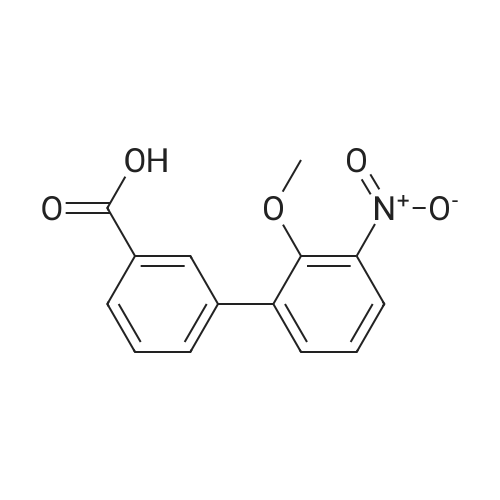
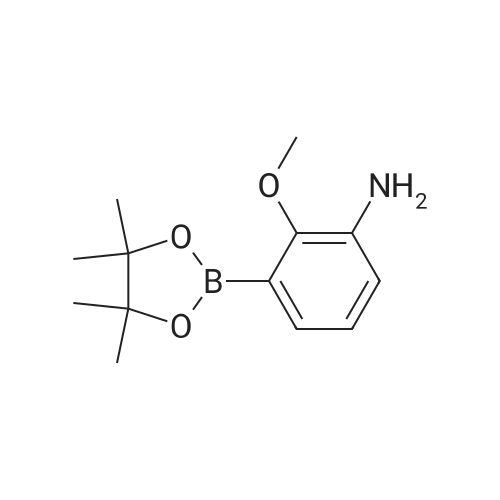
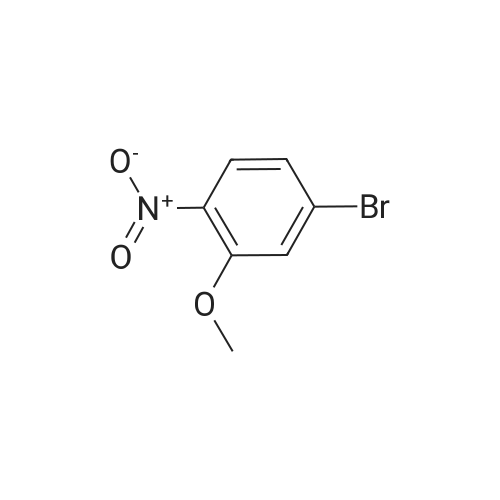
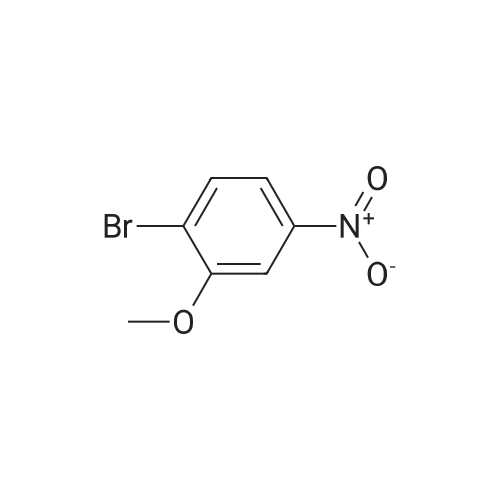
| 95% |
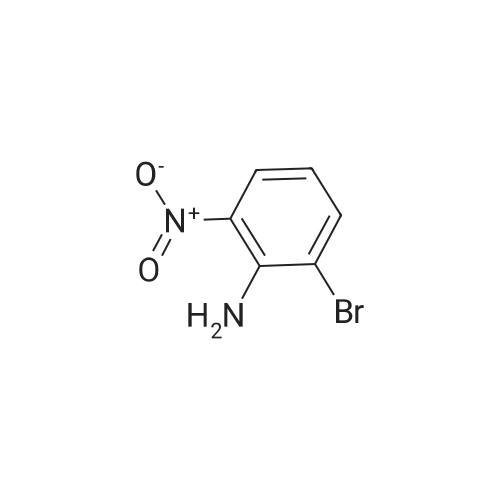
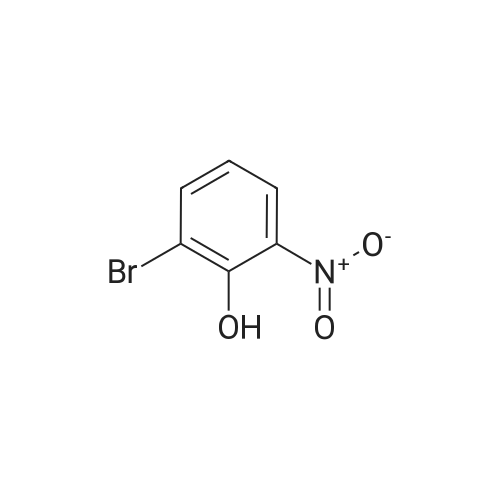
Stage #1: 2-bromo-6-nitro-phenol With potassium carbonate In acetone at 70℃; for 1h;
Stage #2: methyl iodide In acetone for 8h; Reflux; |
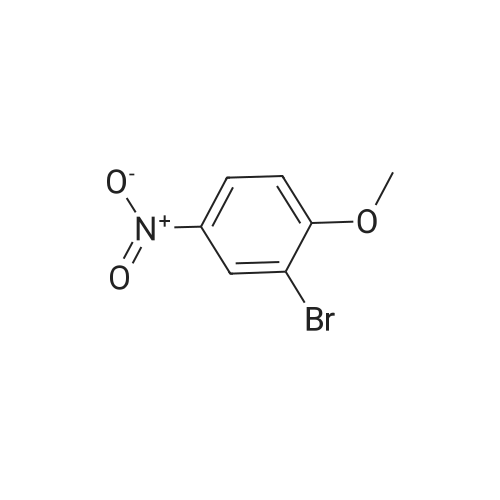
16.1 l-Bromo-2-methoxy-3-nitro-benzene
l-Bromo-2-methoxy-3-nitro-benzene A mixture of 2-Bromo-6-nitro-phenol (43.6 g, 0.2mol), K2CO3 (82.9 g, 0.6 mol), acetone (600mL) is stirred at 70 °C for lh. Then Mel (85.14g, 0.6 mol) is slowly added to the reaction mixture and refluxed for 8h. After reaction, filtered and the filtrate is extracted with ethyl acetate (3 x lOOOmL). The combined SnC12organic phase is washed with water and brine, dried over Na2S04, concentrated in vacuo to obtain the desired product. Yield: 44g (95%) HPLC-MS: M+H=232/234; tRet =2.04 min; AM12 |
| 90% |
With potassium carbonate In acetone at 70℃; for 40h; |
1.2
Step 2 1-Bromo-2-methoxy-3-nitro-benzene; 2-Bromo-6-nitro-phenol 1b (46.55 g, 0.214 mol) was dissolved in 500 mL of acetone followed by addition of potassium carbonate (35.36 g, 0.256 mol) and iodomethane (20.1 mL, 0.32 mol). The reaction mixture was heated to reflux at 70 °C for 40 hours. The reaction was monitored by TLC until the disappearance of the starting materials. The reaction mixture was concentrated under reduced pressure and diluted with 1300 mL of ethyl acetate and 500 mL of water. The aqueous layer was extracted with ethyl acetate (300 mL×2). The combined organic extracts were washed with 4 N hydrochloric acid and saturated aqueous sodium bicarbonate and then dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 1-bromo-2-methoxy-3-nitro-benzene 1c (44.59 g, yield 90.0%) as a brown solid. MS m/z (ESI): 234 [M+1] |
| 90% |
With potassium carbonate In acetone at 70℃; for 40h; |
1.2
Step 2 1-Bromo-2-methoxy-3-nitro-benzene; 2-Bromo-6-nitro-phenol 1b (46.55 g, 0.214 mol) was dissolved in 500 mL of acetone followed by addition of potassium carbonate (35.36 g, 0.26 mol) and iodo- methane (20.1 mL, 0.32 mol). The reaction mixture was heated to reflux at 70°C for 40 hours. The reaction mixture was concentrated under reduced pressure and diluted with 1300 mL of ethyl acetate and 500 mL of water. The aqueous layer was extracted with ethyl acetate (300 mLx2). The combined organic extracts were washed with 4 M hydrochloric acid and saturated sodium bicarbonate solution and then dried over anhydrous magnesium sulfate, filtered and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 1-bromo-2-methoxy-3-nitro-benzene 1c (44.59 g, yield 90.0%) as a brown solid. MS m/z (ESI): 234 [M+1] |
| 90% |
With potassium carbonate In acetone at 70℃; for 40h; |
1.2 Step two 1-bromo-2-methoxy-3-nitrobenzene
2-bromo-6-nitrophenol 1b (46.55g, 0.214mol) was dissolved in 500mL acetone. To this was added potassium carbonate (35.36g, 0.26mol) and iodomethane (20.1mL, 0.32mol) then the solution was heated at reflux 70°C for 40 hours. The reaction solution was concentrated under reduced pressure. Added 1300mL ethyl acetate and 500mL water. The aqueous phase was extracted with ethyl acetate (300mL × 2). The combined organic phases were washed with 4M hydrochloric acid and saturated sodium bicarbonate solution, dried over anhydrous magnesium sulfate, and filtered. The filtrate was concentrated under reduced pressure. The result was purified by column chromatography on silica gel to give 1-bromo-2-methoxy-3-nitrobenzene 1c (44.59g, brown solid). Yield: 90.0%. |
| 89% |
With potassium carbonate In N,N-dimethyl-formamide at 20℃; |
12.1 Preparation 12
Preparation 12 To a solution of 2-bromo-6-nitrophenol (5 g, 22.94 mmol) in DMF (18 ml) was added potassium carbonate (9.51 g, 68.8 mmol) and the resulting mixture was stirred for 15 min, then iodomethane (2.87 ml, 45.9 mmol) was added. The resulting mixture was stirred at rt overnight. HPLC and LCMS indicated complete conversion to product. Cold water added (75 mL), stir/sonicate, solid was collected by filtration. This material was then dissolved in EtOAc (150 mL). This solution was washed lx 10% LiCl, lx brine. dried over sodium sulfate, then filtered and concentrated. Loaded onto a 120g silica gel cartridge, then purified by flash chromatography eluting with 0-50% EtOAc in hexanes. Fractions containing the product were concentrated to afford a pale yellow solid as the product l-bromo-2-methoxy-3 -nitrobenzene (4.997 g, 20.46 mmol, 89% yield). LCMS ave very weak MH+. |
| 89% |
Stage #1: 2-bromo-6-nitro-phenol With potassium carbonate In N,N-dimethyl-formamide for 0.25h;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 20℃; |
12.1 step 1
To a solution of 2-bromo-6-nitrophenol (5 g, 22.94 mmol) in DMF (18 ml) was added potassium carbonate (9.51 g, 68.8 mmol) and the resulting mixture was stirred for 15 min and then iodomethane (2.87 ml, 45.9 mmol). The resulting mixture was stirred overnight at rt. HPLC and LCMS instructions were fully converted to product. Add cold water (75 mL), perform agitation / sonic processing, and collect solids by filtration. The material was then dissolved in EtOAc (150 mL). The solution was washed with 10% LiCl, washed 1x with brine, dried over sodium sulfate, filtered and concentrated. Was loaded onto a 120 g silica gel cartridge and then purified by flash chromatography eluting with 0-50% EtOAc in hexanes. Concentrate the fractions containing the product to afford the product as a pale yellow solid 1-bromo-2-methoxy-3-nitrobenzene (4.997 g, 20.46 mmol, 89% yield). |
| 89% |
Stage #1: 2-bromo-6-nitro-phenol With potassium carbonate In N,N-dimethyl-formamide for 0.25h;
Stage #2: methyl iodide In N,N-dimethyl-formamide at 20℃; |
422.1 Step 1 :
To a solution of 2-bromo-6-nitrophenol (5 g, 22.94 mmol) in DMF (18 ml) was added potassium carbonate (9.51 g, 68.8 mmol). The resulting mixture was stirred for 15 minutes, then iodomethane (2.87 ml,45.9 mmol) was added. The resulting mixture was stirred at room temperature overnight. HPLC and LC-MS indicated complete conversion to product. Cold water was added (75 mL), and the resulting mixture was stirred and sonicated. Next, the solid was collected with filtration. This material was then dissolved in EtOAc (l50mL). This solution was washed lx with 10% LiCl and lx with brine. The reaction mixture was dried over sodium sulfate, then filtered and concentrated. This was loaded onto a l20g ISCO column, then purified by flash chromatography eluting with 0-50% EtOAc in hexanes. The reaction afforded a pale yellow solid, l-bromo-2-methoxy-3-nitrobenzene (4.997 g, 20.46 mmol, 89 % yield) HPLC /R 0.92 min (analytical HPLC Method A). |
| 86% |
With potassium carbonate In N,N-dimethyl-formamide at 20℃; |
10.2 Second step
To a solution of compound 10c (10.80 g, 49.54 mmol) and potassium carbonate (10.40 g, 75.25 mmol) in 150 mL N,N-dimethylformamide was added methyl iodide (10.70 g, 75.38 mmol). The resulting reaction solution was reacted overnight at room temperature. After the reaction is over, filter, add ethyl acetate (100mL) to dilute, wash the organic phase with water (50mL×2), dry with anhydrous sodium sulfate, filter, the filtrate was concentrated under reduced pressure to obtain compound 10d (10.00 g), yield: 86%. |
| 83.6% |
With potassium carbonate In acetone Heating / reflux; |
17
To a mixture of 2-bromo-6-nitrophenol (5.0 g, 22.9 mmol) and potassium carbonate (6.3 g,45.8 mmol) in acetone (100 mL) was added iodomethane (2.85 mL, 45.8 mmol) under stirring and the mixture was then heated to reflux overnight. The mixture was cooled to room temperature, filtered through Celite, the filter cake washed with ethyl acetate, and the filtrate concentrated under reduced pressure. The residue was taken up in ethyl acetate, washed 3x with IN NaOH, once with brine, dried over sodium sulfate and concentrated under reduced pressure. The residue was recrystallized twice from 2-propanol / water to give 4.45 g (83.6%) of the title compound as beige, very fine needles.MS (ESI: 232 / 234 (M+H+) for C7H6BrNO1H-NMR (DMSO-d^ δ: ppm 3.98 (s, 3H); 7.39 (t, IH); 8.02 (dd, IH); 8.08 (dd, IH). |
|
With potassium carbonate In acetone at 80℃; for 6h; |
Description for D312-bromo-6-nitrophenyl methyl ether (D31)To a mixture of K2C03 (2.54 g) and 2-bromo-6-nitrophenol (2g) in acetone (15 mL) at room temperature was added Mel (3.442 mL). The reaction mixture was heated to 80 °C for 6 h, after cooling the reaction, the mixture was filtered and the filtrate was concentrated to afford 2-bromo-6-nitrophenyl methyl ether (D31) (2.1 g). δΗ (CDCI3, 400MHz): 3.96 (3H, s), 4.91 (1H, m), 7.06 (1 H, t), 7.19 (1H, s), 7.72 (1H, m). |
| 2.1 g |
With potassium carbonate In acetone at 80℃; for 6h; |
D31 Description for D31 [0202] 2-bromo-6-nitrophenyl methyl ether (D31)
To a mixture of K2CO3 (2.54 g) and 2-bromo-6-nitrophenol (2 g) in acetone (15 mL) at room temperature was added Mel (3.442 mL). The reaction mixture was heated to 80° C. for 6 h, after cooling the reaction, the mixture was filtered and the filtrate was concentrated to afford 2-bromo-6-nitrophenyl methyl ether (D31) (2.1 g). δH (CDCl3, 400 MHz): 3.96 (3H, s), 4.91 (1H, m), 7.06 (1H, t), 7.19 (1H, s), 7.72 (1H, m). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping