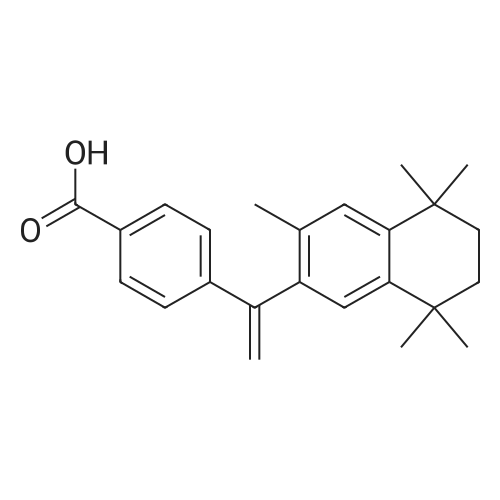
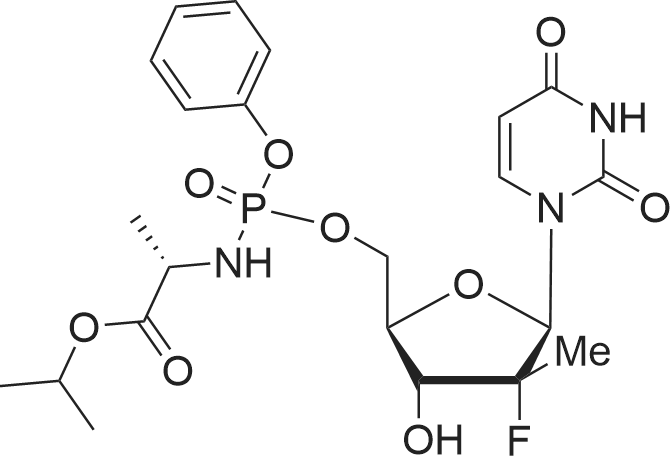
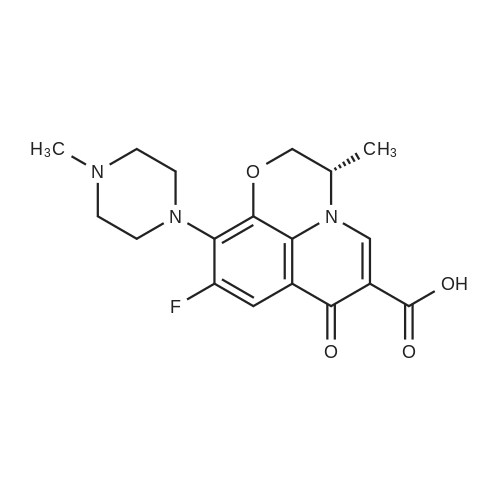
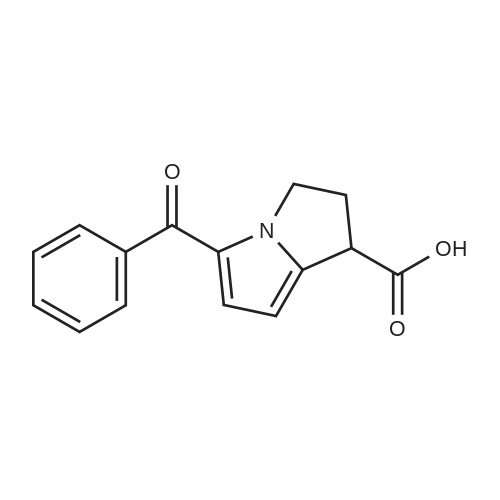
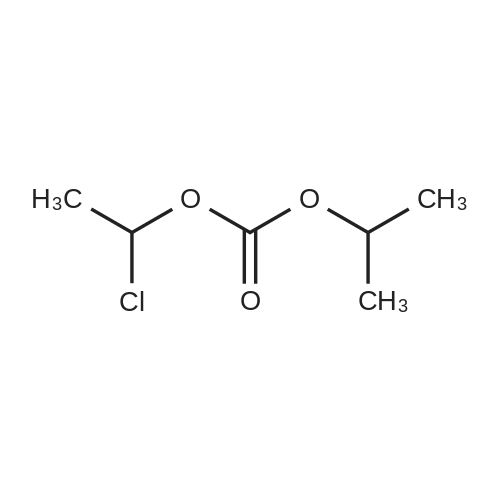
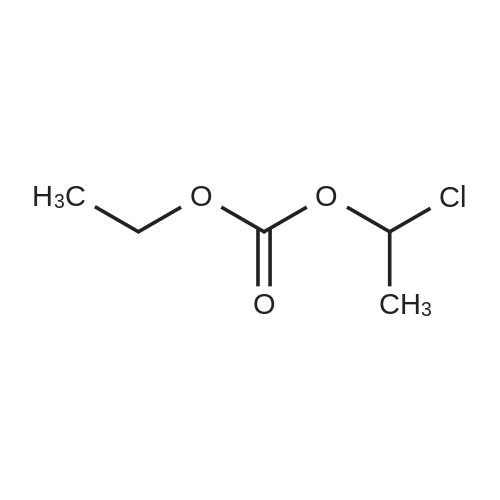
| 66% |
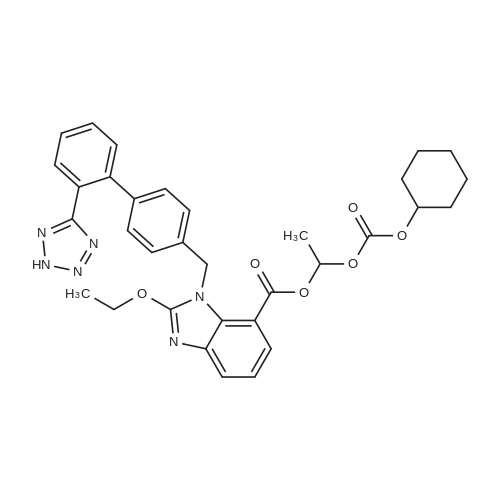
With potassium carbonate; sodium iodide; In N,N-dimethyl-formamide; at 20 - 40℃; |
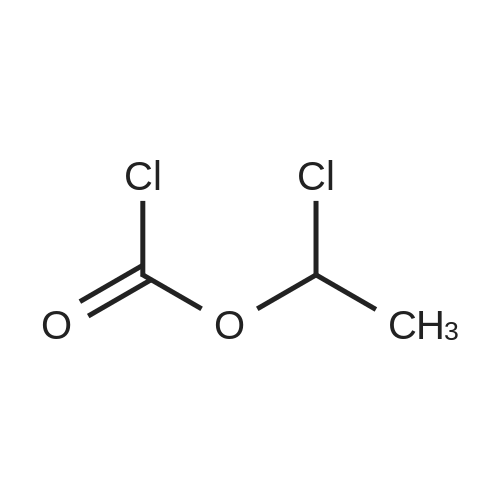
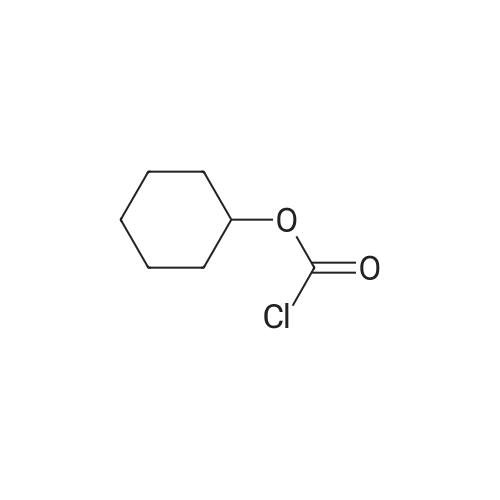
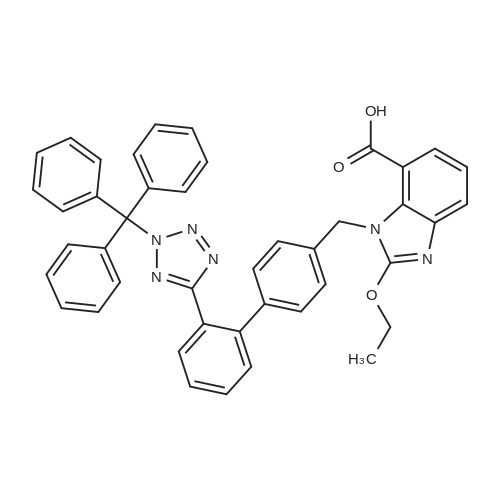
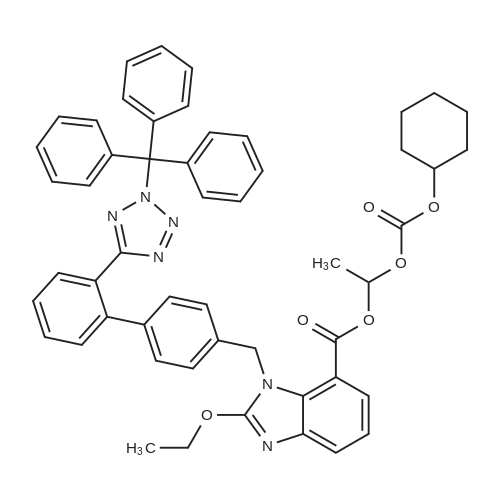
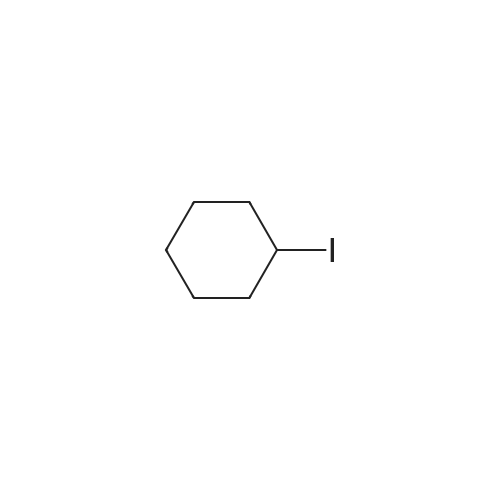
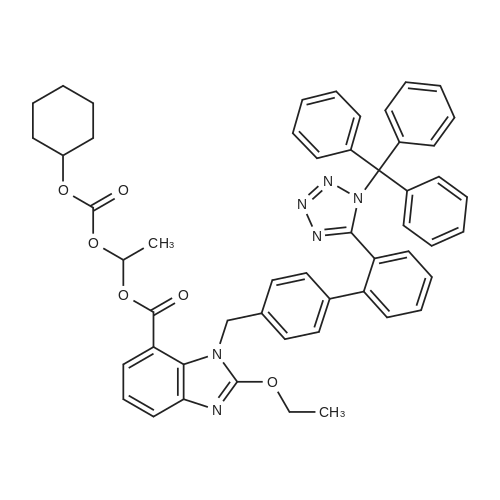
Example 19 Preparation of H-D-Glu(D-Trp-0-CH(CH3)0-CO-0-cyclohexyl)-0-Et hydrochloride salt, Apo854.HCI Cbz-D-Glu(OH)-0-Et (12.1 g, 39.1 mmol), HOSu (4.60 g, 40.0 mmol) andEDCI.HCI (7.67 g, 40.0 mmol) were mixed in DMF (100 mL) under ice-water bath temperature. The reaction mixture was allowed to warm to RT then stirred for overnight. The reaction mixture was cooled again in an ice-water bath and D-Trp- OH (8.17 g, 40.0 mmol) was added. The mixture was stirred at room temperature for overnight. The mixture was poured into a beaker containing 0.5N HCI (200 mL) and ice pellets. The mixture was extracted with ethyl acetate (2x200 mL + 1x100 mL). The organic layers were combined and washed with a 0.5N HCI solution (100 mL), water (2x100 mL) and brine (100 mL), dried over MgS04, then filtered. The filtrate was concentrated via rotary evaporation under reduced pressure and the resulting solid Cbz-D-Glu(D-Trp-OH)-0-Et was triturated with10% ethyl acetate in hexanes. The precipitated white solid was collected via suction filtration (17.6 g). Yield = 90 %; 1H NMR (DMSO-D6l 400 MHz) delta ppm: 12.58 (br. s, 1 H), 10.82 (s, 1H), 8.12 (d, J = 8.1 Hz, 1 H), 7.71 (d, J = 8.1 Hz, 1 H), 7.52 (d, J = 8.1 Hz, 1H), 7.23 - 7.42 (m, 6H), 7.12 (s, 1 H), 7.06 (t, J = 7.6 Hz, 1 H), 6.97 (t, J = 7.6 Hz, 1H), 4.97 - 5.10 (m, 2H), 4.41 - 4.51 (m, 1H), 3.95 - 4.15(m, 3H), 3.15 (dd, J = 14.1 , 5.1 Hz, 1H), 2.99 (dd, J = 15.2, 8.1 Hz, 1 H), 2.09 - 2.26 (m, 2H), 1.83 - 1.96 (m. 1 H), 1.65 - 1.81 (m, 1 H), 1.16 (t, J - 7.1 Hz, 3H); MS-ESI (m/z): 496 [ +1f. To a mixture of Cbz-D-Glu(D-Trp-OH)-0-Et {4.95 g, 0.0 mmol) with potassium carbonate (4.15 g, 30.0 mmol) and sodium iodide (6.00 g, 40.0 mmol) in Lambda/,/V-dimethylformamide (30 mL) at room temperature, 1-chtoroethylcyclohexyl carbonate (6.20 g, 30.0 mmol) was added. After being stirred at room temperature for overnight, additional W,/V-dimethylformamide (30 mL) was added and the reaction mixture was stirred at 40C for overnight. The reaction mixture was diluted with ethyl acetate then washed with water (3x) then with brine. The crude product Cbz-D-Glu(D-Trp-0-CH(CH3)-0-CO-0-cyclohexyl)-0-Et was purified by column chromatography on silica gel using a solvent gradient of a mixture of ethyl acetate in hexanes (20 to 40%) as eluant. Fractions rich in product were combined together and evaporated to dryness. Thus, the desired compound Cbz-D-Glu(D-Trp-0-CH(CH3)-0-CO-0-cyclohexyl)-0-Et (4.43 g) was obtained as a pale-yellow foam. Yield = 66 %; 1H NMR (DMSO-D6> 400 MHz) delta ppm: 10.86 (or. s, 1H), 8.36 (dd, J = 17.2, 7.1 Hz, 1 H), 7.66 - 7.77 (m, 1 H), 7.46(t, J = 8.0 Hz., 1H), 7.22 - 7.42 (m, 6H), 7.10 - 7.20 (m, 1 H), 7.02 - 7.10 (m, 1 H), 6.90 - 7.02 (m, 1 H), 6.58 - 6.70 (m, 0.5H), 6.46 - 6.58 (m, 0.5H), 5.04 (br. s, 2H), 4.38 - 4.61 (m, 2H), 3.93 - 4.15 (m, 3H), 2.90 - 3.17 (m, 2H), 2.20 (br. s, 2H), 1.54 - 1.96 (m, 6H), 1.02 - 1.53 (m, 12H); MS-ESI (m/z): 666 [M+1f. Cbz-D-Glu(D-Trp-0-CH(CH3)-0-CO-0-cyclohexyl)-0-Et (2.0 g, 3.0 mmol) and 10 % Pd/C (wet, 0.6 g) was mixed in ethanol (50 mL) and 2 HCI in ether (1.7 mL, 3.4 mmol). The reaction mixture was hydrogenated in a Parr apparatus at 20-25 psi of hydrogen pressure for an hour. The mixture was filtered through Celite and the cake was washed with ethanol. The filtrate was concentrated by rotary evaporation and the residue was triturated with a mixture of ether and hexanes. Thus, H-D-Glu(D-Trp-O-CH(CH3)-0-CO-0-cyclohexyl)-0-Ethydrochloride salt (Apo854.HCI, 0.80 g) was obtained as a pink solid foam. Yield = 47%; *H NMR (DMSO-D6, 400 MHz) delta ppm: 0.94 (br. s, 1 H), 8.57 (br. s, 4H), 7.47 (t, J = 8.1 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.19 (s, 1 H), 7.07 (t, J = 7.6 Hz, 1 H), 6.88 - 7.03 (m, 1 H), 6.58 - 6.72 (q, J = 5.1 Hz, 0.5H), 6.53 (q, J = 5.1 Hz,0.5H), 4.39 - 4.63 (m, 2H), 4.00 - 4.26 (m, 2H), 3.78 - 4.00 (m, 1 H), 2.93 - 3.18 (m, 2H), 2.18 - 2.41 (m, 2H), 1.88 - 2.02 (m, 2H), 1.82 (br. s, 2H), 1.63 (br. s, 2H), 1.13 - 1.53 (m, 12H); MS-ESI (m/z): 532 [M+1]+ (free base). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping