* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 23, p. 7151 - 7154
[2] Journal of Medicinal Chemistry, 2018, vol. 61, # 5, p. 2087 - 2103
5
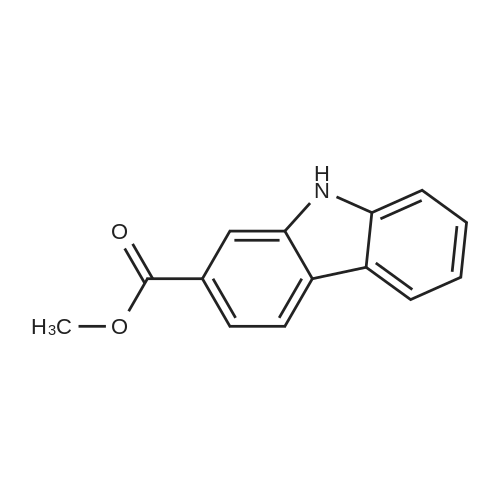
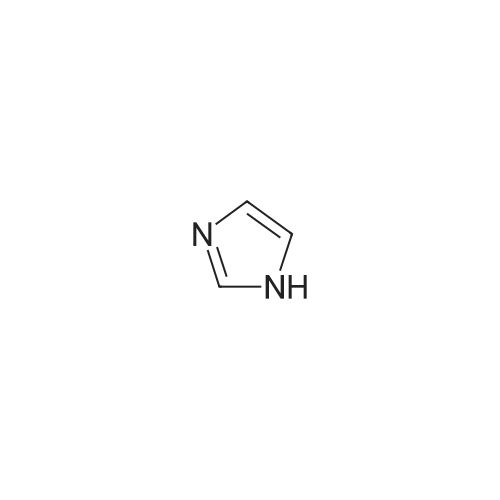
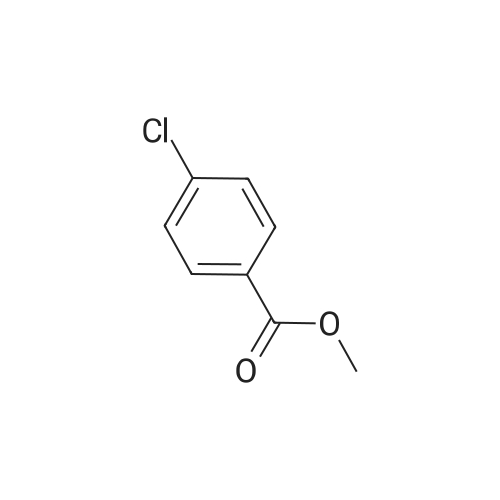
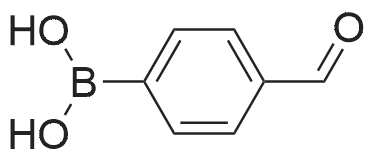
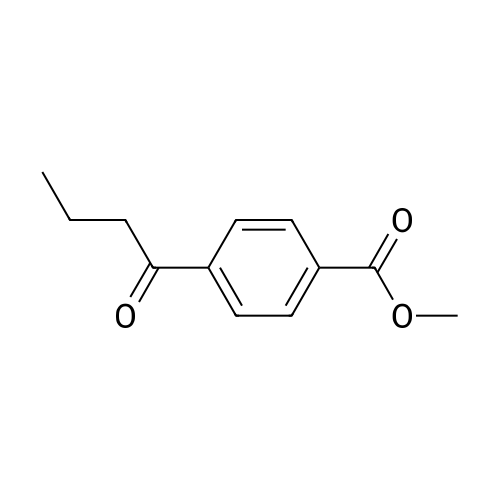
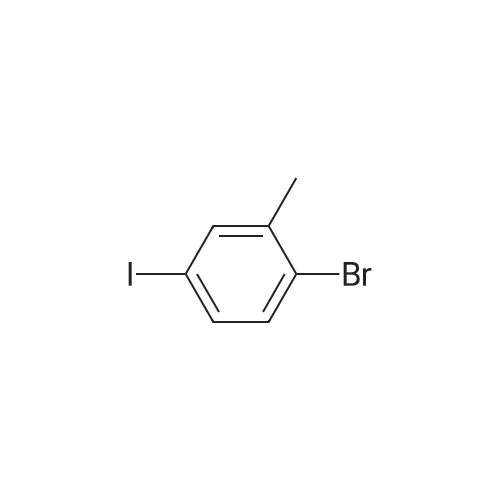
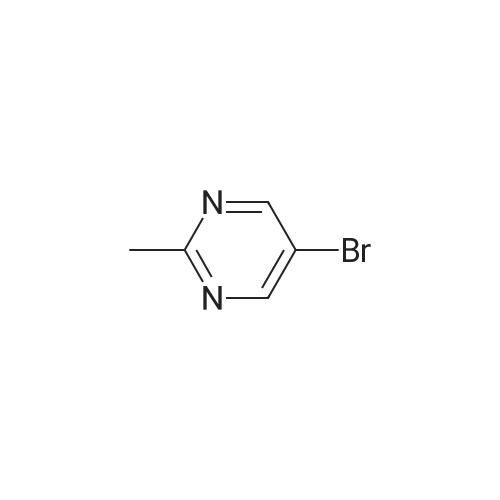
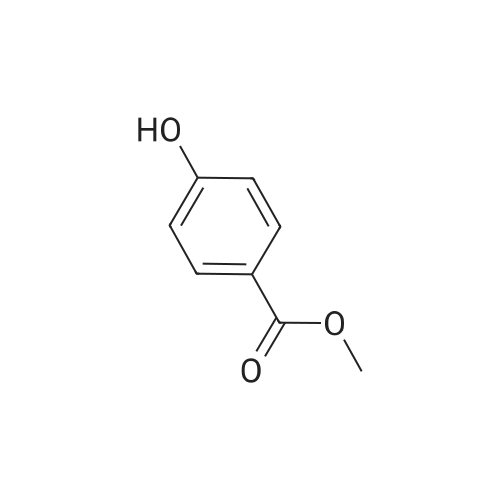
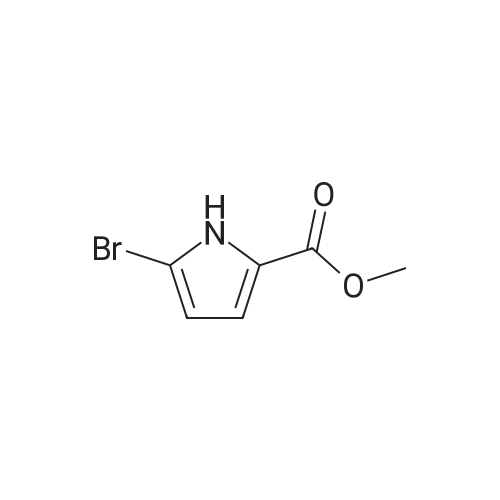
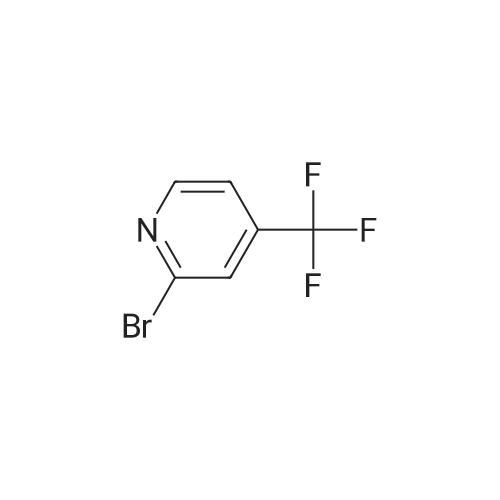
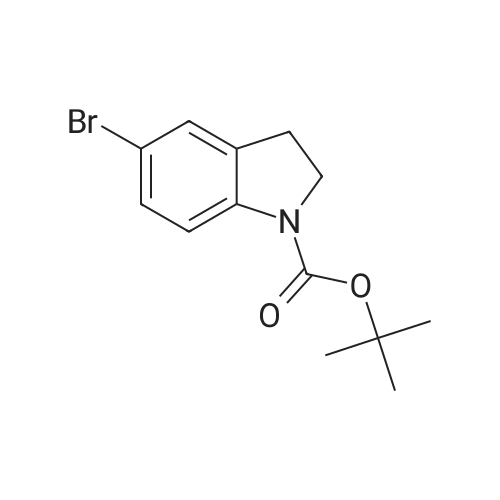
[ 1899-24-7 ]
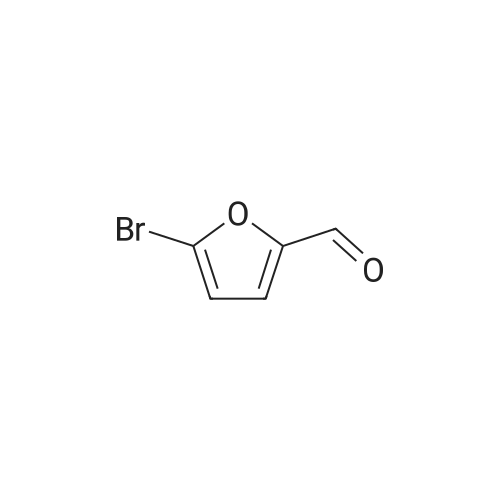
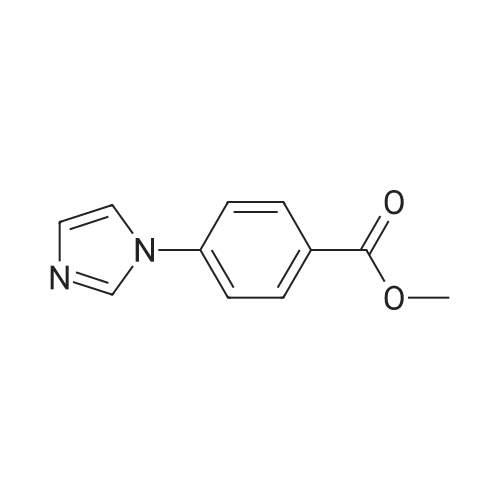
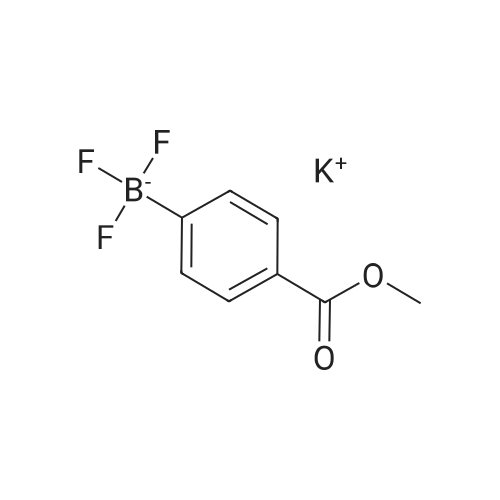
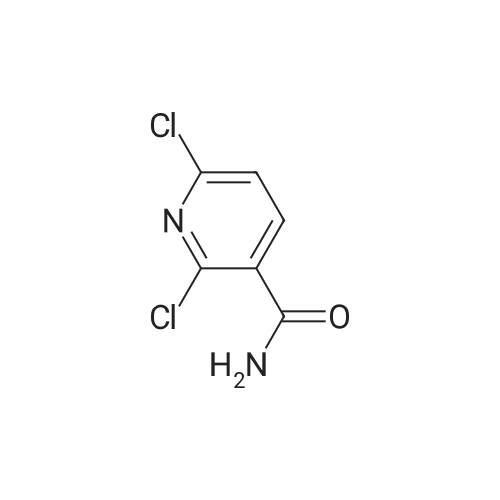
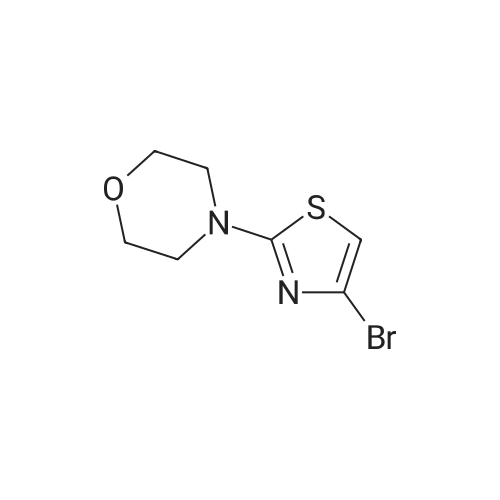
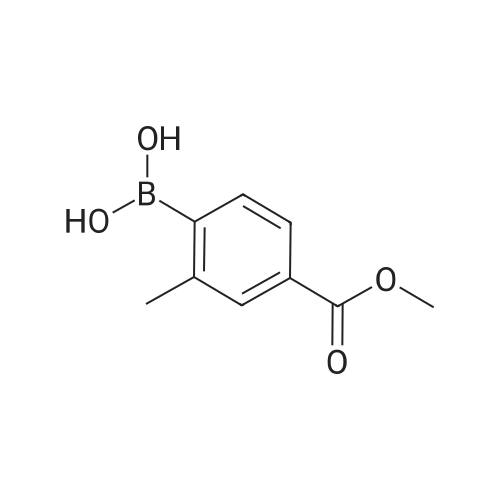
[ 99768-12-4 ]
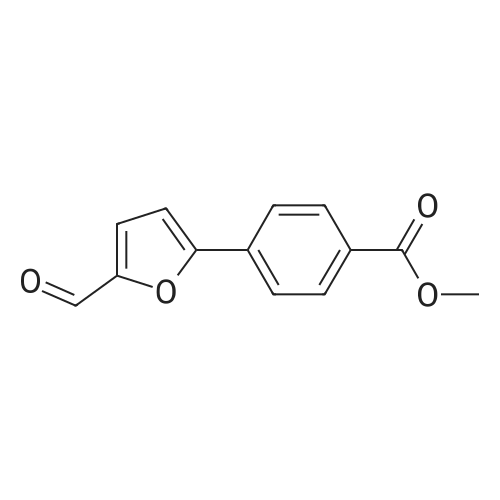
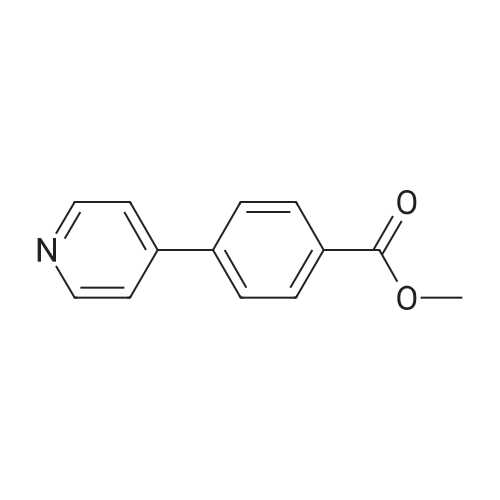
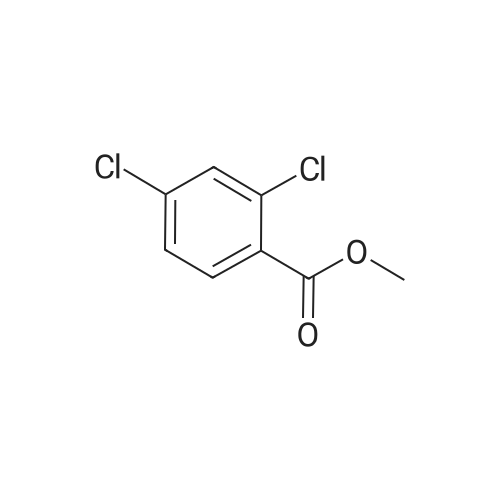
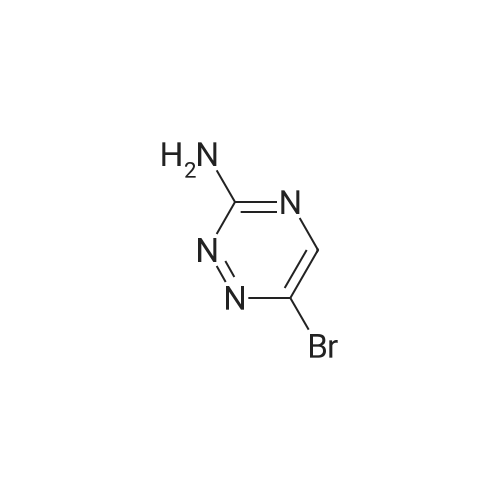
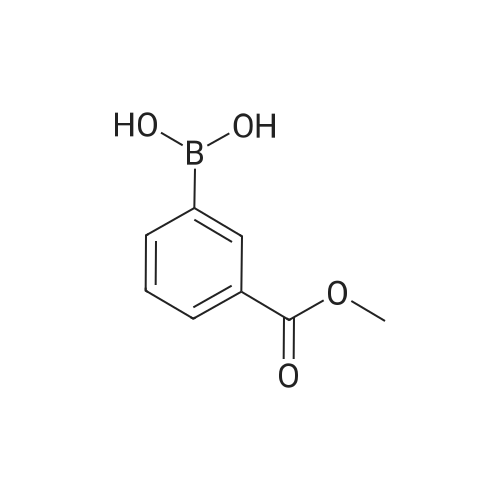
[ 53355-29-6 ]
Yield
Reaction Conditions
Operation in experiment
81%
With potassium fluoride; tri-tert-butyl phosphine In 1,4-dioxane; hexane at 65 - 70℃; for 4 h;
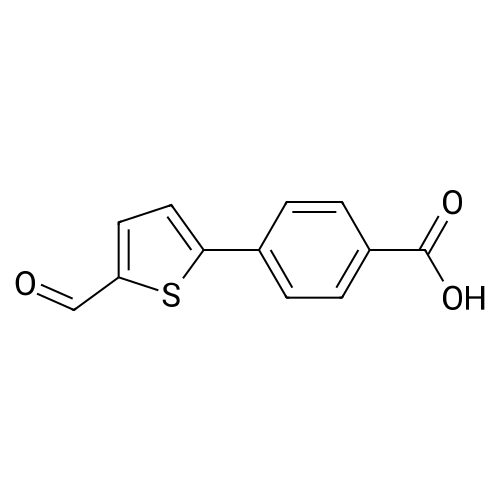
A solution of 5-bromofuraldehyde (2.43 g, 13.9 mmoles), 4-(methoxycarbonyl)phenylboronic acid (2.50 g, 13.9 mmoles), tris(dibenzylideneacetone)palladium (0) (192 mg, 0.2 mmoles) and potassium fluoride (2.42 g, 41.7 mmoles) in 1,4-dioxane (100 ml) was added with a solution of tri-toert-butylphosphine in hexane (10percent by weight, 101 mg, 0.5 mmoles). After heating at 65-70°C for 4 hours, the mixture was cooled to room temperature and treated with methylene chloride (150 ml). After stirring for 10 minutes, the mixture was filtered through Celite and the filtrate was EPO <DP n="13"/>concentrated under reduced pressure. The residue was purified by flash silica gel chromatography eluting with an ethyl acetate-hexane mixture (1 : 1) to give 4-(5-formyl-furan-2-yl)-benzoic acid methyl ester (2.6 g, 81percent yield).1H NMR (300 MHz, CDCl3): δ 9.70 (s, IH), 8.10 (d, 2H), 7.90 (d, 2H), 7.35 (d, IH), 6.95 (d, IH), 3.98 (s, 3H).
78%
With palladium diacetate; sodium carbonate In water; acetone at 20℃; for 0.416667 h;
To a solution of sodium carbonate (588.2 mg, 5.55 mmol) in H2O /acetone (278 mL/ 555 mL) was added 5-bromo-2-furaldehyde (1.9 g, 11 mmol), p-(methoxycarbonyl)phenyl boronic acid (2.0 g, 11 mmol) followed by palladium acetate(24.9 mg, 0.111 mmol) in acetone (278 ml). The reaction mixture was stirred at room temperature for 25 min. The reactionwas quenched by diluting with EtOAc (855 mL) and the organic layer was washed with 1 M HCl (500 mL) twice andwith brine (500 mL) once. The organic layer was dried over Na2SO4 and filtered with silica gel, which was concentratedin vacuo. The residue was purified by flash column chromatography (CH2Cl2/hexane = 9/ 1) to give compound 9 as whitesolid (2.0 g, 8.7 mmol, 78percent)
73%
With Pd(N,N-dimethyl-β-alaninate)2; sodium carbonate In ethanol; water at 120℃; for 0.166667 h; Microwave irradiation; Sealed tube
General procedure: To a solution of the appropriate bromo-substituted heterocyclic aldehydes 17a (1.0 mmol) in EtOH/H2O 5:3 (tot 12 mL) in a 35 mL CEM microwave vessel, the correspondent boronic acids 18c-d (1.2 mmol), Na2CO3 2M (2.0 mmol) and Pd(N,N-Dimethyl β-alaninate)2 (5 molpercent) were added. The vessel was capped and placed in a microwave reactor and the reaction carried out with the following method in dynamic mode: 120° C, 10 min, 50W, with high stirring. After completion the vessel was allowed to cool to room temperature and the mixture was extracted with EtOAc (3 X 10 mL). The organic phase was collected, dried over anhydrous Na2SO4, and the solvent evaporated under vacuum. The crude product (containing a small portion of the ethyl ester as a transesterification product) was then purified via silica gel column chromatography (petroleum ether/EtOAc elution gradient from a 90/10 ratio to a 80/20 ratio) to obtain the pure compounds (yield 40-60percent) (Scheme 1).
Reference:
[1] Patent: WO2006/66846, 2006, A1, . Location in patent: Page/Page column 11; 12
[2] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 9, p. 1562 - 1565
[3] European Journal of Medicinal Chemistry, 2015, vol. 101, p. 63 - 70
6
[ 1899-24-7 ]
[ 13716-12-6 ]
[ 99768-12-4 ]
[ 53355-29-6 ]
Yield
Reaction Conditions
Operation in experiment
81%
With potassium fluoride In hexane; dichloromethane
A. 4-(5-Formyl-furan-2-yl)-benzoic Acid Methyl Ester To a solution of 5-bromofuraldehyde (2.43 g, 13.9 mmol), 4-(methoxycarbonyl)phenyl boronic acid (2.50 g, 13.9 mmol), tris(dibenzylideneacetone)dipalladium(0) (192 mg, 0.21 mmol) and potassium fluoride (2.42 g, 41.7 mmol) in 1,4-doxane (100 ml) was added a solution of tri-t-butylphosphine in hexane (10 weight percent, 1.01 g, 0.5 mmol). After heating at 65-70° C. for 4 hours, the mixture was cooled to room temperature and treated with dichloromethane (150 ml). After stirring for 10 minutes, the mixture was filtered through a pad of celite and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel eluding with ethyl acetate-hexane (1:1) to provide 4-(5-formyl-furan-2-yl)-benzoic acid methyl ester (2.6 g, 81percent yield).
With chloro[tris(para-trifluoromethylphenyl)phosphine]gold (I); cesium fluoride In methanol at 20℃; for 15 h; Irradiation; Inert atmosphere
In a dried Pyrex screwtop reaction tube (4CF3C6H4)3PAuC (0.01 mmo, 10 mo percent)and (4(methoxycarbony)pheny)boronic acid (0.1 mmo, 1.0 equiv.) were dissoved in0.5 mL MeCH, After adding CsF (0.2 mmol, 2.0 equiv.) 4bromobenzenediazoniumbis((trWuoromethy)sufony)amide (0.4 mmoL 0.4 equiv,) the reaction mixture was de gassed under argon by sparging for 51 0 mm. The tube was irradiated with 29 W bue LEDs for 15 hours. The crude mixture was subjected to GCMS analysis, product 3o was detected, The product was purified by pTLC (SiC2, PE/EA, 5:1) to give 10.5 mg of 30 (0.04 mmol, 36percent). This shows, that adding an external fluoride source the reactionsproceeds and the desired product can be formed,
Reference:
[1] Journal of Materials Chemistry A, 2015, vol. 3, # 46, p. 23493 - 23500
13
[ 106-40-1 ]
[ 99768-12-4 ]
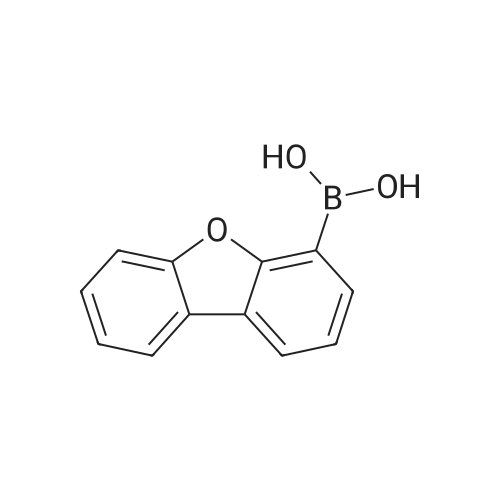
[ 89901-03-1 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2017, vol. 359, # 9, p. 1522 - 1528
14
[ 99768-12-4 ]
[ 69812-51-7 ]
Reference:
[1] Journal of the American Chemical Society, 2013, vol. 135, # 29, p. 10638 - 10641
15
[ 288-32-4 ]
[ 99768-12-4 ]
[ 101184-08-1 ]
Yield
Reaction Conditions
Operation in experiment
84%
With CuI-USY zeolite In methanol at 65℃; for 17 h;
General procedure: In a 10-mL round-bottom flask were successively added CuI-USY (ca. 15 mg, 10 molpercent of copper species), the nucleophile (0.75 mmol, 1.5 equiv), the boronic acid (0.5 mmol, 1.0 equiv), and MeOH (3.0 mL). The mixture was refluxed under air for 17 h and analyzed by LCMS. After cooling to r.t., the solvent was removed and the desired compound was isolated by purification on a short pad of silica gel (cyclohexane/EtOAc mixture).
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate In 1,2-dimethoxyethane at 90℃; for 10 h; Inert atmosphere
Step A: Under nitrogen atmosphere, a mixture of 4-bromopyridine (3.0 g, 19.0 mmol), (4-(methoxycarbonyl)phenyl)boric acid (2.63 g, 14.6 mmol), Pd(PPh3)2Cl2(0.35 g, 0.5 mmol) and sodium carbonate (6.91 g, 65.2 mmol) in 1,2-dimethoxylethane (40 mL) was heated to 90°C and stirred for 10 h. The resultant mixture was concentrated under reduced pressure and the residue was purified with silica gel column chromatography (eluent: petroleum ether/ethyl acetate=6/1-2/1) to give methyl 4-(pyridine-4-yl)benzoate (2.7 g, yield: 86.8percent) as white solid. MS(ESI)m/z:214[M+H+].
86.8%
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate In 1,2-dimethoxyethane at 90℃; for 10 h; Inert atmosphere
A solution of 4-bromopyridine (3.0 g, 19.0 mmol), (4- (methoxycarbonyl) phenyl) boronic acid (2.63 g, 14.6 mmol), Pd (PPh3) 2Cl2 (0.35 g, 0.5 mmol ) And sodium carbonate (6.91 g, 65.2 mmol)1,2-dimethoxyethane (40 ml) was heated to 90 ° C under a nitrogen atmosphere and stirred for 10 hours.The resulting mixture was concentrated under reduced pressure and the residue was purified by silica gel column chromatography (eluent:Petroleum ether / ethyl acetate = 6/1 to 2/1)4- (pyridin-4-yl) benzoate(2.7 g, yield: 86.8percent) as a white solid.
26.4%
With sodium carbonate In 1,4-dioxane; water
2.45a.methyl 4-pyridin-4-yl-benzoate 3.0 g (15 mmol) of 4-bromo-pyridine is dissolved in 50 mL dioxane and 15 mL 2M sodium carbonate solution. 2.7 g (15 mmol) of 4-methoxycarbonylphenyl-boric acid and 1.73 g (2 mmol) of tetrakis-(triphenylphosphine)-palladium are added successively and the reaction is refluxed for 6 h. The hot reaction solution is suction filtered through a glass fibre filter. The solvent is eliminated using the rotary evaporator and the purification is carried out by column chromatography on silica gel (dichloromethane/methanol 9:1). Yield: 845 mg (26.4percent of theory); C13H11NO2 (M=213.238); calc.: molar peak (M+H)+: 214 fnd.: molar peak (M+H)+: 214; Retention time HPLC: 4.1 min (method A).
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
In 500mL three-necked flask,Methyl 4-bromobenzoate (20.2 g, 77.2 mmol) was dissolved in dry THF (200.0 mL)Triisopropyl borate (18.9 g, 100.3 mmol) was added,Cool to -78 ° C,N-Butyllithium (6.8 g, 96.5 mmol) was added dropwise,Maintain the reaction temperature 0.5h.The reaction is completed,Saturated aqueous ammonium chloride solution was quenched,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL × 3), the organic phases were combined,Saturated brine (60 mL × 1), dried over anhydrous sodium sulfate,The solvent was distilled off under reduced pressure, the residue was beaten with n-hexane,The filtrate was filtered to give 4-methoxycarbonyl phenylboronic acid 13.3g, a yield of 95.5percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0063; 0064
21
[ 5419-55-6 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Yield
Reaction Conditions
Operation in experiment
94.8%
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
In 500mL three-necked flask,Methyl 4-iodobenzoate (20.1 g, 76.7 mmol) was dissolved in dry THF (200.0 mL)Triisopropyl borate (18.0 g, 95.9 mmol) was added,Cool to -78 ° C,N-Butyllithium (6.1 g, 95.9 mmol) was added dropwise,Maintain the reaction temperature 0.5h.The reaction was completed, quenched with saturated aqueous ammonium chloride solution,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL x 3)The combined organic phase,Saturated brine (60 mL × 1)Dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure,The residue was beaten with n-hexane, and filtered to obtain 13.1 g of 4-methoxycarbonylphenylboronic acid,Yield 94.8percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0023; 0024; 0079; 0080
22
[ 688-74-4 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Yield
Reaction Conditions
Operation in experiment
91%
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
In 500mL three-necked flask,Methyl 4-iodobenzoate (20.4 g, 77.8 mmol) was dissolved in dry THF (200.0 mL)Tri-n-butyl borate (21.5 g, 93.4 mmol) was added,Cool to -78 ° C,N-Butyllithium (5.5 g, 85.6 mmol) was added dropwise,Maintain the reaction temperature 0.5h.The reaction was completed, quenched with saturated aqueous ammonium chloride solution,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL x 3)The combined organic phases were washed with saturated brine (60 mL × 1)Dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure,The residue was beaten with n-hexane,Filtered to give 4-methoxycarbonyl phenylboronic acid 12.7g, the yield was 91.0percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0039; 0040
23
[ 121-43-7 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Yield
Reaction Conditions
Operation in experiment
94.5%
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
In 500mL three-necked flask,Methyl 4-iodobenzoate (20.3 g, 77.4 mmol) was dissolved in dry THF (200.0 mL)Trimethyl borate (16.1 g, 154.8 mmol) was added,The temperature was lowered to -78 ° C and n-butyllithium (7.4 g, 116.1 mmol) was added dropwise.Maintain the reaction temperature 0.5h.The reaction was completed, quenched with saturated aqueous ammonium chloride solution,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL × 3), the organic phases were combined,Saturated brine (60 mL × 1), dried over anhydrous sodium sulfate,The solvent was distilled off under reduced pressure, the residue was beaten with n-hexane,The filtrate was filtered to give 4-methoxycarbonyl phenylboronic acid 13.2g, a yield of 94.5percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0047; 0048
24
[ 67-56-1 ]
[ 14047-29-1 ]
[ 99768-12-4 ]
Reference:
[1] Journal of the American Chemical Society, 2016, vol. 138, # 39, p. 12767 - 12770
[2] Patent: US2008/76762, 2008, A1, . Location in patent: Page/Page column 44-45
[3] Journal of the American Chemical Society, 2007, vol. 129, # 36, p. 11265 - 11278
[4] Journal of the American Chemical Society, 2007, vol. 129, # 36, p. 11265 - 11278
[5] Angewandte Chemie - International Edition, 2016, vol. 55, # 11, p. 3606 - 3610[6] Angew. Chem., 2016, vol. 128, # 11, p. 3670 - 3674,5
[7] Journal of the American Chemical Society, 2009, vol. 131, # 47, p. 17500 - 17521
[8] Inorganic Chemistry, 2013, vol. 52, # 19, p. 10732 - 10734
25
[ 67-56-1 ]
[ 5720-05-8 ]
[ 99768-12-4 ]
Reference:
[1] Journal of the American Chemical Society, 2009, vol. 131, # 47, p. 17500 - 17521
Reference:
[1] Journal of Organometallic Chemistry, 1989, vol. 367, p. 259 - 266
30
[ 7664-93-9 ]
[ 14047-29-1 ]
[ 99768-12-4 ]
Reference:
[1] Patent: US5866568, 1999, A,
31
[ 14047-29-1 ]
[ 77-78-1 ]
[ 99768-12-4 ]
Reference:
[1] Journal of the American Chemical Society, 1996, vol. 118, # 45, p. 11093 - 11100
32
[ 125971-04-2 ]
[ 99768-12-4 ]
Reference:
[1] Journal of Organometallic Chemistry, 1989, vol. 367, p. 259 - 266
33
[ 619-42-1 ]
[ 99768-12-4 ]
Reference:
[1] Journal of the American Chemical Society, 2012, vol. 134, # 28, p. 11667 - 11673
[2] Journal of Organic Chemistry, 2013, vol. 78, # 13, p. 6427 - 6439
34
[ 13675-18-8 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Reference:
[1] Journal of Organic Chemistry, 2018, vol. 83, # 4, p. 1842 - 1851
35
[ 13675-18-8 ]
[ 1126-46-1 ]
[ 99768-12-4 ]
Reference:
[1] Journal of Organic Chemistry, 2018, vol. 83, # 4, p. 1842 - 1851
36
[ 705254-34-8 ]
[ 93-58-3 ]
[ 1126-46-1 ]
[ 35112-28-8 ]
[ 99768-12-4 ]
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 17, p. 7195 - 7203
37
[ 619-58-9 ]
[ 99768-12-4 ]
Reference:
[1] Patent: CN106565761, 2017, A,
[2] Patent: CN106565761, 2017, A,
[3] Patent: CN106565761, 2017, A,
38
[ 87199-17-5 ]
[ 99768-12-4 ]
Reference:
[1] Angewandte Chemie - International Edition, 2016, vol. 55, # 11, p. 3606 - 3610[2] Angew. Chem., 2016, vol. 128, # 11, p. 3670 - 3674,5
Reference:
[1] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2014, vol. 53, # 5, p. 610 - 618
41
[ 99768-12-4 ]
[ 199678-12-1 ]
Reference:
[1] Patent: US2016/237043, 2016, A1,
42
[ 873-75-6 ]
[ 99768-12-4 ]
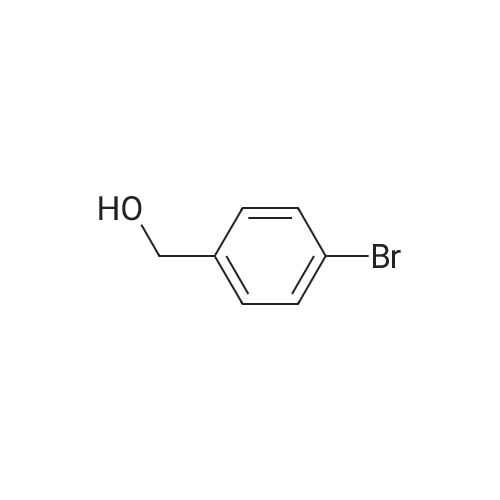
[ 393522-78-6 ]
Yield
Reaction Conditions
Operation in experiment
24%
With palladium diacetate; sodium carbonate In water; N,N-dimethyl-formamide at 70℃;
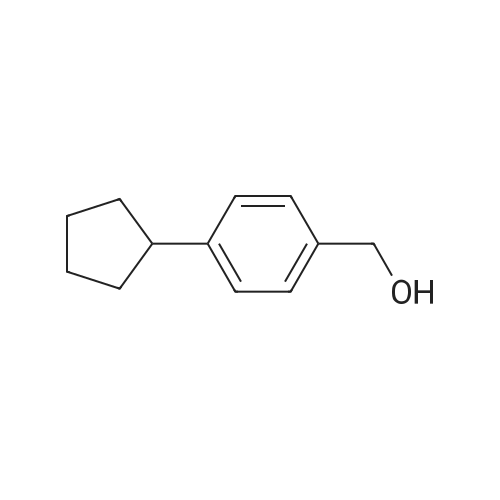
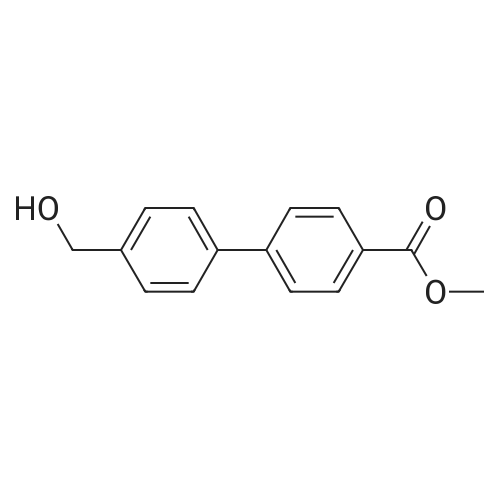
To a solution of (4-bromophenyl)methanol (500 mg, 2.94 mmol) and [4-(methoxycarbonyl) phenyl]boronic acid (550 mg, 3.53 mmol) in DMF (20 mL) was added Pd(OAc)2 (200 mg, 735 μmol) and Na2CO3 (3 g in 20 mL H2O). The reaction mixture was allowed to stir at 70 °C. After 48 h, an extraction with EtOAc was performed; the organic layers were combined and washed with brine, dried over MgSO4, filtrated and concentrated in vacuo. Column chromatography (EtOAc/Hept 1:4) yielded the product (170 mg, 24percent) as white crystals. RF = 0.3 (EtOAc/Hept 1:1). 1H NMR (400 MHz, CDCl3) δ 8.15-8.06 (m, 2H), 7.70-7.58 (m, 4H), 7.52-7.43 (m, 2H), 4.77 (d, J = 3.1 Hz, 2H), 4.07-3.67 (m, 3H), 1.75 (br s, 1H, OH) ppm. FTIR = 3305, 3032, 1718, 1604, 1432, 1399, 1286, 1271, 1220, 1111, 765 cm-1. HRMS (EI) calcd for C15H14O3 (M)+ 242.0942, found 242.0919.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 20, p. 5593 - 5603
43
[ 128-63-2 ]
[ 99768-12-4 ]
[ 933047-52-0 ]
Reference:
[1] Patent: US2016/46738, 2016, A1,
44
[ 99768-12-4 ]
[ 926304-76-9 ]
Yield
Reaction Conditions
Operation in experiment
48%
With oxygen; caesium carbonate In 1,2-dimethoxyethane; water at 20℃; for 10 h;
Examples 64-77 (Table 1) and Examples 78-83 (Table 2) show preparation of representative compounds by a procedure similar to that of Scheme 4, but with the inclusion of water (0.8percent) in the reaction mixture:
With C54H74Cl2N4O2Pd2; In ethanol; at 22℃; for 6h;
Reaction flask (10 mL) equipped with stirring bar was charged with (4-(methoxycarbonyl)phenylboronic acid (66 mg, 0.91 mmol), ethanol (96%) (2 mL), [{Pd(mu-OH)Cl(IPr)}2] (5 10-4 g, 4.55 × 10-7 mol). Reaction mixture was stirred at 22 C for 6 h. Aftercompletion of the reaction solvent was evaporated, the residue was dissolved in hexane (5 mL) andextracted with water (2 × 2 mL). The organic layer was dried over magnesium sulphate, and hexanewas evaporated under reduced pressure. 96 mg of dimethyl[1,1'-biphenyl]-4,4'-dicarboxylate wasobtained as a white solid. Yield = 97%. 1H NMR (403 MHz, CDCl3, ppm) delta: 8.13 (d, J = 8.7 Hz, 4H),7.69 (d, J = 8.7 Hz, 4H), 3.95 (s, 6H); 13C NMR (101 MHz, CDCl3, ppm) delta: 166.79, 153.16, 144.34,130.19, 129.68, 127.23, 124.20, 52.2. MS (EI) m/z (%) = 270 (M+, 48), 255 (11), 239 (100), 211 (9),209 (8), 153 (10), 152 (11), 59 (10).
95%
With Cu2(ophen)2; In N,N-dimethyl-formamide; at 20℃; for 20h;
General procedure: 4.3. Catalytic tests: general procedure I for the Cu2(ophen)2catalyzed homocoupling for Table 2 A solution of the corresponding arylboronic acids (1.0 mmol),Cu2(ophen)2 (1.3 mg, 0.5 mol %), DMF (1.0 mL) in 5 mL round-bottomed ask was stirred under air and the reaction was moni-tored by TLC. After the substrate was consumed, the reaction con-versions were determined bygas chromatography (GC) analysis (FIDfrom AGILENT 7820) using a cross-linked (95%)-dimethyl-(5%)-diphenylpolysil-oxane column (HP-5, 30 m0.32 mm0.25 mm),helium, injector temperature 250 C, detector temperature 300 C,and oven temperature program 45 C (3 min)e20C/mine280 C(2 min). The resulting mixture was poured into brine (10 mL), andextracted with diethyl ether (310 mL). The organic layer waswashed with brine, dried over Mg2SO4, the residue was chromato-graphed via a short column of silica gel (petroleum ether: diethylether15:1) and evaporated under reduced pressure. The productswas determined by 1H NMR spectroscopy. All 1H NMR spectra weremeasured in CDCl3 with TMS as the internal standard.
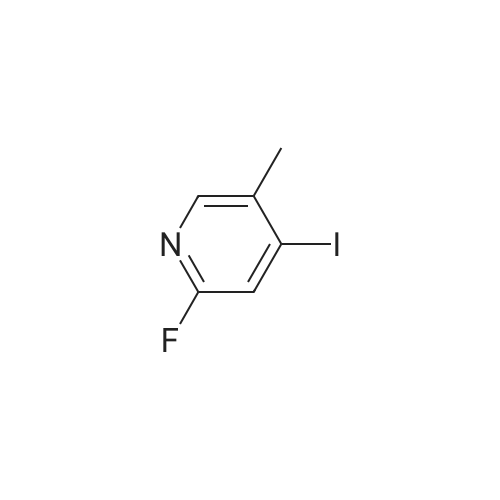
4-(2-fluoro-5-methylpyridin-4-yl)benzoic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
With potassium phosphate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 95℃;
2-Fluoro-4-iodo-5-methyl-pyridine [(0. 90] g, 3.8 mmol), 4- carboxymethyl-phenyl boronic acid (0.72 g, 4.0 mmol), potassium phosphate (2.5 g, 11.8 mmol), and dichloro [1, [1'-BIS] (diphenylphoshino) ferrocene] palladium [(II)] dichloromethane adduct (0.30g, 0.37 mmol) were combined in a screw cap tube and 1.4-dioxane (20 mL) was added. Argon was bubbled through the reaction mixture, which was then sealed and heated to [95C] overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated to a red solid, which was purified by chromatography on silica (EtOAc 0 to 40% in hexanes) to afford [4- (2-FLUORO-5-METHYL-PYRIDIN-4-YL)-BENZOIC] acid methyl ester, 0.62 g, 2.5 mmol, 66% [YIELD. 1H] NMR 500 MHz [(CDC13)] : 8.05 (m, 3H), 7.33 (d, 2H), 6.74 (s, 1H), 3.90 (s, 3H), 2.15 (s, 3H).
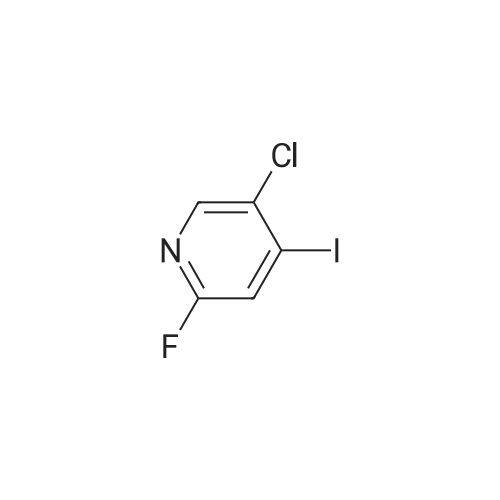
4-(2-fluoro-5-methylpyridin-4-yl)benzoic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
34%
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 85℃;
5-Chloro-2-fluoro-4-iodopyridine, (257 mg, 1 mmol), 4- [CARBOXYMETHYLPHENYL] boronic acid (0.2 g, 1.1 mmol) were dissolved in dimethoxyethane in a screw cap test tube and 1.5 [ML] 2M [NA2C03] was added followed by tetrakis (triphenylphosphine) palladium (50 mg, 0.044 mmol). Argon was bubbled through the reaction mixture for 5 min, the tube was sealed, and then the reaction mixture was heated to [85C] overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated to an oil, which was purified by chromatography on silica (EtOAc 0 to 15% in hexanes) to give 90 mg (0.34 mmol, 34% yield) [OF 4- (5-CHLORO-2-FLUOROPYRIDIN-4-YL)-] benzoic acid methyl [ESTER. 1H NMR] 500 MHz (CDC13) : 8.24 (s, [1EI),] 8.10 (d, 2H), 7. 48 (d, 2H), 6.89 (d, 1H), 3.91 (s, 3H). LCMS: ES+ = 257.9.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 90℃; for 3h;
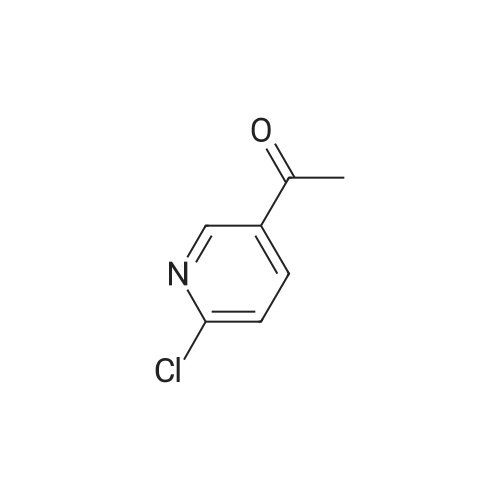
To a stirring solution of <strong>[55676-22-7]1-(6-chloro-pyridin-3-yl)-ethanone</strong> (l.Ommol, CAS No. 55676-22-7) and 4-methoxycarbonylphenyl boronic acid (1.2mmol) in dioxane (0.15M), add tetrakis- (triphenylphosphine) (0.044mmol) and 2M aqueous sodium carbonate (5.0mmol). Heat the reaction to 90C for three hours. After this time, remove the heat and concentrate in vacuo. Purify the title compound via radial chromatography eluting with methanol and dichloromethane. MS (m/e): 256.1 (M+l)
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 90℃; for 3h;
Intermediate Preparation 124-(5-Acetyl-pyridin-2-yl)-benzoic acid methyl ester Procedure U': To a stirring solution of <strong>[55676-22-7]1-(6-chloro-pyridin-3-yl)-ethanone</strong> (1.0 mmol, CAS No.55676-22-7) and 4-methoxycarbonylphenyl boronic acid (1.2 mmol) in dioxane (0.15M), add tetrakis-(triphenylphosphine) palladium (0.044 mmol) and 2M aqueous sodium carbonate (5.0 mmol). Heat the reaction to 90 C. for three hours. After this time, remove the heat and concentrate in vacuo. Purify the title compound via radial chromatography eluting with methanol and dichloromethane. MS (m/e): 256.1 (M+1)
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; for 3.0h;Heating / reflux;
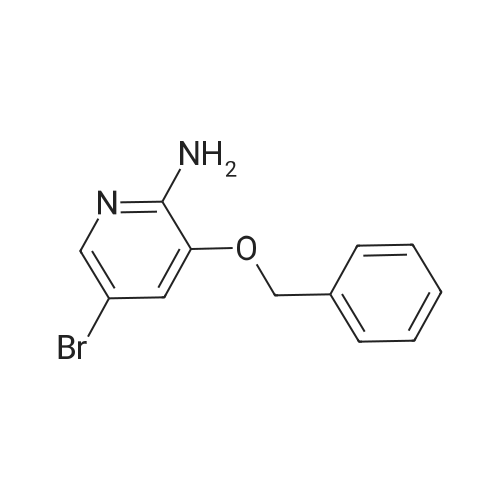
2. To a stirred mixture of <strong>[754230-78-9]3-benzyloxy-5-bromo-pyridin-2-ylamine</strong> (31.0 g, 0.11 mol) in a mixture of DME (600 mL) and H2O (600 mL) was added 4-carboxymethylboronic acid (29.9 g, 0.11 mol), Pd(PPh3)4 (6.4 g, 5.55 mmol), and Na2CO3 (82.0 g, 0.78 mol). The reaction was heated slowly to reflux and allowed to stir for 3 hr. The reaction was cooled to room temperature, then diluted with CH2Cl2 (1.5 L) and partitioned with H2O (700 mL). The organic layer was washed with saturated NaHCO3 (700 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The crude material was purified by column chromatography (silica gel, 1:1 to 4:1 EtOAc:hexanes) and the fractions containing product were combined and concentrated in vacuo to yield 4-(6-amino-5-benzyloxy-pyridin-3-yl)-benzoic acid methyl ester (29.4 g, 0.086 mol, 79%). 1H NMR (CDCl3, 300 MHz) 3.92 (s, 3H), 4.82-4.94 (brs, 2H), 5.15 (s, 2H), 7.22 (d, 1H, J, 1.8 Hz), 7.33-7.42 (m, 5H), 7.54 (d, 2H, J, 8.6), 7.98 (d, 1H, J, 1.8 Hz), 8.06(d, 2H, J, 8.6 Hz).
79%
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; for 3.0h;Heating / reflux;
To a stirred mixture of <strong>[754230-78-9]3-benzyloxy-5-bromo-pyridin-2-ylamine</strong> (31.0 g, 0.1.1 mol) in a mixture of DME (600 mL) and H20 (600 mL) was added 4-carboxymethylboronic acid (29.9 g, 0.11 mol), Pd(PPh3)4 (6.4 g, 5.55 mmol), and Na2C03 (82.0 g, 0.78 mol). The reaction was heated slowly to reflux and allowed to stir for 3 hr. The reaction was cooled to room temperature, then diluted with CH2CI2 (1.5 L) and partitioned with H20 (700 mL). The organic layer was washed with saturated NaHC03 (700 mL), dried (Na2S04), filtered, and concentrated in vacuo. The crude material was purified by column chromatography (silica gel, 1:1 to 4:1 EtOAc:hexanes) and the fractions containing product were combined and concentrated in vacuo to yield 4-(6-amino-5-benzyloxy-pyridin-3-yl)-benzoic acid methyl ester (29.4 g, 0.086 mol, 79%). 1H NMR (CDCI3, 300 MHz) 6 3.92 (s, 3H), 4.82-4.94 (brs, 2H), 5.15 (s, 2H), 7.22 (d, 1H, J, 1.8 Hz), 7.33-7.42 (m, 5H), 7.54 (d, 2H, J, 8.6), 7.98 (d, 1H, J, 1.8 Hz), 8.06(d, 2H, J, 8.6 Hz).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; for 18h;Heating / reflux;
Procedure M: A suspension of <strong>[34259-99-9]<strong>[34259-99-9]4-bromothiazol</strong>e</strong> (1.13 g, 6.89 mmol), 4-methoxycarbonylphenylboronic acid (1.85 g, 10.3 mmol) and Tetrakis (triphenylphosphine) palladium (0) (0.35 g, 0.30 mmol) in dioxane (45 mL) and 2M Na2CC>3 (17.2 mL) is heated to reflux for 18h. The reaction is allowed to cool and filtered. The filtrate is evaporated in vacuo, and the residue is dissolved in ethyl acetate and washed with water (2X) and brine (2X). The combined organic layers are dried over Na2S04, and concentrated in vacuo. The crude material is purified by flash chromatography (100percent hexanes - 40percent ethyl acetate/hexanes) to give 4-thiazol-4-yl-benzoic acid methyl ester as a white solid (0.68g, 45percent) MS (ES+) 220.2
4-(2-[4-(3,4-diethoxyphenyl)-4-oxobutanoyl]amino}-6-phenylpyridin-4-yl)benzoic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
tris-(dibenzylideneacetone)dipalladium(0); In water; toluene;
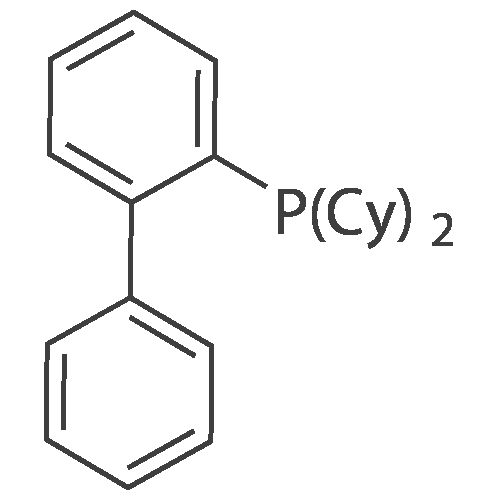
Example 112 methyl 4-(2-[4-(3,4-diethoxyphenyl)-4-oxobutanoyl]amino}-6-phenylpyridin-4-yl)benzoate To a solution of N-(4-chloro-6-phenylpyridin-2-yl)-4-(3,4-diethoxyphenyl)-4-oxobutanamide obtained in Example 43-(3) (190 mg) in toluene (5 mL) were added tris(dibenzylideneacetone)dipalladium (9 mg), biphenyl-2-yl(dicyclohexyl)phosphine (22 mg), aqueous potassium phosphate solution (267 mg) and [4-(methoxycarbonyl)phenyl]boronic acid (151 mg), and the mixture was heated overnight at 100C under nitrogen atmosphere. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with 1N hydrochloric acid, saturated aqueous sodium hydrogencarbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained crystals were recrystallized from ethyl acetate-hexane to give methyl 4-(2-[4-(3,4-diethoxyphenyl)-4-oxobutanoyl]amino}-6-phenylpyridin-4-yl)benzoate (131 mg) as crystals. 1H-NMR (CDCl3) delta: 1.43 - 1.54 (6 H, m, J=8.76, 7.02, 7.02 Hz), 2.90 (2 H, t, J=6.41 Hz), 3.44 (2 H, t, J=6.41 Hz), 3.95 (3 H, s), 4.10 - 4.21 (4 H, m, J=7.16, 7.16, 7.16, 7.16 Hz), 6.89 (1 H, d, J=8.48 Hz), 7.42 - 7.57 (4 H, m), 7.61 - 7.72 (2 H, m), 7.78 (2 H, d, J=8.48 Hz), 7.99 - 8.10 (2 H, m), 8.13 (2 H, d, J=8.48 Hz), 8.35 (1 H, s), 8.44 (1 H, s)
To the stirring solution of 2 (50 g, 287 mmol) in 1, 4-dioxane (1250 mL) was added 4- methoxycarbonyl phenyl boronic acid (2a) (56.89 g, 316 mmol) followed by 1M aqueous solution of potassium carbonate (575 mL, 574 mmol) at RT and purged with N2 gas for 40 minutes. Bis(triphenylphosphine)palladium(II)chloride (14.12 g, 20.12 mmol was added to the reaction mixture. The reaction mixture was heated to reflux temperature for 4h. After completion of the reaction (Confirm by TLC) the reaction mixture was cooled to RT and filtered through a bed of celite. The bed was washed with ethyl acetate (3 X 300 mL). Organic layer was separated and aqueous layer was re extracted with ethyl acetate (2 X 400 mL). The combined organic layer was washed with water, dried over sodium sulfate and concentrated under vacuum. The crude material was purified by column chromatography over silica gel (230-400 mesh) using 3.5% of MeOH in DCM as an eluent to afford 3 (39.1 g, 57%) as yellow solid.
50%
With sodium carbonate;1,1'-bis-(diphenylphosphino)ferrocene; In 1,4-dioxane; water; at 100℃; for 16h;
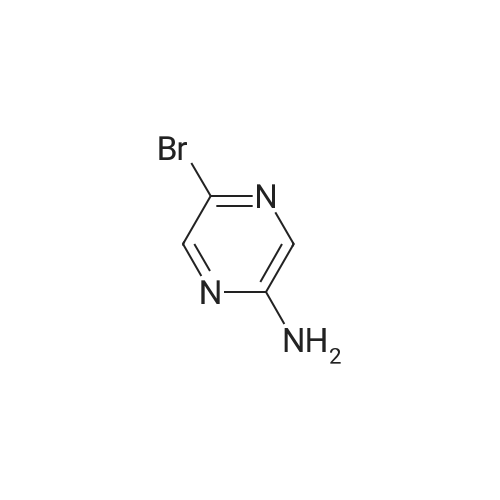
A. Methyl 4-(5-aminopyrazin-2-yl)benzoate. 4-(Methoxycarbonyl) phenylboronic acid (1.8 g, 9.7 mmol), 5-bromopyrazin-2-amine (1.8 g, 9.7 mmol), dichloro[l ,l'-bis(diphenylphosphino) ferrocene]palladium(II) dichloromethane adduct (213 mg, 0.29 mmol), I M sodium carbonate (29 mL, 29 mmol), and dioxane (100 mL) were heated together to 100 C for 16 h under nitrogen. The reaction was filtered through celite and then added with water and ethyl acetate to give a solid. The solid was filtered and dried to give a black solid (1.1 g, 50% yield). 1H NMR (400 MHz, DMSO-c/6) delta 8.63 (s, IH), 8.07 (s, 2H), 7.99 (s, 3H), 6.78 (s, 2H), 3.86 (s, 3H); MS (ESI) m/z 230.4 [M+l]+.
With bis-triphenylphosphine-palladium(II) chloride; potassium phosphate; In 1,4-dioxane; water; at 20℃; for 16h;Inert atmosphere; Reflux;
To the solution of a 5-bromo-pyrazine-2-amine according to formula (ii) (10 g, 57.47 mmol) in 1,4-dioxane (100 ml) was added 4-methoxycarbonylphenylboronic acid (11.377 g, 63.21 mmol) followed by potassium phosphate tribasic (14.638 g, 68.96 mmol) and water 68.96 ml at RT. Reaction mixture was purged with N2 gas for lh. Bis(triphenylphosphine)palladium(II)chloride (2.823 g, 4.02 mmol) was added to the reaction mixture. The reaction mixture was heated to reflux for 16h, cooled to RT and concentrated under vacuum to remove dioxane. Solid was filtered, washed with water ( 20 ml X 3), dried and again washed with MeOH (10 ml X 4) and dried to afford compound a methyl carboxylate intermediate of formula (xiv) (12.035 g, 91.36%, 84% purity) as pale yellow solid.
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; at 150℃;Microwave irradiation;
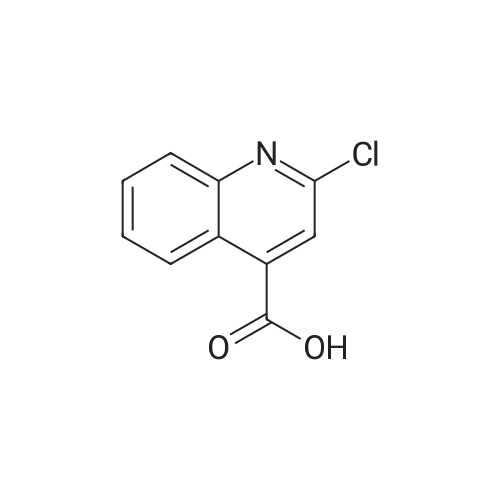
2-Chloro-quinoline-4-carboxylic acid (0.23 g, 1.4 mmol) was dissolved in dioxane (5 mL) and (4-methoxycarbonylphenyl)boronic acid (0.39 g, 2.2 mmol), Pd(PPh3)4 (0.20 g, 0.17 mmol) and K2CO3 (0.73 g, 5.3 mmol) were added. The reaction mixture was degassed, sealed, and heated in the microwave at 150 0C for 30 min. The reaction mixture was filtered and concentrated in vacuo to leave a residue. The residue was purified by flash column chromatography, using a 1:2 mixture of EtO Ac/heptane with 1percent acetic acid as eluent, to give the title compound (0.23 g, 53percent). m/z 308.06 (M+H)+.
53%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 150℃; for 0.5h;Inert atmosphere; Microwave irradiation;
2-Chloro-6-methylisonicotinic acid in place of 2-bromoisonicotinic acid. 2-Chloro-quinoline-4-carboxylic acid (0.23 g, 1.4 mmol) was dissolved in dioxane (5 mL) and (4-methoxycarbonylphenyl)boronic acid (0.39 g, 2.2 mmol), Pd(PPh3)4 (0.20 g, 0.17 mmol) and K2CO3 (0.73 g, 5.3 mmol) were added. The reaction mixture was degassed, sealed, and heated in the microwave at 150 °C for 30 min. The reaction mixture was filtered and concentrated in vacuo to leave a residue. The residue was purified by flash chromatography, using a 1:2 mixture of EtOAc/heptane with 1percent AcOH as eluent, to give the intermediate 2-[4-(methoxycarbonyl)phenyl]quinoline-4-carboxylic acid (0.23 g, 53percent). m/z 308.06 (M+H)+. TBTU (86 mg, 0.27 mmol) and N-methylmorpholine (39 mg, 0.38 mmol) were added to a solution of 2-[4-(methoxycarbonyl)phenyl]quinoline-4-carboxylic acid (29 mg, 0.10 mmol) in DMF (2 mL) and the reaction mixture was stirred at rt for 10 min. tert-Butyl [trans-4-(amino-methyl)cyclohexyl]methyl}carbamate (35 mg, 0.15 mmol) was then added and the reaction mixture was stirred at rt for 2 h. The reaction mixture was concentrated in vacuo to leave a residue, which was dissolved in DCM and washed with a saturated aq. solution of NaHCO3 and dried (phase separator). The mixture was concentrated in vacuo to leave a residue which was dissolved in DMSO and purified by HPLC using a gradient of 30-100percent mobile phase A (100percent CH3CN) over 30 min (mobile phase B = 5percent CH3CN + 95percent 0.1M NH4OAc) to give the intermediate methyl ester (12 mg, 24percent). m/z 530.32 (M+H)+.
With caesium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; water; at 105℃; for 16h;Inert atmosphere;
Example 40 Synthesis of methyl 4-(5-acetylfuran-2-yl)benzoate To a solution of <strong>[3199-50-6]1-(5-bromofuran-2-yl)ethanone</strong> (963 mg, 5.09 mmol) and cesium carbonate (4.98 g, 15.27 mmol) in water/dioxane (5%, 20 mL), flushed with nitrogen for 15 minutes, was added 4-methoxycarbonylphenyl boronic acid (1.01 g, 5.60 mmol), followed by catalyst PdCl2(dppf) (186 mg, 0.255 mmol). The solution was heated to reflux (105 C.) under nitrogen, for 16 hours. Water (50 mL) was added to the mixture after cooling to RT. Filtration gave 1.44 g of black crude material. Purification via silica column chromatography eluding with dichloromethane gave methyl 4-(5-acetylfuran-2-yl)benzoate (580 g, 2.37 mmol, 47% yield) as a yellow solid. LCMS (ES): m/z 245 [M+1]+.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 80℃; for 10h;Inert atmosphere;
General Procedure I-CDTo a solution of compound I-IXa (700 mg, 2.6 mmol) in toluene (15.0 mL) were added EtOH (3.0 mL), aq. Na2CO3 (2.0 M, 1.5 mL) and 4-(methoxycarbonyl)phenylboronic acid, the mixture was stirred under nitrogen atmosphere for 10 minutes, then, Pd(Ph3P)4 (90 mg, 0.08 mmol) was added and the flask was purged with nitrogen for three times. The mixture was stirred at 80 C. for 10 hours. After cooling to room temperature, the reaction mixture was extracted with ethyl acetate and washed with water and brine. The solvent was removed and the residue was purified by column chromatography on silica gel (eluent: PE:EtOAc=6:1) to give compound I-IXb (560 mg, yield 59%) as yellow solid. 1H NMR (400 MHz, CDCl3) delta 8.09 (d, 2H), 7.60 (d, 2H), 7.46 (d, 1H), 6.92 (d, 1H), 6.15 (s, 2H), 3.86 (q, 3H), 1.38 (t, 3H).
59%
To a solution of compound I-IXa (700 mg, 2.6 mmol) in toluene (15.0 mL), EtOH (3.0 mL)Aq. Na2CO3 (2.0 M, 1.5 mL) and 4-(methoxycarbonyl)phenylboronic acid,The mixture was stirred under a nitrogen atmosphere for 10 minutes, then Pd(Ph3P)4 (90 mg, 0.08 mmol) was added and the flask was purged three times with nitrogen.The mixture was stirred at 80 C for 10 hours.After cooling to room temperature, the reaction mixture was extracted with ethyl acetate and washed with water and brine.Solvent removal and purification of the residue on silica gel by column chromatography(eluent: PE: EtOAc = 6:1)Compound I-IXb (560 mg, yield 59%) was obtained as a yellow solid.
4-[5-(4-Hexyloxy-phenyl)-pyridin-2-yl]-benzoic acid Using the same sequence of reactions, the regioisomer was synthesized. Thus, coupling of 2-bromopyridyl-5-boronic acid with 4-O-hexyloxy-iodobenzene followed by reacting with 4-carbomethoxyphenylboronic acid and hydrolysis gave 4-[5-(4-Hexyloxy-phenyl)-pyridin-2-yl]-benzoic acid.
With sodium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; water; at 120℃; for 1h;microwave;
A mixture of <strong>[13676-02-3]2-chloro-6-methoxyquinoline</strong> (Intermediate 1, 200 mg, 1.0 mmol) , 4- (methoxycarbonyl) phenylboronic acid (205 mg, l . l mmol), Pd(dppf)Ci2 (366 mg, 0.5 mmol) and sodium carbonate (212 mg, 2.0 mmol) in 1,4-dioxane/water (3mL /0.6 mL ) was heated to 120C by microwave for 1 h. The precipitates were filtered; washed with EA (10 mL), acetone (10 mL) and water (10 mL) separately; dried to afford product (120 mg, 40.9%).
40.9%
With sodium carbonate;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; water; at 120℃; for 1h;microwave;
A mixture of <strong>[13676-02-3]2-chloro-6-methoxyquinoline</strong> (Intermediate 1, 200 mg, 1.0 mmol) , 4- (methoxycarbonyl) phenylboronic acid (205 mg,l. l mmol), Pd(dppf)Ci2 (366 mg, 0.5 mmol) and sodium carbonate (212 mg, 2.0 mmol) in 1 ,4-dioxane/water (3mL /0.6 mL ) was heated to 120C by microwave for 1 h. The precipitates were filtered; washed with EA (10 mL), acetone (10 mL) and water (10 mL) separately; dried to afford product (120 mg, 40.9%).
With CuI-USY zeolite; In methanol; at 65.0℃; for 17h;
General procedure: In a 10-mL round-bottom flask were successively added CuI-USY (ca. 15 mg, 10 mol% of copper species), the nucleophile (0.75 mmol, 1.5 equiv), the boronic acid (0.5 mmol, 1.0 equiv), and MeOH (3.0 mL). The mixture was refluxed under air for 17 h and analyzed by LCMS. After cooling to r.t., the solvent was removed and the desired compound was isolated by purification on a short pad of silica gel (cyclohexane/EtOAc mixture).
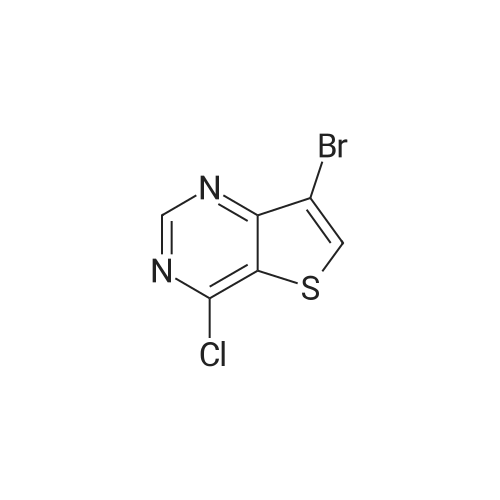
methyl 4-{7-bromothieno[3,2-d]pyrimidin-4-yl}benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; at 80℃; for 16h;Inert atmosphere;
Example 10: 2-(4-(4-(4-((3,4-Dimethylphenyl)carbamoyl)phenyl)thieno[3,2- ^pyrimidin-7-yl)cyclohexyl)acetic acid 1.14 g of 7-bromo-4-cWorotWeno[3,2-(pyrirnidine, 0.82 g of (4- (methoxycarbonyl)phenyl)boronic acid, and 0.316 g of tetrakis(triphenylphosphine)palladium were added to a mixed solvent of 7 mL of a 2 N aqueous sodium carbonate solution and 30 mL of 1 ,4-dioxane, and then stirred at 80 °C for 16 hours under an argon gas atmosphere. After the reaction, the reaction solution was extracted with 30 mL of ethyl acetate and 30 mL of water. The organic layer was dried by using anhydrous magnesium sulfate, filtered and then concentrated to obtain the title compound. This compound was used in the following step without further purification. FontWeight="Bold" FontSize="10" H NMR(300MHz, DMSO- ) S 9.42(s, 1H), 8.82(s, 1H), 8.38~8.18(m, 4H), 3.89(s, 3H).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 90℃; for 72h;Inert atmosphere;
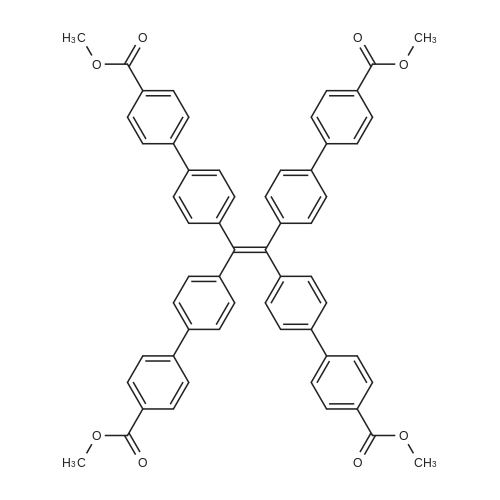
[0068] Synthesis of tcbpe (R, to R4 = H in Scheme 1): To a 250 mL three-neck flask, tetra-(4-bromo-phenyl)ethylene (tpe-Br, 2.85 g, 4.4 mmol), 4-(methoxycarbonyl)phenylboronic acid (5.00 g, 27.8mmol) and Pd(PPh3)4 (0.20 g) were added, then THF (tetrahydrofuran, 100 mL) and K2C03 aqueous solution(3.0 M, 15 mL) were added under nitrogen protection at room temperature, the mixture solution was kept at 90 °C for 3 days. After cooling to room temperature, the reaction solution was extracted with dichloromethane for three times. The organic phase was washed with water, dried with anhydrous magnesium sulfate and then subject to flash chromatography using dichloromethane as fluent. The product tcbpe-ester was obtained as green-yellow solid in 63.6percent yield (2.43 g). ?H NMR (400 MHz, CDC13) oe 8.07 (d, 8H, J = 8.1Hz), 7.64(d, 8H, J = 8.0Hz), 7.45(d, 8H, J = 7.9Hz), 7.21(d, 8H, J = 8.0Hz), 3.93(s, 12H); ?3C NMR (100 MHz, CDC13) oe 167.1, 145.0, 143.6, 140.8, 138.2, 132.2, 130.2, 129.0, 126.9, 52.3; MS (El): calcd. for C58H4408: 868.3036; Found, 868 (M). Anal. Calcd for C58H4408: C, 80.17percent; H, 5.10percent. Found: C, 79.81percent; H, 5.37percent.
53.2%
(2) N2 protection,1 g (1.55 mmol) of tetrakis (4-bromophenyl) ethylene (I) was added successively to a 250 ml two-necked flask, 4-methoxycarbonylphenylboronic acid (1.4 g, 7.8 mmol)2.5 g (16 mmol) of cesium fluoride (CsF)Add 50ml of solvent diethanol dimethyl ether (DME)Stir at room temperature for 30 min.(Pd (PPh3) 4) was added to 100 mg (0.08 mmol) under reflux for 10 h,After cooling, an equal amount of tetrakis (triphenylphosphine) palladium was added,Continue to respond 48h,To obtain a mixture.The reaction ends,Steaming solvent,The residue was sufficiently dissolved with methylene chloride,The insoluble material was removed by vacuum filtration.The filtrate was extracted three times,Collecting organic phase,Dried over anhydrous sodium sulfate,The obtained filtrate was steamed to remove the solvent,The residue was recrystallized from toluene,The yellow compound (II) was filtered off, dried in vacuo,Yield 53.3percent
With pyridine; copper diacetate; In dichloromethane; at 25℃; for 14h;
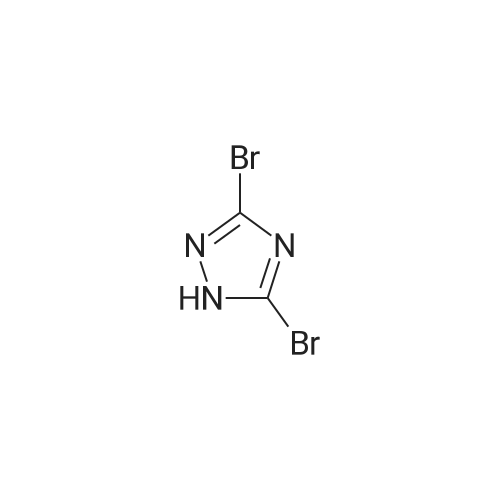
To a stirred solution of <strong>[7411-23-6]3,5-dibromo-1,2,4-triazole</strong>,2 (2.8 g, 12.34 mmol) in dichloromethane (40 mL) was added (4-(methoxycarbonyl)phenyl)boronic acid (2.0 g, 11.1 mmol), copper(II) acetate (4.0 g,22.0 mmol), pyridine (1.75 g, 22.1 mmol) and 4A molecular sieves sequentially. It was stirred for 14 h at 25C during which TLC showed the completion of the reaction. It was filtered through a celite bed and the filtrate was concentrated under reduced pressure to give crude product 3, which was further purified by column chromatography using silica gel (100-200 mesh) using ethyl acetate: hexane (30:70) as the eluent to afford 3 (2.70 g, 60%) as an off-white solid. m.p. 139.2-140.9C; IR (KBr): 2957, 1724, 1607,1510, 1447, 1391, 1311, 1295, 1266, 1118, 1003, 857,769 cm-1; 1H NMR (400 MHz, CDCl3): 3.97 (s, 3H),7.69 (d, 2H), 8.21 (d, 2H); 13C NMR (100 MHz,CDCl3): 52.7, 124.38, 129.2, 130.9, 130.9, 131.4,139.3, 141.9, 165.7; MS (EI): m/z Calcd for C10H7Br2N3O2: 361.89. Found: 361.90 (dibromo pattern).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; at 80℃; for 15h;
Step-2 [0492] Suzuki coupling of Step-1 product with meta/para carbethoxy/methoxy phenyl boronic acid was carried out in presence of Palladium (0) Tetrakis (Triphenyl phosphene) in dioxane and sodium carbonate as base. After completion of reaction, the reaction mixture was filtered through celite pad and filtrate was concentrated under reduced pressure residue was diluted with water and extracted with ethyl acetate to get crude product. Crude products obtained were purified by column chromatography over silica gel using 5-10% ethyl acetate in hexane. The details of the intermediates synthesized are as below in Table 50. Boronic acid (1.2 eq.), Water (5 vol) Dioxane (20 vol), Pd-Tetrakis (10 mol %), Sodium carbonate (2 eq.), 80 C., 15 hrs. Yield 64.8%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; water; at 100℃;Inert atmosphere;
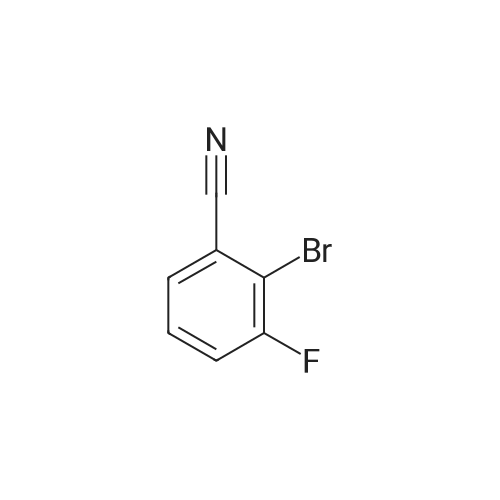
Preparation Example 4 An aqueous solution (3 ml) of tetrakis triphenylphosphine palladium (115 mg) and sodium carbonate (530 mg) was added to a dioxane (20 ml) solution of <strong>[425379-16-4]2-bromo-3-fluorobenzonitrile</strong> (667 mg) and 4-(methoxycarbonyl)phenyl boronic acid (600 mg), followed by stirring overnight at 100 C. in an argon atmosphere, thereafter, cooling to room temperature, diluting with ethyl acetate, washing with saturated brine, and drying over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The solid of the crude product obtained was washed with diisopropylether and dried under reduced pressure, thereby obtaining methyl 2'-cyano-6'-fluorobiphenyl-4-carboxylate (740 mg).
methyl 4-(2-methylpyrimidin-5-yl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85.4%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; at 110℃; for 1h;Microwave irradiation;
To a solution of KR-i4 (637 mg, 3.7 mmol) in i ,4-dixoane was added (4-(methoxycarbonyl)phenyl)boronic acid, R-22, (7i8 mg, 3.7 mmol) and(PPh3)4Pd (46 mg, 0.037 mmol), Na2003 (i .i 7 g, ii .i mmol). The reaction was stirred at ii 0 00 for i h by MW, After TLO (PE/AE i :i) showed thestarting material was consumed, the mixture was filtrated and concentrated under vacuo, and extracted with DOM, the organic layer was washed with brine, dried over anhydrous Na2SO4, The mixture was concentrated to givethe reagent KR-is (720 mg, 85.4percent) as a pale yellow solid. ESI-MS (M+i):229.i calc. for 013H12N202: 228.0.
methyl 4-(3-carbamoyl-6-chloropyridin-2-yl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,2-dimethoxyethane; water; at 90℃;Inert atmosphere;
To a solution of 2,6- dichloronicotinamide 2 (1.91 g, 10.0 mmol), 4-(methoxycarbonyl)phenylboronic acid 3 (1.8 g, 10.0 mmol) and Pd(dppf)Cl2 (816 mg, 1.0 mmol) in DME/H20 (20 mL/2mL) was added Cs2C03 (6.5 g, 20.0 mmol). The resulting solution was degassed with N2 6 times and stirred overnight at 90 C under N2 protection. After the reaction was completed, the solution was concentrated, diluted with ethyl acetate (30 mL) and washed with water (2 x 20 mL) and brine (2 x 20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The residue was purified by silica gel column diluted with 100: 1 to 60: 1 DCM/MeOH to afford the title compound (1.95g, 67%) as a white solid. MS (ESI): m/z = 291.1 [M + H]+.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 110℃; for 2h;Microwave irradiation;
i) Preparation of methyl 3-fluoro-4-(1H-pyrazolo[4,3-b]pyridin-3-yl)benzoate (B-2) A mixture of <strong>[633328-33-3]3-bromo-1H-pyrazolo[4,3-b]pyridine</strong> (B-1) (197 mg, 1.0 mmol), 4-(methoxycarbonyl)phenylboronic acid (198 mg, 1 mmol), Pd(PPh3)4 (115 mg, 0.1 mmol) and K2CO3 (420 mg, 3 mmol) was suspended in 1,4-dioxane (5 ml) and H2O (1 ml). The reaction mixture was heated at 110 C. in a microwave reactor for 2 h. The resultant mixture was diluted with H2O (30 ml) and extracted with ethyl acetate (30 ml*2). The combined organic layers were washed with brine (30 ml), dried over anhydrous Na2SO4 and concentrated to give the title compound B-2 as a brown oil. LCMS (ESI) calc'd for C14H10FN3O2 [M+H]+: 272.08. found: 272.
With Cu2(ophen)2; water; In ethanol; at 20℃;Green chemistry;
General procedure: 4.4. General procedure II for Table 3 A solution of arylboronic acids (1.0 mmol), Cu2(ophen)2 (1.3 mg,0.5 mol percent) in H2O-EtOH (1.8 mL, VH2O:VEtOHn:1) was stirred atroom temperature. After the substrate was consumed, the reactionconversions were determined by GC analysis.
With bis-triphenylphosphine-palladium(II) chloride; sodium hydrogencarbonate; In tetrahydrofuran; water; for 4h;Inert atmosphere; Reflux;
The synthesis has been carried out differing from the literature procedure [15] . A mixture of 4-(methoxycarbonyl)boronic acid (2.04 g, 11.4 mmol) and methyl 4-iodobenzoate (3.00 g, 11.4 mmol) in 75 ml THF and 90 ml H2O was degassed (10 min) under an argon atmosphere followed by the addition of Pd(PPh3)2Cl2 (92.4 mg, 0.132 mmol) and NaHCO3 (9.00 g, 107.1 mmol). After refluxing for 4 h and cooling down to room temperature, the resulting crystalline solid was collected by vacuum filtration (fraction 1). The residue obtained by the evaporation of the filtrate was extracted with 200 ml CH2Cl2. Drying of the organic phase (Na2SO4) and evaporation of the solvent gave a white solid (fraction 2). The two fractions were combined and crystallized from toluene to yield 7 as a white flaky solid (1.96 g, 64percent). Mp: 211-212 °C (lit. 212-213 °C [39] ). 1H NMR (DMSO-d6): deltaH = 3.89 (s, CH3), 7.91 (d, Ar-H, J = 8.55 Hz), 8.07 (d, Ar-H, J = 8.55 Hz). 13C NMR (DMSO-d6): deltaC = 52.2 (CH3), 127.3, 129.2, 129.9, 143.2 (Ar-C), 165.9 (COOMe).
tetramethyl 4,4',4'',4'''-(pyrene-1,3,6,8-tetrayl)tetrabenzoate[ No CAS ]
[ 933047-52-0 ]
Yield
Reaction Conditions
Operation in experiment
82%
In tetrahydrofuran; 1,4-dioxane;
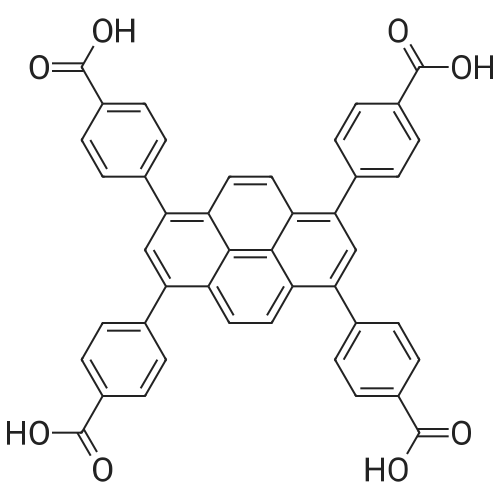
A) Synthesis of 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy) A mixture of (4-(methoxycarbonyl)phenyl)boronic acid (1.040 g, 5.80 mmol), 1,3,6,8-tetrabromopyrene (0.500 g, 0.97 mmol), tetrakis(triphenylphosphine) palladium(O) (0.030 g 0.026 mmol), and potassium tribasic phosphate (1.100 g, 5.30 mmol) in dry dioxane (20 mL) is loaded (in a glovebox) into a 20 mL microwave vial (Biotage) and capped. This mixture is stirred under argon for 72 hours at 130 C. in an oil bath. The reaction mixture is evaporated to dryness and the solid residue is washed with water to remove inorganic salts. The insoluble material is extracted with chloroform (three times by 50 mL), the extract is dried over magnesium sulfate, and the solvent volume is reduced under vacuum. The residue is boiled in tetrahydrofuran for 2 hours and filtered, the resulting filtrate containing mainly impurities. This procedure gives 0.58 g of 1,3,6,8-tetrakis(4-(methoxycarbonyl)phenyl)pyrene (82% yield). 1H NMR (CDCb-d): delta 3.99(s, 12H), 7.75 (d, 8H), 8.01 (s, 2H), 8.15 (s, 4H), 8.23 (d, 8H).
2',5'-difluoro-[1,1':4',1"-terphenyl]-4,4"-dimethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,2-dimethoxyethane; at 85℃; for 36h;Inert atmosphere;
1,4-Dibromo-2,5-difluorobenzene, 4-methoxycarbonylphenylboronic acid, potassium carbonate and tetrakis (triphenylphosphine) palladium were placed in a reaction In the reactor, and ethylene glycol dimethyl ether was added to the reactor, Then under the protection of nitrogen at 85 C for 36h after the first reaction A reaction solution, after the first reaction solution is cooled, The first reaction solution was poured into water and extracted with dichloromethane, and then ethylene glycol dimethyl ether was removed, The remaining material after pumping away ethylene glycol dimethyl ether, separated and purified by silica gel column chromatography to give To the intermediate product 2 ', 5'-difluoro- [1,1': 4 ', 1 "-terphenyl] -4,4" -dimethyl ester;
methyl 4-[(2R)-7-methoxy-4-oxo-3,4-dihydro-2H-1-benzopyran-2-yl]benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64.5%
Example 110A (i?)-methyl 4-(7-methoxy-4-oxochroman-2-yl)benzoate A 20 mL vial was charged with bis(2,2,2-trifluoroacetoxy)palladium (0.264 g, 0.795 mmol), (5)-4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole (0.195 g, 0.954 mmol), ammonium hexafluorophosphate(V) (0.777 g, 4.77 mmol) and (4-(methoxycarbonyl)phenyl)boronic acid (2.86 g, 15.89 mmol). The reaction was stirred in dichloroethane (5 mL) for 5 minutes, and a pale brown color suspension was observed. To this suspension was added Example 5 A (1.4 g, 7.95 mmol) and water (0.716 mL, 39.7 mmol) and the sides of the vial washed with more dichloroethane (5 mL). The vial was capped and the mixture stirred at 60 C overnight. The mixture was filtered through a plug of silica gel and celite and eluted with ethyl acetate to give a red solution. The solvent was removed under reduced pressure and the crude material was chromatographed using a 24 g silica gel cartridge with a gradient of 5-60 % ethyl acetate/heptanes over 20 minutes, a white solid precipitated in the middle of fractions collection and clogged up the line into the IR detection unit. The output line was unclogged and the white solid was filtered, the filtrate was concentrated and chromatographed again using a 12 g cartridge eluting with 100 % dichloromethane to give a white solid which was combined to give the title compound (1.6 g, 5.12 mmol, 64.5 % yield)). 1H NMR (400 MHz, DMSO-d6) delta 8.02 (dd, J = 8.4, 2.1 Hz, 2H), 7.75 - 7.72 (m, 1H), 7.70 (d, J = 8.4 Hz, 2H), 6.72 - 6.66 (m, 2H), 5.77 (dd, J = 12.9, 3.1 Hz, 1H), 3.87 (s, 3H), 3.83 (d, J = 2.0 Hz, 3H), 3.14 (dd, J = 16.8, 12.9 Hz, 1H), 2.82 (dd, J = 16.7, 3.1 Hz, 1H); MS (ESI+) m/z 313 (M+H)+.
64.5%
A 20 mL vial was charged with bis(2,2,2-trifluoroacetoxy)palladium (0.264 g, 0.795 mmol), (S-4-(tert-butyl)-2-(pyridin-2-yl)-4,5 -dihydrooxazole (0.195 g, 0.954 mmol), ammonium hexafluorophosphate(V) (0.777 g, 4.77 mmol) and (4-(methoxycarbonyl)phenyl)boronic acid (2.86 g, 15.89 mmol). The reaction was stirred in dichloroethane (5 mL) for 5 minutes, and a pale brown colored suspension was observed. To the suspension was added <strong>[5751-52-0]7-methoxy-4H-chromen-4-one</strong> (1.4 g, 7.95 mmol) and water (0.716 mL, 39.7 mmol). The sides of the vial were washed with more dichloroethane (5 mL). The vial was capped and the mixture was stirred at 60 C overnight. The mixture was filtered through a plug of silica gel and diatomaceous earth and eluted with ethyl acetate to give a red solution. The solvent was removed under reduced pressure and the crude material was chromatographed using a 24 g silica gel cartridge with a gradient of 5-60 % ethyl acetate/heptanes over 20 minutes. The white solid was collected by filtration and the filtrate was concentrated. The residue was chromatographed using a 12 g cartridge eluting with 100% dichloromethane to give a white solid which was combined with the solid collected by filtration to give the title compound (1.6 g, 5.12 mmol, 64.5 % yield) as a white solid. ?H NMR (400 MHz, DM50-cl6) 6 ppm 8.02 (dd, J = 8.4, 2.1 Hz, 2H), 7.75 - 7.72 (m, 1H), 7.70 (d, J = 8.4 Hz, 2H), 6.72 - 6.66 (m, 2H), 5.77 (dd, J = 12.9, 3.1 Hz, 1H), 3.87 (s, 3H), 3.83 (d, J = 2.0 Hz, 3H), 3.14 (dd, J = 16.8, 12.9 Hz, 1H), 2.82 (dd, J = 16.7, 3.1 Hz, 1H). MS (ESI+) m/z 313 (M+H).
39%
Example 15A methyl 4-[(2R)-7-methoxy-4-oxo-3,4-dihydro-2H-1-benzopyran-2-yl]benzoate A 250-mL round-bottomed flask was charged with (4-(methoxycarbonyl)phenyl)boronic acid (30.6 g, 170 mmol), ammonium hexafluorophosphate(V) (4.16 g, 25.5 mmol), bis(2,2,2-trifluoroacetoxy)palladium (2.123 g, 6.39 mmol), and (S)-4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole (1.565 g, 7.66 mmol), and then 1,2-dichloroethane (85 mL) was added. The resulting suspension was stirred at ambient temperature for 10 minutes, at which point Example 10A (15 g, 85.0 mmol) was added, followed by water (7.67 g, 426 mmol) and an additional 85 mL of 1,2-dichloroethane to rinse the sides of the flask. The reaction was heated at 60 C. in a sand bath (internal temperature) for 36 hours. The flask was cooled to room temperature, and the suspension was filtered through a 1-inch pad of silica, eluted with dichloromethane. The filtrates were concentrated to give a crude solid. tert-Butyl methyl ether (100 mL) and heptanes (100 mL) were added, and the solids were broken up with a spatula. The resulting suspension was heated at 60 C. for 1 hour while stining vigorously. The mixture was then cooled to ambient temperature, and the resulting solid was collected via filtration through a fitted funnel to give 10.54 g of the title compound (39% yield). 1H NMR (400 MHz, CDCl3) delta ppm 8.10 (d, J=8.3 Hz, 2H), 7.87 (d, J=8.9 Hz, 1H), 7.56 (d, J=8.3 Hz, 2H), 6.64 (dd, J=8.8, 2.3 Hz, 1H), 6.52 (d, J=2.4 Hz, 1H), 5.53 (dd, J=13.0, 3.2 Hz, 1H), 3.94 (s, 3H), 3.85 (s, 3H), 2.99 (dd, J=16.9, 13.0 Hz, 1H), 2.86 (dd, J=16.8, 3.2 Hz, 1H); MS (ESI+) m/z 312.8 (M+H)+.
39%
With ammonium hexafluorophosphate; palladium(II) trifluoroacetate; 2-[(S)-4,5-dihydro-4-tert-butyl-1,3-oxazol-2-yl]pyridine; In water; 1,2-dichloro-ethane; at 60℃; for 36h;
A 250-mL round-bottomed flask was charged with (4-(methoxycarbonyl)phenyl)boronic acid (30.6 g, 170 mmol), ammonium hexafluorophosphate(V) (4.16 g, 25.5 mmol), bis(2,2,2-trifluoroacetoxy)palladium (2.123 g, 6.39 mmol), and (S)-4-(tert-butyl)-2-(pyridin-2-yl)-4,5-dihydrooxazole (1.565 g, 7.66 mmol). 1,2-Dichloroethane (85 mL) was added. The resulting suspension was stirred at ambient temperature for 10 minutes, at which point Example 7D (15 g, 85.0 mmol) was added, followed by water (7.67 g, 426 mmol) and an additional 85 mL of 1,2-dichloroethane to rinse the sides of the flask. The reaction mixture was heated at 60 C. (internal temperature) in a sand bath for 36 hours. The flask was cooled to room temperature, and the suspension was filtered through a 1-inch pad of silica, eluting with dichloromethane. The filtrates were concentrated to give a crude solid. tert-Butyl methyl ether (100 mL) and heptanes (100 mL) were added, and the solids were broken up with a spatula. The resulting suspension was heated at 60 C. for 1 hour while stirring vigorously. The mixture was then cooled to ambient temperature, and the resulting solid was collected via filtration through a fritted funnel to give 10.54 g of the title compound (39% yield). 1H NMR (400 MHz, CDCl3) delta ppm 8.10 (d, J=8.3 Hz, 2H), 7.87 (d, J=8.9 Hz, 1H), 7.56 (d, J=8.3 Hz, 2H), 6.64 (dd, J=8.8, 2.3 Hz, 1H), 6.52 (d, J=2.4 Hz, 1H), 5.53 (dd, J=13.0, 3.2 Hz, 1H), 3.94 (s, 3H), 3.85 (s, 3H), 2.99 (dd, J=16.9, 13.0 Hz, 1H), 2.86 (dd, J=16.8, 3.2 Hz, 1H); MS (ESI+) m/z 312.8 (M+H)+.
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate; In 1,4-dioxane; water; at 90℃; for 16h;Inert atmosphere;
Methyl 4-(2-pyrazinyl) benzoate (0432) In a 250-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen were combined a solution of [4-(methoxycarbonyl)phenyl]boronic acid (1 g, 5.56 mmol, 1.20 equiv) in 1,4-dioxane (30 mL), <strong>[56423-63-3]<strong>[56423-63-3]2-bromopyrazin</strong>e</strong> (800 mg, 5.03 mmol, 1.00 equiv), a solution of Na2CO3 (1.5 g, 14.15 mmol, 3.00 equiv) in water (30 mL), and Pd(Ph3P)2Cl2 (330 mg, 0.47 mmol, 0.10 equiv). The resulting solution was stirred for 16 h at 90 C. in an oil bath. The solids were filtered out. The resulting solution was extracted with 3×30 mL of EtOAc, and the organic layers combined and concentrated under vacuum. This resulted in 0.8 g (74%) of the product as a white solid.
With tetrakis(triphenylphosphine) palladium(0); cesium fluoride; In 1,2-dimethoxyethane; for 72h;Reflux; Inert atmosphere;
A mixture of intermediate A (2 g), 4-carbomethoxybenzeneboronic acid (3.4 g), tetrakistriphenylphosphine palladium (0.2 g) and cesium fluoride (3 g) (DME). The reaction mixture was refluxed for 72 hours under nitrogen atmosphere, cooled to room temperature, and 100 ml of distilled water was added thereto. The mixture was extracted three times with dichloromethane, and the organic phase was taken. The organic layer was washed with anhydrous sulfuric acid The magnesium was dried overnight, filtered and the solvent was removed. The crude product was purified on a silica gel column to give intermediate C.
methyl 4’-amino-3‘-chlorobiphenyl-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,4-dioxane; water; at 100℃; for 2h;
Synthesis of methyl 4?-amino-3 ?-chlorobiphenyl-4-carboxylate (65): A mixture of 4-bromo-2-chloroaniline (10 g, 48.5 mmol), 4-(methoxycarbonyl)phenylboronic acid (9.6 g,53.4 mmol), Pd(dppf)C12 (3.6 g, 4.85 mmol) and K2C03 (13.4 g, 97 mmol) in dioxane (100 mL) and water (10 mL) was degassed and heated at 100C for 2 h. After cooling down to room temperature, the reaction mixture was poured into water, extracted with EtOAc (100 mL X 3). The combined organic solvents were dried over anhydrous Na2504, concentrated under reduced pressure and purified by silica gel chromatography (10-50% EtOAc/petroleum ether) to give 11 g of methyl 4?-amino-3?-chlorobiphenyl-4-carboxylate 65 as black solid (86% yield). LCMS: m/z 262.1 [M+H], , tR= 1.98 min
methyl 4’-amino-3‘-(trifluoromethyl)biphenyl-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,4-dioxane; water; at 100℃; for 1h;
Synthesis of methyl 4?-amino-3 ?-(trifluoromethyl)biphenyl-4-carboxylate (80): A mixture of 4-bromo-2-(trifluoromethyl)aniline (16 g, 66.7 mmol), 4- (methoxycarbonyl)phenylboronic acid (14.4 g, 80 mmol), Pd(dppf)C12 (4.9 g, 6.7 mmol) and K2C03 (18.4 g, 133 mmol) in dioxane (100 mL) and water (10 mL) was degassed and heated at 100C for 1 h. After cooling down to room temperature, the reaction mixture was poured into water, extracted with EtOAc (100 mL X 3). The combined organic solvents were dried over anhydrous Na2504, concentrated under reduced pressure and purified by silica gel chromatography (10-50% EtOAc/petroleum ether) to give 17 g of methyl 4?-amino-3?- (trifluoromethyl)biphenyl-4-carboxylate 80 as black solid (58% yield). LCMS: m/z 295.8 [M+H], , tR 1.76 min
With palladium diacetate; potassium carbonate; triphenylphosphine; In 1,2-dimethoxyethane; at 20℃; for 12h;Inert atmosphere;
To a solution of <strong>[106291-80-9]methyl 4-bromo-3-hydroxybenzoate</strong> (347 mg, 1.5 mmol) in (CH3OCH2)2/H2O (10 mL, 1:1) was added (4-(methoxycarbonyl)phenyl)boronic acid (324 mg, 1.8 mmol), Pd(OAc)2 (17 mg, 0.08 mmol), PPh3 (20 mg, 0.08 mmol) and K2CO3 (623 mg, 4.5 mmol). The mixture was stirred at room temperature under argon protection for 12 h. The solution was neutralized by 1N HCl, diluted with EtOAc, wash with H2O, brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel flash column chromatography (hexane/EtOAc=5:1) to afford compound 29 (220 mg, 51% yield). Yellow solid; 1H NMR (500 MHz, CDCl3) delta 8.16 (d, J=8.1 Hz, 2H), 7.69 (dd, J=7.9, 1.1 Hz, 1H), 7.66 (s, 1H), 7.61 (d, J=8.1 Hz, 2H), 7.35 (d, J=7.9 Hz, 1H), 3.96 (s, 3H), 3.94 (s, 3H).
51%
With palladium diacetate; potassium carbonate; triphenylphosphine; In 1,2-dimethoxyethane; water; at 20℃; for 12h;Inert atmosphere;
General procedure: To a solution of methyl 4-bromo-2-hydroxybenzoate (463 mg, 2 mmol) in (CH3OCH2)2/H2O (10 mL, 1:1) was added (4-(methoxycarbonyl)phenyl)boronic acid (432 mg, 2.4 mmol), Pd(OAc)2 (23 mg, 0.1 mmol), PPh3 (26 mg, 0.1 mmol) and K2CO3 (829 mg, 6 mmol). The mixture was stirred at room temperature under argon protection for 12h. The solution was neutralized with 1N HCl and the aqueous phase was extracted with EtOAc (5mL×3). The combined organic layer was wash with H2O, brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel flash column chromatography (hexanes/EtOAc=5:1) to afford compound 1 (537 mg, 94 %).
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In 1,4-dioxane; water; for 2h;Reflux; Inert atmosphere;
ompound 22 is (1.16 g, 5.25mmol), 23 is (710mg, 3.5mmol), Pd (PPh3. 3)2CI2(491 mg,0.7 mmol) were sequentially added to a dioxane (30mL), the state under an argon atmosphere was added K2CO.'s. 3(2.9 g of, 21 mmol) in water(10.5 mL) was heated refluxed for 2h.After completion of the reaction by TLC, the reaction mixture was diluted with 300 mL of ethyl acetate, washed with water, washed withsaturated sodium chloride, dried and evaporated to dryness to give 24 (578 mg, 64%).
With tetrakis(triphenylphosphine) palladium(0); cesium fluoride; In 1,4-dioxane; at 90℃; for 48h;Inert atmosphere;
(1) Under nitrogen protection, 4.33 g (24 mmol) of 4-methoxycarbonylbenzeneboronic acid, 6 g (40 mmol) of cesium fluoride were placed in a 250 ml three-necked flask, and 150 ml of 1,4-dioxane and 2.754 ml (10 mmol) were added. a mixed solution of <strong>[615-59-8]2,5-dibromotoluene</strong>,Then 1.6 g of tetrakis(triphenylphosphine)palladium catalyst was added and refluxed at 90 C for 48 h.After the reaction was completed, an orange solution was obtained with a black precipitate. The system was evaporated to dryness under reduced pressure.A white product of 3.0 g was obtained in a yield of 82%.
81%
With tetrakis(triphenylphosphine) palladium(0); cesium fluoride; In tetrahydrofuran; at 67℃;Inert atmosphere;
(1) <strong>[615-59-8]2,5-dibromotoluene</strong> (10 mmol, 2.5 g), 4-methoxycarbonylbenzeneboronic acid (24 mmol, 4.33 g) under N2 protection.caesium fluoride (40 mmol, 6.0 g) was placed in a 250 mL three-necked flask, and 5% (1.2 to 2.0 g) of Pd (PPh3) catalyst was added.100 mL of anhydrous THF was used as a solvent, and refluxed at 67 C.After completion of the reaction, the product was isolated by silica gel column chromatography (petroleum ether, dichloromethane) to give a pink solid 2.91 g, yield 81%.
methyl 5'-(1H-imidazol-1-yl)-2'-nitro-[1,1'-biphenyl]-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
44%
With bis-triphenylphosphine-palladium(II) chloride; In acetonitrile; at 140℃; for 1.5h;Inert atmosphere; Microwave irradiation;
[0226] Intermediate 1-2 (266 mg), (4-(methoxycarbonyl)phenyl)boronic acid (150 mg), and (Ph3P)2PdCl2, (66 mg, catalyst) were mixed with 2mL acetonitrile and 2mL water in a microwave tube and degassed, and backfilled with argon. A 2-phase system resulted. This mixture was heated at 140 degrees (High) for 3600 sec, then another 1800 sec. The black and tarry looking reaction was filtered, partitioned with EtOAc and water, separated, and dried over magnesium sulfate. After chromatography, as above, 140 mg (44percent) light yellow 85percent pure methyl 5'-(lH-imidazol-l-yl)-2'-nitro-[l, -biphenyl]-4-carboxylate, 1-3, was obtained. (0457) GC/MS: 5.736min, 15percent, m/e: 270, 239(base), dimethyl [l,l '-biphenyl]- 4,4'- dicarboxylate (impurity) and 7.081min (85percent, m/e: 323, 292, 235), 1-3.
With water; sodium hydroxide; In tetrahydrofuran; at 130℃; for 72h;Reflux;
(4-(Methoxycarbonyl)phenyl)boronic acid (1.040 g, 5.80 mmol), 1,3,6,8-tetrabromoindole (0.500 g, 0.97 mmol), tetrakis(triphenylphosphine)palladium ( 0) (0.030g 0.026mmol) and anhydrous potassium phosphate (1.100g,5.30 mmol) was placed (in a glove box) in a 20 mL microwave vial and capped.The mixture was stirred under an argon atmosphere at 130 C for 72 hours in an oil bath. The reaction mixture was evaporated to dryness and the solid residue was washed with water to remove inorganic salts. ChlorineThe insoluble material (50 mL three times) was extracted, and the extract was dried over magnesium sulfate, and the solvent volume was reduced under vacuum. The residue was boiled in tetrahydrofuran for 2 hours and filtered; the filtrate obtained mainly contained impurities.This procedure gave 1,8,6,8-tetrakis(4-(methoxycarbonyl)phenyl)indole 0.58 g (yield 82%). Then, to a 250 mL round bottom flask containing 0.58 g (0.78 mmol) of solid 1,3,6,8-tetrakis(4-(methoxycarbonyl)phenyl)anthracene, 100 mL of 1.5 g (37.5 mmol) NaOH was added. A mixture of tetrahydrofuran/water (ratio 1:1) was added and the resulting suspension was stirred vigorously under reflux overnight. The solvent was removed under vacuum and water was added to a residue that formed a clear yellow solution. The clear yellow solution was stirred at room temperature for 2 hours and the pH was adjusted to 1 using concentrated HCl. The resulting yellow solid was collected by filtration and washed several times with water. The crude product was recrystallized from DMF, filtered, washed with EtOAc. This gave 0.49 g (91%) of pure product H4TBAPy, abbreviated as H4L.
With pyridine; copper diacetate; In N,N-dimethyl-formamide; at 20℃; for 4h;
Compound 2 (2 mmol),4-methoxycarbonylbenzeneboronic acid (2 mmol),Copper acetate (0.4 mmol),Pyridine (0.4 mmol) in the reaction flask,Add 15 mL of DMF and dissolve at room temperature with stirring.Open reaction for about 4 hours,The reaction was complete by TLC.Add appropriate amount of ethyl acetate for extraction.Washed with saturated saline,The organic phase is concentrated,Separation and purification by silica gel column chromatography (ethyl acetate /Petroleum ether = 1/3),Obtained 2.1 g of white solid compound 25,The yield was 80%.
With bis-triphenylphosphine-palladium(II) chloride; sodium carbonate; In 1,4-dioxane; ethanol; at 140℃; for 0.5h;Sealed tube; Microwave irradiation;
General procedure: tert-Butyl 3-bromo 1H-indazole-1-carboxylate (0.5 mmol), Na2CO3(0.5 mmol), boronic acid (1.0 mmol), Pd(PPh3)2Cl2 (1 mol%), and 1,4-dioxane: ethanol = 4:1 (5 mL) were added to10 mL vial. The vial was sealed with a crimp cap and placed in a Biotage initiator microwave cavity. After thereaction vial was irradiated at 140 C for 30 min, themicrowave reactor was cooled with air. Product was separatedwith ethyl acetate and saturated aqueous NH4Cl solution.The organic layer was dried with anhydrous sodiumsulfate, filtered, and concentrated. Products were purifiedby silica gel column chromatography using a hexane: ethylacetate = 4:1.
To a stirred suspension of A-l (700 mg, 5.22 mmol) in DCM (25 mL) is added A-2 (1.88 g, 10.4 mmol), pyridine (843 /iL, 10.43 mmol) and TEA (1.43 mL, 10.43 mmol). The resultant mixture is sparged with oxygen for 10 min. Copper (II) acetate (1.90 g, 10.43 mmol) is added and the reaction mixture is stirred at the ambient temperature under an oxygen atmosphere. After 3 days, the mixture is filtered and the filtrate is washed with water (10 mL), dried over Na2S04, filtered and concentrated. The crude mixture is purified on Si02 eluting with 10% EtOAc in hexane to afford A-3.
methyl 4-(3-formyl-1H-pyrazol-1-yl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
With pyridine; oxygen; copper diacetate; In dichloromethane; at 20℃; for 18h;
To a solution of 1H-pyrazole-3-carbaldehyde (10.0 g, 104 mmol, CAS3920-20-1) and (4-methoxy carbonyl-phenyl) boronic acid (22.5 g, 125 mmol, CAS99768-12-4) in DCM (50 mL) was added Cu(OAc)2 (22.7 g, 125 mmol) and pyridine (32.9 g, 416 mmol). The reaction mixture was stirred at rt for 18 hours under oxygen gas (balloon). On completion, the mixture was concentrated in vacuo. The residue was purified by silica gel chromatography to give the title compound (12.0 g, 50% yield) as a white solid. 1H NMR (400 MHz, CDCl3) delta 10.10 (s, 1H), 8.24-8.14 (m, 2H), 8.06 (d, J=2.4 Hz, 1H), 7.90-7.82 (m, 2H), 7.02 (d, J=2.4 Hz, 1H), 3.95 (s, 3H).
methyl 4-(3-bromo-4-nitropyrazol-1-yl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
With pyridine; oxygen; copper diacetate; In dichloromethane; at 20℃; under 775.743 Torr; for 12h;
To a solution of <strong>[784193-37-9]3-bromo-4-nitro-1H-pyrazole</strong> (4.8 g, 25.0 mmol, CAS784193-37-9) and (4-methoxycarbonylphenyl) boronic acid (6.75 g, 37.5 mmol, CAS99768-12-4) in DCM (100 mL) was added Cu(OAc)2 (4.54 g, 25.0 mmol), and pyridine (7.91 g, 100 mmol) under O2 (15 Psi). The reaction mixture was stirred at 20 C. for 12 hours under O2 (15 Psi) atmosphere. On completion, the mixture was washed with NH3.H2O (50 mL), filtered and extracted with ethyl acetate (3×100 mL). The combined organic layers were washed with 2 N aq.HCl (60 mL) and brine (2×100 mL), dried over anhydrous sulfate sodium, filtered and concentrated in vacuo. The crude product was triturated with PE:EA=10:1 (100 mL) to give the title compound (5.00 g, 61% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) delta 9.82 (s, 1H), 8.11-8.06 (m, 4H), 3.88 (s, 3H).
tert-butyl 5-(4-(methoxycarbonyl)phenyl)indoline-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 80℃; for 20h;
Procedure B: To a mixture of <strong>[261732-38-1]tert-butyl 5-bromoindoline-1-carboxylate</strong> (18.0 g, 50.8 mmol) in dioxane/H2O (135 mL), were added (4-(methoxycarbonyl)phenyl)boronic acid (13.2 g, 76.2 mmol), Pd(dppf)Cl2 (4.2 g, 5.1 mmol) and Cs2CO3 (49.4 g, 152 mmol). The reaction mixture was stirred at 80 for 20 h, then diluted with DCM, and washed with brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography over silica gel (PE/EA 4:1, v/v) to give tert-butyl 5-(4-(methoxycarbonyl)phenyl) indoline-1-carboxylate as a yellow solid (11.2 g, 62%). LC-MS (ESI): m/z (M-56)+ = 298.14.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping