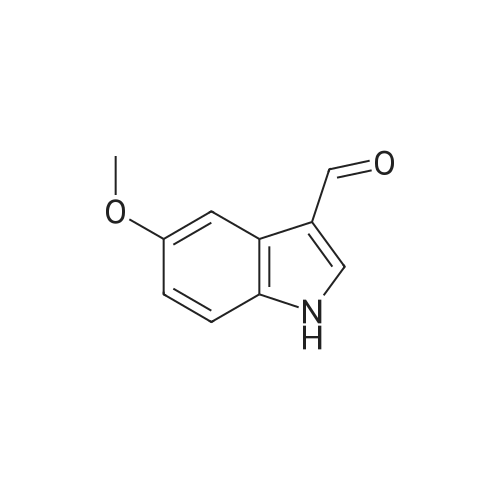
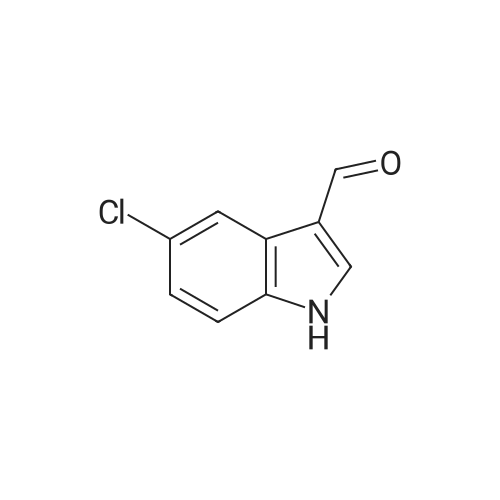
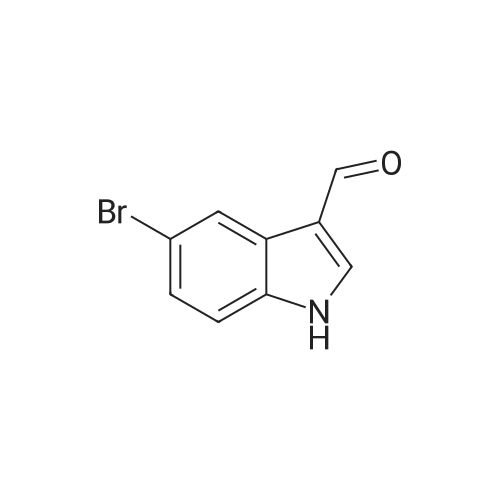
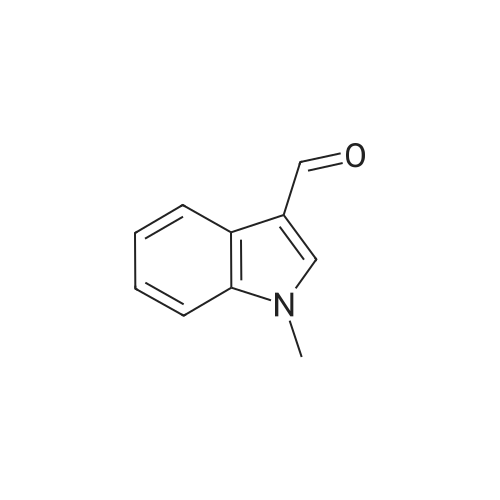
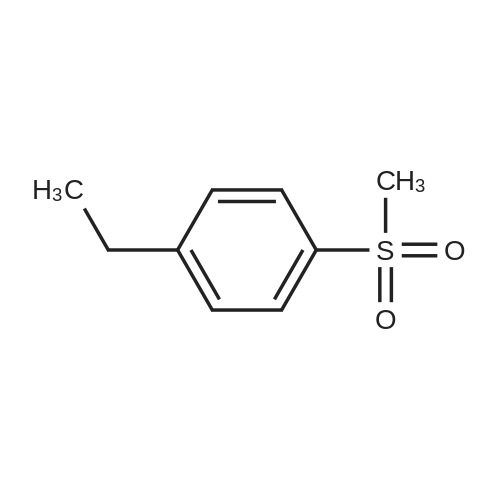
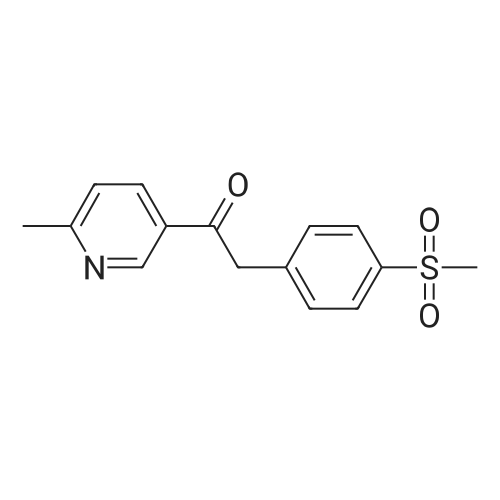
| 87% |
With bromine In chloroform |
29
EXAMPLE 292-Bromo-l-(4-methanesuIfonyl-phenyl)-ethanone): To a solution of l-(4- Methanesulfonyl-phenyl)-ethanone (25 g, 0.12 mol) in 500 mL chloroform was added , drop-wise, bromine (19.2 g, 0.12 mol) in 60 mL chloroform over a period of 3 h. The mixture was stirred over night. Water (200 mL) was carefully added to the reaction mixture, mixed well and the organic layer isolated was washed with saturated sodium hydrogen carbonate in water (200 mL), followed by brine (200 mL), dried over anhydrous sodium sulfate, filtered and filtrate evaporated to get pure product as a light yellow powder (32 g, 87%) |
| 87% |
With hydrogen bromide; bromine; acetic acid In water at 20 - 25℃; for 2 - 3h; |
Synthesis of Bromoketone 2
2 L 3-neck round bottom flask equipped with a mechanical stirrer, temperature probe, addition funnel, and N2 inlet was charged with glacial acetic acid, ketosulfone and aq. 48% HBr. An addition funnel was charged with bromine. A 10% (8.1 g) charge of bromine gave an orange slurry which was aged 30 min at 25°C and was then sampled. The bromination reaction has an induction period of 1-15 min after which bromine was rapidly consumed as it was added. The remainder of the bromine was added over 50 min at 20-25°C. The resulting pale yellow slurry was aged for 2 h at 22-25°C. After ageing the mixture for 2-3 h the batch was filtered. The wet cake was washed once with 200 mL of 1:1 H2O:HOAc and once with H2O (200 mL). The cake was dried in vacuo at 40°C with a N2 sweep to give 126.0 g of bromoketone (87%). |
| 86% |
With aluminium trichloride; bromine In chloroform at -5℃; |
|
| 85% |
Stage #1: 4-(methanesulfonyl)acetophenone With hydrogen bromide; acetic acid at 20℃; for 0.0833333h;
Stage #2: With bromine at 25 - 58℃; for 2.5h; |
4.1.3. 2-Bromo-1-(4-(methylsulfonyl)phenyl)ethanone (9)
To a solution of 4-(methylsulfonyl)acetophenone (8, 2.0 g,0.01 mol) in glacial acetic acid (10 mL) at r.t. was added 48% HBr a drop, well stirred over 5 min, followed by a solution of bromine (0.5 mL, 0.01 mol) dissolved in 5 mL glacial acetic acid was added indrop about 0.5 mL, the solution was orange yellow, raise the temperature to 58 °C, after 0.5 h the solution changed to white. Then it was cooled to 25 °C and the remaining bromine and glacial acid solution was added in drop. The mixture was stirred for 2 h, after completion of the reaction, ice H2O (100 mL) was added and solid precipitated then filtered and the filtrate was dried under vacuum to give 2.35 g of 5 (85%). |
| 83% |
With bromine In chloroform at 20℃; |
α-bromo-4-(methylsulfonyl)acetophenone (4)
Dissolve 2g (10.1 mmol) of 2 in 20 ml CHCl3. The brominewas added drop wise. After the reaction was completed(monitored by TLC), the solvent was evaporated under reducedpressure and the precipitate was recrystallized inEthanol. Yield 83%; white crystalline powder; mp: 125-127C; IR (KBr disk): (cm-1) 1165, 1308 (SO2), 1710(C=O); LC-MS (ESI) m/z: 276.7 (M+1, 100). |
| 77% |
With bromine In chloroform at -5℃; for 1h; |
|
| 73% |
With acetic acid In dichloromethane at 20℃; |
|
| 66% |
With bromine In chloroform at 15℃; for 1h; |
215.1 2-Bromo-1-(4-(methylsulfonyl)phenyl)ethanone
To the solution of 1-(4-(methylsulfonyl)phenyl)ethan-1-one (5.00 g, 25.2 mmol) inCHCI3 (100 mL) was added Br2 (4.0 0 g, 25.2 mmol) in CHCI3 (15 mL) dropwiseover period of 1 h, the mixture was stirred for 1 h at RT (15 00). The solution was washed with saturated NaHCO3 (30 mL) and brine (30 mL), dried over Na2504. After removal of solvent under reduced pressure, the crude product was recrystallized from EtOH (50 mL) at RT to afford a white solid as product 20 (4.60g, 16.6 mmol, 66 %). 1H NMR (300 MHz, CDCI3): O = 3.12 (5, 3 H), 4.48 (5, 2 H),8.10 (m, 2 H), 8.18 (m, 2 H) ppm. |
| 66% |
With bromine In chloroform at 15℃; |
661.1 Step 1 : 2-Bromo-1-(4-(methylsulfonyl)phenyl)ethanone
Step 1 : 2-Bromo-1-(4-(methylsulfonyl)phenyl)ethanone To the solution of 1-(4-(methylsulfonyl)phenyl)ethan-1-one (5.00 g, 25.2 mmol) in CHCIs (100 mL) was added Br2 (4.0 0 g, 25.2 mmol) in CHCI3 (15 mL) dropwise over period of 1 h, the mixture was stirred for 1 h at RT (15 °C). The solution was washed with saturated NaHC03 (30 mL) and brine (30 mL), dried over Na2S04. After removal of solvent under reduced pressure, the crude product was recrystallized from EtOH (50 mL) at RT to afford a white solid as product 20 (4.60 g, 16.6 mmol, 66 %). 1 H NMR (300 MHz, CDCI3): δ = 3.12 (s, 3 H), 4.48 (s, 2 H), 8.10 (m, 2 H), 8.18 (m, 2 H) ppm. |
| 60% |
With N-Bromosuccinimide; toluene-4-sulfonic acid In acetonitrile at 50℃; for 24h; |
25.1 Step 1: Compound P025-b
At room temperature, compound P025-a (5.00 g, 25.25 mmol, 1.00 eq.),N-bromosuccinimide (NBS) (4.587g, 25.57mmol, 1.02eq.) and p-toluenesulfonic acid monohydrate (4.80g, 25.25mmol, 1.00eq.) are dissolved in anhydrous acetonitrile (15.00 mL, 3.00eqv.) at 50°C for 24 hours. The reaction solution was concentrated under reduced pressure, mixed with 40 mL of saturated sodium bicarbonate solution, extracted with dichloromethane (30 mL×3), combined the organic phases, washed with water until neutral, washed with saturated brine, dried with anhydrous sodium sulfate, filtered, Concentrate under reduced pressure to obtain crude product. The crude product was purified by silica gel chromatography column (petroleum ether: ethyl acetate=95:5) to obtain compound P025-b. Yield: 60%; |
|
With bromine; acetic acid |
|
|
With chloroform; bromine |
|
| 15.7 g |
With hydrogenchloride; bromine In acetic acid |
|
|
With hydrogenchloride; bromine In acetic acid; pentane |
2 2-bromo-1-(4-methanesulphonylphenyl)-ethanone
EXAMPLE 2 2-bromo-1-(4-methanesulphonylphenyl)-ethanone 38.5 ml of bromine dissolved in 110 ml of acetic acid are added dropwise onto a suspension of 159 g of the product of Example 1 in 1.6 l of acetic acid and 1.6 ml of hydrochloric acid, the reaction mixture is then stirred for 3 h at ambient temperature. The precipitate obtained is filtered, rinsed with water and then dissolved in dichloromcthane and dried over magnesium sulphate. The dichloromethane is evaporated off, the residue is taken up in pentane and filtered to give 100 g of the expected product. A second batch is obtained by pouring the filtrate of the reaction mixture onto an icc/water mixture, then by filtering off and by treating the precipitate as previously. The two batches are then combined and recrystallized from acetic acid. Yield 143 g, 64%, melting point 130° C. |
|
With hydrogenchloride; bromine In water; acetic acid |
1 Stem 3:Preparation of 2-bromo-4'-(methylsulfonyl) acetophenone
Stem 3:Preparation of 2-bromo-4'-(methylsulfonyl) acetophenone To a stirred solution of 11.91 g (60.5 mmol) of 4-(methylsulfonyl)acetophenone (prepared in Step 2) in 133 mL of glacial acetic acid and 0.11 mL of hydrochloric acid at ambient temperature was added a solution of 8.22 g (51.4 mmol) of bromine in 9.3 mL of glacial acetic acid over a period of three hours. The reaction mixture was diluted with 500 mL of water and extracted with chloroform. The combined extracts were dried (MgSO4) and concentrated in vacuo to give 15.7 g of crude 2-bromo-(4'-methylsulfonyl)acetophenone as a solid: NMR (CDCl3) δ 3.10 (s, 3H), 4.45 (s, 2H), 8.08 (d, J=9 Hz, 2H), 8.17 (d, J=9 Hz, 2H). |
|
With bromine In hexane; chloroform; water; ethyl acetate |
1.1 Step 1
To a cooled (-5° C.) solution of 174 g of 4-(methyl-sulfonyl)acetophenone in 2.5 L of CHCl3 was added 20 mg of AlCl3, followed by a solution of 40 mL of Br2 in 300 mL CHCl3. The reaction mixture was then treated with 1.5 L of water and the CHCl3 was separated. The aqueous layer was extracted with 1 L of EtOAc. The combined extracts was dried over Na2 SO4 and concentrated. The crude product was recystalized from 50/50 EtOAc/hexane to give 210 g of the title compound as a white solid. |
|
With hydrogenchloride; bromine In water; acetic acid |
12.3 Preparation of 2-bromo-4'-(methylsulfonyl) acetophenone
Step 3 Preparation of 2-bromo-4'-(methylsulfonyl) acetophenone To a stirred solution of 11.91 g (60.5 mmol) of 4-(methylsulfonyl)acetophenone (prepared in Step 2) in 133 mL of glacial acetic acid and 0.11 mL of hydrochloric acid at ambient temperature was added a solution of 8.22 g (51.4 mmol) of bromine in 9.3 mL of glacial acetic acid over a period of three hours. The reaction mixture was diluted with 500 mL of water and extracted with chloroform. The combined extracts were dried (MgSO4) and concentrated in vacuo to give 15.7 g of crude 2-bromo-(4'-methylsulfonyl)acetophenone as a solid: NMR (CDCl3) δ 3.10 (s, 3H), 4.45 (s, 2H), 8.08 (d, J=9 Hz, 2H), 8.17 (d, J=9 Hz, 2H). |
|
With aluminum (III) chloride; bromine In chloroform at -5℃; |
1
A solution of 197 g of 4-(methylthio)acetophenone (ref: JACS, 1952, 74, p. 5475) in 700 mL of MeOH and 3500mL of CH2Cl2 was added 881 g of MMPP over a period of 30 min. After 3 h at room temperature the reaction mixture was filtered and the filtrate was washed with 2 L of saturated aqueous solution of NaHCO3 and 1 L of brine. The aqueous phase was further extracted with 2 L of CH2Cl2. The combined extracts was dried over Na2SO4 concentrated to give 240 g of 4(methylsulfonyl)acetophenone as a white solid. To a cooled (-5° C.) solution of 174 g of 4(methylsulfonyl)acetophenone in 2.5 L of CHCl3 was added 20 mg of AlCl3, followed by a solution of 40 mL of Br2 in 300 mL CHCl3. The reaction mixture was then treated with 1.5 L of water and the CHCl3 was separated. The aqueous layer was extracted with 1 L of EtOAc. The combined extracts was dried over Na2SO4 and concentrated. The crude product was recrystalized from 50/50 EtOAc/hexane to give 210 g of 2-bromo-1-(4-(methylsulfonyl)phenyl)ethanone as a white solid. |
|
With bromine |
|
|
With aluminum (III) chloride; bromine In chloroform at 0℃; |
6
FIG. 7 outlines the synthesis of a no-carrier-added iodine-123 labeled analogue of rofecoxib 1. Thioanisole 2 was converted to the requisite potassium trifluoroboronate salt 8 in six steps. Ketone 3 was prepared by Friedel-Crafts acylation of thioanisole 2. Oxidation of 3 using MMPP (magnesium monoperoxyphthalate hexahydrate) afforded sulfone 4 which was allowed to react with bromine in chloroform at 0° C. in the presence of a trace amount of AlCl3 to generate 5. Bromoketone 5 was then coupled with 4-iodophenylacetic acid and then cyclized in the presence of triethylamine and DBU to produce 6 (the non radioactive analogue of the target molecule 1) in 52% yield. Compound 6 was converted to boronic ester 7 using Suzuki chemistry. Addition of KHF2 then generated the trifluoroborate 8. Compound 8 was then reacted according to the method of the invention to generate the no-carrier-added radioiodinated 1. The radiochemical purity of the product exceeded 98% as revealed by radio TLC and the radiochemical yield was 42%. |
|
With bromine In tetrahydrofuran at 20℃; for 26h; |
1
1.51 mmol of substituted acetophenone are dissolved in 4 mL of THF in a 20 mL flask, then 1.81 mmol (1.2 eq) of bromine supported on Amberlyst A26 are added to the solution. After 6 hours of reaction, a further 0.1 eq of supported bromine is added then the mixture is stirred for 20 hours at room temperature. The resin is subsequently filtered then washed with THF, the filtrate is evaporated to give the crude reaction products which are purified either by flash chromatography over silica gel or by recrystallisation.- 2-bromo-l-(4-methanesulphonyl-phenyl)-ethanone (Method 1)Method 1 above was used to prepare the aforementioned product. EPO 1H NMR (400 MHz, CDCl3): δ 8.20 (d, 2H, aromatic H); 8.10 (d, 2H, aromatic H); 4.48 (s,2H5 CH2); 3.12 (s, 3H, CH3).MS: 277.4+ (M+H)+Rf= 0.14 (silica, 2/1 hept/AcOEt) |
|
With bromine; acetic acid |
|
|
With bromine; 1,2-dichloro-ethane |
|
|
With copper(ll) bromide In ethyl acetate Reflux; Sonication; |
Preparation of α-Cyanoketones
General procedure: 20 mmol of the appropriate acyl ketone was dissolved in 40 mL of ethyl acetate to make a 0.5 M solution. 2.6 equivalents of CuBr2 was added and the mixture refluxed with a condenser for 3-5 h until the starting ketone was fully consumed as indicated by TLC or 1H NMR. Once the reaction was complete, the mixture was cooled, and then the ethyl acetate was removed under reduced pressure. Hexanes were added to the crude solid and the mixture was sonicated for 5 min. The hexanes were decanted and a fresh volume was added to the remaining solids, again sonicating for 5min. This process was repeated once more, for a total of 3 extractions. The combined hexane layers, which typically appeared as an amber or light yellow solution, were evaporated under reduced pressure to yield the crude product which typically appeared as yellow or orange oil. If a sonicator is not available, a Soxhlet extraction with hexanes gives similar yields. The crude bromoketone oil was next dissolved in a 5:1 mixture of ethanol/water to give a 0.4 M solution overall. The solution was cooled over ice, and then 3 equivalents of NaCN were added. The reaction was stirred overnight (16 h), allowing the ice to melt. The solution was then diluted with enough water to roughly double the initial volume. This solution was filtered through a Celite pad to remove suspended solids. This solution was then acidified by adding concentrated HCl to a stirring solution. CAUTION This will cause the evolution of HCN gas Only perform in a well-ventilated fume hood The acidified solution was allowed to stir for 15 minutes. The solution was checked by pH paper to ensure that it was acidic (pH ≤ 2). If not, additional HCl was added, taking the same precautions noted above. Once the solution was acidic, it was transferred to a separatory funnel and extracted 3x with DCM. The organic layers were combined, dried over magnesium sulfate, filtered, and then evaporated under reduced pressure to obtain the final product. Purity was confirmed by 1H NMR. If significant impurities are observed, the product can be recrystallized, most typically in isopropanol or toluene. |
|
With bromine In chloroform at 20℃; |
|
|
With hydrogen bromide; bromine; acetic acid at 0℃; for 20h; |
4.1.1.1 Synthesis of 2-bromo-1-(4-(methylsulfonyl)phenyl)ethan-1-one (1)
Bromine (0.019mol, 1mL) in acetic acid (10mL) was added to a solution of substituted 1-(4-(methylsulfonyl)phenyl)ethan-1-one (0.016mol, 3.168 gr) and 3-5 drops of hydrogen bromide in acetic acid (10mL) at 0°C. The reaction was processed under magnetic stirring for 20h. After completion of the reaction the mixture was poured in an ice-bath, precipitated product was filtered, dried, and recrystallized from EtOH. |
|
With copper(ll) bromide In ethyl acetate Reflux; |
|
|
With N-Bromosuccinimide; toluene-4-sulfonic acid In water; acetonitrile Reflux; |
|
|
With N-Bromosuccinimide; toluene-4-sulfonic acid In 1,4-dioxane for 6h; Reflux; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping