* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Rapid Communications in Mass Spectrometry, 2010, vol. 24, # 5, p. 634 - 642
[2] Biochimie, 2012, vol. 94, # 3, p. 741 - 747
2
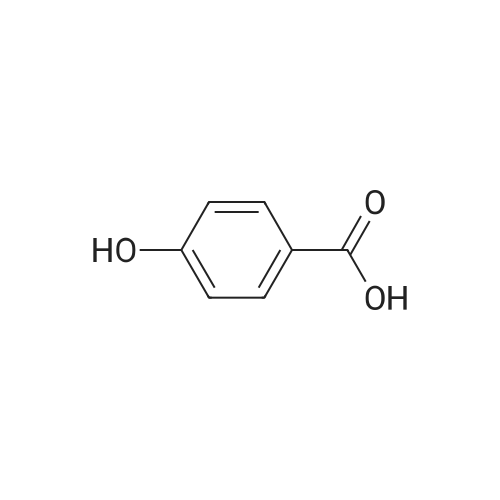
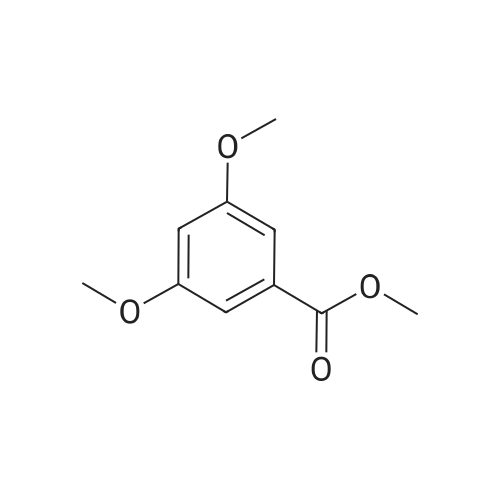
[ 99-10-5 ]
[ 14615-72-6 ]
Reference:
[1] Russian Chemical Bulletin, 2004, vol. 53, # 4, p. 830 - 833
[2] Synthetic Communications, 2002, vol. 32, # 20, p. 3149 - 3158
[3] Bulletin of the Chemical Society of Japan, 1983, vol. 56, # 6, p. 1889 - 1890
[4] Journal of Organic Chemistry, 1983, vol. 48, # 12, p. 1941 - 1944
[5] Journal of Heterocyclic Chemistry, 1973, vol. 10, p. 649 - 654
3
[ 99-10-5 ]
[ 19520-75-3 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2013, vol. 63, p. 415 - 422
4
[ 99-10-5 ]
[ 77-78-1 ]
[ 19520-75-3 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 1658 - 1666
5
[ 99-10-5 ]
[ 95-64-7 ]
[ 96-22-0 ]
[ 56038-89-2 ]
Reference:
[1] Patent: US4261926, 1981, A,
6
[ 67-56-1 ]
[ 99-10-5 ]
[ 2150-44-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
for 16 h; Acidic conditions; Heating
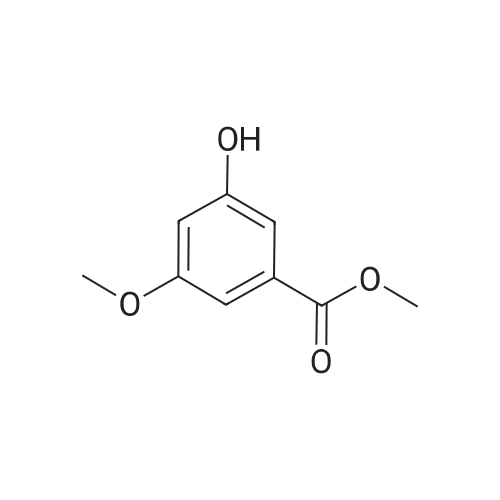
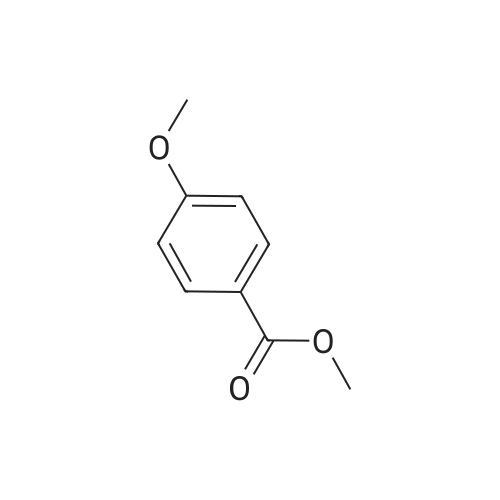
The approach described by Brouwer et al. (ibid.) for the convergent synthesis of amino acid based dendrimers was adapted in order to obtain dendrimers with surface propargyl groups to enable a 1,3-dipolar cycloaddition ("click") reaction with amino acid /peptide derived azides. First generation dendrimer 1 was synthesized from 3,5-dihydroxybenzoic acid in an overall yield of 81percent as shown in Scheme 1. In detail, 3,5-dihydroxymethylbenzoate (21.4 g, 130 mmol) was dissolved in dry DMF (250 mL) and anhydrous K2CO3 (45 g, 330 mmol, 2.5 equiv.) was added. To this suspension, a solution of propargylbromide in toluene (35 mL, 314 mmol, 2.5 equiv.) was added dropwise. The reaction mixture was stirred for 48 h at room temperature. Then, DMF was removed by evaporation and the residue was redissolved in EtOAc (400 mL) and the organic phase was washed with H2O (3 x 100 mL) in KHSO4 (3 x 100 mL) and brine (3 x 100 mL), dried (Na2SO4) and evaporated in vacuo. The residue was recrystallized from EtOAc/hexane to obtain 1 as off-white crystals in 81percent yield (25.2 g). Rf(EtOAc/hexane 4:1 v/v): 0.76; Rf (DMC/MeOH 98:2 v/v): 0.87; Rf(CHCl3/MeOH/AcOH 95:20:3 v/v): 0.83; 1H-NMR (CDCl3) δ 2.55 (t (J 2.47 Hz), 2H), 3.91 (s, 3H), 4.72 (d (J 2.47 Hz), 4H), 6.81 (t (J 2.20 Hz), 1H), 7.29 (d (J 2.20 Hz), 2H); 13C-NMR (CDCl3) δ 52.4, 56.0, 76.0, 77.9, 107.5, 108.8, 132.0, 157.8, 158.4; Elemental analysis: calculated for C14H12O4 C 68.83, H 4.95, found C 68.76, H 4.95.
98%
at 0℃; Reflux; Inert atmosphere
General procedure: Thionyl chloride (1.16 g, 1.5 equiv) was added drop-wise to a solution of acid (1.0 g, 1.0 equiv) in the corresponding alcohol (15 ml) at 0&d eg;C. The solution was refluxed under a nitrogen atmosphere until all starting material was consumed (TLC monitoring). Then the solvent was removed under vacuo and the residue was purified by silica gel column chromatography eluting with ethyl acetate/n-hexane to afford the corresponding carboxylic esters.#10;
98%
for 8 h; Reflux
To a solution of 8a (10.00 g, 64.9 mmol) in methanol (50 mL) was added concentrated sulfuric acid(750 μL in 10 mL methanol), and the solution was allowed to reflux for 8 h. After cooled to ambient temperature, methanol was evaporated under reduced pressure and the residue was dissolved in ethylacetate (50 mL), which was then washed with saturated NaHCO3 solution and water. The organic layer was dried over anhydrous Na2SO4 and ethyl acetate was removed in vacuo to afford of methyl3,5-dihydroxylbenzoate 8a′ (10.74 g, 98percent) as a white powder.
95%
for 20 h; Reflux
A solution of 3,5-dihydroxybenzoic acid (20.0 g, 129.9 mmol) in dry methanol (100 mL) and H2SO4 (1 mL) was refluxed for 20 h. The volatile product was removed in vacuo and the residue was redissolved in ethyl acetate (EA) and washed with aqueous NaHCO3, water and brine. The organic phase was dried with anhydrous sodium sulphate and the solvent was evaporated to yield methyl 3,5-dihydroxybenzoate as a white colored solid (95 percent yield, m.p.= 17O0C). 1H-NMR (300 MHz, CDCl3) δ 7.1 (d, 2H), 6.6 (t, IH), 4.9 (s, OH), 3.9 (s, OCH3).
95%
Reflux
Methyl 3,5-dihydroxybenzoate (8)To a solution of 3,5-dihydroxybenzoic acid (5.0 g, 32 mmol) in methanol (170 ml) was added a catalytic amount of sulphuric acid (0.3 ml). After stirring at reflux overnight, the mixture was cooled and neutralized with 4M NaOH (aq. solution). After concentration, the residue was dissolved in ethyl acetate and washed with water and brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. Compound 2 was isolated as a white solid (5.13 g, 95percent); m.p. 164-165° C. (ethyl acetate); 1H-NMR (500 MHz, d6-DMSO) δ: 9.65 (2H, s), 6.81 (2H, d, J 2.3 Hz, CHar), 6.43 (1H, d, J 2.3 Hz, CHar), 3.78 (3H, s, CH3); 13C-NMR (125 MHz, d6-DMSO) δ: 166.2 (C═O), 158.4 (C x2), 131.2 (C), 107.1 (CH), 107.0 (CH x2), 51.9 (CH3); MS (ES)− m/z: 167 [M−H]−; HPLC tR=3.11; IR (neat) v (cm−1): 3229, 1688, 1600, 1486, 1305, 1161, 1102, 995, 765. Data were in good agreement with the literature22.
90%
for 17 h; Reflux; Inert atmosphere
1. Preparation of 3,5-dihydroxybenzoic acid methyl ester (1); To a solution of 3,5-dihydroxybenzoic acid (10 g, 64.9 mmol) in anhydrous methanol (150 mL) acetyl chloride (2 mL, 28.1 mmol) was added dropwise. The mixture was heated at reflux under N2 for 17 hours. The solvent was evaporated and the residue was dissolved in ethyl acetate (100 mL), then washed with saturated sodium hydrogen carbonate (3 x 100 mL). The combined organic layers were washed with water (2 x 100 mL), dried over sodium sulfate, filtered and concentrated in vacuo to afford 2 as a colourless solid (9.80 g, 90percent): mp 167-168°C (lit.1 mp 168-169°C); 1H NMR (400 MHz, Acetone-flfe) δ 8.66 (s, 2H), 7.00 (d, J = 2.4 Hz, 2H), 6.59 (t, J = 2.4 Hz, 2H), 3.83 (s, 3H).
90%
for 17 h; Reflux; Inert atmosphere
To the solution of anhydrous methanol (150 mL) was added slowly acetyl chloride (2 mL, 28.1 mmol) followed by the solution of 3,5-dihydroxybenzoic acid (10 g, 64.9 mmol). The mixture was refluxed under nitrogen for 17 hours. The solvent was evaporated and the residue was dissolved in ethyl acetate (100 mL), and washed with saturated sodium hydrogen carbonate (3 × 100 mL). The organic layer was washed with distilled water (2 × 100 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure to afford 2 as an off white solid (9.80 g, yield 90percent), mp 167-168°C (lit.[32] mp 168-169°C). 1H NMR (400 MHz, Acetone-d6) δ (ppm) 8.66 (2H, s, 2 × OH), 7.00 (2H, d, J=2.4 Hz, H-2,6), 6.59 (1H, t, J=2.4 Hz, H-4), 3.83 (3H, s, COOCH3). The 1H NMR spectrum was in good agreement with previously reported data[33].
89%
at 60℃; for 2 h; Inert atmosphere
Then, under an argon atmosphere, in a 300 mL flask,3,5-dihydroxybenzoic acid (8.0 g, 51.9 mmol, 1 eq.) Was dissolved in dry methanol (40 mL).Subsequently, thionyl chloride (7.4 g, 4.5 mL, 62 mmol, 1.2 eq.) Was added dropwise, and the flask was heated on an oil bath and reacted at 60 ° C. with stirring for 2 hours.The mixture was then cooled to room temperature and the volatiles evaporated under reduced pressure. Then, the solid precipitate was suspended in toluene,The volatiles were evaporated to give compound (A-4) as a beige solid (yield 7.79 g, 46.3 mmol, yield 89percent).
86%
for 24 h; Reflux
Conc. H2SO4 (80 mL) was added slowly to a stirred solution of 3,5-dihydroxybenzoic acid 301 (50 g, 0.33 mol) in CH3OH (660 mL) at rt and this solution was heated to reflux for 24 h. The reaction mixture was cooled to rt and H2O (500 mL) was added to the solution. The solution was extracted with EtOAc (3*300 mL), and the combined organic extracts were washed with a saturated aq NaHCO3 solution (2*300 mL), The organic layer was dried (Na2SO4), and concentrated under reduced pressure to afford a white crude powder. The crude solid was purified by flush column chromatography (FCC) (10percent ethyl acetate in hexane) to afford a white powdered ester 302 (48 g, 86percent): 1H NMR (300 MHz, CDCl3) δ 7.10 (2H, d, J=2.4 Hz HAr), 6.57 (1H, t, J=2.0 Hz, HAr), 4.99, (2H, br, s, HO), 3.84 (31-1, s, H3COO).
30 g
at 0℃; for 2 h; Reflux
A) methyl 3,5-dihydroxybenzoate To a solution of 3,5-dihydroxybenzoic acid (30.0 g) in methanol (300 mL) was added dropwise thionyl chloride (20 mL) at 0°C, and the mixture was heated under reflux for 2 hr. The reaction mixture was concentrated, and the obtained crystals were washed with diethyl ether to give the title compound (30.0 g) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 3.79 (3H, s), 6.44 (1H, s), 6.81 (2H, s), 9.60 (2H, s).
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1981, p. 2825 - 2829
[2] Chemistry Letters, 1999, # 11, p. 1193 - 1194
[3] Chemical Communications, 2005, # 36, p. 4581 - 4583
[4] Patent: EP1733742, 2006, A1, . Location in patent: Page/Page column 5-6; 12
[5] Journal of the American Chemical Society, 2007, vol. 129, # 10, p. 2774 - 2776
[6] Tetrahedron, 2010, vol. 66, # 26, p. 4745 - 4759
[7] Synlett, 2012, vol. 23, # 20, p. 2919 - 2922
[8] Heterocycles, 2014, vol. 8, # 2, p. 1275 - 1285
[9] Journal of Organic Chemistry, 1995, vol. 60, # 20, p. 6389 - 6396
[10] Tetrahedron, 2000, vol. 56, # 13, p. 1873 - 1882
[11] Organic Letters, 2002, vol. 4, # 17, p. 2833 - 2836
[12] Beilstein Journal of Organic Chemistry, 2012, vol. 8, p. 732 - 737
[13] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 6, p. 1564 - 1569
[14] Synlett, 2016, vol. 27, # 10, p. 1587 - 1591
[15] Journal of Organic Chemistry, 1992, vol. 57, # 20, p. 5462 - 5469
[16] Chemistry - A European Journal, 2007, vol. 13, # 12, p. 3346 - 3353
[17] Angewandte Chemie - International Edition, 2006, vol. 45, # 45, p. 7609 - 7611
[18] Chemistry - A European Journal, 2005, vol. 11, # 11, p. 3389 - 3404
[19] Patent: WO2010/12710, 2010, A1, . Location in patent: Page/Page column 13
[20] Organic and Biomolecular Chemistry, 2013, vol. 11, # 26, p. 4414 - 4418
[21] Patent: US2014/134110, 2014, A1, . Location in patent: Paragraph 0087; 0090; 0091
[22] Journal of the Chemical Society - Series Chemical Communications, 1987, vol. No. 19, p. 1457 - 1459
[23] Synthesis, 2010, # 7, p. 1150 - 1158
[24] Chemical Communications, 2010, vol. 46, # 25, p. 4616 - 4618
[25] Tetrahedron Letters, 2013, vol. 54, # 16, p. 2093 - 2096
[26] Synthetic Communications, 2002, vol. 32, # 20, p. 3149 - 3158
[27] European Journal of Organic Chemistry, 2001, # 10, p. 1903 - 1915
[28] Patent: WO2012/149608, 2012, A1, . Location in patent: Page/Page column 33-34
[29] European Journal of Medicinal Chemistry, 2013, vol. 63, p. 415 - 422
[30] Chemistry - An Asian Journal, 2014, vol. 9, # 7, p. 1830 - 1840
[31] Synlett, 2015, vol. 26, # 6, p. 751 - 754
[32] Synthetic Communications, 2010, vol. 40, # 7, p. 1082 - 1087
[33] Collection of Czechoslovak Chemical Communications, 2010, vol. 75, # 2, p. 175 - 186
[34] Patent: JP2018/12651, 2018, A, . Location in patent: Paragraph 0252; 0254; 0263
[35] Patent: US2017/342047, 2017, A1, . Location in patent: Paragraph 0176
[36] Russian Chemical Bulletin, 2004, vol. 53, # 4, p. 830 - 833
[37] Zeitschrift fur Naturforschung - Section B Journal of Chemical Sciences, 2004, vol. 59, # 7, p. 829 - 835
[38] European Journal of Inorganic Chemistry, 2016, vol. 2016, # 27, p. 4524 - 4529
[39] Inorganic Chemistry, 2011, vol. 50, # 11, p. 4882 - 4891
[40] Journal of Materials Chemistry, 2012, vol. 22, # 21, p. 10852 - 10859
[41] Chemistry - A European Journal, 2010, vol. 16, # 27, p. 8062 - 8071
[42] Chemistry - A European Journal, 2013, vol. 19, # 33, p. 11051 - 11061
[43] Monatshefte fuer Chemie, 1908, vol. 29, p. 663
[44] Journal of the Chemical Society, 1942, p. 368
[45] Journal of Medicinal Chemistry, 1976, vol. 19, p. 1359 - 1362
[46] Tetrahedron Letters, 1987, vol. 28, # 4, p. 455 - 458
[47] Tetrahedron, 1997, vol. 53, # 6, p. 2163 - 2176
[48] Carbohydrate Research, 1987, vol. 171, p. 317 - 328
[49] Journal of Chemical Research, Miniprint, 1985, # 2, p. 451 - 466
[50] Tetrahedron Letters, 1997, vol. 38, # 30, p. 5245 - 5248
[51] Xenobiotica, 1995, vol. 25, # 9, p. 951 - 961
[52] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 2, p. 463 - 466
[53] Journal of the American Chemical Society, 2006, vol. 128, # 10, p. 3324 - 3334
[54] Organic and Biomolecular Chemistry, 2007, vol. 5, # 6, p. 935 - 944
[55] Journal of Materials Chemistry, 2007, vol. 17, # 14, p. 1399 - 1411
[56] Journal of Physical Chemistry B, 2005, vol. 109, # 12, p. 5700 - 5706
[57] Journal of the American Chemical Society, 2008, vol. 130, p. 13079 - 13094
[58] Patent: EP2048140, 2009, A1, . Location in patent: Page/Page column 32
[59] Journal of the American Chemical Society, 2009, vol. 131, p. 1294 - 1304
[60] Chemistry - A European Journal, 2010, vol. 16, # 8, p. 2427 - 2441
[61] Journal of Polymer Science, Part A: Polymer Chemistry, 2010, vol. 48, # 11, p. 2498 - 2508
[62] Journal of Organometallic Chemistry, 2012, vol. 715, p. 102 - 112
[63] Chemistry - An Asian Journal, 2012, vol. 7, # 11, p. 2547 - 2550,4
[64] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 10, p. 2941 - 2944
[65] Journal of the American Chemical Society, 2013, vol. 135, # 24, p. 9055 - 9077
[66] Journal of Asian Natural Products Research, 2014, vol. 16, # 5, p. 511 - 521
[67] Patent: EP2816032, 2014, A1, . Location in patent: Paragraph 0357; 0358
[68] Angewandte Chemie - International Edition, 2016, vol. 55, # 27, p. 7821 - 7825[69] Angew. Chem., 2016, vol. 128, p. 7952 - 7956,5
[70] RSC Advances, 2017, vol. 7, # 14, p. 8550 - 8560
Reference:
[1] Journal of Organic Chemistry, 1992, vol. 57, # 20, p. 5462 - 5469
[2] European Journal of Medicinal Chemistry, 2013, vol. 63, p. 415 - 422
[3] Journal of Organic Chemistry, 2013, vol. 78, # 11, p. 5587 - 5603
[4] Patent: EP2816032, 2014, A1,
[5] Patent: WO2015/24905, 2015, A1,
[6] Patent: US2015/80391, 2015, A1,
[7] Synlett, 2016, vol. 27, # 10, p. 1587 - 1591
20
[ 99-10-5 ]
[ 74-88-4 ]
[ 19520-74-2 ]
[ 1132-21-4 ]
Reference:
[1] Monatshefte fuer Chemie, 1887, vol. 8, p. 429
21
[ 99-10-5 ]
[ 29654-55-5 ]
Yield
Reaction Conditions
Operation in experiment
84%
Stage #1: With sodium tetrahydroborate In 1,2-dimethoxyethane at 25 - 30℃; for 2.5 h; Inert atmosphere Stage #2: With boron trifluoride diethyl etherate In 1,2-dimethoxyethane at 25 - 30℃; for 7 h; Inert atmosphere
The reaction should be carried out under nitrogen, keeping the reaction system as dry as possible.5 g of sodium borohydride (132 mmol)And 54.5 gEthylene glycol dimethyl ether was added to the reactor, and stirred to uniformly disperse sodium borohydride in the ethylene glycol dimethyl ether solution.(44 mmol) of 3,5-dihydroxybenzoic acid was dissolved in 34 g of ethylene glycol dimethyl ether solution,This was slowly added dropwise to a solution of sodium borohydride in ethylene glycol dimethyl ether (dropwise over an hour)Keep the temperature between 25 - 30 ° C. After completion of the dropwise addition, stirring was continued at room temperature for 1.5 hours.then,To the reaction system, 25 g (172 mmol) of boron trifluoride diethyl ether solution was added dropwise slowly,It was also added for approximately one hour and maintained at a temperature between 25 and 30 ° C.After completion of the dropwise addition, the reaction was carried out at room temperature for 6 hours. then,The temperature was raised to 35 ° C. 44.3 g of n-butanol was added to the reaction mixture. After stirring for half an hour, the temperature was lowered and the mixture was filtered. The filtrate was removed under reduced pressure at 70/30 mmHg to give 5.3 g of product in about 84percent yield. This product was used in the next step without further purification.
Reference:
[1] Journal of the American Chemical Society, 2003, vol. 125, # 3, p. 650 - 651
[2] Patent: CN105622350, 2016, A, . Location in patent: Paragraph 0030; 0033; 0034
[3] Journal of the American Chemical Society, 1996, vol. 118, # 18, p. 4354 - 4360
[4] Tetrahedron Letters, 2008, vol. 49, # 20, p. 3260 - 3263
[5] Patent: CN105884580, 2016, A,
22
[ 99-10-5 ]
[ 16534-12-6 ]
Reference:
[1] European Journal of Organic Chemistry, 2007, # 1, p. 125 - 135
[2] Organic Letters, 2013, vol. 15, # 17, p. 4588 - 4591
[3] Journal of Organic Chemistry, 2014, vol. 79, # 11, p. 5269 - 5281
[4] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 20, p. 5483 - 5489
[5] Justus Liebigs Annalen der Chemie, 1872, vol. 164, p. 118
[6] Bioscience, biotechnology, and biochemistry, 2001, vol. 65, # 1, p. 167 - 171
[7] Journal of the American Chemical Society, 2005, vol. 127, # 38, p. 13144 - 13145
[8] Patent: US4172960, 1979, A,
23
[ 99-10-5 ]
[ 16534-12-6 ]
Reference:
[1] Journal of Organic Chemistry, 2008, vol. 73, # 17, p. 6849 - 6852
24
[ 99-10-5 ]
[ 877-88-3 ]
Reference:
[1] Journal of Labelled Compounds and Radiopharmaceuticals, 2004, vol. 47, # 3, p. 167 - 174
[2] Journal of Asian Natural Products Research, 2011, vol. 13, # 4, p. 290 - 296
[3] Synlett, 2013, vol. 24, # 9, p. 1109 - 1112
[4] Asian Journal of Chemistry, 2014, vol. 26, # 7, p. 2092 - 2098
[5] Patent: CN105503652, 2016, A,
[6] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2016, vol. 55B, # 8, p. 1035 - 1038
[7] Patent: US2017/342047, 2017, A1,
25
[ 99-10-5 ]
[ 77-78-1 ]
[ 877-88-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2017, vol. 27, # 1, p. 81 - 85
26
[ 99-10-5 ]
[ 54915-31-0 ]
Reference:
[1] Patent: WO2012/149608, 2012, A1,
[2] European Journal of Medicinal Chemistry, 2013, vol. 63, p. 415 - 422
[3] Patent: JP2018/12651, 2018, A,
27
[ 99-50-3 ]
[ 89-86-1 ]
[ 149-91-7 ]
[ 99-10-5 ]
[ 303-38-8 ]
[ 490-79-9 ]
[ 99-06-9 ]
[ 69-72-7 ]
[ 74-88-4 ]
[ 99-96-7 ]
[ 2150-41-6 ]
[ 606-45-1 ]
[ 5368-81-0 ]
[ 121-98-2 ]
[ 2150-38-1 ]
[ 1916-07-0 ]
[ 2150-42-7 ]
[ 2150-40-5 ]
[ 2150-37-0 ]
Reference:
[1] Chemical Communications, 2014, vol. 50, # 14, p. 1694 - 1697
28
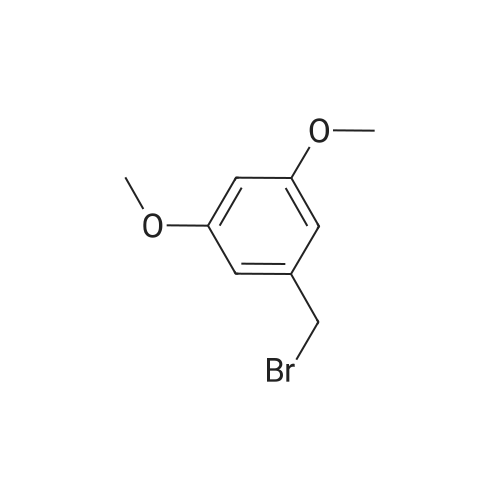
[ 99-10-5 ]
[ 76045-71-1 ]
Reference:
[1] Tetrahedron, 1983, vol. 39, # 24, p. 4189 - 4192
[2] Organic Process Research and Development, 2004, vol. 8, # 4, p. 571 - 575
29
[ 99-10-5 ]
[ 52189-63-6 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 1658 - 1666
30
[ 99-10-5 ]
[ 75996-29-1 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 1658 - 1666
31
[ 67-56-1 ]
[ 99-10-5 ]
[ 2150-44-9 ]
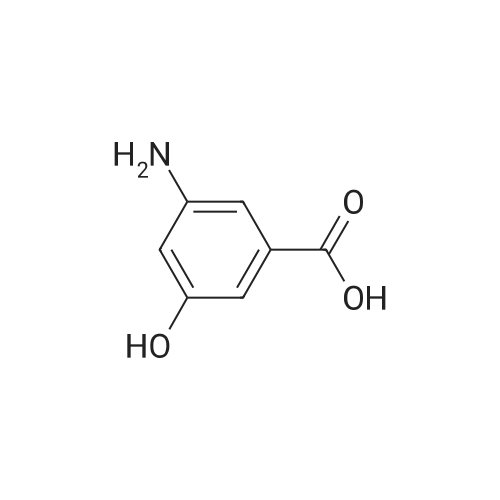
[ 67973-80-2 ]
Reference:
[1] Journal of Organic Chemistry, 2013, vol. 78, # 11, p. 5587 - 5603
Stage #1: at 180℃; for 72 h; Autoclave Stage #2: With hydrogenchloride In water at 100℃; for 24 h;
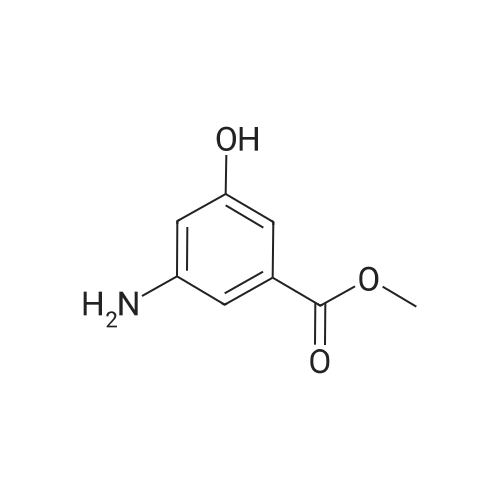
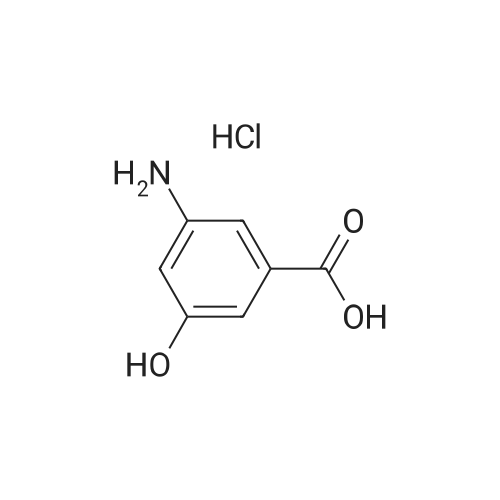
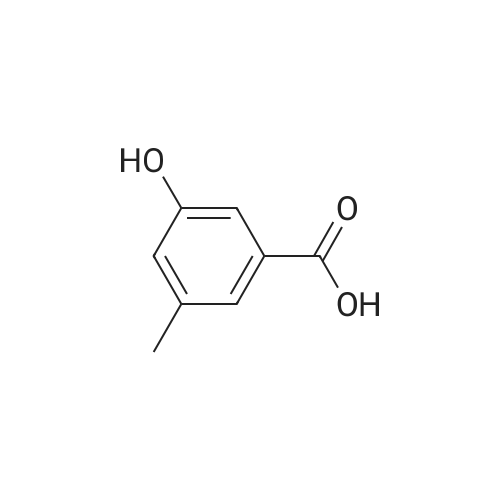
Step 1: 3-amino-5-hydroxy-benzoic acid hydrochloride A mixture of 3,5-dihydroxybenzoic acid (250.0 g), ammonium chloride (213.0 g), and 28percent ammonium hydroxide (750.0 mL) was stirred at 180° C. for 3 days under autoclave. The reaction was cooled to room temperature and then concentrated under reduced pressure. The resulting residue was dissolved in a 6 N hydrochloric acid solution (3.0 L). The reaction mixture was heated at 100° C. for 24 hours and then cooled to 70° C.~80° C. Active carbon (30.0 g) was added to the reaction mixture, which was then filtered with celite pad. The filtrate was concentrated under reduced pressure. The resulting residue was washed with a 6 N hydrochloric acid solution twice and then dried under reduced pressure to obtain 192.0 g of the titled compound (Yield: 62.4percent). 1H-NMR (d6-DMSO) δ 7.81 (s, 1H), 7.69 (s, 1H), 7.56 (s, 1H)
Reference:
[1] Journal of the American Chemical Society, 2010, vol. 132, # 36, p. 12757 - 12765
[2] Patent: US2012/178765, 2012, A1, . Location in patent: Page/Page column 11
[3] Synthetic Communications, 1999, vol. 29, # 8, p. 1379 - 1382
[4] Tetrahedron, 1983, vol. 39, # 24, p. 4189 - 4192
[5] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 7, p. 2242 - 2252
[6] Organic and Biomolecular Chemistry, 2007, vol. 5, # 10, p. 1615 - 1620
1
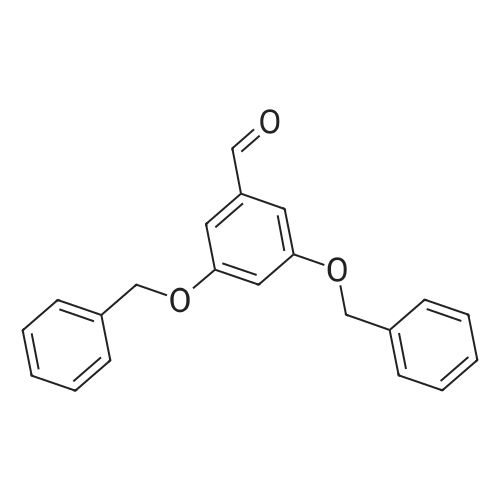
The approach described by Brouwer et al. (ibid.) for the convergent synthesis of amino acid based dendrimers was adapted in order to obtain dendrimers with surface propargyl groups to enable a 1,3-dipolar cycloaddition ("click") reaction with amino acid /peptide derived azides. First generation dendrimer 1 was synthesized from 3,5-dihydroxybenzoic acid in an overall yield of 81% as shown in Scheme 1. In detail, 3,5-dihydroxymethylbenzoate (21.4 g, 130 mmol) was dissolved in dry DMF (250 mL) and anhydrous K2CO3 (45 g, 330 mmol, 2.5 equiv.) was added. To this suspension, a solution of propargylbromide in toluene (35 mL, 314 mmol, 2.5 equiv.) was added dropwise. The reaction mixture was stirred for 48 h at room temperature. Then, DMF was removed by evaporation and the residue was redissolved in EtOAc (400 mL) and the organic phase was washed with H2O (3 x 100 mL) in KHSO4 (3 x 100 mL) and brine (3 x 100 mL), dried (Na2SO4) and evaporated in vacuo. The residue was recrystallized from EtOAc/hexane to obtain 1 as off-white crystals in 81% yield (25.2 g). Rf(EtOAc/hexane 4:1 v/v): 0.76; Rf (DMC/MeOH 98:2 v/v): 0.87; Rf(CHCl3/MeOH/AcOH 95:20:3 v/v): 0.83; 1H-NMR (CDCl3) δ 2.55 (t (J 2.47 Hz), 2H), 3.91 (s, 3H), 4.72 (d (J 2.47 Hz), 4H), 6.81 (t (J 2.20 Hz), 1H), 7.29 (d (J 2.20 Hz), 2H); 13C-NMR (CDCl3) δ 52.4, 56.0, 76.0, 77.9, 107.5, 108.8, 132.0, 157.8, 158.4; Elemental analysis: calculated for C14H12O4 C 68.83, H 4.95, found C 68.76, H 4.95.
99%
With thionyl chloride for 2h; Heating;
99%
With thionyl chloride at 20 - 97℃; for 2.41667h; Inert atmosphere;
99%
With thionyl chloride at 20℃; Reflux;
99%
With toluene-4-sulfonic acid
98%
With sulfuric acid for 20h; Heating;
98%
With sulfuric acid Heating;
98%
With hydrogen cation
98%
With sulfuric acid for 18h; Reflux;
98%
With thionyl chloride at 0℃; Reflux; Inert atmosphere;
5.2. General procedure for preparation of esters
General procedure: Thionyl chloride (1.16 g, 1.5 equiv) was added drop-wise to a solution of acid (1.0 g, 1.0 equiv) in the corresponding alcohol (15 ml) at 0&d eg;C. The solution was refluxed under a nitrogen atmosphere until all starting material was consumed (TLC monitoring). Then the solvent was removed under vacuo and the residue was purified by silica gel column chromatography eluting with ethyl acetate/n-hexane to afford the corresponding carboxylic esters.
98%
With sulfuric acid for 8h; Reflux;
2.1 Synthesis and analytical data of compound 8a′.
To a solution of 8a (10.00 g, 64.9 mmol) in methanol (50 mL) was added concentrated sulfuric acid(750 μL in 10 mL methanol), and the solution was allowed to reflux for 8 h. After cooled to ambient temperature, methanol was evaporated under reduced pressure and the residue was dissolved in ethylacetate (50 mL), which was then washed with saturated NaHCO3 solution and water. The organic layer was dried over anhydrous Na2SO4 and ethyl acetate was removed in vacuo to afford of methyl3,5-dihydroxylbenzoate 8a′ (10.74 g, 98%) as a white powder.
98%
With sulfuric acid for 6h; Reflux;
96%
With sulfuric acid for 5h; Heating;
96%
With toluene-4-sulfonic acid for 24h; Heating;
96%
With sulfuric acid for 5h; Heating;
96%
With sulfuric acid Reflux;
95%
With sulfuric acid Heating;
95%
With sulfuric acid for 20h; Reflux;
4
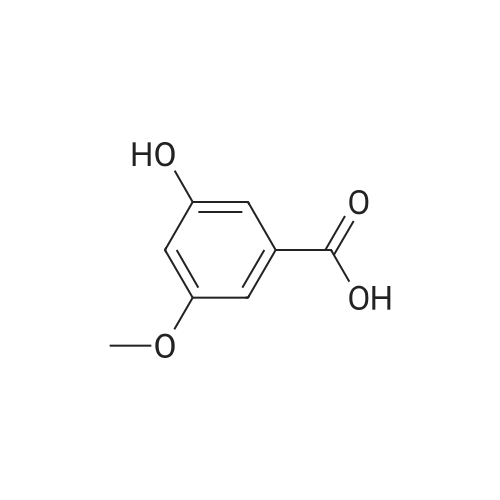
A solution of 3,5-dihydroxybenzoic acid (20.0 g, 129.9 mmol) in dry methanol (100 mL) and H2SO4 (1 mL) was refluxed for 20 h. The volatile product was removed in vacuo and the residue was redissolved in ethyl acetate (EA) and washed with aqueous NaHCO3, water and brine. The organic phase was dried with anhydrous sodium sulphate and the solvent was evaporated to yield methyl 3,5-dihydroxybenzoate as a white colored solid (95 % yield, m.p.= 17O0C). 1H-NMR (300 MHz, CDCl3) δ 7.1 (d, 2H), 6.6 (t, IH), 4.9 (s, OH), 3.9 (s, OCH3).
95%
With sulfuric acid Reflux;
95%
With sulfuric acid Reflux;
Methyl 3,5-dihydroxybenzoate (8)
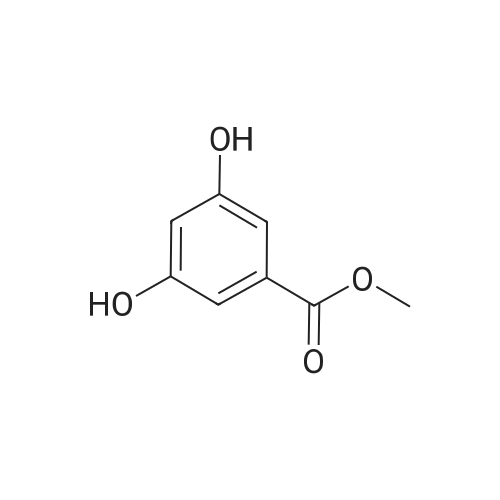
Methyl 3,5-dihydroxybenzoate (8)To a solution of 3,5-dihydroxybenzoic acid (5.0 g, 32 mmol) in methanol (170 ml) was added a catalytic amount of sulphuric acid (0.3 ml). After stirring at reflux overnight, the mixture was cooled and neutralized with 4M NaOH (aq. solution). After concentration, the residue was dissolved in ethyl acetate and washed with water and brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. Compound 2 was isolated as a white solid (5.13 g, 95%); m.p. 164-165° C. (ethyl acetate); 1H-NMR (500 MHz, d6-DMSO) δ: 9.65 (2H, s), 6.81 (2H, d, J 2.3 Hz, CHar), 6.43 (1H, d, J 2.3 Hz, CHar), 3.78 (3H, s, CH3); 13C-NMR (125 MHz, d6-DMSO) δ: 166.2 (C═O), 158.4 (C x2), 131.2 (C), 107.1 (CH), 107.0 (CH x2), 51.9 (CH3); MS (ES)- m/z: 167 [M-H]-; HPLC tR=3.11; IR (neat) v (cm-1): 3229, 1688, 1600, 1486, 1305, 1161, 1102, 995, 765. Data were in good agreement with the literature22.
95%
With (1S)-10-camphorsulfonic acid Reflux;
95%
With sulfuric acid for 4h; Reflux; Cooling;
94%
With sulfuric acid for 2h; Reflux;
93%
With hydrogen cation
92%
With sulfuric acid at 68℃; for 72h; Inert atmosphere;
92%
With sodium hydrogensulfate monohydrate for 20h; Reflux;
92%
With sulfuric acid for 18h; Reflux;
92.7%
With sulfuric acid for 12h; Reflux;
90%
With sulfuric acid
90%
With sulfuric acid Heating;
90%
With acetyl chloride for 17h; Reflux; Inert atmosphere;
B.1
1. Preparation of 3,5-dihydroxybenzoic acid methyl ester (1); To a solution of 3,5-dihydroxybenzoic acid (10 g, 64.9 mmol) in anhydrous methanol (150 mL) acetyl chloride (2 mL, 28.1 mmol) was added dropwise. The mixture was heated at reflux under N2 for 17 hours. The solvent was evaporated and the residue was dissolved in ethyl acetate (100 mL), then washed with saturated sodium hydrogen carbonate (3 x 100 mL). The combined organic layers were washed with water (2 x 100 mL), dried over sodium sulfate, filtered and concentrated in vacuo to afford 2 as a colourless solid (9.80 g, 90%): mp 167-168°C (lit.1 mp 168-169°C); 1H NMR (400 MHz, Acetone-flfe) δ 8.66 (s, 2H), 7.00 (d, J = 2.4 Hz, 2H), 6.59 (t, J = 2.4 Hz, 2H), 3.83 (s, 3H).
90%
With acetyl chloride for 17h; Reflux; Inert atmosphere;
Preparation of 3,5-dihydroxybenzoic acid methyl ester (2)
To the solution of anhydrous methanol (150 mL) was added slowly acetyl chloride (2 mL, 28.1 mmol) followed by the solution of 3,5-dihydroxybenzoic acid (10 g, 64.9 mmol). The mixture was refluxed under nitrogen for 17 hours. The solvent was evaporated and the residue was dissolved in ethyl acetate (100 mL), and washed with saturated sodium hydrogen carbonate (3 × 100 mL). The organic layer was washed with distilled water (2 × 100 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure to afford 2 as an off white solid (9.80 g, yield 90%), mp 167-168°C (lit.[32] mp 168-169°C). 1H NMR (400 MHz, Acetone-d6) δ (ppm) 8.66 (2H, s, 2 × OH), 7.00 (2H, d, J=2.4 Hz, H-2,6), 6.59 (1H, t, J=2.4 Hz, H-4), 3.83 (3H, s, COOCH3). The 1H NMR spectrum was in good agreement with previously reported data[33].
90%
With sulfuric acid Reflux;
90%
With thionyl chloride
89%
With bismuth(lll) trifluoromethanesulfonate for 18h; Reflux;
89%
With sulfuric acid
89%
With thionyl chloride at 60℃; for 2h; Inert atmosphere;
1 [Production of Compound (A-7)]
Then, under an argon atmosphere, in a 300 mL flask,3,5-dihydroxybenzoic acid (8.0 g, 51.9 mmol, 1 eq.) Was dissolved in dry methanol (40 mL).Subsequently, thionyl chloride (7.4 g, 4.5 mL, 62 mmol, 1.2 eq.) Was added dropwise, and the flask was heated on an oil bath and reacted at 60 ° C. with stirring for 2 hours.The mixture was then cooled to room temperature and the volatiles evaporated under reduced pressure. Then, the solid precipitate was suspended in toluene,The volatiles were evaporated to give compound (A-4) as a beige solid (yield 7.79 g, 46.3 mmol, yield 89%).
88%
With sulfuric acid for 16h; Reflux;
86%
With sulfuric acid for 24h; Reflux;
3, 5-Dihydroxy Methylbenzoate (302)
Conc. H2SO4 (80 mL) was added slowly to a stirred solution of 3,5-dihydroxybenzoic acid 301 (50 g, 0.33 mol) in CH3OH (660 mL) at rt and this solution was heated to reflux for 24 h. The reaction mixture was cooled to rt and H2O (500 mL) was added to the solution. The solution was extracted with EtOAc (3*300 mL), and the combined organic extracts were washed with a saturated aq NaHCO3 solution (2*300 mL), The organic layer was dried (Na2SO4), and concentrated under reduced pressure to afford a white crude powder. The crude solid was purified by flush column chromatography (FCC) (10% ethyl acetate in hexane) to afford a white powdered ester 302 (48 g, 86%): 1H NMR (300 MHz, CDCl3) δ 7.10 (2H, d, J=2.4 Hz HAr), 6.57 (1H, t, J=2.0 Hz, HAr), 4.99, (2H, br, s, HO), 3.84 (31-1, s, H3COO).
84%
With sulfuric acid at 80℃; for 24h;
82%
With sulfuric acid for 10h; Heating;
80%
With sulfuric acid In dichloromethane Heating;
80%
With thionyl chloride
78%
With sulfuric acid In water for 18h; Reflux;
Synthesis of methyl 3,5-dihydroxybenzoate 4.
In a flask, 2 g (12.98 mmol) of 3,5-dihydroxybenzoic acid were dissolved in 25 mL of MeOH atroom temperature. Once dissolved 0.22 mL of concentrated H2SO4 sulfuric acid were slowlyadded. The mixture was refluxed for 18 h and in constant stirring. The reaction was monitored bythin layer chromatography (TLC). When the absence of one of the reactants was observed, thereaction was stopped by adding sodium carbonate until neutral pH. The reaction mixture wasfiltered, the obtained liquid was brought to dryness in a rotary evaporator, and finally it waspurified in a column of silica (230-400), obtaining a white solid 1.71 g (10.14 mmol) with a yieldof 78%.
60%
With sulfuric acid at 68℃; for 24h;
60%
With sulfuric acid In water at 68℃; for 24h;
56%
With toluene-4-sulfonic acid for 20h; Reflux; Inert atmosphere;
56%
With toluene-4-sulfonic acid for 20h; Reflux; Inert atmosphere;
With hydrogenchloride
With sulfuric acid
With hydrogenchloride
With sulfuric acid Heating;
for 17h; Heating;
With acid
With hydrogenchloride at 24℃; for 12h;
With hydrogenchloride at 20℃;
With sulfuric acid at 70℃; for 20h;
With sulfuric acid Heating;
for 16h; Heating;
With sulfuric acid
With toluene-4-sulfonic acid
for 23h; Heating / reflux;
X.a
Example X This example illustrates the preparation of [(4-Methoxy-4-(3-phosphoryloxy-5-methoxy(D3)phenyl)] spiro [1,2-dioxetane-3,2'-adamantane], disodium salt (72).; The sequence of the reaction in accordance herewith: (a). Synthesis of Methyl 3,5-dihydroxybenzoate (66), (a starting ester). In a 1000mL round bottom flask, 91.0g of 3,5-dihydroxybenzoic acid (65), 400mL of methanol and 15mL of concentrated sulfuric acid were heated at reflux for 23 hours. TLC on silica gel plates showed formation of the product. The solvent was evaporated under reduced pressure and the residue was stirred with deionized water (500ml) for 30 minutes. The solid was filtered off and air-dried for 24 hours yielding 81 parts of a crude product. A 31 parts of the crude product was purified by chromatography on silica gel column eluting with a 35% ethyl acetate-hexanes solution. Product-containing fractions were checked by TLC on silica gel plates and combined. The solvent was evaporated under reduced pressure giving 30.3 parts of the titile compound (66) as an off-white solid. The structure was confirmed by 1H NMR. The reaction proceeded as follows :
With sulfuric acid for 20h; Reflux;
With sulfuric acid
With sulfuric acid for 24h; Reflux;
With sulfuric acid
With sulfuric acid Reflux;
With sulfuric acid Reflux;
30 g
With thionyl chloride at 0℃; for 2h; Reflux;
15.A A) methyl 3,5-dihydroxybenzoate
A) methyl 3,5-dihydroxybenzoate To a solution of 3,5-dihydroxybenzoic acid (30.0 g) in methanol (300 mL) was added dropwise thionyl chloride (20 mL) at 0°C, and the mixture was heated under reflux for 2 hr. The reaction mixture was concentrated, and the obtained crystals were washed with diethyl ether to give the title compound (30.0 g) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 3.79 (3H, s), 6.44 (1H, s), 6.81 (2H, s), 9.60 (2H, s).
With sulfuric acid for 5h; Reflux;
With sulfuric acid at 120℃;
With sulfuric acid
1 Example 1. Exemplary Benzoic Acid Syntheses
While certain benzoic acids are commercially available, others may be synthesized using an esterification, etherification, and deprotection sequence as shown below.
With sulfuric acid In water at 80℃;
Synthesis of Methyl 3, 5-dihydroxybenzoate (2)
The reaction was started with 3, 5-dihydroxybenzoic acid (1g, 0.00649mol), in methanol (10mL), and concentrated sulfuric acid (96% pure in water, 0.5mL, 0.0093mmol) slowly added to the above solution. The reaction mixture was heated to reflux for 3h. The reaction mixture was cooled to room temperature and then solvent removed under reduced pressure. The residue was extracted with ethyl acetate (50mL) and washed with water (10mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated. The crude residue so obtained was purified by column chromatography (hexane-ethyl acetate 14%), which afforded compound 2 as a white solid in 94% yield.
With thionyl chloride Inert atmosphere;
With sulfuric acid Reflux;
With thionyl chloride at 0℃; for 1h; Reflux; Inert atmosphere;
Methyl 3,5-Dihydroxybenzoate
3,5-Dihydroxybenzoic acid (60.00 g, 389 mmol) was suspended in MeOH (780 mL, 0.5 M) and cooled to 0 °C. To this mixture was added dropwise SOCl2 (113 mL, 4.0 equiv). The mixture was allowed to warm to rt and reflux for 1 h. The solvent was then removed in vacuo. The crude product was used without further purification in the next step (an aliquot was purified for analytical purposes). IR (film): 3365, 3228, 1686, 1600, 1509, 1485, 1439, 1297, 1176,1009, 764, 686 cm-1. 1H NMR (600 MHz, DMSO-d6): δ = 9.61 (s, 2 H), 6.81 (d, J = 2.2 Hz, 2H), 6.44 (t, J = 2.2 Hz, 1 H), 3.79 (s, 3 H). 13C NMR (151 MHz, DMSO-d6): δ = 166.2, 158.5, 131.3, 107.2, 107.1, 51.9. HRMS (ESI+): m/z [M + Na]+ calcd for C8H8O4Na: 191.0320; found: 191.0314.
Synthesis of Compounds 7a and b.
Compounds 7a and b were synthesized using similar procedures. The procedure for the synthesis of 7a, used as an example, was performed as follows. 3,5-Hydroxybenzoic acid 6a (5.0 g, 32.5 mmol) was dissolved in anhydrous ethanol (50 mL). After addition of concentrated H2SO4 (0.5 mL), the solution was refluxed for 6 h. The ethanol was removed, and the mixture was extracted with dichloromethane and 10% NaHCO3 aqueous solution. Combined organic extracts were washed with water and brine, subsequently dried over anhydrous MgSO4, and concentrated to yield a white solid (yield, 95%).
95%
With sulfuric acid for 48h; Reflux;
94%
With sulfuric acid for 24h; Inert atmosphere; Reflux;
94%
With sulfuric acid for 22h; Reflux;
90%
With sulfuric acid for 4h; Heating;
90%
With sulfuric acid at 90℃; for 8h;
90%
With sulfuric acid for 24h; Reflux;
85%
With sulfuric acid for 12h; Reflux;
81%
With hydrogen cation
77.2%
With sulfuric acid at 65℃; for 96h;
3 Reference Example 3
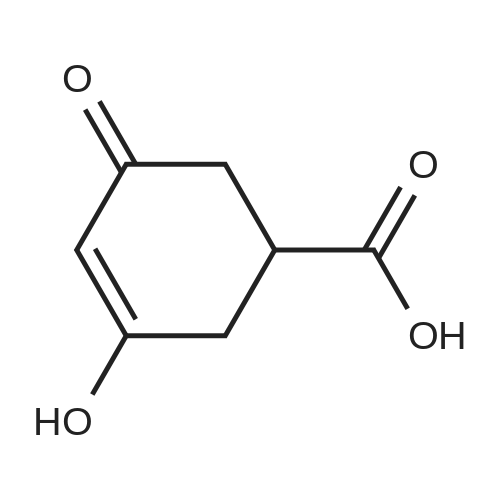
Reference Example 3 Synthesis of ethyl 3-hydroxy-5-oxo-cyclohexa-3-ene carboxylate To an ethanol solution (200 mL) of 3,5-dihydroxybenzoic acid (25 g, 162.2 mmol) was added sulfuric acid (3 mL) and stirred over night at room temperature and then under heating at 65°C for 4 days. The reaction liquid was concentrated under reduced pressure and poured into ice water (about 300 mL) while stirring to filter off white crystal, 3,5-dihydroxybenzoic acid ethyl ester (22.8 g, 77.2%). 3,5-Dihydroxybenzoic acid ethyl ester (10 g, 54.89 mmol) was dissolved in ethanol (15 mL), followed by adding sodium formate (4.48 g, 65.87 mmol), replacing inside a reactor with nitrogen at 30°C for 15 minutes, adding palladium on carbon (364 mg) and reacting at 30°C for 3 hours then at 40°C over night. Catalyst was filtered off, followed by neutralization with a 1N HCl solution, concentration under reduced pressure and purification of thus obtained residue with silica gel column chromatography (hexane/ethyl acetate=1/1 to 0/1) to obtain an objective compound (1.53 g, 15.1%). 1H-NMR (200 MHzFT,TMS,CDCl3) 1.26(3H,dt,J=1.8,7.1Hz), 2.66(2H,d,J=2.7Hz), 2.83(1H,dd,J=1.8,6.6Hz), 3.01-3.19(1H,m), 3.32-3.55(1H,m), 4.18(2H,q,J=7.2Hz), 5.51(1H,s), 5.80-6.10(1H,br) MS(ESI) m/z 185 [M+H]+
76%
With sulfuric acid
75%
With sulfuric acid for 8h; Heating / reflux;
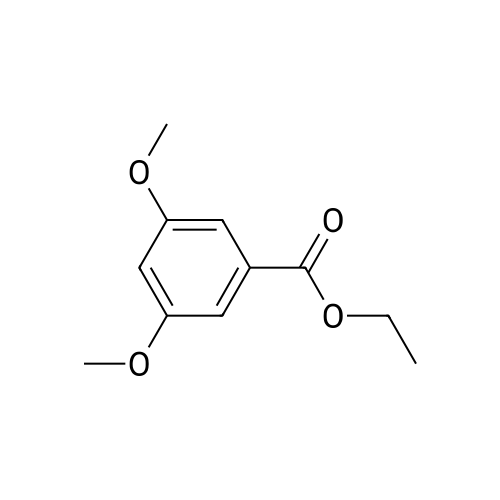
Examples; Ethyl-3,5-dihydroxy-benzoate (3).; To a solution of 3,5-dihydroxy benzoic acid, 1 (19 g, 123 mmol) in dry ethanol (250 mL) was added 2-3 niL of concentrated sulphuric acid drop wise and refluxed for 8 h. Ethanol was evaporated on rotavapor, diluted with water and extracted with ether. Combined ethereal layers were again washed with saturated sodium bicarbonate solution, water and dried over sodium sulphate, filtered and evaporated to dryness to afford 3. Yield 16.8 g (75%); mp 128-1300C; MS (FAB) 183 (M+ +1); IR (KBr) 3496, 1819, 1687; 1H NMR (200 MHz, DMSO-d6) δ 9.89 (s, 2H), 7.06 (d, / = 2.2 Hz, 2H), 6.69 (t, J = 2.2 Hz, IH), 4.48 (q, J = 7.1 Hz, 2H), 1.52 (t, J = 7.1 Hz, 3H).
75%
With sulfuric acid for 8h; Reflux;
75%
With thionyl chloride at 0℃; Reflux; Inert atmosphere;
5.2. General procedure for preparation of esters
General procedure: Thionyl chloride (1.16 g, 1.5 equiv) was added drop-wise to a solution of acid (1.0 g, 1.0 equiv) in the corresponding alcohol (15 ml) at 0&d eg;C. The solution was refluxed under a nitrogen atmosphere until all starting material was consumed (TLC monitoring). Then the solvent was removed under vacuo and the residue was purified by silica gel column chromatography eluting with ethyl acetate/n-hexane to afford the corresponding carboxylic esters.
69%
With sulfuric acid at 20℃; for 36h; Reflux;
16
Example 16. Preparation of 2-[4-(2-hydroxy-ethoxy)-3,5-dimethyl-phenyl]-5,7-diisopropoxy-3H-quinazolin-4-one [0210] To a solution of 3,5-dihydroxybenzoic acid (10.0 g, 64.9 mmol) in anhydrous ethanol (100 ml_) at room temperature was slowly added concentrated sulfuric acid (10 ml_). The resulting mixture was stirred at reflux for 36 hours. The reaction was cooled to room temperature, diluted with water (200 ml_), extracted with CH2Cb (3 x 100 ml_), and concentrated on a rotary evaporator, to afford 3,5- dihydroxybenzoic acid ethyl ester as a colorless oil. Yield: 8.2 g (69%).[0211] A solution of 3,5-dihydroxybenzoic acid ethyl ester (6.0 g, 33 mmol) and 2-iodo-propane (9.9 ml_, 99 mmol) in DMF (200 ml_) was mixed with potassium carbonate (13.7 g, 98.9 mmol) and the mixture was stirred at room temperature for 14 hours. The reaction mixture was then diluted with water (300 ml_), and extracted with ethyl acetate (3 x 100 ml_). The residue obtained upon concentration was subjected to column chromatography (SiO2, hexanes / ethyl acetate = 3:1) to afford 3,5-diisopropoxybenzoic acid ethyl ester. Yield: 8.8O g (100%).[0212] A solution of 3,5-diisopropoxybenzoic acid ethyl ester (8.80 g, 33.1 mmol) and lithium hydroxide (3.18 g, 132 mmol) in water (100 ml_), methanol (50 ml_), and THF (50 ml_) was stirred at reflux for 3 hours. It was then cooled to room temperature, diluted with water (200 mL), acidified with 2 N hydrochloric acid, to pH approximately 2, extracted with CH2Cb (3 χ 100 mL), and concentrated on a rotary evaporator, to afford 3,5-diisopropoxybenzoic acid as a white solid. Yield: 7.60 g (97%).[0213] A solution of 3,5-diisopropoxybenzoic acid (7.60 g, 31.9 mmol), triethylamine (5.3 mL, 38 mmol), and diphenylphosphoroyl azide (8.3 mL, 38 mmol) in 1,4-dioxane (120 mL) and tert-butanol (30 mL) was stirred at reflux for 16 hours. The reaction mixture was then cooled to room temperature, diluted with 0.2 N sodium bicarbonate aqueous (200 mL), extracted with CH2CI2 (3 x 100 mL), and concentrated on a rotary evaporator. The residue obtained was subjected to column chromatography (SiO2, hexanes / ethyl acetate = 3:1) to afford 3,5-diisopropoxyphenyl)-carbamic acid tert-butyl ester as a white solid. Yield: 5.60 g (57%). [0214] A solution of 3,5-diisopropoxyphenyl)-carbamic acid tert-butyl ester (5.60 g, 18.2 mmol) in trifluoroacetic acid (30 mL) was stirred at reflux for 30 minutes and concentrated on a rotary evaporator to dryness to afford 3,5-diisopropoxyphenylamine trifluoroacetic acid salt as an oil. Yield: 5.27 g (90%).[0215] To a round-bottomed flask contained 3,5-diisopropoxyphenylamine trifluoroacetic acid salt (5.27 g, 16.4 mmol) was slowly added oxalyl chloride (20 mL) and the mixture was stirred at reflux for 1 hour. Extra oxalyl chloride was removed by distillation and methanol (100 mL) was added to the residue. It was then stirred at room temperature for 30 minutes and concentrated to dryness on a rotary evaporator to afford 4,6-diisopropoxy-1H-indole-2,3-dione as a semi-solid. Yield: 4.33 g (100%).[0216] A solution of potassium hydroxide (15.3 g, 273 mmol) in water (60 mL) was mixed with 4,6-diisopropoxy-1H-indole-2,3-dione (4.33 g, 16.4 mmol). To this mixture was slowly added hydrogen peroxide. The resulting mixture was stirred at 700C for 30 minutes and cooled to 00C. The mixture was acidified at 0°C with 2 N hydrochloric acid to pH approximately 4, extracted with CH2CI2 (3 x 100 mL), and concentrated on a rotary evaporator to afford 2-amino- 4,6-diisopropoxy-benzoic acid as a semi-solid. Yield: 2.91 g (70%). [0217] A solution of 2-amino-4,6-diisopropoxybenzoic acid (2.91 g, 11.5 mmol), N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (3.20 g, 16.7 mmol), HOBt (3.10 g, 23.0 mmol), and triethylamine (4.2 ml_, 30 mmol) in THF (200 ml_) was stirred at room temperature for 20 minutes. 50% (v/v) ammonia aqueous (20 ml_) was then added. The resulting solution was stirred at room temperature for 14 hours, diluted with water (200 ml_), extracted with CH2CI2 (3 x 100 ml_), and concentrated on a rotary evaporator. The residue obtained was subjected to column chromatography (SiO2, ethyl acetate / dichloromethane / methanol = 6:2:1) to afford 2-amino-4,6- diisopropoxybenzamide. Yield: 1.2 g (41%).[0218] A solution of 2-amino-4,6-diisopropoxybenzamide (0.30 g, 1.2 mmol), 4-(2-hydroxy-ethoxy)-3,5-dimethylbenzaldehyde (0.28 g, 1.4 mmol), sodium bisulfite (0.25 g, 1.4 mmol), and p-toluenesulfonic acid (20 mg, 0.11 mmol) in dimethyl acetamide (10 ml_) was stirred at 150°C for 12 hours. Extra solvent was evaporated on a rotary evaporator and the residue was diluted with saturated sodium bicarbonate aqueous solution (100 ml_) and extracted with CH2CI2 (3 x 100 ml_). The residue obtained upon concentration was subjected to column chromatography (SiO2, ethyl acetate / dichloromethane / hexanes / methanol = 4:4:4:1) to afford the title compound as a light yellow solid. Yield: 35 mg (6.9%). 1H NMR (400 MHz, CDCI3): δ 9.78 (br s, 1 H), 7.66 (s, 2H), 6.78 (d, 1 H), 6.42 (d, 1H), 4.72 (m, 1H), 4.63 (m, 1H), 3.97 (t, 3H), 3.92 (t, 2H), 2.33 (s, 6H), 1.45 (d, 3H), 1.41 (d, 3H). MS (ES+) m/z: 427.13 (M+1).
69%
With sulfuric acid for 36h; Reflux;
15
To a solution of 3,5-dihydroxybenzoic acid (10.0 g, 64.9 mmol) in anhydrous ethanol (100 mL) at room temperature was slowly added concentrated sulfuric acid (10 mL). The resulting mixture was stirred at reflux for 36 hours. The reaction was cooled to room temperature, diluted with water (200 mL), extracted with CH2Cl2 (3*100 mL), and concentrated on a rotary evaporator, to afford 3,5-dihydroxybenzoic acid ethyl ester as a colorless oil. Yield: 8.2 g (69%)
With hydrogenchloride
With sulfuric acid
With sulfuric acid Heating;
With sulfuric acid
With sulfuric acid; acetic anhydride Heating;
With sulfuric acid for 12h; Heating;
With sulfuric acid Heating;
With sulfuric acid
Heating / reflux;
1.1a
Example 1; With reference to FIG. 1, the synthesis of monomer 1a and 1b: Example 1a; Synthesis of Compound 5; The amphiphilic monomer was synthesized from commercially available 3,5-dihydroxybenzoic acid (4) using ethanol and catalytic amount of fuming sulfuric acid.
With sulfuric acid for 12h; Reflux;
With sulfuric acid In water for 8h;
3.1 Synthesis of 5,17-Di(ethoxycarbonyl)Tetranitrooxacalix[4]arene (DEOC) (4)
3,5-dihydroxy ethyl benzoate (2) was prepared from 3,5-dihydroxybenzoic acid (1), as per the reported procedure [30]. Compound (2) (4 gm, 21.9 mmol), compound (3),1,5-difluoro-2,4-dinitrobenzene (21.9 mmol) and finelyground anhydrous K2CO3 (54.9 mmol) were stirred inDMSO (80 mL) in a 250 mL round bottom flask at roomtemperature for 30 min. The reaction was monitored byTLC. The reaction mixture was added drop wise to 1 MHCl (400 mL). The aqueous layer was extracted withEtOAc (500 mL 9 2). The combined organic layers werewashed with brine (500 mL), dried over anhydrous Na2-SO4, filtered, and concentrated in vacuo. The DEOC (4,Scheme 1) was purified using 30 % ethyl acetate in hexaneas the eluent, with a yield of 80 %.
With sulfuric acid at 90℃; for 48h; Inert atmosphere;
With sulfuric acid In hexane; chloroform for 10h; Inert atmosphere; Reflux; Dean-Stark;
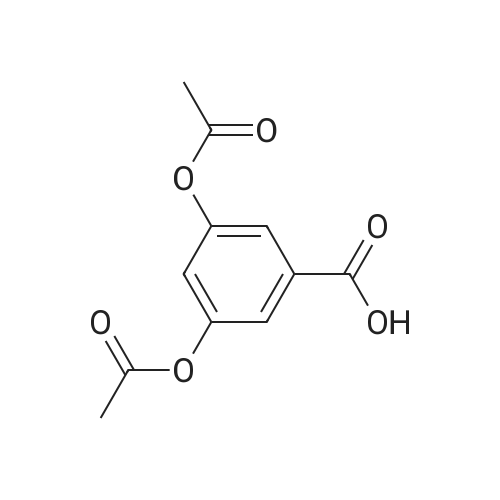
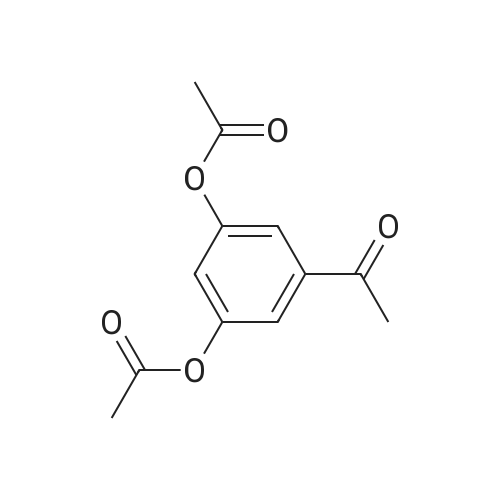
A mixture of 7.7 g (0.05 mol) of 3,5-dihydroxybenzoic acid (1), 25 mL of acetic anhydride and 2 mL of triethylamine was mixed and stirred for 10 min and heated at 90 to 100 C. TLC was monitored. Product into the ice water, a large number of white precipitate generated, filtered to obtain a white solid is the crude product,Recrystallization from ethanol gave 11.0 g of white crystals (2) in 92% yield.
91%
at 130℃; for 4h;
General procedure: Garlic acid (2000 mg, 11.763 mmol) was dissolved in acetic anhydride(7205 mg, 70.578 mmol). The solution was then heatedunder reflux at 130 C for 4 h, after which the deionized water(4 ml) was add to the solution to remove the unreacted acetic anhydride.The solvent was removed using a rotary evaporator underreduced pressure to yield white solid. The product was recrystallizedin CH2Cl2 and dried under vacuum. Yielding 95% compound 4a(3308 mg) as a white solid. Compounds 4b-c were synthesizedfollowing the procedure of preparation 4a.
90%
Preparation of 3,5-diacetyloxybezoic acid: Reaction Scheme 1-iTo 200 ml of tetrahydrofuran, 15.4 g (0.09 mol) of 3,5-dihydroxybenzoic acid and 38 ml (0.27 mol) of triethylamine were added, and the reaction mixture was stirred for 10 minutes. To the reaction mixture, 23 ml (0.24 mol) of acetic anhydride was added dropwise, and then the resultant mixture was refluxed for 3 hours. The reaction mixture was cooled to room temperature and allowed to evaporate under reduced pressure. Then, dichloromethane and water were added thereto, and the organic layer was washed with water and 1N aqueous HCl solution many times, and allowed to evaporate under reduced pressure. Hexane was added to the remaining oil to form precipitate. The precipitate was filtered under reduced pressure to obtain 15 g (90%) of the target product.
90%
[Example 1] Preparation of 3,5-dihydroxy-N-(4-hydroxyphenyl)benzamide; Preparation of 3,5-diacetyloxybezoic acid:; Reaction Scheme l-i>To 200ml of tetrahydrofuran, 15.4g (0.09mol) of 3,5-dihydroxybenzoic acid and 38ml (0.27mol) of triethylamine were added, and the reaction mixture was stirred for 10 minutes. To the reaction mixture, 23ml (0.24mol) of acetic anhydride was added dropwise, and then the resultant mixture was refluxed for 3 hours. The reaction mixture was cooled to room temperature and allowed to evaporate under reduced pressure. Then, dichloromethane and water were added thereto, and the organic layer was washed with water and IN aqueous HCl solution many times, and allowed to evaporate under reduced pressure. Hexane was added to the remaining oil to form precipitate. The precipitate was filtered under reduced pressure to obtain 15g (90%) of the target product.
88.4%
With pyridine; at 100℃;
Step 1: Acetoxybenzoic Acid A 100 ml three-necked flask with magnetic stirrer, thermometer and condenser was charged with 30 g (0.195 mol) of 3,5-dihydroxybenzoic acid, 47 ml (0.497 mol) of acetic anhydride and 2.4 ml (29.8 mmol) of pyridine. The mixture was heated to 100 C. and kept under stirring for 3-4 hours then cooled down, poured into 400 ml of ice water and filtered. The filter cake was washed with ice water and dried at 45 C. under 1 mbar vacuum. 41 g of 3,5-diacetoxybenzoic acid as a white solid was obtained. The yield was 88.4%.
88.4%
With pyridine; at 100℃;
Step 1: Acetoxybenzoic acid A 100 ml three-necked flask with magnetic stirrer, thermometer and condenser was charged with 30 g (0.195 mol) of 3,5-dihydroxybenzoic acid, 47ml (0.497 mol) of acetic anhydride and 2.4 ml (29.8 mmol) of pyridine. The mixture was heated to 100 C and kept under stirring for 3 to 4 hours then cooled down, poured into 400 ml of ice water and filtered. The filter cake was washed with ice water and dried at 45 C under 1 mbar vacuum. 41 g of 3,5-diacetoxybenzoic acid as a white solid was obtained. The yield was 88.4 %.
72%
With pyridine; dmap; In ethyl acetate; at 0 - 20℃;
A suspension of 3,5-dihydroxybenzoic acid (15.40 g, 0.100 mol) in ethyl acetate (220 mL) wascooled in an ice-bath. Acetic anhydride (24.52 mL, 0.2421 mol), pyridine (16.16 mL, 0.1998 mol) and4-(dimethylamino)pyridine (100 mg, 0.8186 mmol) were added and the reaction stirred at 0 C for60 min and then at room temperature overnight. Formic acid (5.12 mL, 0.1357 mmol) was added andthe reaction poured onto ice (ca. 500 g). The volume was increased by the addition of further ethylacetate (300 mL) and the organic phase separated and washed with water (2 × 200 mL), sat. aq. NaHCO3(100 mL), further water (2 × 200 mL), and then dried and evaporated to a white solid. Recrystallization ofthe product from 5:1 (v/v) EtOAc/hexane (120 mL) gave 2 crops of 3,5-diacetoxybenzoic acid (combinedweight 17.07 g, 72%) as a white powder. Rf 0.20 (1:1 EtOAc/hexane), 0.39 (3:1 EtOAc/hexane); mp161-162 C (lit. [27] mp: 157-159 C); δH (CDCl3) 2.29 (s, 6H, 2 × OAc), 7.18 (pseudo t, 1H,J = 2.1 Hz, 4-H) and 7.70 (pseudo d, 2H, J = 2.1 Hz, 2-H, 6-H); δC (CD3OD) 18.43, 118.94, 119.02,131.70, 150.19, 165.46 and 168.17; m/z (ESI) 261 ([M + Na]+, 100%).
34%
for 6h;Reflux;
A 500 mL round bottom flask was charged with 3,5-dihydroxybenzoic acid (77.00 g,0.50 mol) and acetic anhydride (200 mL). The reaction mixture was heated to reflux, asthe temperature increased the dihydroxy acid gradually dissolved into solution and themixture was left to reflux for 6 h. A brown solution was obtained containing a small amountof insoluble material; the excess acetic anhydride and acetic acid by-product were removedunder reduced pressure, the compound dissolved in refluxing chloroform (200 mL) andfiltered hot. Petroleum ether (70 mL) was then added to the mother liquor, precipitating awhite solid. The mixture was left overnight, and a white product was isolated by filtration,which was thoroughly washed with petroleum ether. Yield: 40.60 g, 34%; 1H NMR (CDCl3) 10.19 (br s, 1H, -COOH), 7.76 (d, 2H, Ar o-CH), 7.22 (t, 1H, Ar p-CH), 2.31 (s, 6H, -CH3);13C NMR (CDCl3, 300 MHz) 170.0 (COOH), 168.8 (C=O), 151.3 (m-Ar), 131.4 (i-Ar), 122.1(p-Ar), 121.0 (o-Ar), 21.0 (CH3); IR (cm1) 3400-2400, 1765 (COOR), 1687 (COOH), 1603;ES-MS, 237 (M+); MP 160-162 C.
Stage #1: 3,5-Dihydroxybenzoic acid; acetic anhydride With sodium hydroxide In water for 0.666667h;
Stage #2: With sulfuric acid; water at 0℃;
10
10. Preparation of 3-acetoxy-5-hydroxybenzoic acid To 3,5-dihydroxybenzoic acid (20 g, 129 mmol) was added NaOH (15.6 g, 390 mmol) in 100 mL water. After the mixture was cooled to 0° C., Ac2O (13.2 g, 129 mmol) was added. The solution was stirred for 40 min and then acidified with 10% H2SO4 at 0° C. The precipitate was filtered and washed with cold water. The crude product was recrystallized from water to give 3-acetoxy-5-hydroxybenzoic acid (15.2 g, 60%). Data are: 1H NMR (Aceton-d6, 300 MHz) δ 8.96 (s, 1H), 7.40 (dd, 1H, J=1.5 Hz), 7.26 (dd, 1H, J=1.5 Hz), 6.86 (t, 1H, 2.1H), 2.27 (s, 3H); HRMS (EI+) found 196.0380 M+, calcd 196.0372 for C9H8O5.
With sodium hydroxide In water at 0℃; for 0.833333h;
2 Step i: 3 -Acetoxy- 5 -hydroxy-benzoic acid (Intermediate Gen-41-a)
Intermediate Gen-44-a: 3-Hydroxy-5-methoxy-benzoic acid methyl ester Step i: 3 -Acetoxy- 5 -hydroxy-benzoic acid (Intermediate Gen-41-a) To a solution of Gen-40-a (4.12 g, 26.7 mmol, 1 eq.) in suspension in water (20 mL) is added NaOH (3.20 g, 80.2 mmol, 3 eq.). The reaction mixture is cooled down to 0°C and acetic anhydride (2.50 mL, 26.7 mmol, 1 eq.) is added slowly. After 50 min, HC1 6N is added until pH 1. Then the mixture is extracted with EtOAc. The organic layer is washed with water, dried over Na2S04, filtered, concentrated to dryness to give Gen-41-a which is used as such in the next step.
With sodium hydroxide In water at 0℃; for 0.833333h;
2.ii 3-Acetoxy-5-hydroxy-benzoic acid (Intermediate Gen-41-a)
To a solution of Gen-40-a (4.12 g, 26.7 mmol, 1 eq.) in suspension in water (20 mL) is added NaOH (3.20 g, 80.2 mmol, 3 eq.). The reaction mixture is cooled down to 0° C. and acetic anhydride (2.50 mL, 26.7 mmol, 1 eq.) is added slowly. After 50 min, HCl 6N is added until pH 1. Then the mixture is extracted with EtOAc. The organic layer is washed with water, dried over Na2SO4, filtered, concentrated to dryness to give Gen-41-a which is used as such in the next step.
1.1 (1) Preparation of intermediate 2 (ie compound 2): 3,5-dimethoxybenzoic acid
Add to 100mL single-mouth flask3,5-dihydroxybenzoic acid (1.54 g, 10 mmol),Dissolve it by adding 20 mL of acetone.Potassium carbonate (4.14 g, 30 mmol) was added at room temperature.At the same time, 3.5 mL of dimethyl sulfate was added dropwise.Heated to 55 ° C,The reaction was refluxed overnight.After the reaction is complete,Concentrate and recover acetone under reduced pressure.Add 30mL of water,Then add a 30% sodium hydroxide solution to adjust the pH to 14,React at 75 ° C for 4 hours,After the hydrolysis is complete, the system is cooled to room temperature.Adjust the pH to about 6 with concentrated hydrochloric acid.There are a lot of white solids,Filtered,Washed,The title compound 2 (1.78 g, yield 98%) was obtained.
98%
With potassium carbonate In acetone at 55℃;
96%
With sodium hydroxide In water
84%
With sodium hydroxide heating;
75%
With sodium hydroxide
With alkali
With potassium hydroxide; ethanol; water
With sodium hydroxide
With sodium hydroxide In water at 50℃; for 6.6h;
1
To prepare a N205 / CH2C12 solution at a concentration of 0. lg / mL, 50 mL of the solution was added to a three-necked flask with a cooling jacket. To the flask was added 0. 5 g of ZBS catalyst. After warming to 70 ° C, Benzene dropwise with constant pressure dropping funnel dropping to the bottle, after the completion of mixing continued stirring 20min, and then adding saturated sodium bicarbonate solution for washing, washing and centrifugal separation to obtain solid is the resorcinol70 parts of resorcinol, 10 parts of anhydrous potassium carbonate and 20 parts of carbon dioxide gas were respectively added to a 10 L autoclave equipped with an electric stirring device, and dimethylformamide was added as a solvent at 100 ° C and 110 MPa After 10 h reaction, the autoclave was cooled down to 30 ° C and depressurized. The material was poured out, The filtrate was evaporated under reduced pressure, and dimethylformamide was distilled off. To the filtrate was added 120 mL of water, stirred and dissolved, and 38% of concentrated hydrochloric acid was added dropwise to adjust the concentration of the solution The pH value was 4, the insoluble matter was removed by filtration, the zinc powder was decolorized in the filtrate, the excess zinc powder and the unreacted resorcin were removed, the aqueous phase was acidified to pH 1 with 40% concentrated hydrochloric acid, ° C, and filtered to give 3,5-dihydroxybenzoic acid. 70 g of 3,5-dihydroxybenzoic acid was added to a 1 L four-necked flask equipped with a stirrer, a thermometer, a constant-pressure dropping funnel and a condenser tube. To the bottle, 150 mL of dimethyl sulfate was added and 200 mL of a 20% aqueous sodium hydroxide solution was added dropwise with stirring And then heated to 50 ° C under stirring conditions continue to reaction 40min, refluxing 2h. then added to the bottle 100mL mass fraction of 40% hydrogen peroxide Then refluxed for 4h, refluxed to room temperature and then acidified to pH 1 with 40% concentrated hydrochloric acid. The mixture was allowed to stand at 0 ° C for 4 hours, filtered under reduced pressure, and the solid was washed twice with ice water Drying at 105 ° C for 1.5 hours to obtain 3,5-dimethoxybenzoic acid; A dding the solid of 3,5-dimethoxybenzoic acid obtained above to a beaker, adding solid-liquid mass The ratio of 1: 1 ethanol to boil, during boiling slowly dropping to the concentration of 90% concentrated sulfuric acid, boiled for 20min after filtration to remove solids, the filtrate was cooled to 0 ° C after filtration solid, with ice Water rinse, rinse placed at 80 ° C under dry 2h, that 3,5 - dimethoxy benzoate
With (S)-N-(E)-3',4',5'-trimethoxy-3-phenylprop-2-enoicserinecyclohexylamide In N,N-dimethyl-formamide for 1h; Reflux;
3.4
To a solution of 3,5-dihydroxybenzoic acid (2.00 g, 13.0 mmol) in DMF were added K2CO3 (27.0 g, 0.195 mol), CH3I (20.2 ml, 0.325 mol) and the mixture was stirred under reflux for 1h. The mixture was filtered and the filtrated solvent was evaporated in vacuo. The residue was dissolved in CH2Cl2, and the organic layer was washed with H2O and then dried over MgSO4. The solvent was evaporated in vacuo and the resulting precipitate was purified by silica-gel column chromatography (hexane:AcOEt=4:1) to give methyl ester (2.11 g, 10.7 mmol). To a solution of methyl ester (2.11 g, 10.7 mmol) in THF was added to LiAlH4 (0.812 g, 21.4 mmol) at 0. After being stirred for 1h at room temperature, H2Oand 1M NaOH added to the solution. The mixture was filtered and the organic layer was evaporated in vacuo to yield alcohol (1.40 g, 8.32 mmol). To a solution of alcohol (1.40 g, 8.32 mmol) in CH2Cl2 was added PDC (4.96 g, 8.32 mmol) and the mixture was stirred at room temperature for 5h. The mixture was filtered and the solvent was evaporated in vacuo. The residue was purified by silica-gel column chromatography (hexane:AcOEt=4:1) to give benzaldehyde (1.25 g, 7.52 mmol). To a solution of benzaldehyde (1.25 g, 7.52 mmol) were added ethyldiethylphosphonoacetate (3.01 ml, 15.4 mmol) and NaH (0.330 g, 8.27 mmol) at 0. After being stirred for 1h at room temperature, AcOEt and H2O were added to the solution. The organic layer was evaporated in vacuo and the residue was purified by silica-gel column chromatography (hexane:AcOEt=4:1) to give unsaturated ethyl ester (0.540 g, 2.03 mmol). To a solution of unsaturated ethyl ester (0.540 g, 2.03 mmol) in MeOH was added LiOH-H2O (1.22 g, 29.1 mmol).After being stirred under reflux for 2h, 1M HCl aq. and AcOEt were added to the mixture. The organic layer was washed with H2O and brine. The solvent was evaporated in vacuo to yield unsaturated carboxyl acid (1.35 g, 6.48 mmol, 89 %) as a white powder.
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
1 Methyl 3,5-dimethoxybenzoate (B)
[0081] 3,5-Dihydroxybenzoic acid (A) (10 gm, 1.0 equiv), potassium carbonate (40.3 gm, 4.5 equiv) and methyl iodide (30.4 gm, 3.3 equiv) were stirred in dimethylformamide (100 mL) overnight at Rt. The reaction wasmonitored by TLC. After completion of the reaction, water (600 mL) was added, and the reaction mixture was extracted with ethyl acetate (3 × 350 mL). The combined ethyl acetate layers were dried over anhydrous MgSO4, filtered, and the solvent was evaporated under reduced pressure to afford methyl 3,5-dimethoxybenzoate (B) (86%). Methyl 3,5-dimethoxybenzoate (B) was used next step without any purification.
1
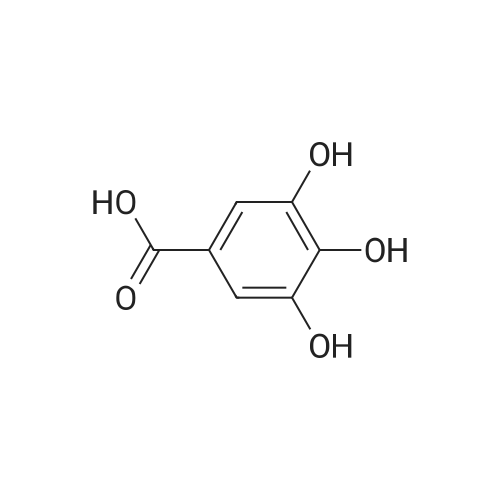
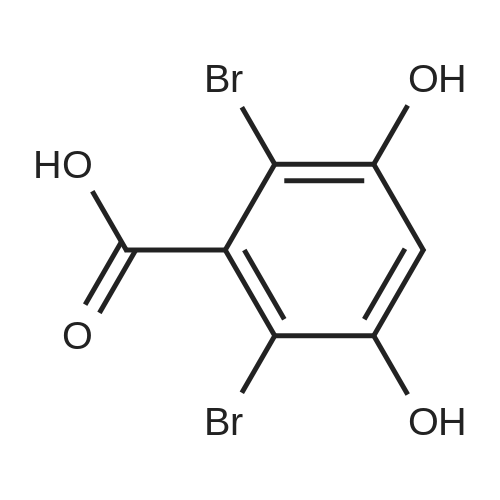
Example 1 4-Formyl-3-hydroxy-8-methoxy-1,9-dimethyl-11-oxo-11H-dibenzo[b,e][1,4]dioxepine-6-carboxylic acid (psoromic acid) (Compound No: F7) 1mmol of 2,4-dihydroxybenzoic acid was taken in 2ml of chloroform, and a solution of 2mmol of bromine in 0.5 ml of chloroform was added from a pressure-equalizing dropping funnel at room tempreture over a period of 1.5 h. A NaOH trap system was provided on top of the addition funnel. The mixture was then stirred for 5 h and filtered. The filter cake was washed with chloroform and then with water. The precipitate was 2,6-Dibromo-3,5-dihydroxybenzoic Acid (97%). 1HNMR (DMSO-d6) δ 6.69 (s, 1H).
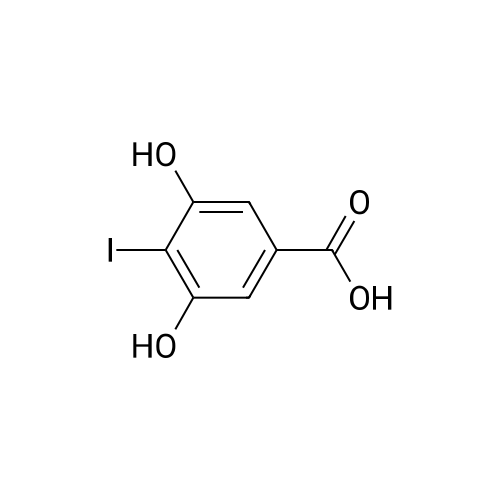
With N-iodo-succinimide In methanol at 1 - 60℃; for 2.5h;
1.a EXAMPLE 1; Preparation of compound (I) wherein R1 is ethyl (key intermediate product-equation 1):5-(3,5-Diethoxy-4-iodo-benzyl)-pyrimidine-2,4-diamine
This compound can be obtained by the following reaction sequence (stages a)-g)): Stage a) 3,5-Dihydroxy-4-iodo-benzoic acid: [00156] 3,5 Dihydroxybenzoic acid (500 g; 3.24 mol) is dissolved in methanol (2,000 ml), while stirring and gassing with argon. After cooling to 1° C., a solution of N-iodosuccinimide (730 g; 3.24 mol) in methanol (2,000 ml) is added dropwise at 1-60° C. in the course of 90 minutes. After one hour the reaction solution is poured on to ice-water (1,500 ml) and a 5% sodium thiosulphate solution in water (500 ml) is added until decolourization has occurred. The mixture is extracted with tert-butyl methyl ether (4,050 ml). The organic phase is then washed twice with saturated aqueous sodium chloride solution (2,000 ml) and the aqueous phases are re-extracted with tert-butyl methyl ether (2×3,240 ml). The combined organic phases are dried with sodium sulphate (160 g), filtered and concentrated. The residue is crystallized from water. Yield: 816 g (90%) as pale beige crystals. M.p. 245-248° C. [00157] MS: 280 (M)
90%
With N-iodo-succinimide In methanol at 0 - 20℃;
4
Example 4 Preparation of 3,5-dihydroxy-4-iodobenzoic Acid (6) A solution of N-iodosuccinimide (5 g; 22.2 mmol; 1.05 eq.) in methanol (15 ml) is added dropwise to a solution of 3,5-dihydroxybenzoic acid (3.26 g; 21.2 mmol; 1 eq.) cooled to 0° C. in methanol (13 ml). The reaction mixture is allowed to stir at room temperature for 3 h. The mixture is poured into cold water (13 ml) and then into a 5% aqueous solution of Na2SO3 (20 ml). The aqueous phase is extracted with ether (3*100 ml). The organic phase is washed with NaCl, dried on MgSO4, filtered and evaporated to yield 5.37 g of a white solid. Yield: 90% Empirical formula: C7H5IO4
68%
With iodine; silver sulfate In sulfuric acid at 55 - 60℃; for 2.5h;
28%
With iodine; sodium hydrogencarbonate In tetrahydrofuran; water at 0 - 20℃; for 52h;
4.2.2. Synthesis of 3,5-dihydroxy-4-iodobenzoic acid (4)
4.2.2. Synthesis of 3,5-dihydroxy-4-iodobenzoic acid (4). 3,5-Dihydroxybenzoate(5.045 g, 30 mmol) was dissolved in THF and H2O(70 mL:70 mL) at 0 C, to which iodine (8.009 g, 31.5 mmol) andNaHCO3 (2.660 g, 31.5 mmol) were added in four portions, respectively,in 4 h. The mixture was stirred for 1 day at room temperature.The mixture of iodine (3.20 g, 12.6 mmol) and NaHCO3(1.064 g, 12.6 mmol) was added and stirred for another 2 days atroom temperature. The mixture was diluted with ether and stirredwith K2SO3 before the red colour disappeared and then extractedwith ether (50 mL3). After evaporation, the residue was recrystallizedfrom methanol to afford 4 as a yellow crystal. Yield: 2.000 g,28%. 1H NMR (600 MHz, DMSO-d6): 10.51 (br s, 2H), 6.93 (s, 2H), 3.79(s, 6H). 13C NMR (150MHz,DMSO-d6): 166.0,158.3,130.6,105.8, 81.8,52.2. MS (EI, m/z): calcd: 279.9; found: 280 (M).
With iodine; sodium hydrogencarbonate
With N-iodo-succinimide
With sodium iodide In aq. phosphate buffer at 30℃; for 0.5h;
2.2 Methods for halogenation assays and productanalysis
The TiaM-catalyzed chlorination of 1 was carried out accordingto our previous report [23]. Halogenation reactionswere carried out in 1.5 mL Eppendorf microtubes at 30 °C ina water bath (Yuhua, China) or an Eppendorf ThermoMixerC (Germany). The enzyme assay mixtures contained 40 μM1, 3.0 μM TiaM, 0.3 μM SsuE (a flavin reductase from E.coli [27]), 0.2 mM FAD, 8 mM NADH, 100 mM NaCl (or0.1 mM to 1 M NaI) in 100 mM potassium phosphate buffer(pH 6.0-8.0) or citrate buffer (pH 3.0-6.0) at a total volumeof 200 μL. After a period of incubation time (ranging from5 min to 3 h), products were extracted by adding 400 μLethyl acetate. The ethyl acetate extracts were evaporated todryness by vacuum. The dried reaction products were dissolvedin 250 μL methanol and subjected to HPLC analysison an Agilent 1260 Workstation. HPLC was carried out usinga reversed phase column (ACE SuperC18, 250×4.6 mm,UK) with UV detection at 270 nm under the following program:solvent system (solvent A, 10% acetonitrile in water supplementing with 0.08% formic acid; solvent B, 90%acetonitrile in water); 5% B to 100% B (linear gradient,023 min), 100% B (2325 min), 100% B to 5% B(2526 min), 5% B (2630 min); flowrate at 1 mL/min.
Step 1: 3-amino-5-hydroxy-benzoic acid hydrochloride A mixture of 3,5-dihydroxybenzoic acid (250.0 g), ammonium chloride (213.0 g), and 28% ammonium hydroxide (750.0 mL) was stirred at 180 C. for 3 days under autoclave. The reaction was cooled to room temperature and then concentrated under reduced pressure. The resulting residue was dissolved in a 6 N hydrochloric acid solution (3.0 L). The reaction mixture was heated at 100 C. for 24 hours and then cooled to 70 C.~80 C. Active carbon (30.0 g) was added to the reaction mixture, which was then filtered with celite pad. The filtrate was concentrated under reduced pressure. The resulting residue was washed with a 6 N hydrochloric acid solution twice and then dried under reduced pressure to obtain 192.0 g of the titled compound (Yield: 62.4%). 1H-NMR (d6-DMSO) delta 7.81 (s, 1H), 7.69 (s, 1H), 7.56 (s, 1H)
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 2h;
100%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 20h;
100%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 24h;
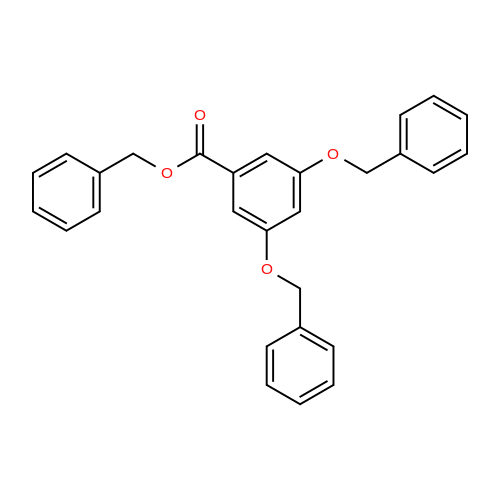
Benzyl Ester 8
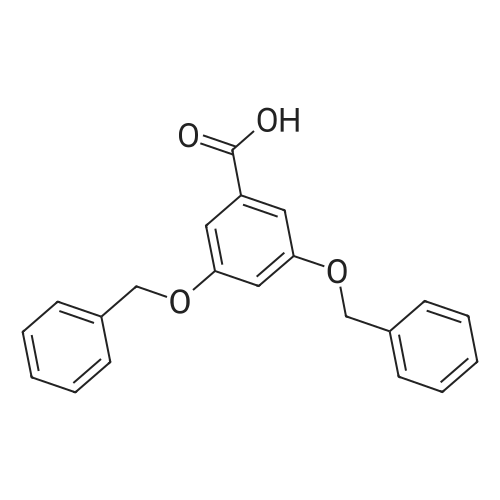
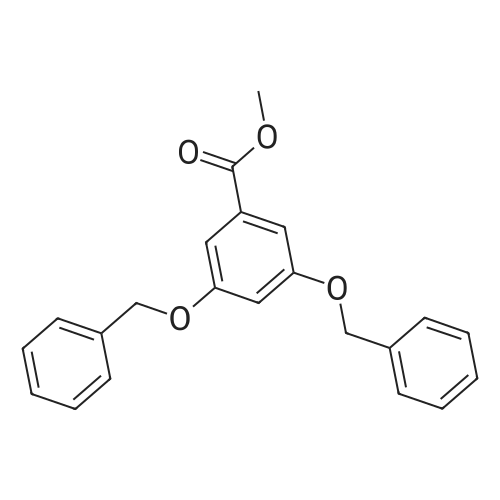
To a solution of 3,5-dihydroxybenzoic acid (7; 2.01 g, 13.0 mmol) inDMF (26 mL) were successively added K 2 CO 3 (10.8 g, 77.9 mmol) andBnBr (4.8 mL, 40 mmol) at rt. After stirring for 24 h at this tempera-ture, the reaction was quenched with 2 M aq HCl at 0 °C. The productswere extracted with Et 2 O (×3) and the combined extracts werewashed with sat. aq NaHCO 3 and brine, and dried (Na 2 SO 4 ). Concen-tration and purification by column chromatography (silica gel, hex-ane/EtOAc 95:5 to 9:1) gave benzyl ester 8 (5.53 g, quant) as a whitesolid; mp 65-66 °C.IR (ATR): 1715, 1596, 1455, 1347, 1297, 1220, 1164, 1030, 771, 696cm -1 .1 H NMR (CDCl 3 , 500 MHz): = 7.30-7.44 (m, 17 H), 6.80 (t, J = 2.2 Hz,1 H), 5.34 (s, 2 H), 5.06 (s, 4 H).13 C NMR (CDCl 3 , 125 MHz): = 166.1, 159.7, 136.4, 135.9, 132.0,128.61, 128.57, 128.2, 128.13, 128.11, 127.6, 108.5, 107.2, 70.3, 66.8.HRMS (ESI): m/z [M + Na] + calcd for C 28 H 24 NaO 4 : 447.1572; found:447.1565.
97%
With potassium carbonate In acetone for 24h; Reflux; Inert atmosphere;
81%
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 18h;
19%
With potassium carbonate In acetone Reflux;
With 18-crown-6 ether; potassium carbonate In acetone for 24h; Heating;
With potassium carbonate In acetone for 72h; Heating;
With sodium hydroxide
With potassium carbonate In N,N-dimethyl-formamide at 60℃; for 14h;
With potassium carbonate
With potassium carbonate In acetone
With caesium carbonate In acetonitrile for 16h; Reflux; Inert atmosphere;
4.1.2.1. 3-Benzyloxybenzyl bromide (3c)
General procedure: 3-Hydroxybenzaldehyde (5.00 g, 0.041 mol) was dissolved in anhydrous acetonitrile (130 mL) under argon atmosphere. Cesium carbonate (20.01 g, 0.061 mol) was added and the suspension stirred for 5 min. Benzyl bromide (11.69 mL, 0.102 mol) was then added and the solution heated at reflux for 16 h. The solution was concentrated on rotary evaporator, water was added and the mixture was extracted with EtOAc. The organic phase was washed twice with water, once with brine, dried over MgSO4, filtered and concentrated. Water was added and the mixture was extracted with CH2Cl2. The crude product was purified by flash chromatography on silica gel with hexanes/EtOAc (80/20) to yield 3a as a white solid (8.59 g).
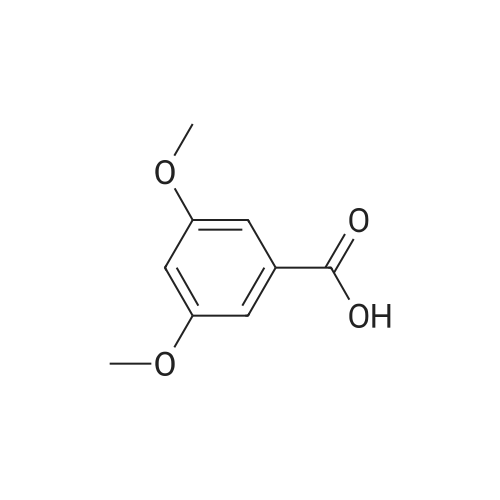
To 3,5-dihydroxybenzoic acid (15.4 g, 0.1 mol) dissolved in acetone (150 mL) was added potassium carbonate (42.8 g, 0.3 mol,3.1 equiv.) followed by dimethyl sulphate (19 mL, 2 equiv.). The mixture was then heated to reflux for 4 h with vigorous stirring. Upon cooling to room temperature, water was added so as to completely dissolve all the potassiumcarbonate and the mixture extracted with diethyl ether (3 x 30 mL). The combined organic extracts were dried over magnesium sulphate anhydrous andconcentrated under reduced pressure to afford the title compound without the needfor further purification. Yield 98%, Beige solid; Mp 43-44 °C {lit. [i]42-44 °C}; TLC (EtOAc/Hexane, 1:4 v/v): Rf = 0.38; 1H-NMR[CDCl3, 600 MHz] δ: 7.16 (d, J= 2.3 Hz, 2H), 6.62 (t, J = 2.3 Hz,1H), 3.89 (s, 3H), 3.80 (s, 6H); 13C-NMR [CDCl3, 150 MHz]δ: 166.9, 160.7, 132.1, 107.2, 105.8, 55.6, 52.3
96%
With potassium carbonate In acetone for 4h; Heating;
96%
With potassium carbonate In acetone for 4h; Reflux;
93.6%
With potassium carbonate In acetone for 3h; Heating;
93%
With potassium carbonate In acetone for 3h; Heating;
93.6%
With potassium carbonate In acetone for 3h; Reflux;
92%
With potassium carbonate In acetone for 4h; Inert atmosphere; Reflux;
92%
Stage #1: 3,5-Dihydroxybenzoic acid In acetone at 40℃; for 0.25h;
Stage #2: With potassium carbonate at 60℃; for 0.166667h;
Stage #3: dimethyl sulfate In acetone at 60 - 80℃; for 6h;
91%
With potassium carbonate In acetone for 4h; Reflux;
91%
Stage #1: 3,5-Dihydroxybenzoic acid With potassium carbonate In acetone at 40 - 60℃; for 0.416667h;
Stage #2: dimethyl sulfate In acetone Reflux;
90%
With potassium carbonate In acetone for 4h; Heating;
90.7%
With potassium carbonate In acetone for 4.5h; Reflux;
1 Synthesis of methyl 3,5-dimethoxybenzoate 2
14.231g (0.092mol) 3,5- dihydroxybenzoic acid 1,Potassium carbonate 26.021g (0.189mol) in 500mL acetone and 100mL two-necked flask,With uniform constant pressure dropping funnel was slowly added dropwise 17mL dimethyl sulfate;About 30min after the start of heating drops,Reflux 4h;TLC tracking and detection,Completion of the reaction was cooled to room temperature;filter,The combined filtrate and filter residue was washed with acetone,Dried over anhydrous sodium sulfate,The solvent was distilled off under reduced pressure,dry,To give compound 2;90.7% yield,
87%
With potassium carbonate In acetone at 60℃; for 4.5h;
85%
With potassium carbonate In acetone Reflux;
Methyl 3,5-dimethoxybenzoate (3)
A two-necked, round-bottomed flask (3 L) equipped with mechanical stirrer and reflux condenser was charged with 3,5-dihydroxybenzoic acid (70.7 g, 0.5 mol), freshly pulverized anhydrous K2CO3 (200 g, 1.4 mol) and anhydrous acetone (1 L). An addition funnel, containing dimethylsulfate (150 mL, 1.58 mol), was connected at the top of the condenser. The mixture was stirred under reflux while the addition was made in a gentle stream over 2 h. After refluxing overnight the reaction was not yet complete. A further addition of freshly pulverized K2CO3 (100 g, 0.72 mol) followed by dimethylsulfate (75 mL, 0.79 mol) was added. After refluxing 2 h, the reaction was then complete. The mixture was partitioned between H2O (2 L) and Et2O (2 L). The organic layer was washed with H2O (2 X 1L) then washed with saturated aqueous NaHCO3 (1 L) and then washed with brine (1 L). After drying over anhydrous MgSO4, the solvent was rotary evaporated leaving a crude solid. Reduced pressure distillation (0.1 torr) gave compound 3 as a colorless oil that solidified to a white, low melting solid (83 g, 85%). 1H NMR (CDCl3): δ 7.17 (d, J = 2.4 Hz, 2H), 6.63 (t, J = 2.4 Hz, 1H), 3.88 (s, 3H), 3.81 (s, 6H); 13C NMR (CDCl3): δ 166.91, 160.73, 132.08, 107.18, 105.79, 55.59, 52.27. Elemental analysis calculated for C10H12O4: C, 61.22; H, 6.16. Found: C, 61.00; H, 6.09.
84%
Stage #1: 3,5-Dihydroxybenzoic acid With potassium carbonate In acetonitrile at 40℃; for 0.5h;
Stage #2: dimethyl sulfate for 5h; Reflux;
1.16 g
With potassium carbonate In acetone for 4h; Heating;
With potassium carbonate
With sodium hydroxide In water Reflux;
With potassium carbonate In acetone for 4h; Reflux;
With sodium tetrahydroborate; Trimethyl borate; dimethyl sulfate; In tetrahydrofuran; at 20℃; for 5.5h;
step 1,Under the protection of nitrogen,Dimethyl sulfate (2 kg) was added dropwise to a solution of sodium borohydride (615 g) in tetrahydrofuran (1 kg) at 0 C for 1 hour.Stirring at 0 C for 1 hour, then stirring at room temperature for 4 hours until the mixture was free of gas;Further, a solution of 3,5-dihydroxybenzoic acid (1 kg) and trimethyl borate (2 kg) in tetrahydrofuran (1 kg) was added dropwise thereto, and the mixture was added dropwise for 0.5 hour, and stirred at room temperature for 5 hours;After the reaction is over, keep the system at 0 C.Deionized water (1 kg) was added thereto and stirred for 0.5 hours.Tetrahydrofuran was distilled off at 50 C using a rotary evaporator, followed by extraction with ethyl acetate (2 kg).Then, wash back three times with saturated sodium bicarbonate solution (2 kg each time), and wash the backwash 3 times with saturated brine (2 kg each time).Finally, it was dried and concentrated at 45 C using a rotary evaporator.Obtained white solid B (770 g), yield 85%
84%
The reaction should be carried out under nitrogen, keeping the reaction system as dry as possible.5 g of sodium borohydride (132 mmol)And 54.5 gEthylene glycol dimethyl ether was added to the reactor, and stirred to uniformly disperse sodium borohydride in the ethylene glycol dimethyl ether solution.(44 mmol) of 3,5-dihydroxybenzoic acid was dissolved in 34 g of ethylene glycol dimethyl ether solution,This was slowly added dropwise to a solution of sodium borohydride in ethylene glycol dimethyl ether (dropwise over an hour)Keep the temperature between 25 - 30 C. After completion of the dropwise addition, stirring was continued at room temperature for 1.5 hours.then,To the reaction system, 25 g (172 mmol) of boron trifluoride diethyl ether solution was added dropwise slowly,It was also added for approximately one hour and maintained at a temperature between 25 and 30 C.After completion of the dropwise addition, the reaction was carried out at room temperature for 6 hours. then,The temperature was raised to 35 C. 44.3 g of n-butanol was added to the reaction mixture. After stirring for half an hour, the temperature was lowered and the mixture was filtered. The filtrate was removed under reduced pressure at 70/30 mmHg to give 5.3 g of product in about 84% yield. This product was used in the next step without further purification.
75%
With Trimethyl borate; dimethylsulfide borane complex; In tetrahydrofuran; for 24h;Reflux;
To the stirring BH^-MeiS (lOml, 2M solution) and B(OMe)3 in THF was slowly added a solution of 3,5-dihydroxy benzoic acid (l .5g, leq) in THF at RT. After addition, reaction was refluxed for 24 hours. Upon completion, reaction was quenched with methanol. Solvent was removed under reduced pressure. Methanol was again added to the residue and removed under reduced pressure to get crude product which was then purified using silica gel column chromatography using acetone / chloroform to afford 3,5-dihydroxybenzyl alcohol (1 02g, 75%) as a white solid.
Stage #1: 3,5-Dihydroxybenzoic acid With sodium hydroxide; hydrogen In water at 60℃; for 120h;
Stage #2: With hydrogenchloride In water at 0℃;
II.A
A. S-Hydroxy-δ-oxocyclohex-S-ene-i -carboxylic acid50.0 g (0.32 mol) 3,5-dihydroxybenzoic acid and 10.0 g Raney-Nickel were suspended in 175 ml 4N aqueous sodium hydroxide and hydrogenated for 5 d (60 0C, 100-150 bar hydrogen). The mixture was filtered through Celite and the filtrate was acidified with cone, hydrochloric acid at 0 0C. The resulting precipitate was collected, washed with ice-cold water and dried to give 25.2 g (50 %) of the title compound.
With potassium hydrogencarbonate In N,N-dimethyl-formamide at 20℃; for 4h;
11.52 Step 52
3,5-Dihydroxybenzoic acid 86 (manufactured by Tokyo Chemical Industry Co., Ltd., 2.11 g, 13.69 mmol) was dissolved in N,N′-dimethylformamide (35 mL). To the solution, potassium bicarbonate (1.716 g, 17.14 mmol) and benzyl bromide (3.51 g, 2.439 mL, 20.54 mmol) were added, and the mixture was stirred at room temperature for 4 hours. Saturated ammonium chloride was added to the reaction solution, followed by extraction with dichloromethane. Then, the organic layer was washed with water and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (heptane/ethyl acetate=50/50) to quantitatively obtain compound 87.ESI-MS m/z: 245 (M+H)+
99%
With potassium hydrogencarbonate In N,N-dimethyl-formamide at 40℃; Inert atmosphere;
95%
With potassium hydrogencarbonate In N,N-dimethyl-formamide at 40℃;
89.3%
With sodium carbonate In N,N-dimethyl-formamide at 25℃; for 12h; Inert atmosphere;
84%
With sodium hydrogencarbonate; 3-butyl-1-methyl-1H-imidazol-3-ium hexafluorophosphate at 40℃; for 0.583333h;
76%
With sodium carbonate In N,N-dimethyl-formamide at 20℃;
76%
With potassium hydrogencarbonate In N,N-dimethyl-formamide at 40℃; for 5h; Inert atmosphere;
1 [Production of Compound (A-3)]
Under an argon atmosphere, 3,5-dihydroxybenzoic acid (4.00 g, 26.0 mmol, 1 eq.)And a solution prepared by suspending potassium hydrogen carbonate (3.11 g, 31.1 mmol, 1.2 eq.) In 30 mL of dry N, N-dimethylformamide (DMF) was placed in a 200 mL two-necked flask .Benzyl bromide (5.34 g, 3.7 mL, 31.2 mmol, 1.2 eq.) Was then slowly added and reacted at 40 ° C. with stirring for 5 h.The mixture was then quenched with water to quench the reaction and extracted with ethyl acetate.The organic layer was then washed with saturated sodium bicarbonate solution, water, and brine and dried over anhydrous sodium sulfate.Subsequently, the obtained crude composition was concentrated under reduced pressure and allowed to stand to crystallize.Then, recrystallization from toluene gave Compound (A-1) as a white solid (Yield 4.82 g, 19.7 mmol, yield 76%).
73%
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
With sodium hydrogencarbonate In N,N-dimethyl-formamide
A magnetically stirred suspension of resorcylic acid (616 mg, 4.0 mmol) and K2CO3 (2.76 g, 20 mmol) in DMF (15 imL) was treated with benzyl bromide (0.96 imL, 16 mmol) and stirring continued for 3 h. The reaction volume was diluted with EtOAc (75 imL), washed with water (3 x 50 imL), brine (100 imL) and the organic phase dried (MgSO4), filtered and concentrated under reduced pressure. The resultant ester was used immediately in step ii. magnetically stirred solution of the benzyl ester (1 .27 g, 3.0 mmol) in MeOH (10 imL) was treated with NaOH (10 mL of a 2 M aq. solution) and brought to reflux for 1 h. The resultant reaction volume was concentrated under reduced pressure and washed with ether (50 mL). The aq. phase was acidified to pH 2 (HCI, 4M aq. solution) and extracted with CH2CI2 (2 x 25 mL). The combined chlorinated layers were washed with brine (100 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give 3,5-Bis(benzyloxy)benzoic acid (1 g, quant.) as colourless crystals. The spectral data for this compound, is identical in all aspects with those previously reported in Fuse S, Otake Y, Mifune Y, Tanaka H: A Facile Preparation of alpha-Aryl Carboxylic Acid via One-Flow Arndt-Eistert Synthesis. Aust J Chem 2015, 68(1 1 ):1657-1661 (a copy of which is hereby incorporated by reference).
(3S)-3-([{(9H-fluoren-9-yl)methoxy}carbonyl]amino)-2-hydroxy-4-phenylbutanoic acid[ No CAS ]
Fmoc-Leu-OHFmoc-Val-OH[ No CAS ]
3-[(2R,3S)-3-((S)-2-{(S)-2-[(S)-2-Amino-3-(3,5-dihydroxy-benzoylamino)-propionylamino]-3-methyl-butyrylamino}-4-methyl-pentanoylamino)-2-hydroxy-4-phenyl-butyrylamino]-benzoic acid[ No CAS ]
With caesium carbonate In N,N-dimethyl-formamide at 23℃; for 1h;
93%
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 12h; Inert atmosphere;
3 Example 3 Preparation of compound of formula S2
3,5-Dihydroxybenzoic acid (5.0 g, 32.4 mmol)Was dissolved in N, N-dimethylformamide (100 ml).Cesium carbonate (53.0 g, 162 mmol) was further added thereto,After stirring at room temperature for 5 minutes,Allyl bromide (11.2 ml, 129.6 mmol)Slowly added dropwise to the reaction system.Under nitrogen, the reaction was carried out for 12 hours at room temperature.After the reaction was completed, it was filtered through celite and the filtrate was collected.Concentration under reduced pressure removed most of the N, N-dimethylformamide, and the remaining sample was dissolved in ethyl acetate (50 ml) and washed three times with 50 mL each.The organic phase is washed once more with saturated brine (50 ml).The organic phase was collected and concentrated under reduced pressure to give the compound of formula S2 (8.28 g, yield 93%) as a light yellow oil.
92%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 3h; Inert atmosphere;
3,5-Bis(allyloxy)benzoic acid (14).
To a stirred suspension of 3,5-dihydroxybenzoic acid (13) (0.2 g, 1.30 mmol) and K2CO3 (0.90 g, 6.49 mmol) in anhydrous DMF (8 mL) was added allyl bromide (0.34 mL, 3.80 mmol) slowly under nitrogen atmosphere at room temperature. The reaction mixture was stirred for 3 h. After completion of the reaction, filtered through Celitepad and washed with ether (30 mL). The filtrate was washed with water (3 × 15 mL), brine (3 × 15 mL), dried over anhyd. Na2SO4 and concentrated in vacuo. The crude was purified by column chromatography (EtOAc/hexane = 1/2) to yieldallyl 3,5-bis(allyloxy)benzoate (0.33 g, 92%) as colorless liquid. R f = 0.80 (EtOAc/hexane = 1/2);1H NMR(300 MHz, CDCl 3 ) δ 7.20 (2H, d, J = 2.4 Hz), 6.68 (1H, t,J = 2.4 Hz), 6.02 (3H, m), 5.40 (3H, dd, J = 17.1, 1.5 Hz),5.28 (3H, dd, J = 10.2, 1.5 Hz), 4.80 (2H, dt, J = 5.4,1.5 Hz), 4.54 (4H, dt, J = 4.8, 1.5 Hz);13C NMR(75 MHz, CDCl 3 ) δ 166.0, 159.7, 133.0, 132.3, 132.1,118.4, 118.1 108.5, 107.3, 69.4, 66.0.
92%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 3h; Inert atmosphere;
3,5-bis (allyloxy) benzoic acid{3,5-Bis (allyloxy) benzoicacid} (Compound14)
To a suspension of3,5-dihydroxybenzoic acid (Compound13) (0.2 g, 1.298 mmol) and K2CO3(0.897 g, 6.489 mmol) in anhydrous DMF (8 mL) was added allyl bromide (0.34 mL, 3.793 mmol) was gradually added to the room temperature in a nitrogen atmosphere.The reaction mixture was stirred for three hours.After completion of the reaction, the reaction mixture was filtered through a Celite pad and washed with ether (30 mL).The filtrate was washed with water (3 x 15 mL) and brine (3 x 15 mL), dried over anhydrous Na2SO4and concentrated in vacuo.The crude compound was purified by column chromatography (EtOAc / hexane = 1/2) to obtain colorless liquid allyl 3,5-bis (allyloxy) benzoate (0.327 g, 92%).The above ester (0.175 g, 0.638 mmol)l) was dissolved in methanol (5 mL) and stirred. 1.0 N NaOH (1.91 mL) was added and refluxed for two hours.The mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The approximate pH was adjusted to 2 with 1N HCl and extracted with EtOAc (2 x 25 mL). spiritThe combined organic solvent layers were washed with brine (2 x 30 mL), dried over anhydrous N2SO4 and concentrated in vacuo. An unrefined white solid acid compound 14 (0.127 g, 85%) was obtained which was used in the next step without further purification.
92%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 3h; Inert atmosphere;
3,5-Dihydroxybenzoic acid (Compound 13) (0.2 g, 1.298 mmol) and Allyl bromide (0.34 mL, 3.793 mmol) was added slowly to a suspension of K2CO3 (0.897 g, 6.489 mmol) in anhydrous DMF (8 mL) at room temperature under a nitrogen atmosphere.The reaction mixture was stirred for three hours.After completion of the reaction, the reaction mixture was filtered through a Celite pad and washed with ether (30 mL). The filtrate was washed with water (3 x 15 mL) and brine (3 x 15 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude compound was purified by column chromatography (EtOAc / hexane = 1/2) to obtain colorless liquid allyl 3,5-bis (allyloxy) benzoate (0.327 g, 92%).
92%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 3h; Inert atmosphere;
3,5-Dihydroxybenzoic acid (Compound 13) (0.2 g, 1.298 mmol) and K2CO3(0.897 g, 6.489 mmol) in anhydrous DMF (8 mL) was added allyl bromide (0.34 mL, 3.793 mmol) slowly to the room temperature under a nitrogen atmosphere.The reaction mixture was stirred for three hours. After completion of the reaction, the mixture was filtered through a pad of Celite (R) and washed with ether (30 mL). The filtrate was washed with water (3 x 15 mL) and brine (3 x 15 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude compound was purified by column chromatography (EtOAc / hexane = 1/2) to give a colorless liquidAllyl 3,5-bis (allyloxy) benzoate (0.327 g, 92%) was obtained.
80%
With potassium carbonate In N,N-dimethyl-formamide at 22 - 57℃; for 2.25h; Inert atmosphere;
1a Example 1 a: Compound 2, 3,5-diallyloxy allylbenzoate.
3, 5-di hydroxy benzoic acid (1 ) (100 g, 0.65 mol) was dissolved in DMF (500 ml), under nitrogen, in a 2 L reactor stirred with a 4 blade stainless steel stirrer. Potassium carbonate (345 g, 2.5 mol, Fw= 138.2 g/mol) was added by spoon, trying to avoid caking. The temperature rose from 22 to 34 °C and the mixture got thick. The reactor was fitted with a spiral condenser and the jacket temperature was set to 50 °C. Allyl bromide (344 g, 246 ml, 2.85 mol, d=1 .40 g/ml, Mw=121 g/mol) was filled in a 250 ml pressure equalizing dropping funnel and fitted to the reactor. When the inner temperature had reached 47 °C, addition was started with 25 ml portions for the first 150 ml and then 50 ml portions. The inner temperature was allowed to peak (50 to 57 °C and fall back a degree or to 55 °C before the next addition. Total addition time 2 h 15 min. The reaction mixture was left stirring with a jacket temperature of 50 °C until next day. The slurry was removed through the bottom valve (washed out with 60 ml DMF) and collected in a 1 L bottle and passed through a glass fiber filter on a Buchner funnel. The cake was washed with 3 x 60 ml DMF. The reactor was cleaned with water and the DMF solution was reintroduced. The reactor was fitted with a distillation head with an efficient vertical spiral condenser. The pressure was reduced in the reactor (central vacuum, about 20 mbar) and the DMF was distilled off at a brisk pace with a jacket temperature gradually increased from 70 to 85 °C, liquid temperature from 53- 75 °C and a vapor temperature of 50-53 °C. The byproduct allyl potassium carbonate precipitates as an oil as the DMF is removed. The residue was drained through the bottom valve while still warm and collected in a 500 ml RB flask. The allyl carbonate solidifies to lumps when left in the fridge overnight. The oil/solids were transferred to 4 x 50 ml centrifuge tubes and centrifuged. The supernatant product was decanted. Yield 142 g, 518 mmol, 80 %.1H NMR (400 MHz, CDCI3): δ = 7.23 (s, 2H), 6.70 (s, 2H), 6.05 (m, 3H), 5.42 (m, 3H), 5.30 (m, 3H), 4.81 (d, J = 5.58 Hz, 2H), 4.55 (d, J = 5.20 Hz, 4H).
23.1 g (87%)
With potassium carbonate In chloroform; N,N-dimethyl-formamide
78.a a.
a. 2-Propenyl 3,5-bis-(2-propenyloxy)-benzoate 3,5-Dihydroxybenzoic acid (15.0 g, 97.3 mmol) and potassium carbonate (44.0 g, mol) were mixed in N,N-dimethylformamide (60 ml) and heated to 80° C. Allyl bromide (42.0 g, 0.35 mol) was added dropwise during 1 h and efficient stirring was continued for 24 h. The reaction mixture was then evaporated to a semisolid residue, which was treated with chloroform (500 ml) and filtered. The filtrate was washed with water (3*150 ml), dried (Na2SO4) and filtered through a short column of alumina. Evaporation of the solvent gave 23.1 g (87%) of the product as a colorless oil. 1H NMR (CDCl3): 7.22 (d, 2H, J=2 Hz), 6.69 (t, 1H, J=2 Hz), 5.95-6.12 (m, 3H), 5.34-5.47 (m, 3H), 5.23-5.33 (m, 3H), 4.80 (d, 2H, J=5 Hz), 4.55 (d, 4H, J=5 Hz).
23.1 g (87%)
With potassium carbonate In chloroform; N,N-dimethyl-formamide
78.a a.
a. 2-Propenyl 3,5-bis-(2propenyloxy)benzoate 3,5-Dihydroxybenzoic acid (15.0 g, 97.3 mmol) and potassium carbonate (44.0 g, mol) were mixed in N,N-dimethylformamide (60 ml) and heated to 80° C. Allyl bromide (42.0 g, 0.35 mol) was added dropwise during 1 h and efficient stirring was continued for 24 h. The reaction mixture was then evaporated to a semisolid residue, which was treated with chloroform (500 ml) and filtered. The filtrate was washed with water (3*150 ml), dried (Na2SO4) and filtered through a short column of alumina. Evaporation of the solvent gave 23.1 g (87%) of the product as a colorless oil. 1H NMR (CDCl3): 7.22 (d, 2H, J=2 Hz), 6.69 (t, 1H, J=2 Hz), 5.95-6.12 (m, 3H), 5.34-5.47 (m, 3H), 5.23-5.33 (m, 3H), 4.80 (d, 2H, J=5 Hz), 4.55 (d, 4H, J=5 Hz).
With potassium carbonate In N,N-dimethyl-formamide at 60℃; for 3h;
3,5-bis(allyloxy) benzoic acid
3,5-dihydroxybenzoic acid (1.54 g, 10 mmol) was dissolved inDMF (30 mL). Powdered K2CO3 (8.3 g, 60 mmol, 6 eq.) and allyl bromide (5.2 mL, 60 mmol, 6eq.) were added and the formed suspension was heated to 60°C and vigorously stirred for 3hours. Then, the flask was cooled to RT and the reaction was transferred to a separationfunnel using 250 mL ether and 100 mL H2O. Aqueous phase was dropped and the organicphase was washed again with 100 mL H2O. Combined aqueous phases were extracted with100 mL ether, the organic phases were combined and further washed with brine (2x100 mL).Organic phase was dried (MgSO4), filtered and evaporated to dryness. Hydrolysis of theallylic ester was performed by dissolving the obtained oily residue in dioxane:H2O 4:1v/v (50mL) and adding NaOH 4N (5 mL, 20 mmol, 2 eq.). Flask was heated to 80°C and stirred for 1hour and then cooled to RT. The solution was acidified to pH 2-3 with a solution of HCl 1Nand the dioxane was evaporated. The remaining aqueous solution was transferred to aseparation funnel with ethyl acetate (250 mL) and this organic phase was washed with HCl 1N (100 mL), brine (100 mL), dried (MgSO4), evaporated to dryness and dried under highvacuum. The product was obtained as an off-white solid in 90% yield (2.1 g).1H-NMR (CDCl3): δ 7.27 (d, J = 2.4 Hz, 2H, arom H), 6.75 (t, J = 2.4 Hz, 1H, arom H), 6.01-6.10(m, 2H, vinylic H), 5.43 (dd, J = 17.3 Hz, 1.6 Hz, 2H, trans vinylic H), 5.31 (dd, J = 10.5 Hz, 1.5Hz, 2H, cis vinylic H), 4.57 (dt, J = 5.3 Hz, 1.6 Hz, 4H, allylic H); 13C-NMR (CDCl3): δ 171.7,159.8, 132.9, 131.2, 118.2, 108.8, 108.2, 69.3.
(2R*,4S*)-2-benzyl-N-(4-quinolylmethyl)-N-trifluoroacetyl-4-piperidinamine[ No CAS ]
[ 99-10-5 ]
[ 68641-49-6 ]
(2R*,4S*)-2-benzyl-1-(3,5-dihydroxybenzoyl)-N-(4-quinolylmethyl)-N-trifluoroacetyl-4-piperidinamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
With triethylamine;
(2R*,4S*)-2-Benzyl-1-(3,5-dihydroxybenzoyl)-N-(4-quinolylmethyl)-N-trifluoroacetyl-4-piperidinamine 200 mg (0.467 mmol) of (2R*,4S*)-2-benzyl-N-(4-quinolylmethyl)-N-trifluoroacetyl-4-piperidinamine are reacted in analogy to Example 4a with 87 mg (0.562 mmol) of 3,5-dihydroxybenzoic acid, 143 mg (0.561 mmol) of bis-(2-oxo-3-oxazolidinyl)phosphinic chloride and 144 mul (1.03 mmol) of triethylamine. The title compound (99 mg, 38%) is obtained as white foam. TLC: methylene chloride/methanol/conc. ammonia (700:50:1) Rf =0.41, FD-MS: M+ =563.
EXAMPLE 2 Preparation of 3,5-Diacetoxybenzoic Acid from 3,5-Dihydroxybenzoic Acid (Branching Monomer) 3,5-Dihydroxybenzoic acid (446.42 g, 2.89 moles) was charged to a 2000 mL round bottom 3-neck flask with a Friedrich condenser and thermocouple under a blanket of nitrogen. Acetic arkhydride (1515 g, 14.85 moles) was added slowly. The suspension was stirred and refiuxed (-135 C.) for 3 hours. After cooling, the cream solid was filtered off and washed with toluene. The acetic anhydride was removed under reduced pressure 2 more times to yield additional cream solids. Yield of 3,5-diacetoxybenzoic acid (347.02 g, 50%, HPLC 97.5% purity). DSC m.p. 155 C. 1H NMR (400 MHz, d6 -DMSO) δ 13.31, 7.59, 7.28, 2.29. 13C NMR (100 MHz, d6 -DMSO)δ 169.20, 166.01, 151.15, 133.12, 120.53, 120.32, 20.70 MS (DIP) m/e 238 (M+).
50%
With acetic anhydride;
EXAMPLE 2 Preparation of 3.5-Diacetoxybenzoic Acid from 3.5-Dihydroxybenzoic Acid (Branching Monomer) 3,5-Dihydroxybenzoic acid (446.42 g, 2.89 moles) was charged to a 2000 mL round bottom 3-neck flask with a Friedrich condenser and thermocouple under a blanket of nitrogen. Acetic anhydride (1515 g, 14.85 moles) was added slowly. The suspension was stirred and refluxed (~ 135 C.) for 3 hours. After cooling, the cream solid was filtered off and washed with toluene. The acetic anhydride was removed under reduced pressure 2 more times to yield additional cream solids. Yield of 3,5-diacetoxybenzoic acid (347.02 g, 50%, HPLC 97.5% purity). DSC m.p. 155 C. 1H NMR (400 MHz, d6 -DMSO) δ 13.31, 7.59, 7.28, 2.29. 13C NMR (100 MHz, d6 -DMSO)δ 169.20, 166.01, 151.15, 133.12, 120.53, 120.32, 20.70 MS (DIP) m/e 238 (M+).
With hydrogenchloride; sulfuric acid; In acetic anhydride;
EXAMPLE 7 6-Amino-5-(3,5-diacetoxyphenyl)carboxyamido-3-methyl-1-phenyluracil 3,5-Dihydroxybenzoic acid (7.2 g, 50 mM) was dissolved in 11 ml of acetic anhydride. To the resulting solution were added 5 drops of sulfuric acid, followed by stirring at room temperature for 20 minutes. Subsequently, 10 g of ice was added and 12 ml of 1N hydrochloric acid was then added. After sonication, a precipitated crystal was recovered by filtration. Washing the crystal with toluene gave 3,5-diacetoxybenzoic acid (10.23 g).
With pyridine; formic acid; acetic anhydride; In ethyl acetate;
Example 10 3,5-Diacetoxybenzoic acid (10). A suspension of 3,5-dihydroxybenzoic acid in ethyl acetate was cooled in an ice-bath. Acetic anhydride and pyridine were added and the reaction allowed proceeding for 60 minutes. The homogenous solution was stirred overnight at room temperature. Formic acid was added and the solution then poured onto ice-water. Further ethyl acetate was added and the organic phase separated and successively washed twice each with sat. NaHCO3 and water, then dried, filtered and rotary evaporated to give a white solid. Recrystallization from EtOAc/hexane produced 3,5-diacetoxybenzoic acid as a white powder. Rf 0.20 (1:1 hexane/EtOAc), 0.39 (3:1 hexane/EtOAc); mp 161-162 C., lit mp: 157-159 C. Turner et al, Macromolecules, 1993, 26, 4617-4623; 1H NMR (CDCl3): δ 2.29 (s, 6H, 2*OAc), 7.18 (pseudo t, 1H, J=2.1 Hz, H4), 7.70 (pseudo d, 2H, J=2.1 Hz, H2, H6); 13C JMOD NMR (CD3OD): δ 18.43 (2*CH3), 118.94 (C4), 119.02 (C2,6), 131.70 (C1), 150.19 (C5), 165.46 (COOH), 168.17 (2*OCOCH3); LRESI positive ion mass spectrum; m/z 261 (MNa+, 100%).
With pyridine; dmap; formic acid; acetic anhydride; sodium hydrogencarbonate; In water; ethyl acetate;
3,5-Diacetoxybenzoic acid A suspension of 3,5-dihydroxybenzoic acid (15.40 g, 0.100 mol) in ethyl acetate (220 mL) was cooled in an ice-bath. Acetic anhydride (24.52 mL, 0.2421 mol), pyridine (16.16 mL, 0.1998 mol) and 4-(dimethylamino)pyridine (100 mg, 0.81855 mmol) were added and the reaction stirred at 0 C. for 60 minutes and then at room temperature overnight. Formic acid (5.12 mL, 0.1357 mmol) was added and the reaction poured onto ice (ca. 500 g). Further ethyl acetate (300 mL) was added and the organic phase separated and washed with water (2*200 mL), sat. aq. NaHCO3 (100 mL), further water (2*200 mL), and then dried and evaporated to a white solid. Recrystallization from 5:1 EtOAc/hexane (120 mL) gave 2 crops of 3,5-diacetoxybenzoic acid (combined weight 17.07 g, 72%) as a white powder. Rf 0.20 (1:1 EtOAc/hexane), 0.39 (3:1 EtOAc/hexane); mp 161-162 C. (from EtOAc/hexane) (lit. mp: 157-159 C.); δH(CDCl3) 2.29 (s, 6H, 2*OAc), 7.18 (pseudo t, 1H, J 2.1, 4-H) and 7.70 (pseudo d, 2H, J 2.1, 2-H, 6-H); δC(CD3OD) 18.43, 118.94, 119.02, 131.70, 150.19, 165.46 and 168.17; m/z (ESI) 261 (MNa+, 100%).
The starting material can be manufactured, for example, as follows: 50 g of 3,5-dihydroxybenzoic acid are suspended in 132.2 g of acetic anhydride and heated to 50. After the addition of 15 drops of concentrated H2 SO4, the resulting solution is stirred for 1 hour at 60. The reaction mixture is poured onto ice and extracted with CH2 Cl2. The combined organic phases are washed 3 times with H2 O, dried over Na2 SO4 and concentrated by evaporation. The residue is crystallized from ether and yields 3,5-diacetoxybenzoic acid having a melting point of 140-145.
With thionyl chloride; acetic anhydride; In pyridine; CH2 Cl; 5,5-dimethyl-1,3-cyclohexadiene; hydrogen; benzene;
EXAMPLE 1 3,5-Diacetoxybenzaldehyde A total of 82.4 ml of acetic anhydride (89.8 g, 0.88 mole) was added in small portions to a stirring mixture of 61.7 g of 3,5-dihydroxybenzoic acid (0.4 mole) in 130 ml of pyridine. After the addition was complete, the reaction mixture was left stirring for 15 hours at ambient temperature. The pyridine was removed by evaporation in vacuo. The residue was taken up in CH2 Cl 2 and washed several times with 6N HCl and water and dried (Na2 SO4). Removal of solvent under reduced pressure left 68.2 g (72%) of <strong>[35354-29-1]3,5-diacetoxybenzoic acid</strong> which was used without further purification. A stirring mixture of 68.2 g (0.29 mole) of <strong>[35354-29-1]3,5-diacetoxybenzoic acid</strong> and 21 ml (0.29 mole) of thionyl chloride in 150 ml of dry benzene was heated in an oil bath at 80-90 C. for 2 hours. After cooling the benzene was removed in vacuo to leave 3,5-diacetoxybenzoyl chloride as a tan, solid which was recrystallized from cyclohexane as colorless crystals, mp 87-89 C., 61.8 g (83% yield). A mixture of 61.8 g (0.24 mole) of 3,5-diacetoxybenzoyl chloride and 6 g of 5% palladium on BaSO4 in 200 ml of dry xylene was efficiently stirred while bubbling in hydrogen gas. The reaction mixture was slowly heated to 115 C. in an oil bath and the heating was continued until the evolution of HCl gas ceased (approximately 5 hours). After cooling, the mixture was filtered and the xylene was removed in vacuo to leave 48 g (90%) of 3,5-diacetoxybenzaldehyde which was used without further purification in subsequent reactions.
5. Preparation of 3,5-diacetoxybenzoic acid To a solution of 3,5-dihydroxybenzoic acid (7.71 g, 50 mmol) in EtOAc (10 mL) were added acetic anhydride (12.25 mL, 130 mmol) and pyridine (8.08 mL, 100 mmol) in an ice-water bath under a nitrogen atmosphere. The mixture was stirred at 0 C. for 40 min and then stirred at ambient temperature for 4 h. 98% Formic acid (2.36 mL, 60 mmol) was added and the resulting mixture was stirred for 1 h. Then, it was poured into water and extracted with EtOAc. The organic phase was washed with water and brine, dried over Na2SO4, filtered, and evaporated in vacuo. Purification of the residue via recrystallization from n-heptane/EtOAc provided 3,5-diacetoxybenzoic acid (8.97 g, 75.3%) as white solid. Data are: 1H NMR (CDCl3, 300 MHz) δ 12.1 (bs, 1H), 7.73 (d, 2H), 7.21 (t, 1H), 2.32 (s, 6H); 13C NMR (CDCl3, 75 MHz) δ 170.4, 169.0, 151.2, 131.5, 121.3, 121.0, 21.2; mp=161-162 C.; HRMS (EI+) found 238.0475 M+, calcd 238.0477 for C11H10O6; Anal. Calcd for C11H10O6: C, 55.47; H, 4.23. Found: C, 55.62; H, 4.37.
Bis(N-imidazolyl)butane (0.0190 g, 0.1 mmol) was dissolved in 2 mL ethanol. To this solution was added 3,5-dihydroxybenzoic acid (0.0150 g, 0.1 mmol) in 6 mL ethanol. Colorless block crystals were afforded after several days by slow evaporation of the solvent (yield: 0.0265 g, 75%, based on L2). mp 185-187 C. Elemental analysis: Calc. for C34H42N8O9(706.76): C, 57.73; H, 5.94; N, 15.85. Found: C, 57.68; H, 5.86; N, 15.82. Infrared spectrum (KBr disk, cm-1): 3662s(br, ν(OH)), 3454s(multiple, νas(NH)), 3342s(νs(NH)), 3136 m, 3056 m, 2986 m, 2916 m, 1592s(νas(COO-)), 1542 m, 1488s, 1406s(νs(COO-)), 1358 m, 1300 m, 1240 m, 1196 m, 1130 m, 1060 m, 1020 m, 936 m, 853 m, 800 m, 738 m, 680 m, 620 m.
<strong>[106941-25-7]Adefovir</strong> (54.60 mg, 0.2 mmol) and 3, 5-dihydroxybenzoic acid(30.80 mg, 0.2 mmol) were suspended in 10 mL distilled water, and stirred at 90 C for about 3 h. After refluxing, the resulting mixtures were filtered, and the filtrate was placed in an oven at 60 C for 4days, colorless crystals of 2 were appeared in 90% yield.
6,6"-oxybis(3',6'-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9'-xanthene]-1',8'-dicarboxylic acid)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With ammonium chloride; In neat (no solvent); at 180 - 190℃; for 1.5h;
General procedure: General Procedure:In a 250 ml round bottom flask a well mixed mixture of 0.308 gm(0.001mol) of 4,4?-oxydiphthalic anhydride (1) and substituted phenols (2a-eScheme 1) 0.004 mol were taken and fused in oil bath at 180 oC. The fused melt was added 0.2 g of NH4Cl, and stirred the reaction mixture mechanicallyat 180-190oC until the solid mass obtained. The solid mass was dissolvedin 10 ml of 5 % sodium hydroxide solution. The solution was filtered toremove any insoluble impurities and filtrate was treated with 3 ml of 30%hydrochloric acid and precipitated the dyes (3a-e). Similarly the derivativesof 1,4,5,8-Naphthalenetetracarboxylic dianhydride (6a-e) with substitutedphenols (2a-e, Scheme 2) were synthesized by following the same procedure.6,6??-oxybis(3?,6?-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9?-xanthene]-1?,8?-dicarboxylic acid) (3a)Yellowish brown, m.p> 250oC; Rf: 0.34 (ethyl acetate: ethanol 1: 1);lambdamax (nm): 453; FTIR (Neat) nu: 3300-3500 (br, COOH, OH), 3130 (C=C-H,str), 1782 (lactone C=O), 1753 (carboxylic C=O), 1642 (C=C), 1588 (C=C),1128 (C-O), 853 (Ar-H, bend), 810 (Ar-H, bend) cm-1. 1H-NMR . 1H-NMR(DMSO-d6, 300 MHz) delta (ppm): 12.24 (s, 4H, COOH), 7.90 (d, 2H), 7.30 (m,2H), 7.13 (d, 2H), 6.90 (s, 4H), 6.75 (s, 4H), 5.03 (s, 2H, 4OH). 13C-NMR(DMSO 75 MHz) delta (ppm): 172.98, 168.92, 161.54, 156.45, 156.31, 151.17,132.70, 130.85, 123.46, 119.69, 116.12, 113.98, 110.13, 107.96. Anal. Calcd.For C44H22O19: C, 61.84 H, 2.59; Found: C, 61.96; H, 2.46.
2.2.1 Synthesis of [(C4H8N5+)·(C7H5O4-)]·2H2O hydrous salt (1) The <strong>[1004-38-2]TAP</strong>I (0.20 mmol) and HDHBA (0.40 mmol) were dissolved in 10 ml of methanol-distilled water (H2O) mixed solution. The mixed solution was stirred for 30 min to receive a colorless solution. After seven days of slow evaporation, crystals with good diffraction quality were obtained at 20-25 C. The resulting crystals were filtered and dried after rinsed with methanol-H2O mixed solution. Yield: 62%. Analysis calculated for C11H17N5O6: C, 41.85; H, 5.39; N, 22.20%. Found: C, 42.11; H, 5.42; N, 22.31%. Infrared spectrum (KBr disc, cm-1): 3429s, 3375s, 3315s, 3192s, 1683s, 1635s, 1610s, 1583s, 1516s, 1481m, 1424s, 1331m, 1303m, 1267m, 1245m, 1200w, 1163m, 1103w, 1007m, 982w, 940m, 920w, 853w, 871w, 797m, 767m, 730m, 698m, 665m, 470m.
Stage #1: 3,5-Dihydroxybenzoic acid With potassium carbonate In acetone at 20℃; for 0.25 - 0.333333h;
Stage #2: dimethylsulfide In acetone at 50 - 55℃; for 5 - 6h;
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 6h;
27 Example 27. Preparation of 3,5-dimethoxybenzoic acid methyl ester
In a two-neck round flask, 10 mmol of 3,5-dihydroxy benzoic acid was added at room temperature, then 50 ml of dimethylformamide was added. 36 mmol of potassium carbonate (K2CO3) was added as a catalyst, and 36 mmol of bromomethane was slowly added dropwise. After addition, the reaction was allowed to proceed at room temperature for 6 hours. 500 ml of purified water was added to the reaction mixture and extracted with 100 ml of n-hexane. The hexane layer was dried under reduced pressure then Chromatography of the silica gel column using a mixed solvent of n-hexane and ethyl acetate as a developing solvent gave 3,5-dimethoxybenzoic acid methyl ester and High Speed atomic impact mass spectrometry (hereinafter referred to as FAB-MS).
3,5-bis((fluorosulfonyl)oxy)benzoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With fluorosulfonyl fluoride; N-ethyl-N,N-diisopropylamine In dichloromethane; water at 20℃; Sealed tube;
1(J) II. Representative procedure for biphasic reaction conditions.
3,5-Bis(fluorosulfonate)benzoic acid (12-30). 3,5-Dihydroxybenzoic acid (0.77 g, 5 mmol) was dissolved in a 3:2 CH2Cl2:water mixture (0.5 M). Diisopropylethylamine (4.5 mL, 25 mmol) was added, the reaction flask was sealed with a septum, air was evacuated, and a balloon filled with SO2F2 gas was introduced. The reaction mixture was vigorously stirred under SO2F2 atmosphere at room temperature overnight. Upon completion, the volatiles were removed by rotary evaporation, and the residue was acidified and extracted with CH2Cl2. The desired product was obtained as a white solid (1.45 g, 91% yield). mp 127-128° C. 1H NMR (400 MHz, CDCl3) δ 10.8 (br s, 1H), 8.19 (d, J=2.3 Hz, 2H), 7.68 (t, J=2.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 168.1, 150.1, 133.8, 123.3, 120.3; 19F NMR (376 MHz, CDCl3) δ +39.1; ESI-MS (m/z): 317 [M]-.
85%
With [(4-acetamidophenyl)(fluorosulfonyl)amino]sulfonyl fluoride; 1,8-diazabicyclo[5.4.0]undec-7-ene In tetrahydrofuran at 20℃; for 0.166667h;
Weigh 63.3 mg of penciclovir and 308.2 mg of 3,5-dihydroxybenzoic acid, add 1 mL of 50% ethanol (volume ratio of ethanol and water is 1: 1) to obtain a suspension, and place the suspension in Stir at room temperature for 24h, suction filter, wash the obtained white solid with an appropriate amount of 50% ethanol, and then dry at 40 C for 48h to obtain a solid sample of penciclovir co-crystallized with 3,5-dihydroxybenzoic acid.
With N-benzyl-N,N,N-triethylammonium chloride; at 110℃; for 8h;Inert atmosphere;
15.4 g of 3,5-dihydroxy benzoic acid, 10.81 g of <strong>[17557-23-2]neopentyl glycol diglycidyl ether</strong>, 0.01 g of benzyl triethylammonium chloride were mixed in 26 g of 1 -Methoxy-2-propanyl acetate (PGMEA) solvent. The reaction proceeded at H0C under nitrogen for 8 hours. After cooling down to room temperature, the reaction mixture was transferred to a bottle for use. The GPC (using polystyrene standards) shows it has a weight average molecular weight of Mw=736 and a number average molecular weight Mn=505. The dissolution rate in AZ 300 MIF developer is: 5500 A/sec. The GPC data also confirmed the absence of any residual starting materials.
With benzotriazol-1-ol; O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate; triethylamine In N,N-dimethyl-formamide at 25℃; for 16h; Inert atmosphere;
Compound16-2
Under argon atmosphere,HOBT (1.62 g, 12.0 mmol), HBTU (4.55 g, 12.0 mmol),and triethylamine (1.52 g, 15.0 mmol) were successively added toa solution ofcompound16-1(1.54 g, 10.0 mmol) and dimethylamine hydrochloride (815 mg, 10.0 mmol) in DMF (50 mL). The reaction mixture was stirred at room temperature for 16 hrs. Upon completion, the reaction solvent wasevaporated to dryness in vacuo. The residue was purifiedon silica gel column elution (2-10% MeOH in dichloromethane) afforded compound16-2(1.65 g, 91.0% yield) as a whitesolid.1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 2H), 6.23 (t,J= 2.0Hz, 1H), 6.14 (d,J= 2.0Hz, 2H), 2.90 (s, 3H), 2.87 (s, 3H).
91%
With benzotriazol-1-ol; O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate; triethylamine In N,N-dimethyl-formamide at 25℃; for 16h; Inert atmosphere;
Compound16-2
Under argon atmosphere,HOBT (1.62 g, 12.0 mmol), HBTU (4.55 g, 12.0 mmol),and triethylamine (1.52 g, 15.0 mmol) were successively added toa solution ofcompound16-1(1.54 g, 10.0 mmol) and dimethylamine hydrochloride (815 mg, 10.0 mmol) in DMF (50 mL). The reaction mixture was stirred at room temperature for 16 hrs. Upon completion, the reaction solvent wasevaporated to dryness in vacuo. The residue was purifiedon silica gel column elution (2-10% MeOH in dichloromethane) afforded compound16-2(1.65 g, 91.0% yield) as a whitesolid.1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 2H), 6.23 (t,J= 2.0Hz, 1H), 6.14 (d,J= 2.0Hz, 2H), 2.90 (s, 3H), 2.87 (s, 3H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping