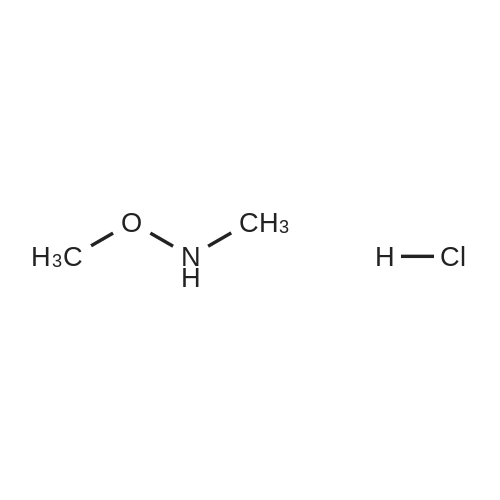
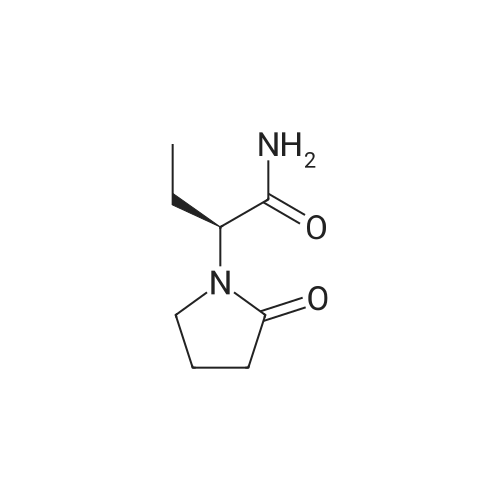
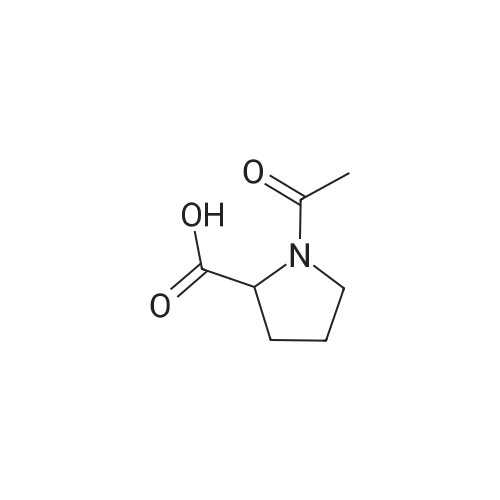
| 56% |
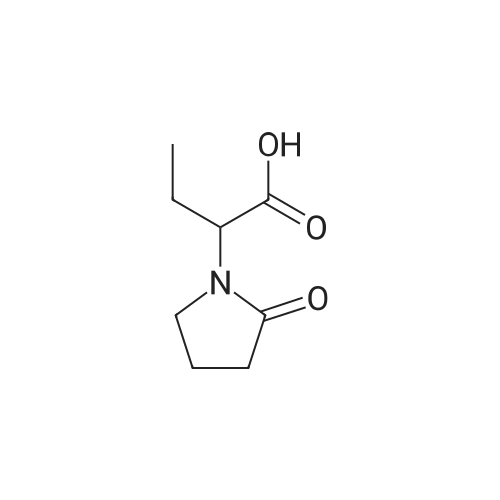
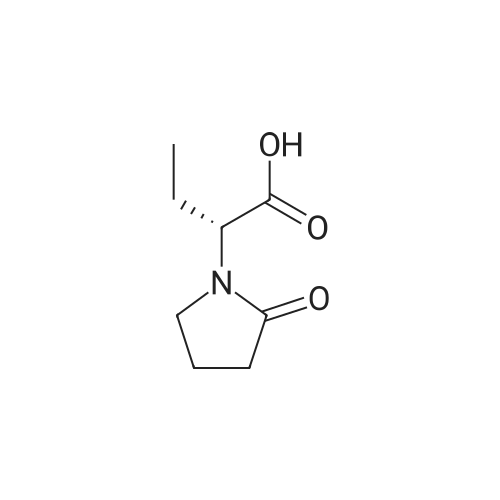
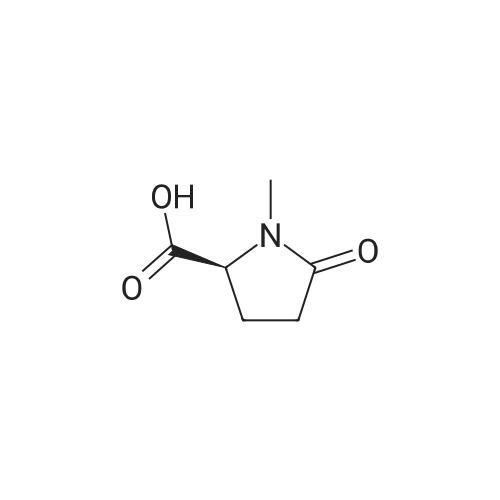
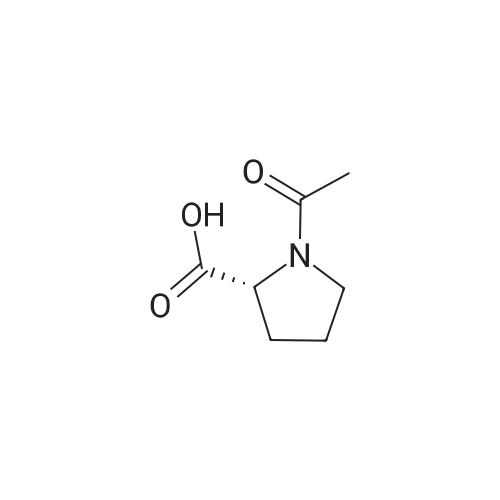
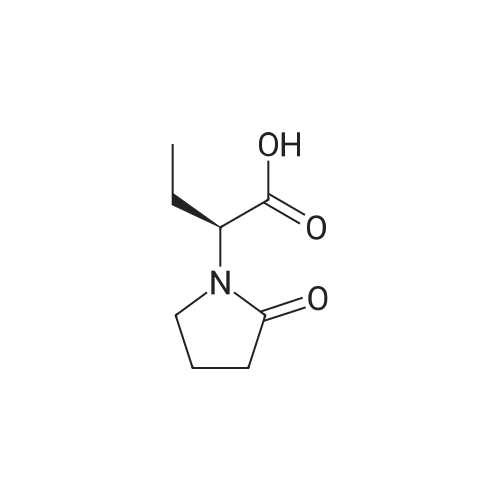
Stage #1: chloroformic acid ethyl ester; (2S)-2-(2-oxopyrrolidin-1-yl)butanoic acid With triethylamine In dichloromethane at 0℃; for 0.5h;
Stage #2: (+)-(7R,11R)-7-[(3-chloro-6,7,10,11-tetrahydro-9-methyl-7,11-methanocycloocta[b]quinolin-12-yl)amino]heptan-1-amine In dichloromethane at 20℃; for 72h; |
9 Example 9: Preparation of (+)-(αS,7R,11R)-N-{7-[(3-chloro-6,7,10,11-tetrahydro-9-methyl-7,11-methanocycloocta[b]quinolin-12-yl)amino]heptyl}-2-(2-oxopyrrolidin-1-yl)butanamide (αS,7R,11R)-(Ia)
Example 9 Preparation of (+)-(αS,7R,11R)-N-{7-[(3-chloro-6,7,10,11-tetrahydro-9-methyl-7,11-methanocycloocta[b]quinolin-12-yl)amino]heptyl}-2-(2-oxopyrrolidin-1-yl)butanamide (αS,7R,11R)-(Ia) A solution of (S)-2-(2-oxopyrrolidin-1-yl)butanoic acid (IIa) (99 mg, 0.58 mmol) in anhyd. CH2Cl2 (6 mL) was cooled to 0 °C with an ice bath, and treated dropwise with anhyd. Et3N (0.16 mL, 117 mg, 1.16 mmol) and ethyl chloroformate (0.05 mL, 62.8 mg, 0.58 mmol). The resulting mixture was stirred at 0 °C for 30 min and treated with a solution of amine (+)-(7R,11R)-(IIIa) (230 mg, 0.58 mmol) in anhyd. CH2Cl2 (5 mL). The reaction mixture was stirred at room temperature for 3 days and treated with 10% aq Na2CO3 (40 mL). The phases were separated and the aqueous phase was further extracted with CH2Cl2 (3 x 30 mL). The combined organic extracts were dried with anhyd. Na2SO4, filtered, and evaporated under reduced pressure to give a crude product (343 mg), which was subjected to column chromatography [40-60 µm silica gel (10 g), CH2Cl2/MeOH/50% aq NH4OH mixtures]. On elution with CH2Cl2/MeOH/50% aq NH4OH 99.95:0.05:0.2, the compound of the title (176 mg, 56% yield) was isolated as a yellowish solid. The isolated compound of the title was transformed into the corresponding hydrochloride as follows: A solution of the free base (176 mg, 0.32 mmol) in CH2Cl2 (15 mL) was filtered through a 0.2 µm NYL filter and treated with a 0.53 N methanolic solution of HCl (2.7 mL, 1.43 mmol). The solution was concentrated in vacuo to dryness and the solid residue was washed with pentane and dried at 65 °C/2 Torr for 7 days to give (αS,7R,11R)-(Ia)•HCl (168 mg) as a yellowish solid. Characterization: (αS,7R,11R)-(Ia): Rf = 0.70 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.05). (αS,7R,11R)-(Ia)•HCl: mp 102-105 °C (CH2Cl2/MeOH 85:15); [α]20D = +152 (c = 0.48, MeOH); IR (KBr) ν: 3500-2500 (max at 3256, 3055, 2925, 2860, 2796, N-H,+N-H, and C-H st), 1657, 1649, 1630, 1581, 1565, 1514 (C=O, ar-C-C and ar-C-N st) cm-1; HRMS (ESI) calcd for (C32H4335ClN4O2 + H+) 551.3147, found 551.3138. Elemental analysis, calcd for (C32H43ClN4O2•HCl•0.5H2O) C 64.42, H 7.60, N 9.39, Cl 11.88; found C 64.86, H 7.98, N 8.64, Cl 11.00. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping