There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
Structure of 99614-02-5 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium hydroxide In ethanol; waterHeating / reflux
680 g of ondansetron hydrochloride dihydrate was dissolved in 4000 ml of ethanol at reflux. A solution of 82 g of NAOH in 1000ML of water was added. A solid was formed. 3000 ml of water was added and the mixture was cooled to ambient temperature. The solid was filtered off and washed with 2*500 ml of water. The solid was dried at 50°C under vacuum for 2 days. The product exhibited the DSC curve shown in fig. 1 and the XRPD pattern of fig. 2. Yield : 527 g (96percent)80 g of ondansetron hydrochloride dihydrate was suspended in 500 ml of ethanol and heated to reflux until a clear solution was obtained. To this solution, 250 ml of a 1 M NAOH solution was added. During addition a solid started to form. 250 ml of water was added and the mixture was slowly cooled to room temperature. The mixture was cooled to 10- 15°C and the solid was filtered off. The solid was washed with 2X200 ml of water. After drying in a vacuumoven at 40°C for 3 days. 59.3 g of a white solid was obtained. The product exhibited the DSC curve shown in fig. 3 and the XRPD pattern of fig. 4. Yield : 59.3 g (92percent)
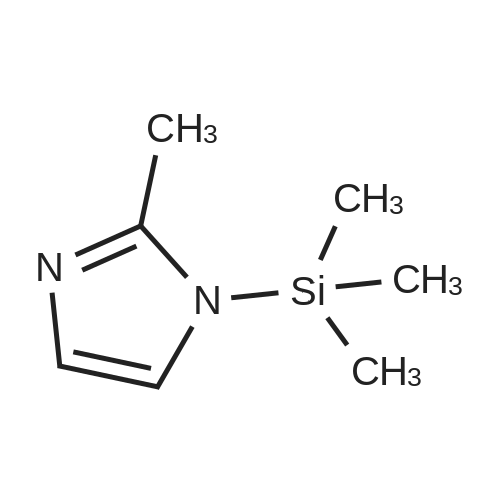
Example 17 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and 11 g of 2-methyl-l-trimethylsilyl imidazole were suspended in 35 ml of acetonitrile. The solution of 6.2 g of tetra-n- butylammonium fluoride hydrate in 15 ml of acetonitrile was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, the reaction solvent was distilled off. 100 ml of water was added to the residue, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.61 g of the title compound as a white solid (yield 98percent).; Example 20 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and 22 g of 2-methyl-l-trimethylsilyl imidazole were suspended in 35 ml of acetonitrile. The solution of 6.2 g of tetra-n- butylammonium fluoride hydrate in 15 ml of acetonitrile was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, the reaction solvent was distilled off. 100 ml of water was added to the residue, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.75 g of the title compound as a white solid (yield 100percent).
96.7%
for 2.16667 h; Heating / reflux
Example 19 The same procedures as described in Example 17 were repeated, except that ethyl acetate was employed instead of acetonitrile, to give 6.72 g of the title compound (yield 96.7percent).
96.1%
at 80℃; for 2 h;
Example 21 To the mixture of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7 and 22 g of 2-methyl-1- trimethylsilyl imidazole was added 6.2 g of tetra-n-butylammonium fluoride hydrate at 80 °C, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.48 g of the title compound as a white solid (yield 96. 1percent).
94.5%
for 2.16667 h; Heating / reflux
Example 18 The same procedures as described in Example 17 were repeated, except that tetrahydrofuran was employed instead of acetonitrile, to give 6.57 g of the title compound (yield 94.5percent).
92%
at 80℃; for 2.16667 h; Heating / reflux
Example 15 The mixture of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7 and 11 g of 2-methyl-1- trimethylsilyl imidazole was heated to 80 °C. 23.7 ml of IN tetra-n- butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.2 g of the title compound as a white solid (yield 92percent). Example 16 The mixture of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7 and 22 g of 2-methyl-1- trimethylsilyl imidazole was heated to 80°C. 2. 37ml of IN tetra-n-butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.61 g of the title compound as a white solid (yield 98percent).
87%
for 2.16667 h; Heating / reflux
Example 14 5g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and llg of 2-methyl-l-trimethylsilyl imidazole were suspended in 25 ml of acetonitrile. 1. 9ml of IN tetra-n- butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 5.85 g of the title compound as a white solid (yield 87percent).
With acetyl chloride In toluene; acetonitrileHeating / reflux
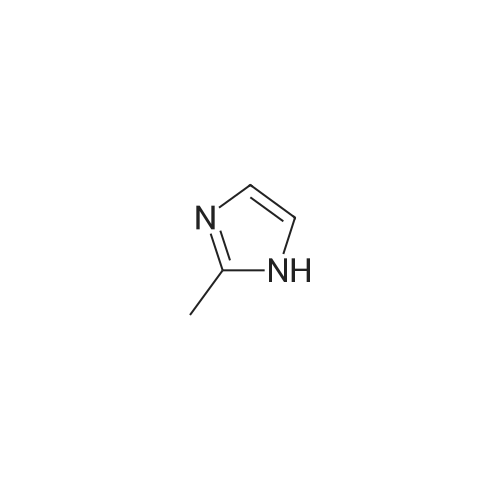
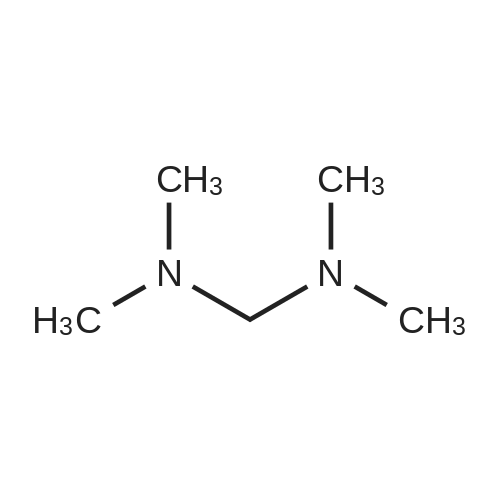
Example 11: Synthesis of ls239-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl methyl]-4H-carbazol-4-one To the solution of 36 ml of acetyl chloride in 50 ml of actonitrile and 750 ml of toluene, was slowly added 51 ml of N, N, N', N'- tetramethyldiaminomethane at 0°C, which was then stirred for 10min. 50 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one and 100 g of 2-methyl imidazole was subsequently added to the reaction mixture, which was then stirred under reflux. After completion of reaction, the reaction mixture was evaporated, and then 150 ml of water was added to the resulting residue. The resulting solid was filtered and washed, and then dried under a reduced pressure. The resulting solid was suspended in 350 ml of methanol, and then 0.7g of active carbon was added to thereto, which was then stirred for 1 hour under reflux. The resulting mixture was filtered and washed with methanol, which was then stirred for 3 hours at room temperature. The resulting solid was filtered and dried to give 60 g of pure white title compound (yield 81.5percent).
75%
Stage #1: at 120 - 130℃; for 8 h; Stage #2: With sodium hydroxide In water
Example 8: Synthesis of 12s3 " 9-tetrahydro-9-methyl-3-[(2-methel-lH- imidazole-1-vl) methyl]-4H-carbazol-4-one At-10°C, 4 ml of N, N, N', N'-tetramethyldiaminomethane was slowly added to 46 ml of trifluoroacetic acid, which was then stirred for 30 min. 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one and 8 g of 2-methyl imidazole was added to the reaction mixture, which was then stirred for about 8 hours at 120-130°C. After completion of reaction, the reaction mixture was cooled at room temperature, and then IN aq. sodium hydroxide was added thereto. The resulting solid was filtered and washed with water, and then dried. The resulting solid was suspended in 80 ml of methanol, and then 0.28 g of active carbon was added thereto, which was then stirred under reflux for 1 hour. The resulting mixture was filtered and washed with methanol, and then evaporated to give 2.2 g of 1, 2,3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazol-1-yl) methyl]- 4H-carbazol-4-one (yield 75percent).
66%
With chloro-trimethyl-silane In DMF (N,N-dimethyl-formamide) at 90℃; for 10 h;
Example 2: Synthesis of 1, 23*9-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-1yl) methyll-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2 ml of N, N, N', N'-tetramethyldiaminomethane were suspended in 20 ml of N, N-dimethylformamide, and then 4 ml of chlorotrimethylsilane was slowly added thereto. The reaction mixture was stirred at 90 °C for 10 hours. 100ml of water was added to the reaction mixture. The resulting solid was filtered and dried, which was then suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.93 g of title compound (yield 66percent).
60%
With chloro-trimethyl-silane In acetonitrile for 10 h; Heating / reflux
Example 1 : Synthesis of 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methyll-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2 ml of N, N, N', N'-tetramethyldiaminomethane were suspended in 30 ml of acetonitrile, and then 4 ml of chlorotrimethylsilane was slowly added thereto. The reaction mixture was stirred under reflux for 10 hours. The reaction mixture was concentrated to remove the solvent, and then 40 ml of water was added to the resulting residue. The resulting solid was filtered and dried, which was then suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.76 g of title compound (yield 60percent).
58%
With acetyl chloride In toluene for 10 h; Heating / reflux
Example 7: Synthesis of 123*9-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-1-yl methyl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 2 ml of N, N, N', N'-tetramethyl- diaminomethane were suspended in 30 ml of toluene, and then 1.4 ml of acetyl chloride was slowly added thereto, which was then stirred for 10 min. 1.65g of 2-methyl imidazole was added to the reaction mixture, which was then stirred under reflux for 10 hours. The reaction mixture was concentrated to remove the solvent, and then 150ml dichloromethane and 50ml of IN aq. sodium hydroxide were added to the resulting residue. The resulting organic layer was separated and dried over MgS04, and then evaporated. The resulting solid was suspended in acetone, which was then stirred for 3 hours, and then filtered and dried to give 1.70 g of title compound (yield 58percent).
55%
With aluminum (III) chloride In acetonitrile for 10 h; Heating / reflux
Example 4: Synthesis of 123, 9-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-1-yDmethyl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2 ml of N, N, N', N'-tetramethyldiaminomethane were suspended in 30 ml of acetonitrile, and then 1.4 g of aluminum chloride was slowly added thereto. The reaction mixture was stirred under reflux for 10 hours. 150 ml dichloromethane and 50 ml of IN aq. sodium hydroxide were added to the reaction mixture. The resulting organic layer was separated and dried over MgS04, and then evaporated. The resulting solid was suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.61 g of title compound (yield 55percent).
Preparation of 1, 23*9-Tetrahydro-9-methYl-3- [ (2-methyl-1 H-imidazol-1-yPmethyl]-4H-carbazol-4-onc; Example 1; 3- [ (Dimethylamino) methyl]-1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (10 g, 34 mmol) and 2-methylimidazole (16.8 g, 205 mmol, 6.0 eq. ) were suspended in mixture of water (75 ml) and dimethyl formamide (37.5 ml). The reaction mixture was heated to reflux (102-103°C) and stirred for a further 6 hours at this temperature. The reaction mixture was cooled to 5-10°C and stirred for half an hour at this temperature. The precipitated crude ondansetron base was filtered and washed with cold water (3x 90 ml) and dried under vacuum at 60 °C to give ondansetron base (9.68 g, 96.4 percent yield) in 98.9percent HPLC purity. Example 2; 3- [(Dimethylamino) methyl]-1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (12 kg, 41 mol) and 2-methylimidazole (20.2 kg, 246 mol, 6 eq. ) were suspended in mixture of water (120 L) and DMF (30 L). The reaction mixture was heated to reflux (100-102°C) and stirred for 5 hours at this temperature. The reaction mixture was cooled to 5-10 °C and stirred for half an hour. The precipitated crude ondansetron base was filtered and washed with cold water (2x110 L) and dried under vacuum at 60 °C to give ondansetron base (10 kg, 83percent yield) in 97.3percent HPLC purity. The major impurity was the exomethylene carbazolone which amounted to 2.6percent of the crude ondansetron mixture.
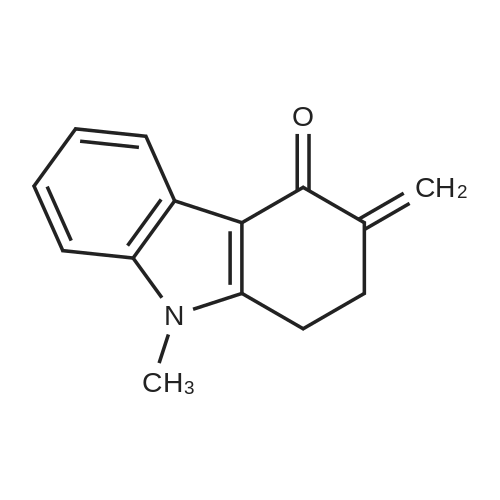
Example 8 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7,5. 89 g of 2-methyl imidazole and 1 g of montmorillonite K10 were added to 100ml of toluene, and then the reaction mixture was stirred under reflux for 6 hours. After completion of reaction, the solvent was distilled off, and then chloroform was added to the resulting residue and the catalyst was filtered off. The filtrate was washed with water, dried over anhydrous magnesium sulfate, and evaporated. The resulting solid was purified with ethyl acetate to give 6.94 g of the title compound as white solid (yield 99percent).
94%
for 3 - 4 h; Heating / reflux
Example 4 Synthesis of Ondansetron Without Using Alumina In this reaction, we examined the feasibility of producing ondansetron from 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one without alumina as a catalyst. The crude 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one from Example 3 (145 grams, 0.69 mol) and 2-methylimidazole (71 grams; 0.86 mol) were added to toluene (800 mL), and the mixture was heated to reflux temperature. After about 3-4 hours (TLC indicated that 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one had been substantially consumed), the reaction was cooled to room temperature. The resulting precipitate was isolated by filtration to provide 190 grams (0.65 mol; 94percent yield) of crude ondansetron. Accordingly, ondansetron was prepared from 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one in a yield of about 94percent after heating for about 3-4 hours. Note that alumina was not used as a catalyst. In addition, the ondansetron was isolated from the reaction mixture quickly and efficiently by filtering the reaction mixture.
91%
for 2 h; Heating / reflux
Example 13 The same procedures as described in Example 10 were repeated, except that toluene was employed instead of acetonitrile, to give 6.32 g of the title compound (yield 91percent).
90%
for 6 h; Heating / reflux
Example 9 The suspension of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7,5. 89 g of 2-methyl imidazole and 1 g of montmorillonite KSF in 100ml of toluene was stirred under reflux for 6 hours. After completion of reaction, the reaction solvent was distilled off, and then chloroform was added to the resulting residue, and then the catalyst was filtered off. The filtrate was washed with water, dried over anhydrous magnesium sulfate, and evaporated to dryness. The resulting solid was purified with ethyl acetate to give 6.3 g of the title compound as white solid (yield 90percent).
88.3%
for 2 h; Heating / reflux
Example 11 The same procedures as described in Example 10 were repeated, except that tetrahydrofuran was employed instead of acetonitrile, to give 6.14 g of the title compound (yield 88.3percent).
84.2%
for 2 h; Heating / reflux
Example 10 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and 11 g of 2-methyl-1-trimethylsilyl imidazole was suspended in 25 ml of acetonitrile. 23. 7ml of 1N tetra-n- butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 5.85 g of the title compound as a white solid (yield 84.2percent).
80%
for 2 h; Heating / reflux
Example 12 The same procedures as described in Example 10 were repeated, except that 1.4-dioxane was employed instead of acetonitrile, to give 5.56 g of the title compound (yield 80percent).
With aluminum (III) chloride In acetonitrile for 10 h; Heating / reflux
Example 6: Synthesis of 123s9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methvl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 274 g of dipiperidinomethane were suspended in 30 ml of acetonitrile, and then 2 g of aluminum chloride was slowly added thereto. The reaction mixture was stirred under reflux for 10 hours. 150 ml dichloromethane and 50 ml of IN aq. sodium hydroxide were added to the reaction mixture. The resulting organic layer was separated and dried over MgS04, and then evaporated. The resulting solid was suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 2.05 g of title compound (yield 70percent).
68%
With chloro-trimethyl-silane In DMF (N,N-dimethyl-formamide) at 90℃; for 8 h;
Example 5: Synthesis of 1239-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-l-yl . methyl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2.74 g of dipiperidinomethane were suspended in 20 ml of N, N-dimethylformamide, and then 4 ml of chlorotrimethylsilane was slowly added thereto. The reaction mixture was stirred for 8 hours at 90 °C. 100 ml of water was added to the reaction mixture. The resulting solid was filtered and dried, which was then suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.99 g of title compound (yield 68percent).
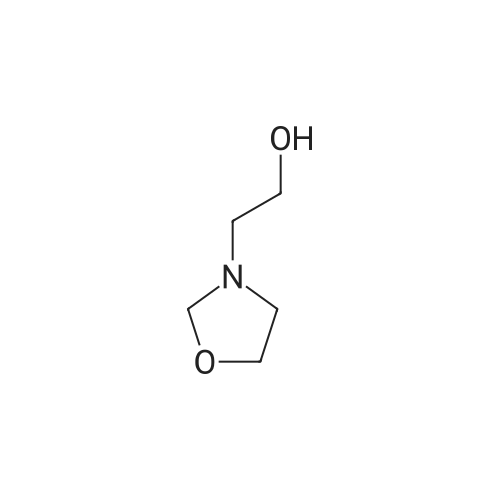
Step 1 9-Methyl-3- (2-methyl-imidazol-1ylmethyl)-1,2,3,9-tetrahydro-carbazol-4-one: At about 70° C., a solution of 2-(oxazolidin-3-yl)ethanol (372 mg, 3.18 mmol, 1.50 equiv) and n-butanol (50 mL) was added to a mixture of 9-methyl-2,3-dihydro-1H-carbazol-4(9H)-one (420 mg, 2.11 mmol, 1.00 equiv), methanesulfonic acid (324 mg, 3.38 mmol, 1.60 equiv) and n-butanol (50 mL). The resulting mixture was stirred at about 80° C. for about 30 minutes, and then stirred at about 120° C. for about 2.5 hours. 2-Methyl-1H-imidazole (870 mg, 10.6 mmol, 5.00 equiv) was then added to the mixture. After stifling the mixture at about 120° C. for about 6 hours, the mixture was concentrated in vacuo and the resulting residue was resuspended in methanol. The resulting solids were collected by filtration and washed with water and methanol. The solids were then purified by silica gel column chromotagraphy (ethyl acetate/petroleum ether (1:1)), to give the title product as a light yellow solid (200 mg, yield=32percent).
Reference:
[1] Patent: US2010/119623, 2010, A1,
11
[ 693-98-1 ]
[ 99614-02-5 ]
Reference:
[1] Patent: US4695578, 1987, A,
12
[ 108-74-7 ]
[ 693-98-1 ]
[ 27387-31-1 ]
[ 99614-02-5 ]
Yield
Reaction Conditions
Operation in experiment
54%
With chloro-trimethyl-silane In toluene; acetonitrileHeating / reflux
Example 10: Synthesis of 1*23*9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methel]-4H-carbazol-4-one To the solution 19.5 g of 1, 3, 5-trimethylhexahydro-1, 3,5-triazine in 20 ml of actonitrile and 150 ml of toluene, was slowly added 19 ml of chlorotrimethylsilane, which was then stirred for lOmin. lOg of 1,2, 3,9- tetrahydro-9-methyl-4H-carbazol-4-one and 8.2g of 2-methyl imidazole was subsequently added to the reaction mixture, which was then stirred under reflux. After completion of reaction, the reaction mixture was evaporated, and then 200 ml of water was added to the resulting residue, which was then stirred for 2 hours at room temperature. The resulting solid was filtered and washed with water to give 7.95 g of light yellow title compound (yield 54percent).
51%
Heating / reflux
Example 9: Synthesis of 12s39-tetrahydro-9-methel-3-[(2-methyl-lH- imidazole-1-yl) methyll-4H-carbazol-4-one To the solution of 7.1 ml of 1, 3, 5-trimethylhexahydro-1, 3,5-triazine in 150ml of toluene, was slowly added 4 ml of trifluoroacetic acid, which was then stirred for 10min. lOg of 1,2, 3,9-tetrahydro-9-methyl-4H-carbazol-4-one and 20 g of 2-methyl imidazole were subsequently added to the reaction mixture, which was then stirred under reflux. After completion of reaction, the reaction mixture was evaporated, and then 200 ml of water was added to the resulting residue, which was then stirred for 2 hours at room temperature. The resulting solid was filtered and washed with excess water to give 7.5 g of light yellow title compound (yield 51percent).
8
Example 8 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7,5. 89 g of 2-methyl imidazole and 1 g of montmorillonite K10 were added to 100ml of toluene, and then the reaction mixture was stirred under reflux for 6 hours. After completion of reaction, the solvent was distilled off, and then chloroform was added to the resulting residue and the catalyst was filtered off. The filtrate was washed with water, dried over anhydrous magnesium sulfate, and evaporated. The resulting solid was purified with ethyl acetate to give 6.94 g of the title compound as white solid (yield 99%).
94%
In toluene for 3 - 4h; Heating / reflux;
4 Synthesis of Ondansetron Without Using Alumina
Example 4 Synthesis of Ondansetron Without Using Alumina In this reaction, we examined the feasibility of producing ondansetron from 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one without alumina as a catalyst. The crude 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one from Example 3 (145 grams, 0.69 mol) and 2-methylimidazole (71 grams; 0.86 mol) were added to toluene (800 mL), and the mixture was heated to reflux temperature. After about 3-4 hours (TLC indicated that 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one had been substantially consumed), the reaction was cooled to room temperature. The resulting precipitate was isolated by filtration to provide 190 grams (0.65 mol; 94% yield) of crude ondansetron. Accordingly, ondansetron was prepared from 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one in a yield of about 94% after heating for about 3-4 hours. Note that alumina was not used as a catalyst. In addition, the ondansetron was isolated from the reaction mixture quickly and efficiently by filtering the reaction mixture.
91%
In water; toluene for 2h; Heating / reflux;
13
Example 13 The same procedures as described in Example 10 were repeated, except that toluene was employed instead of acetonitrile, to give 6.32 g of the title compound (yield 91%).
90%
In toluene for 6h; Heating / reflux;
9
Example 9 The suspension of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7,5. 89 g of 2-methyl imidazole and 1 g of montmorillonite KSF in 100ml of toluene was stirred under reflux for 6 hours. After completion of reaction, the reaction solvent was distilled off, and then chloroform was added to the resulting residue, and then the catalyst was filtered off. The filtrate was washed with water, dried over anhydrous magnesium sulfate, and evaporated to dryness. The resulting solid was purified with ethyl acetate to give 6.3 g of the title compound as white solid (yield 90%).
88.3%
In tetrahydrofuran; water for 2h; Heating / reflux;
11
Example 11 The same procedures as described in Example 10 were repeated, except that tetrahydrofuran was employed instead of acetonitrile, to give 6.14 g of the title compound (yield 88.3%).
84.2%
In water; acetonitrile for 2h; Heating / reflux;
10
Example 10 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and 11 g of 2-methyl-1-trimethylsilyl imidazole was suspended in 25 ml of acetonitrile. 23. 7ml of 1N tetra-n- butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 5.85 g of the title compound as a white solid (yield 84.2%).
80%
In 1,4-dioxane; water for 2h; Heating / reflux;
12
Example 12 The same procedures as described in Example 10 were repeated, except that 1.4-dioxane was employed instead of acetonitrile, to give 5.56 g of the title compound (yield 80%).
With aluminum oxide In toluene for 4h; Heating; Yield given;
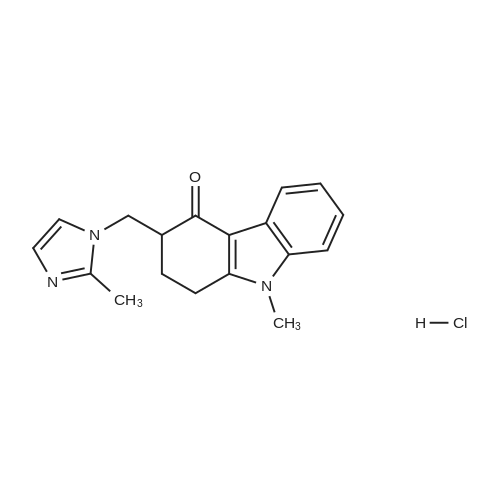
With hydrogenchloride; In ethanol; water; at -10 - 20℃; for 2h;Product distribution / selectivity;
Example 12: Synthesis of 12*3n9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methyll-4H-carbazol-4-one hydrochloride dihydrate 29.4 g of 1, 2,3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH-imidazol-l- yl) methyl]-4H- carbazol-4-one obtained in Example 9 was suspended in 300ml of ethanol and 14ml of water, which was then cooled at-10C. 12. 5g of conc. HC1 was slowly added to the reaction mixture, which was then stirred at room temperature for 2 hours. The resulting solid was filtered and washed with cold ethanol, and then dried in vacuous at 35 C to give 32.6 g of title compound (yield 90%).
87%
With hydrogenchloride; pyrographite; In water; at 0 - 100℃; for 5.16667h;pH 1 - 6;
Under nitrogen, 10 g of crude ondansetron base (prepared according to the method described in example 3b of the HU-Pat. 212 785, and in example 3 of the Hungarian patent application Number of P-00-01287; impurity content measured by HPLC: 3.8 %, colour brownish red) is suspended in 150 ml of desalinated water, the pH is adjusted to 6 with concentrated hydrochloric acid at [95-100 C,] 2 g of charcoal is added to the solution, and the mixture is stirred at this temperature for 1 h. The charcoal is filtered off at [95-100 C,] the pH of the filtrate is adjusted to 1-2 with concentrated hydrochloric acid and the solution is cooled slowly, at a rate of not more, than 0. 3 C/min to 20- [25 C,] then after cooling to [0-5 C] the product is filtered off, washed with cooled [(0-5 C)] desalinated water [(2X5] ml), and dried below [35 C.] The obtained dried ondansetron hydrochloride dihydrate is 10.8 g (87 %) (water content: 9.5 %, which is equivalent to 2 mol of water). Additional measured data: the purity of the product is 99.90 w/w % according to HPLC after system peak correction, the total amount of the impurities is 0.10 w/w %, they are 0.03 w/w % (RT 6.76), 0.02 w/w % (RT 7.10) and 0.05 w/w % (RT [8.] 65), respectively. Colour: white, AE = 0.5 on the range of colours Granule size results: <63 [PM 36 %] [<150UM 72%] <250 [URN 95 %] The above results were obtained by the following analytical methods: A. HPLC measurements: Type of the apparatus: Spectra [SYSTEM/TSP] (manufacturer: Thermo Separation Products, USA) Column: LICHROCART 250-4, LICHROSPHER 100 CN (5 [URN)] Eluent : 70 % of 0.02 M [KH2PO4] 30 % of acetonitrile Flow rate: 1.00 ml/min Wavelength: 216 nm Temperature: [25 C] Dissolution of samples: in the eluent B. Colour measurement: Type of the apparatus: Minolta chromometer with CR-200 measuring head. Before measuring the sample the apparatus is calibrated to standard white CR- A43. Measured parameters: L lightness factor [A) CHROMAPARAMETER] b [AE,] the colour deviation from the standard, is calculated from the measured data according to the following equation: AEab = [[(DL) 2] + [(DA)] [2] + (db) [2 % 2] If AEab value is between 0-0.5, the deviation from the white colour is not visible; if tE value is between 0.5-1. 5, in some cases the deviation can be visible to good eyes, this value fulfils a very good colour requirement. C. Granule size: the granule size was determined by sieve analysis, using Alpine [AIRJCT] sieve
85%
With hydrogenchloride; In ethanol; water; at 20 - 70℃;
Example 22 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH-imidazol-1-yl) methyl]- 4H-carbazol-4-one obtained in Example 8 was suspended in 50 ml of ethanol and 5 ml of water. 1.9 g of conc. HCl was slowly added to the suspension, which was then stirred for 30 min at room temperature. The reaction mixture was dissolved at 70 C, and then filtered to remove insoluble impurities. The reaction mixture was cooled to room temperature, and solidified for 3 hours. The resulting solid was filtered, washed with cold ethanol, and then dried in vacuous at 25 C to give 5.3 g of a pure title compound as white solid (yield 85%). Example 23 6 g of 1, 2,3, 9-tetrahydro-9-methyl-3- [ (2-methyl-1H-imidazol-1-yl) methyl] - 4H-carbazol-4-one obtained in Example 21 was suspended in 60 ml of ethanol and 6 ml of water. 2.1 g of conc. HCl was slowly added to the suspension, which was then stirred at room temperature for 30min. The reaction mixture was dissolved at 70 C, and then filtered to remove insoluble impurity. The mixture was cooled to ambient temperature, and solidified for 3 hours. The resulting solid was filtered, washed with cold ethanol, and then dried in vacuous at 25 C to give 6.51 g of a pure title compound as a white solid (yield 87%).
49%
With hydrogenchloride; In water; isopropyl alcohol; for 1h;pH 1 - 2;Heating / reflux;
Example 5 Synthesis of Ondansetron Hydrochloride Dihydrate The crude ondansetron from Example 4 (190 grams; 0.65 mol) was suspended in a mixture of isopropanol (1.2 L) and water (0.24 L). Concentrated hydrochloric acid (32% (w/w)) was added until the pH of the mixture was about 1-2 (about 70 mL). The solution was heated to reflux temperature for one hour and then cooled to room temperature. The precipitated crystals were collected by filtration, and dried to provide 117 grams (0.32 mol; 49% yield) of crude ondansetron hydrochloride dihydrate. The purity was determined to be about 98-99% by HPLC. The crude ondansetron hydrochloride dihydrate was recrystallized from a mixture of isopropanol and water (6:1 v/v) to provide a crystalline product having a purity of at least 99.8% by HPLC.
With methanesulfonic acid; In butan-1-ol; at 80 - 120℃; for 2.86667h;
1,2, 3, 9-TETRAHYDRO-9-METHYL-4H-CARBAZOL-4-ONE (13.26 G =-66. 5 MMOLE) and methane- sulfonic acid (10.23 g = 106. 4 MMOLE) in 1-butanol (45 mi) were heated to 90C. In 2 minutes 3-ETHYL-OXAZOLIDINE (10.09 g = 99. 9 mmole) in 1-butanol (8 ml) was added. After 50 minutes at 80C 2-methylimidazole (27.32 9 = 332. 5 mmole) and 1-butanol (8 ML) were added. After 2 hours at 120C 180 ML of toluene and 120 ML of water were added at 80C. The layers were separated. The water layer was extracted with 180 ML of toluene and 60 ml of 1-butanol. The combined organic layers were washed twice with 240 ml of water. The organic layer was evaporated to dryness. 150 mi of 1-butanol and 10 ml of 36% M/M hydrochloric acid were added to the residue. At 0 C crystallization soon occurred. After 1 hour at 0 C the formed crystals were filtered off, washed with 1butanol and MTBE and subsequently dried: 15.67 g (71.4%) of 1,2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one hydrochloride was isolated. HPLC: 295%. NMR and MS: see Example 8. The mother liquor contained 2.06 G (9.4%) of product.
With methanesulfonic acid; In butan-1-ol; at 80 - 120℃; for 2.86667h;
1,2, 3, 9-tetrahydro-9-methyl-4H-carbazol-4-one (13.26 g-66. 5 mmole) and methanesulfonic acid (10. 23 G =-106. 4 mmole) in 1butanol (45 ml) were heated to 90C. In 2 minutes 11. 68 g (99.8 mmole) of 3-OXAZOLIDINEETHANOL in 1-butanol (11 ml) was added. After 50 minutes at 80C 2-METHYLIMIDAZOLE (27.32 G--_ 332. 5 mmole) and 1 butanol (8 ml) were added. After 2 hours at 120C 180 ml of toluene and 120 ml of water were added at 80C. The layers were separated. The water layer was extracted with 180 mi of toluene and 60 ml of 1-butanol. The combined organic layers were washed twice with 240 ML of water. The organic layer was evaporated to dryness. 150 ml of 1-butanol and 10 ml of 36% m/m hydrochloric acid were added to the residue. At 0 C crystallization soon occurred. After 1 hour at 0C the formed crystals were filtered off, washed with 1butanol and MTBE and subsequently dried: 15.39 G (70. 1%) of 1,2, 3, 9-tetrahydro-9-methyl-3- [ (2-METHYL-1 H-IMIDAZOL-1-YL) METHYL]-4H-CARBAZOL-ONE hydrochloride was isolated. HPLC: 2 95%.'H NMR [200 MHz, DMSO-d6:CDCl3 4: 1] 8 2.00 (1H, m), 2.20 (1H, m), 3.69 (3H, s), 3.09 (3H, m), 3.75 (3H, s), 4.30 (1H,dd), 4.67 (1H, dd), 7.23 (2H, m), 7.53 (2H, m), 7.69 (1H, d), 8.01 (1H, d). MS [ESI] MH* = 294. The mother liquor contained 3.19 G (14.5%) of product.
With methanesulfonic acid; In butan-1-ol; at 80 - 120℃; for 2.86667h;
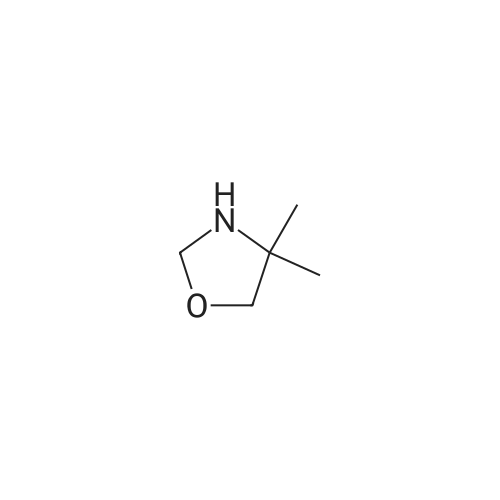
1,2, 3, 9-TETRAHYDRO-9-METHYL-4H-CARBAZOL-4-ONE (13.26 g = 66. 5 mmole) and methane- SULFONIC acid (10.23 g = 106. 4 mmole) in 1-butanol (45 ML) were heated to 90C. In 2 minutes 4, 4-DIMETHYL-OXAZOLIDINE (10.09 G = 99. 9 mmole) in 1-butanol (8 ML) was added. After 50 minutes at 80C 2-METHYLIMIDAZOLE (27.32 G--_ 332. 5 mmole) and 1-butanol (8 ml) were added. After 2 hours at 120C 180 ML of toluene and 120 ml of water were added at 80C. The layers were separated. The water layer was extracted with 180 ml of toluene and 60 ml of 1-butanol. The combined organic layers were washed twice with 240 ml of water. The organic layer was evaporated to dryness. 150 ml of 1-butanol and 10 ml of 36% M/M hydrochloric acid were added to the residue. At 0C crystallization soon occurred. After 1 hour at 0 C the formed crystals were filtered off, washed with 1butanol and MTBE and subsequently dried : 10.02 G (45.7%) of 1,2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one hydrochloride. The mother liquor contained 2.70 g (12.3%) of product. HPLC: 2 95%. NMR and MS: see Example 8.
With sodium hydroxide; In ethanol; water;Heating / reflux;
680 g of <strong>[99614-01-4]ondansetron hydrochloride</strong> dihydrate was dissolved in 4000 ml of ethanol at reflux. A solution of 82 g of NAOH in 1000ML of water was added. A solid was formed. 3000 ml of water was added and the mixture was cooled to ambient temperature. The solid was filtered off and washed with 2*500 ml of water. The solid was dried at 50C under vacuum for 2 days. The product exhibited the DSC curve shown in fig. 1 and the XRPD pattern of fig. 2. Yield : 527 g (96%)80 g of <strong>[99614-01-4]ondansetron hydrochloride</strong> dihydrate was suspended in 500 ml of ethanol and heated to reflux until a clear solution was obtained. To this solution, 250 ml of a 1 M NAOH solution was added. During addition a solid started to form. 250 ml of water was added and the mixture was slowly cooled to room temperature. The mixture was cooled to 10- 15C and the solid was filtered off. The solid was washed with 2X200 ml of water. After drying in a vacuumoven at 40C for 3 days. 59.3 g of a white solid was obtained. The product exhibited the DSC curve shown in fig. 3 and the XRPD pattern of fig. 4. Yield : 59.3 g (92%)
With ammonia; In methanol; water; at 30℃; for 2h;pH 9;
Example 5; Preparation of Base ondansetron Form E (industrial plant method); A stirred suspension of 16 kg of <strong>[99614-01-4]ondansetron hydrochloride</strong> in 80 L of methanol and 80 L of water is heated at 30 C to total dissolution. 6 L of 25% aqueous ammonia is added over the course of 2 hours, until pH 9 is reached. Base ondansetron precipitates out and the resulting suspension is heated to 35 C and stirred at that temperature for 1 hour. It is then cooled to 22-25 C and the suspension is centrifuged. The resulting cake is washed with water (2 x 40 L) and suspended again in 60 L of water. The suspension is stirred at 35 C for 30 minutes, cooled to 22-25C and centrifuged again, washing finally with water (2 x 40 L). The water-moistened solid is suspended in 180 L of methanol and the mixture brought to reflux with stirring for 1 hour. The suspension fluidises but does not reach dissolution. It is cooled to 20-22 C and the suspension is stirred for 30 minutes. It is cooled to 0-5 C and the suspension is stirred for 1 hour at that temperature. The suspension is centrifuged and the cake washed with 20 L of cold methanol. The product is dried at 60C in vacuo for 15 hours. 10.8 kg of base ondansetron Form E (84%) is obtained.
With chloro-trimethyl-silane; In toluene; acetonitrile;Heating / reflux;Product distribution / selectivity;
Example 10: Synthesis of 1*23*9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methel]-4H-carbazol-4-one To the solution 19.5 g of 1, 3, 5-trimethylhexahydro-1, 3,5-triazine in 20 ml of actonitrile and 150 ml of toluene, was slowly added 19 ml of chlorotrimethylsilane, which was then stirred for lOmin. lOg of 1,2, 3,9- tetrahydro-9-methyl-4H-carbazol-4-one and 8.2g of 2-methyl imidazole was subsequently added to the reaction mixture, which was then stirred under reflux. After completion of reaction, the reaction mixture was evaporated, and then 200 ml of water was added to the resulting residue, which was then stirred for 2 hours at room temperature. The resulting solid was filtered and washed with water to give 7.95 g of light yellow title compound (yield 54%).
51%
With trifluoroacetic acid;Heating / reflux;Product distribution / selectivity;
Example 9: Synthesis of 12s39-tetrahydro-9-methel-3-[(2-methyl-lH- imidazole-1-yl) methyll-4H-carbazol-4-one To the solution of 7.1 ml of 1, 3, 5-trimethylhexahydro-1, 3,5-triazine in 150ml of toluene, was slowly added 4 ml of trifluoroacetic acid, which was then stirred for 10min. lOg of 1,2, 3,9-tetrahydro-9-methyl-4H-carbazol-4-one and 20 g of 2-methyl imidazole were subsequently added to the reaction mixture, which was then stirred under reflux. After completion of reaction, the reaction mixture was evaporated, and then 200 ml of water was added to the resulting residue, which was then stirred for 2 hours at room temperature. The resulting solid was filtered and washed with excess water to give 7.5 g of light yellow title compound (yield 51%).
With acetyl chloride; In toluene; acetonitrile;Heating / reflux;Product distribution / selectivity;
Example 11: Synthesis of ls239-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl methyl]-4H-carbazol-4-one To the solution of 36 ml of acetyl chloride in 50 ml of actonitrile and 750 ml of toluene, was slowly added 51 ml of N, N, N', N'- tetramethyldiaminomethane at 0C, which was then stirred for 10min. 50 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one and 100 g of 2-methyl imidazole was subsequently added to the reaction mixture, which was then stirred under reflux. After completion of reaction, the reaction mixture was evaporated, and then 150 ml of water was added to the resulting residue. The resulting solid was filtered and washed, and then dried under a reduced pressure. The resulting solid was suspended in 350 ml of methanol, and then 0.7g of active carbon was added to thereto, which was then stirred for 1 hour under reflux. The resulting mixture was filtered and washed with methanol, which was then stirred for 3 hours at room temperature. The resulting solid was filtered and dried to give 60 g of pure white title compound (yield 81.5%).
75%
Example 8: Synthesis of 12s3 « 9-tetrahydro-9-methyl-3-[(2-methel-lH- imidazole-1-vl) methyl]-4H-carbazol-4-one At-10C, 4 ml of N, N, N', N'-tetramethyldiaminomethane was slowly added to 46 ml of trifluoroacetic acid, which was then stirred for 30 min. 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one and 8 g of 2-methyl imidazole was added to the reaction mixture, which was then stirred for about 8 hours at 120-130C. After completion of reaction, the reaction mixture was cooled at room temperature, and then IN aq. sodium hydroxide was added thereto. The resulting solid was filtered and washed with water, and then dried. The resulting solid was suspended in 80 ml of methanol, and then 0.28 g of active carbon was added thereto, which was then stirred under reflux for 1 hour. The resulting mixture was filtered and washed with methanol, and then evaporated to give 2.2 g of 1, 2,3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazol-1-yl) methyl]- 4H-carbazol-4-one (yield 75%).
66%
With chloro-trimethyl-silane; In DMF (N,N-dimethyl-formamide); at 90℃; for 10h;Product distribution / selectivity;
Example 2: Synthesis of 1, 23*9-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-1yl) methyll-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2 ml of N, N, N', N'-tetramethyldiaminomethane were suspended in 20 ml of N, N-dimethylformamide, and then 4 ml of chlorotrimethylsilane was slowly added thereto. The reaction mixture was stirred at 90 C for 10 hours. 100ml of water was added to the reaction mixture. The resulting solid was filtered and dried, which was then suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.93 g of title compound (yield 66%).
60%
With chloro-trimethyl-silane; In acetonitrile; for 10h;Heating / reflux;Product distribution / selectivity;
Example 1 : Synthesis of 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methyll-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2 ml of N, N, N', N'-tetramethyldiaminomethane were suspended in 30 ml of acetonitrile, and then 4 ml of chlorotrimethylsilane was slowly added thereto. The reaction mixture was stirred under reflux for 10 hours. The reaction mixture was concentrated to remove the solvent, and then 40 ml of water was added to the resulting residue. The resulting solid was filtered and dried, which was then suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.76 g of title compound (yield 60%).
58%
With acetyl chloride; In toluene; for 10h;Heating / reflux;Product distribution / selectivity;
Example 7: Synthesis of 123*9-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-1-yl methyl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 2 ml of N, N, N', N'-tetramethyl- diaminomethane were suspended in 30 ml of toluene, and then 1.4 ml of acetyl chloride was slowly added thereto, which was then stirred for 10 min. 1.65g of 2-methyl imidazole was added to the reaction mixture, which was then stirred under reflux for 10 hours. The reaction mixture was concentrated to remove the solvent, and then 150ml dichloromethane and 50ml of IN aq. sodium hydroxide were added to the resulting residue. The resulting organic layer was separated and dried over MgS04, and then evaporated. The resulting solid was suspended in acetone, which was then stirred for 3 hours, and then filtered and dried to give 1.70 g of title compound (yield 58%).
55%
With aluminum (III) chloride; In acetonitrile; for 10h;Heating / reflux;Product distribution / selectivity;
Example 4: Synthesis of 123, 9-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-1-yDmethyl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2 ml of N, N, N', N'-tetramethyldiaminomethane were suspended in 30 ml of acetonitrile, and then 1.4 g of aluminum chloride was slowly added thereto. The reaction mixture was stirred under reflux for 10 hours. 150 ml dichloromethane and 50 ml of IN aq. sodium hydroxide were added to the reaction mixture. The resulting organic layer was separated and dried over MgS04, and then evaporated. The resulting solid was suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.61 g of title compound (yield 55%).
With aluminum (III) chloride; In acetonitrile; for 10h;Heating / reflux;Product distribution / selectivity;
Example 6: Synthesis of 123s9-tetrahydro-9-methyl-3-[(2-methyl-lH- imidazole-1-yl) methvl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 274 g of dipiperidinomethane were suspended in 30 ml of acetonitrile, and then 2 g of aluminum chloride was slowly added thereto. The reaction mixture was stirred under reflux for 10 hours. 150 ml dichloromethane and 50 ml of IN aq. sodium hydroxide were added to the reaction mixture. The resulting organic layer was separated and dried over MgS04, and then evaporated. The resulting solid was suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 2.05 g of title compound (yield 70%).
68%
With chloro-trimethyl-silane; In DMF (N,N-dimethyl-formamide); at 90℃; for 8h;Product distribution / selectivity;
Example 5: Synthesis of 1239-tetrahydro-9-methyl-3-r (2-methyl-lH- imidazole-l-yl . methyl]-4H-carbazol-4-one 2.0 g of 1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one, 1.65 g of 2- methyl imidazole and 2.74 g of dipiperidinomethane were suspended in 20 ml of N, N-dimethylformamide, and then 4 ml of chlorotrimethylsilane was slowly added thereto. The reaction mixture was stirred for 8 hours at 90 C. 100 ml of water was added to the reaction mixture. The resulting solid was filtered and dried, which was then suspended in acetone and stirred for 3 hours, and then filtered and dried under a reduced pressure to give 1.99 g of title compound (yield 68%).
17; 20
Example 17 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and 11 g of 2-methyl-l-trimethylsilyl imidazole were suspended in 35 ml of acetonitrile. The solution of 6.2 g of tetra-n- butylammonium fluoride hydrate in 15 ml of acetonitrile was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, the reaction solvent was distilled off. 100 ml of water was added to the residue, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.61 g of the title compound as a white solid (yield 98%).; Example 20 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and 22 g of 2-methyl-l-trimethylsilyl imidazole were suspended in 35 ml of acetonitrile. The solution of 6.2 g of tetra-n- butylammonium fluoride hydrate in 15 ml of acetonitrile was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, the reaction solvent was distilled off. 100 ml of water was added to the residue, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.75 g of the title compound as a white solid (yield 100%).
96.7%
In ethyl acetate for 2.16667h; Heating / reflux;
19
Example 19 The same procedures as described in Example 17 were repeated, except that ethyl acetate was employed instead of acetonitrile, to give 6.72 g of the title compound (yield 96.7%).
96.1%
at 80℃; for 2h;
21
Example 21 To the mixture of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7 and 22 g of 2-methyl-1- trimethylsilyl imidazole was added 6.2 g of tetra-n-butylammonium fluoride hydrate at 80 °C, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.48 g of the title compound as a white solid (yield 96. 1%).
94.5%
In tetrahydrofuran for 2.16667h; Heating / reflux;
18
Example 18 The same procedures as described in Example 17 were repeated, except that tetrahydrofuran was employed instead of acetonitrile, to give 6.57 g of the title compound (yield 94.5%).
92%
In water at 80℃; for 2.16667h; Heating / reflux;
15; 16
Example 15 The mixture of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7 and 11 g of 2-methyl-1- trimethylsilyl imidazole was heated to 80 °C. 23.7 ml of IN tetra-n- butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.2 g of the title compound as a white solid (yield 92%). Example 16 The mixture of 5 g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H- carbazol-4-one prepared in any one of Examples 1 to 7 and 22 g of 2-methyl-1- trimethylsilyl imidazole was heated to 80°C. 2. 37ml of IN tetra-n-butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to ambient temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 6.61 g of the title compound as a white solid (yield 98%).
87%
In water; acetonitrile for 2.16667h; Heating / reflux;
14
Example 14 5g of 1, 2,3, 9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one prepared in any one of Examples 1 to 7 and llg of 2-methyl-l-trimethylsilyl imidazole were suspended in 25 ml of acetonitrile. 1. 9ml of IN tetra-n- butylammonium fluoride solution was added dropwise for 10 min under reflux, and then the reaction mixture was stirred for 2 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture, and then stirred for 30 min. The resulting solid was filtered and washed with 100 ml of water to give 5.85 g of the title compound as a white solid (yield 87%).
In water; N,N-dimethyl-formamide at 5 - 103℃; for 5.5 - 6.5h;
1; 2
Preparation of 1, 23*9-Tetrahydro-9-methYl-3- [ (2-methyl-1 H-imidazol-1-yPmethyl]-4H-carbazol-4-onc; Example 1; 3- [ (Dimethylamino) methyl]-1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (10 g, 34 mmol) and 2-methylimidazole (16.8 g, 205 mmol, 6.0 eq. ) were suspended in mixture of water (75 ml) and dimethyl formamide (37.5 ml). The reaction mixture was heated to reflux (102-103°C) and stirred for a further 6 hours at this temperature. The reaction mixture was cooled to 5-10°C and stirred for half an hour at this temperature. The precipitated crude ondansetron base was filtered and washed with cold water (3x 90 ml) and dried under vacuum at 60 °C to give ondansetron base (9.68 g, 96.4 % yield) in 98.9% HPLC purity. Example 2; 3- [(Dimethylamino) methyl]-1, 2,3, 9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (12 kg, 41 mol) and 2-methylimidazole (20.2 kg, 246 mol, 6 eq. ) were suspended in mixture of water (120 L) and DMF (30 L). The reaction mixture was heated to reflux (100-102°C) and stirred for 5 hours at this temperature. The reaction mixture was cooled to 5-10 °C and stirred for half an hour. The precipitated crude ondansetron base was filtered and washed with cold water (2x110 L) and dried under vacuum at 60 °C to give ondansetron base (10 kg, 83% yield) in 97.3% HPLC purity. The major impurity was the exomethylene carbazolone which amounted to 2.6% of the crude ondansetron mixture.
In water
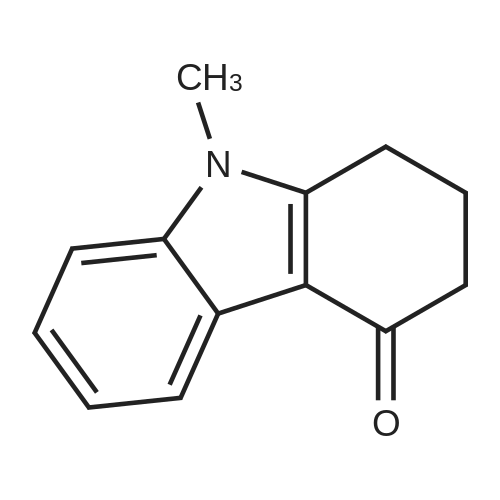
1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
EXAMPLE 1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7 g) in water (17 ml) was treated with 2-methylimidazole (1.4 g) and then heated under reflux for 20 h. The cooled mixture was filtered and the residue washed with water (3*15 ml) to give a product (1.7 g) m.p. 221-221.50. This material was recrystallized from methanol to give the title compound (1.4 g) m.p. 231-2320.
In water
2 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
EXAMPLE 2 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7 g) in water (17 ml) was treated with 2-methylimidazole (1.4 g) and then heated under reflux for 20 h. The cooled mixture was filtered and the residue washed with water (3*15 ml) to give crude product (1.7 g) m.p. 221°-221.5°. This material was recrystallized from methanol to give the title compound (1.4 g) m.p. 231°-232°.
In water
1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
EXAMPLE 1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7 g) in water (17 ml) was treated with 2-methylimidazole (1.4 g) and then heated under reflux for 20 h. The cooled mixture was filtered and the residue washed with water (3*15 ml) to give a product (1.7 g) m.p. 221°-221.5°. This material was recrystallized from methanol to give the title compound (1.4 g) m.p. 231°-232°.
In water
2 1,2,3,9-Tetrahydro-9-methyl-3-[2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
EXAMPLE 2 1,2,3,9-Tetrahydro-9-methyl-3-[2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7 g) in water (17 ml) was treated with 2-methylimidazole (1.4 g) and then heated under reflux for 20 h. The cooled mixture was filtered and the residue washed with water (3*15 ml) to give a product (1.7 g) m.p. 221°-221.5°. This material was recrystallized from methanol to give the title compound (1.4 g) m.p. 231°-232°.
In water
1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
Example 1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7g) in water (17ml) was treated with 2-methylimidazole (1.4g) and then heated under reflux for 20h. The cooled mixture was filtered and the residue washed with water (3x15ml) to give a product (1.7g) m.p. 221-221.5°. This material was recrystallized from methanol to give the title compound (1.4g) m.p. 231-232°.
In water
7 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
EXAMPLE 7 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7 g) in water (17 ml) was treated with 2-methylimidazole (1.4 g) and then heated under reflux for 20 h. The cooled mixture was filtered and the residue washed with water (3*15 ml) to give crude product (1.7 g) m.p. 221°-221.5°. This material was recrystallized from methanol to give the title compound (1.4 g) m.p. 231°-232°, identical by t.l.c. with product from Example 4.
In water
1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one
Example 1 1,2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one A solution of 3-[(dimethylamino)methyl]-1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one hydrochloride (1.7g) in water (17ml) was treated with 2-methylimidazole (1.4g) and then heated under reflux for 20h. The cooled mixture was filtered and the residue washed with water (3x15ml) to give a product (1.7g) m.p. 221-221.5°. This material was recrystallized from methanol to give the title compound (1.4g) m.p. 231-232°.
Preparation of Ondansetron Form A Monohydrate from Ondansetron Hydrochloride Form A Dihydrate Example 12 Ondansetron hydrochloride Form A dihydrate (5 g) in 70 ml of a 96% aqueous solution of EtOH was heated to reflux temperature for 22 hrs. The reaction mixture was then allowed to cool to room temperature and then was cooled to 0 C. The solid that precipitated was filtered and dried at 65 C. for 20 hrs, yielding 1.2 g of ondansetron hydrochloride Form A monohydrate, KF =5.4%. Example 13 Ondansetron hydrochloride Form A dihydrate (5.0 g) in 70 ml of a 90% aqueous solution of EtOH was heated to reflux temperature for 22 hrs. The reaction mixture was allowed to cool to room temperature and then cooled to 0 C. The solid was then filtered, dried at 65 C. for 20 hrs. to give 4.0 g of ondansetron hydrochloride Form A monohydrate; KF =5.0%. Example 14 Ondansetron hydrochloride Form A dihydrate (5.0 g) was slurried in 70 ml of a 90% aqueous solution of EtOH at room temperature for 22 hrs. The solid was then filtered, dried at 65 C. for 20 hrs. to give 3.5 g of ondansetron hydrochloride Form A monohydrate; KF =5.2%. Example 15 Ondansetron hydrochloride Form A dihydrate (5 g) was slurried in 70 ml of a 50% aqueous solution of EtOH at room temperature for 22 hrs. Methyl ethyl ketone (100 ml) was then added to preciptate the ondansetron hydrochloride. The mixture was cooled to 0 C. and the precipitate was filtered and dried at 65 C. for 20 hrs. to give 0.4 g of ondansetron hydrochloride Form A monohydrate; KF =5.2%. Example 16 Ondansetron hydrochloride Form A dihydrate (5 g) was slurried in 70 ml of a 50% aqueous solution of EtOH at room temperature for 22 hrs. The solid was then filtered, dried at 65 C. for 20 hrs. to give 0.4 g of ondansetron hydrochloride Form A monohydrate; KF =5.7%. Some of the compound was recovered from the mother liquor by adding 125 ml of MEK for precipitation and filtering under vacuum. The solid was dried at 65 C. for 20 hrs. to give 1.7 g of ondansetron hydrochloride Form A monohydrate; KF 5.4%. Example 17 Ondansetron hydrochloride Form A dihydrate (5 g) was slurried in 70 ml of a 96% aqueous solution of EtOH at room temperature for 22 hrs. The solid was then filtered and dried at 65 C. for 20 hrs. to give 3.8 g of ondansetron hydrochloride Form A; KF =6.1 %.
13
[ 123-91-1 ]
[ 27387-31-1 ]
[ 18162-48-6 ]
[ 99614-02-5 ]
Yield
Reaction Conditions
Operation in experiment
In chloroform;
EXAMPLE 4 Preparation of 1 2,3,9-Tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one Hydrochloride Dihydrate 3.0 g of <strong>[27387-31-1]1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one</strong> and 3.8 g of 1-(N-morpholinylmehyl)-2-methylimidazole obtained in Preparation Example 3 were suspended in 30 ml of chloroform, and 6.0 ml of t-butyldimethylchlorosilane was added thereto. The suspension was heated to 110 C., and the reaction solvent was distilled off for 3 hrs. Then, 20 ml of 1,4-dioxane was added thereto and the reaction solvent was further distilled off for 3 hrs. The reaction mixture was cooled and dissolved in 40 ml of chloroform, and the resulting solution was washed with 20 ml of saturated sodium bicarbonate. The organic layer was dried over magnesium sulfate and vacuum-distilled, to obtain 3.5 g of crude 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one.
In chloroform;
Example 4 Preparation of 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one hydrochloride dihydrate 3.0 g of <strong>[27387-31-1]1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one</strong> and 3.8 g of 1-(N-morpholinylmehyl)-2-methylimidazole obtained in Preparation Example 3 were suspended in 30 ml of chloroform, and 6.0 ml of t-butyldimethylchlorosilane was added thereto. The suspension was heated to 110C, and the reaction solvent was distilled off for 3 hrs. Then, 20 ml of 1,4-dioxane was added thereto and the reaction solvent was further distilled off for 3 hrs. The reaction mixture was cooled and dissolved in 40 ml of chloroform, and the resulting solution was washed with 20 ml of saturated sodium bicarbonate. The organic layer was dried over magnesium sulfate and vacuum-distilled, to obtain 3.5 g of crude 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one.
Example 10 Preparation of 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)methyl]-4H-carbazol-4-one hydrochloride dihydrate 3.0 g of <strong>[27387-31-1]1,2,3,9-tetrahydro-9-methyl-4H-carbazol-4-one</strong>, 2.1 g of 1-(N,N-dimethylaminomethyl)-2-methylimidazole obtained in Preparation Example 1 and 2.5 g of 2-methylimidazole were suspended in a mixture of 15 ml of chloroform and 15 ml of acetonitrile, and 3.8 ml of chlorotrimethylsilane was added thereto. The suspension was heated at 70C for 1 hr. Then, 15 ml of water and 15 ml of 1,4-dioxane were added thereto and the reaction solvent was further distilled off for 6 hrs. The reaction mixture was cooled to room temperature, and the resulting crystalswere filtered, to obtain 3.62 g of crude 1,2,3,9-tetrahydro-9-methyl-3-[(2-methyl- 1H-imidazol-1-yl)methyl]-4H-carbazol-4-one.
With pyridine; hydroxylamine hydrochloride In methanol at 80℃; for 24h;
1
To a slurry of Ondansetron hydrochloride (5g, 13.6 mmoles) in a mixture of pyridine-.methanol (1:2,20ml,) was added hydroxylamine hydrochloride (5g, 71 mmoles).The resulting slurry was stirred at 80° C for 24 hours. Subsequently the reaction mixture was cooled and filtered to yield, a white crystalline solid, which on drying at high vacuum gave the desired compound (3.5g, 83.5%). R/0.7(9:l, dichloromethane: methanol), HPLC(purity): 97 %, mp 231- 233 0C; 1H-NMR (CDCl3) ? (ppm): 8.01 (d, IH), 7.33-7.25 (m,3H), 7.21-7.17 (m, 2H), 6.95 (d, IH), 4.18-4.01 (m, 3H), 3.71 (s, 3H), 2.62 (s, 3H), and2.25-2.20 (m, 2H); IR (cm"1) 3138.1, 2934.6, 2835.9, 1621.9, 1474.8, and 1278.1; MS m/z:309.2 [M+l]
2
To a solution of <9-Benzyl hydroxylamine hydrochloride (0.3g, 1.87 mmoles) in a mixture of pyridine :methanol (1:1, 10ml) was added Ondansetran hydrochloride in portions (0.3g, 0.82 mmoles). The resulting slurry was stirred at 120 0C for 36 hours. After complete conversion, the solvent was evaporated and resulting residue was re-dissolved in 20ml of chloroform, followed by 20ml of water. The organic layer was separated, dried over anhydrous sodium sulfate and evaporated at reduced pressure to yield a glassy brown solid, which was then subjected to silica gel column chromatography using a gradient of methanol in ethyl acetate (0-11%), to yield the product (168 mg, 51.9%), R/ = 0.5(9:1 chloroform: methanol); ?PLC (purity): 94.5 %; 1H-NMR (CDCl3) ? (ppm): 8.16 (d, IH), 7.46 (d, IH), 7.38-7.25 (m, 7H), 6.92 (s, IH), 6.76 (s, IH), 5.23 (s, 2H), 4.13-4.08 (m, 2H), 3.92-3.89 (m, IH), 3.69 (s, 3H), 2.87-2.83 (m, 2H), 2.29 (s, 3H), and 1.98-1.90 (m, 2H); MS m/z: 399.3 [M+l]
Step 1 9-Methyl-3- (2-methyl-imidazol-1ylmethyl)-1,2,3,9-tetrahydro-carbazol-4-one: At about 70 C., a solution of 2-(oxazolidin-3-yl)ethanol (372 mg, 3.18 mmol, 1.50 equiv) and n-butanol (50 mL) was added to a mixture of <strong>[27387-31-1]9-methyl-2,3-dihydro-1H-carbazol-4(9H)-one</strong> (420 mg, 2.11 mmol, 1.00 equiv), methanesulfonic acid (324 mg, 3.38 mmol, 1.60 equiv) and n-butanol (50 mL). The resulting mixture was stirred at about 80 C. for about 30 minutes, and then stirred at about 120 C. for about 2.5 hours. 2-Methyl-1H-imidazole (870 mg, 10.6 mmol, 5.00 equiv) was then added to the mixture. After stifling the mixture at about 120 C. for about 6 hours, the mixture was concentrated in vacuo and the resulting residue was resuspended in methanol. The resulting solids were collected by filtration and washed with water and methanol. The solids were then purified by silica gel column chromotagraphy (ethyl acetate/petroleum ether (1:1)), to give the title product as a light yellow solid (200 mg, yield=32%).
In methanol; dichloromethane; for 1h;Sonication; Reflux;
Weigh 0.147g Ondansetron racemate and0.219 g O, O'-di-p-anisal-L-tartaric acid resolving agentInto a 50ml round bottom flask,18 ml of dichloromethane and 5 ml of methanol were added,Ultrasound assisted heating reflux for 1 hour,Solids dissolve quickly, the solution is clear,Standing or stirring for three hours cooling, suction filtration,0.163 g of O, O'-di-p-anisalyl-L-tartrate as a white powdery solid levododeoxytron,The optical purity was 90.5percent ee and the yield was 44.5percent.
With aluminum (III) chloride; dihydrogen peroxide In water at 140℃; for 8h;
1.1; 1.2
Step S1, hydrothermal reaction: first weigh 100 mg of ondansetron raw material and an appropriate amount of aluminum chloride, and add an aqueous solution of hydrogen peroxide to prepare an ondansetron solution. The mass fraction of hydrogen peroxide in the aqueous hydrogen peroxide solution is 12%. ,The concentration of ondansetron is 5mg/mL,The concentration of aluminum chloride is 0.5 mg/mL;The solution was transferred to a 50 mL reaction vessel with a Teflon liner, sealed and placed in an oven at 140 ° C for 8 h; the reaction was completed.After cooling to room temperature, ethyl acetate was extracted.The ethyl acetate fraction is collected and concentrated to obtain a hydrothermal reaction extract;Step S2, HSCCC is separated and purified in one step:Two-phase solvent system selects n-hexane/vinegar with a volume ratio of 5:7:5:1:5Ethyl acetate / methanol / ethanol / water,The respective solvents are thoroughly mixed according to the volume ratio, and the layers are allowed to stand, on,The lower phase is separated, and the ultrasonication is used; the hydrothermal reaction extract is ultrasonically dissolved and mixed with 10 mL of the upper phase and 10 mL of the lower phase to prepare a sample solution;The upper phase of the prepared solvent system is pumped into the HSCCC (Tongtian TEB300B) spiral tube as a stationary phase, and after the upper phase is filled with the spiral tube,Open HSCCC, set the speed of 850r/min, volume flow rate of 1.3mL/min, detection wavelength of 230nm, pump the lower phase of the solvent systemThe spiral tube acts as a mobile phase, and when the mobile phase begins to flow continuously from the tail end of the detector, it indicates that the two-phase solvent in the spiral tube reaches a hydrodynamic equilibrium;Inject 20 mL of sample solution into the injection valve, start recording the chromatogram and continue pumping into the mobile phase.Collect the elution fraction corresponding to impurity A according to the chromatogram (Figure 1).It was obtained by concentration and drying, and the purity was 98.9%, and the yield was 56.8%.
With dihydrogen peroxide; manganese(ll) chloride In water at 170℃; for 6h;
2.1; 2.2
Step S1, hydrothermal reaction: first weigh 100 mg of ondansetron raw material and appropriate amount of manganese chloride, add hydrogen peroxide solutionDissolved into an ondansetron solution, the mass fraction of hydrogen peroxide in the aqueous hydrogen peroxide solution was 17%, and the concentration of ondansetron was5mg/mL, manganese chloride concentration is 0.5mg/mL; transfer the solution to 50mL reactor with PTFE liner, sealedAfter being placed in an oven and reacted at 170 ° C for 6 h; the reaction was completed, and after cooling to room temperature, ethyl acetate was extracted to collect ethyl acetate.And concentrated and dried to obtain a hydrothermal reaction extract;Step S2, one-step separation and purification of HSCCC: two-phase solvent system selects n-hexane/vinegar with a volume ratio of 5:6:3:2:6Ethyl acetate / methanol / ethanol / water, the solvent is thoroughly mixed according to the volume ratio, standing layering, separation of the upper and lower phases, ultrasonic treatmentThe hydrothermal reaction extract is ultrasonically dissolved in a mixed solvent of 10 mL of the upper phase and 10 mL of the lower phase to prepare a sample solution;The upper phase of the good solvent system is pumped into the spiral tube of HSCCC (Tongtian TEB300B) as the stationary phase. After the upper phase is filled with the spiral tube,Open HSCCC, set the speed of 850r/min, volume flow rate of 1.3mL/min, detection wavelength of 230nm, pump the lower phase of the solvent systemThe spiral tube acts as the mobile phase, and when the mobile phase begins to flow continuously from the tail end of the detector, it indicates that the two-phase solvent in the spiral tube reachesHydrodynamic equilibrium; the injection valve injects 20 mL of sample solution, begins to record the chromatogram and continues to pump into the mobile phase separation, according toThe chromatogram (Fig. 2) was used to collect the elution fraction corresponding to the impurity D, and concentrated and dried to obtain a purity of 98.5% and a yield of 47.2%.
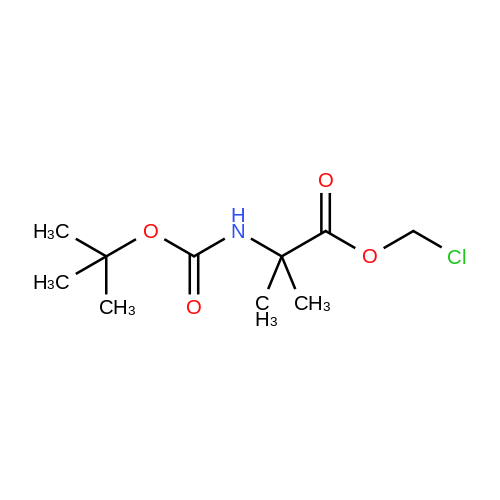
43 Reference Example 43 3-[[2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-2-ethyl-1-oxobutoxy]methyl]-2-30 methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
379 mg (1.29 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 190 mg (0.65 mmol) of 2-ethyl-2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]butanoic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C., and then the residue was purified by silica gel column chromatography (15→20% methanol/chloroform) to obtain 297 mg (78%) of the title compound. 1H-NMR (CDCl3, δ):0.77 (6H, t, J=8 Hz), 1.38 (9H, s), 1.55-1.65 (4H, m), 2.05 (1H, qd, J=12, 5 Hz), 2.80-2.82 (1H, m), 3.06 (3H, s), 3.13-3.37 (5H, m), 3.70 (3H, s), 4.57 (1H, dd, J=14, 5 Hz), 4.67 (1H, br-s), 4.76 (1H, dd, J=14, 7 Hz), 6.12 (1H, d, J=11 Hz), 6.18 (1H, d, J=11 Hz), 7.25-7.33 (3H, m), 7.36 (1H, d, J=2 Hz), 7.87 (1H, d, J=2 Hz), 8.09-8.11 (1H, m)
55 Reference Example 55 3-[[2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-1-oxobutoxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
543 mg (1.85 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 287 mg (0.93 mmol) of 2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-2-methylbutanoic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C. The residue was purified by silica gel column chromatography (12% methanol/chloroform) to obtain 517 mg (99%) of the title compound. 1H-NMR (CDCl3, δ):0.89-0.90 (3H, m), 1.37(9/2H, s), 1.39(9/2H, s), 1.54-1.55 (211, m), 2.08(1H, br-s), 2.64-2.76 (2H, m), 3.03-3.39 (8H, m), 3.71 (3H, s), 4.56-4.60 (1H, m), 4.74(1H, dd, J=14, 7 Hz), 5.13 (1H, br-s), 6.14 (1H, d, J=14 Hz), 6.17 (1H, d, J=14 Hz), 7.26-7.33 (3H, m), 7.41-7.42 (1H, m), 7.83-7.85 (1H, m), 8.08(1H, br-s)
59 Reference Example 59 3-[[3-phenyl-2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-1-oxobutoxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
575 mg (1.96 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 320 mg (0.98 mmol) of 2-benzyl-3-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]propanoic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C. The residue was purified by silica gel column chromatography (13% methanol/chloroform) to obtain 371 mg (61%) of the title compound. 1H-NMR (CDCl3, δ):1.39(9/2H, s), 1.41(9/2H, s), 1.98-2.07 (1H, m), 2.74-2.86 (7H, m), 2.95-3.45 (5H, m), 3.69(3/2H, s), 3.70(3/2H, s), 4.41-4.52(1H, m), 4.65-4.74 (1H, m), 5.12(1H, br-s), 5.98-6.07 (2H, m), 7.05-7.09 (2H, m), 7.17-7.33 (7H, m), 7.72-7.74 (1H, m), 8.09 (1H, d, J=7 Hz)
60 Reference Example 60 3-[[[7-[[(1,1-dimethylethoxy)carbonyl]amino]-1-oxoheptyl]oxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
379 mg (1.29 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 253 mg (0.86 mmol) of 7-[[(1,1-dimethylethoxy)carbonyl]amino]heptanoic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C. The residue was purified by silica gel column chromatography (13-420% methanol/chloroform) to obtain 387 mg (82%) of the title compound. 1H-NMR (CDCl3, δ):1.29-1.31 (5H, m), 1.43 (9H, s), 1.58-1.64 (3H, m), 2.25-2.30(1H, m), 2.39 (2H, t, J=8 Hz), 2.74-2.77(1H, m), 2.95-3.33 (8H, m), 3.69 (3H, s), 4.56-4.62 (2H, m), 4.75 (1H, dd, J=14, 7 Hz), 6.12 (1H, d, J=12 Hz), 6.14 (1H, d, J=12 Hz), 7.19-7.34 (3H, m), 7.42 (1H, d, J=2 Hz), 7.88(1H, d, J=2 Hz), 8.08-8.10(1H, m)
54 Reference Example 54 3-[[2-[[(1,1-dimethylethoxy)carbonyl]amino]-2-methyl-1-oxopropoxy]methyl]-2-methy 1-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
466 mg (1.59 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 200 mg (0.79 mmol) of 2-[[(1,1-dimethylethoxy)carbonyl]amino]-2-methylpropane acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C., and then the residue was purified by silica gel column chromatography (15% methanol/chloroform) to obtain 248 mg (58%) of the title compound. 1H-NMR (CDCl3, δ):1.32 (9H, s), 1.45 (3H, s), 1.46 (3H, s), 2.03(1H, qd, J=13, 5 Hz), 2.70-2.75 (1H, m), 3.03 (3H, s), 3.09-3.33 (3H, m), 3.69 (3H, s), 4.48 (1H, dd, J=14, 6 Hz), 4.75 (1H, dd, J=14, 6 Hz), 4.90(1H, br-s), 6.21 (1H, d, J=12 Hz), 6.27(1H, d, J=12 Hz), 7.26-7.33 (3H, m), 7.43(1H, d, J=2 Hz), 7.73(1H, d, J=2 Hz), 8.11-8.13 (1H, m)
49 Reference Example 49 3-[([[1-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]cyclopentyl]carbonyl]oxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
500 mg (1.70 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 249 mg (0.85 mmol) of 1-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]cyclopentane carboxylic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C., and then residue was purified by silica gel column chromatography (15% methanol/chloroform) to obtain 408 mg (82%) of the title compound. 1H-NMR (CDCl3, δ):1.38 (9H, s), 1.60-1.97 (8H, m), 2.06(1H, qd, J=12, 5 Hz), 2.76-2.78 (1H, m), 3.03 (3H, s), 3.12-3.34 (5H, m), 3.70 (3H, s), 4.57 (1H, dd, J=14, 5 Hz), 4.75(1H, dd, J=14, 7 Hz), 5.03(1H, br-s), 6.14(1H, d, J=1 Hz), 6.18(1H, d, J=11 Hz), 7.25-7.33 (3H, m), 7.43(1H, br-s), 7.88(1H, br-s), 8.09 (1H, d, J=7 Hz)
44 Reference Example 44 3-[[[[1-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]cyclopropyl]carbonyl]oxy]meth yl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
779 mg (2.66 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 350 mg (1.33 mmol) of 1-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]cyclopropanecarboxylic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C., and then the residue was purified by silica gel column chromatography (15% methanol/chloroform) to obtain 487 mg (66%) of the title compound. 1H-NMR (CDCl3, δ):1.11 (2H, br-s), 1.30 (2H, br-s), 1.42 (9H, s), 2.06 (1H, qd, J=12, 6 Hz), 2.76-2.77 (1H, m), 3.04 (3H, s), 3.12-3.32 (5H, m), 3.70 (311, s), 4.58 (1H, dd, J=14, 5 Hz), 4.77 (1H, dd, J=14, 7 Hz), 5.16 (1H, br-s), 6.13 (1H, d, J=12 Hz), 6.19 (1H, d, J=12 Hz), 7.26-7.33 (3H, m), 7.42 (1H, br-s), 7.87 (1H, br-s), 8.11 (1H, d, J=7 Hz)
45 Reference Example 45 3-[[2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-3,3-dimethyl-1-oxobutoxy]methy 1]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium Chloride
360 mg (1.22 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 180 mg (0.61 mmol) of 2-[[[(1,1-dimethylethoxy)carbonyl]amino]methyl]-3,3-dimethyl-butanoic acid chloromethyl ester and stirred at 100° C. overnight. The reaction solution was condensed at a water bath set to 40° C., and then the residue was purified by silica gel column chromatography (15% methanol/chloroform) to obtain 233 mg (69%) of the title compound. 1H-NMR (CDCl3, δ):0.94 (9/2H, s), 0.96 (9/2H, s), 1.35 (9/2H, s), 1.39 (9/2H, s), 2.02-2.17 (1H, m), 2.60-2.67 (1H, m), 2.75-2.78 (1H, m), 3.01 (3/2H, s), 3.05 (3/2H, s), 3.11-3.48 (5H, m), 3.70 (3/2H, s), 3.72 (3/2H, s), 4.56-4.63 (1H, m), 4.72 (1H, dd, J=15, 8 Hz), 5.05 (1/2H, br-s), 5.15 (1/2H, br-s), 6.13-6.16 (2H, m), 7.24-7.33 (3H, m), 7.37 (1/2H, d, J=2 Hz), 7.39 (1/2H, d, J=2 Hz), 7.85 (1/2H, d, J=2 Hz), 7.90 (1/2H, br-s), 8.06-8.09 (1H, m)
9 3-[[[[1-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-2,2-dimethylpropoxy]carbonyl]oxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium chloride
701 mg (2.39 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 644 mg (1.99 mmol) of carbonic acid chloromethyl 1-[[2-[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-2,2,-dimethylpropyl ester and stirred at 85° C. overnight. The reaction solution was condensed in a water bath set to 40° C. The residue was purified by silica gel column chromatography (8→20% methanol/chloroform) to obtain 806 mg (66%) of the title compound. 1H-NMR (CDCl3, δ): 0.89 (9H, s), 1.42 (9/2H, s), 1.43 (9/2H, s), 1.61-1.68 (1H, m), 1.83-2.09 (2H, m), 2.74-2.77 (1H, m), 2.91-3.35 (8H, m), 3.68 (3H, s), 4.54-4.58 (2H, m), 4.75-4.80 (2H, m), 6.15-6.23 (2H, m), 7.24-7.32 (3H, m), 7.47-7.48 (1H, m), 7.94-7.95 (1H, m), 8.09-8.10 (1H, m)
27 3-[[[[(3R)-3-[[(1,1-dimethylethoxy)carbonyl]amino]-1-piperidinyl]carbonyl]oxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium chloride
377 mg (1.28 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 373 mg (1.27 mmol) of (3R)-3-[[(1,1-dimethylethoxy)carbonyl]amino]-1-piperidine carboxylic acid chloromethyl ester and stirred at 85° C. overnight. The reaction solution was condensed in a water bath set to 40° C. The residue was purified by silica gel column chromatography (10% methanol/chloroform) to obtain 697 mg (92%) of the title compound. (0222) 1H-NMR (CDCl3, δ): 1.32-1.57 (12H, m), 1.88-1.92 (1H, m), 2.03-2.11 (1H, m), 2.77-3.07 (5H, m), 3.12-3.35 (3H, m), 3.49-3.65 (2H, m), 3.70-3.95 (5H, m), 4.56-4.82 (3H, m), 6.06-6.21 (2H, m), 7.26-7.32 (3H, m), 7.39-7.45 (1H, m), 7.87 (1/2H, d, H=8 Hz), 7.96-7.80 (1/2H, m), 8.09-8.11 (1H, m)
With sodium iodide; calcium(II) chloride In dichloromethane; acetonitrile at 45℃; for 6h;
39 3-[[[[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethoxy]carbonyl]oxy]methyl]-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium iodide
924 mg (3.15 mmol) of ondansetron, 944 mg (6.30 mmol) of sodium iodide, and 105 mg (0.95 mmol) of calcium chloride were added at room temperature to a mixed solution of dichloromethane and acetonitrile of 800 mg (3.15 mmol) of carbonic acid chloromethyl 2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl ester and stirred at 45° C. for 6 hours. The reaction solution was filtered, and then the reaction solution was condensed in a water bath set to 40° C. The residue was purified by silica gel column chromatography (10→15% methanol/chloroform) to obtain 480 mg (24%) of the title compound, 1H-NMR (CDCl3, δ): 1.42 (9H, s), 2.04 (1H, qd, J=12, 5 Hz), 2.74-2.79 (1H, m), 3.05-3.11 (4H, m), 3.27-3.43 (4H, m), 3.69 (3H, s), 4.26 (2H, t, J=5 Hz), 4.36 (1H, dd, J=14, 6 Hz), 4.86 (1H, dd, J=14, 6 Hz), 4.95 (1H, br-s), 6.11 (1H, d, J=12 Hz), 6.15 (1H, d, J=12 Hz) 7.26-7.32 (3H, m), 7.46 (1H, d, J=2 Hz), 7.68 (1H, d, J=2 Hz), 8.10-8.12 (1H, m)
56 3-[[[[[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl][2-(methylthio)ethyl]amino]carbonyl]oxy]methyl]-2-methyl-1-[(2,3,4,9-tetrahydro-9-methyl-4-oxo-1H-carbazol-3-yl)methyl]-1H-imidazolium chloride
285 mg (0.97 mmol) of ondansetron was added at room temperature to an acetonitrile solution of 317 mg (0.97 mmol) of N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[2-(methylthio)ethyl]carbamic acid chloromethyl ester and stirred at 85° C. overnight. The reaction solution was condensed in a water bath set to 40° C. The residue was purified by silica gel column chromatography (10→20% methanol/chloroform) to obtain 444 mg (72%) of the title compound. (0333) 1H-NMR (CDCl3, δ): 1.40 (9/2H, s), 1.41 (9/2H, s), 2.03-2.08 (1H, m), 2.09 (3/2H, s), 2.10 (3/2H, s), 2.16-2.77 (3H, m), 3.03 (3/2H, s), 3.06 (3/2H, s), 3.11-3.51 (9H, m), 3.70 (3H, s), 4.56-4.59 (1H, m), 4.74 (1/2H, dd, J=15, 7 Hz), 4.77 (1/2H, dd, J=15, 7 Hz), 5.16 (1/2H, br-s), 5.43 (1/2H, br-s), 6.12 (1H, d, J=12 Hz), 6.17 (1H, d, J=12 Hz), 7.25-7.33 (3H, m), 7.46 (1/2H, d, J=2 Hz), 7.62 (1/2H, br-s), 7.85 (1H, br-s), 8.10 (1H, d, J=8 Hz)






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +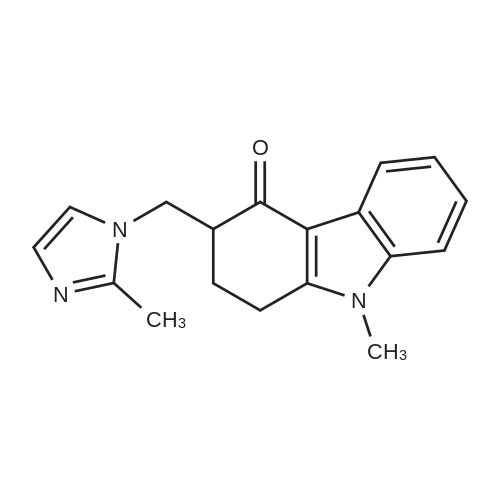

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping