* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the American Society for Mass Spectrometry, 2016, vol. 27, # 5, p. 897 - 907
3
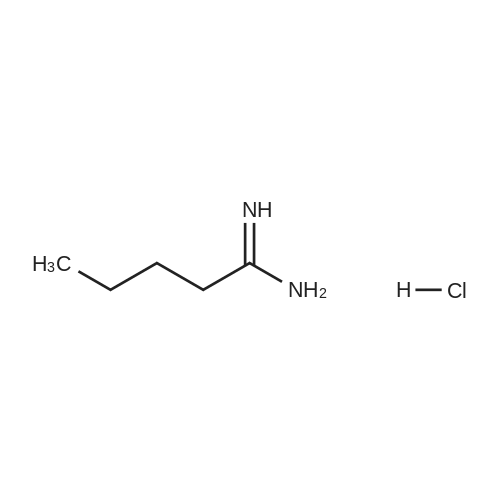
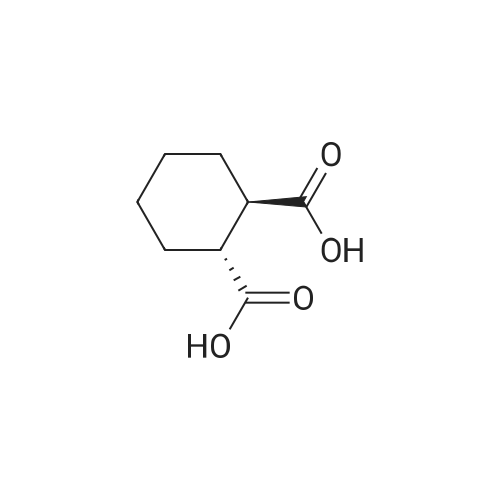
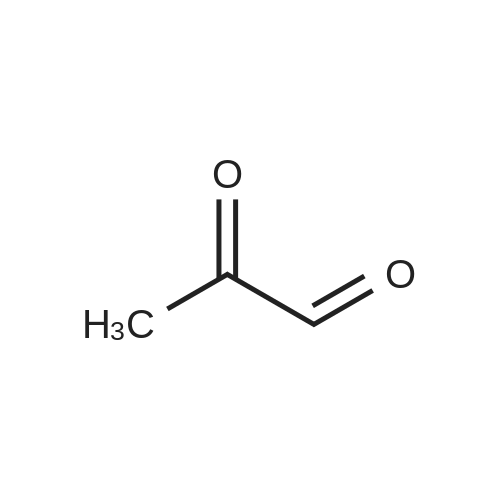
[ 96-26-4 ]
[ 38993-84-9 ]
Reference:
[1] Patent: US6268363, 2001, B1,
4
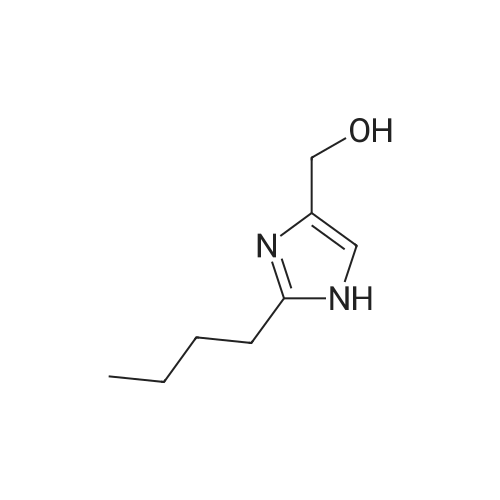
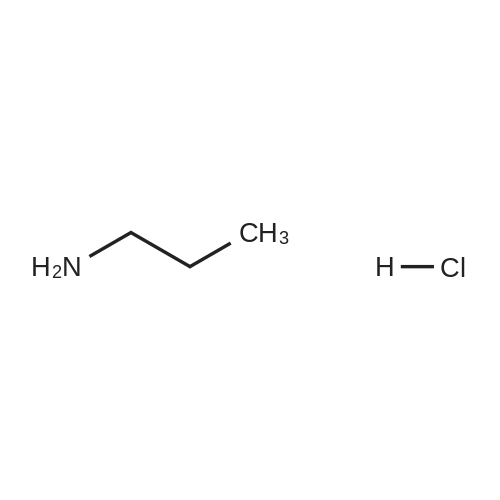
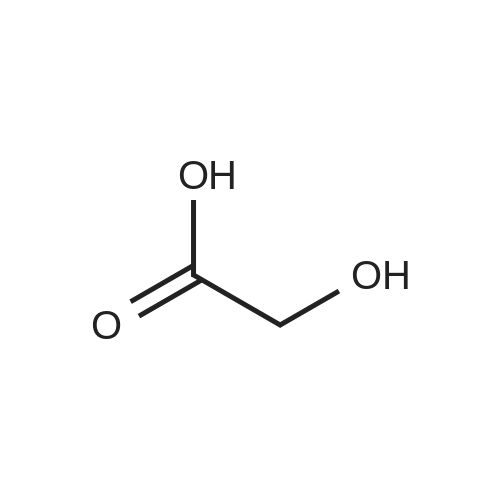
[ 96-26-4 ]
[ 26177-45-7 ]
[ 38993-84-9 ]
[ 226931-22-2 ]
Reference:
[1] Patent: US6268363, 2001, B1,
5
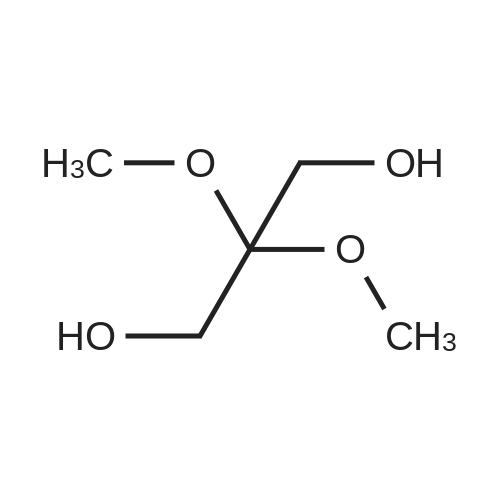
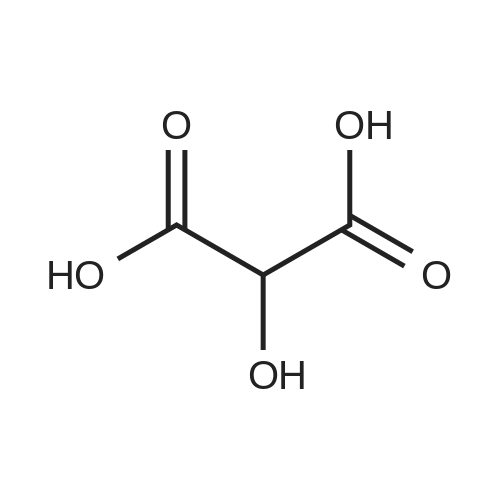
[ 96-26-4 ]
[ 38993-84-9 ]
[ 66973-95-3 ]
Reference:
[1] Patent: US6268363, 2001, B1,
6
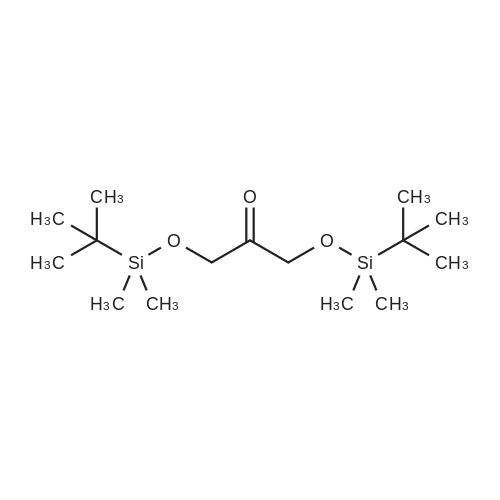
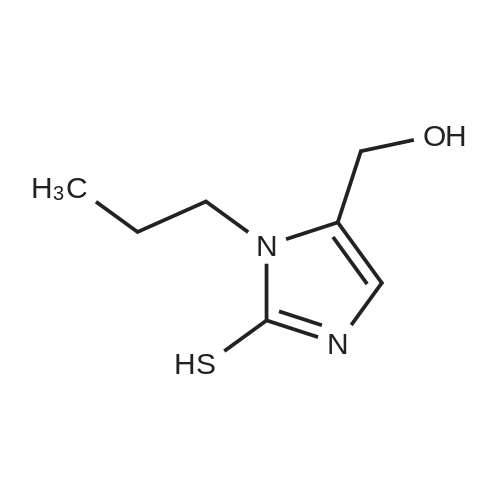
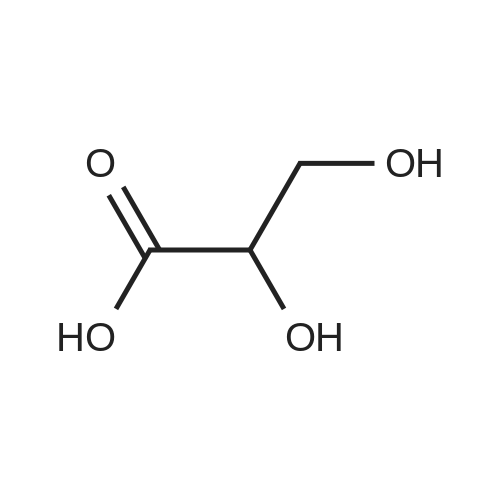
[ 96-26-4 ]
[ 18600-40-3 ]
[ 38993-84-9 ]
[ 915922-45-1 ]
Reference:
[1] Patent: US6268363, 2001, B1,
7
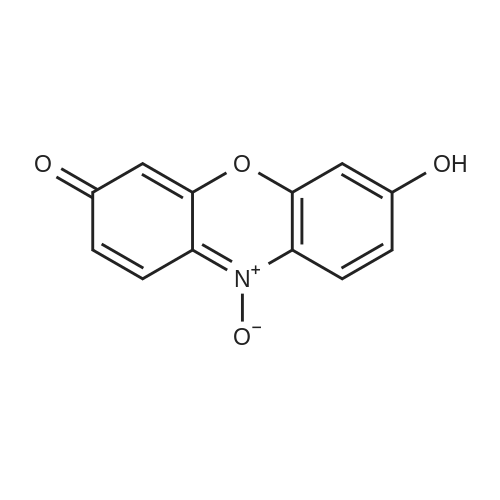
[ 96-26-4 ]
[ 38993-84-9 ]
[ 226931-06-2 ]
Reference:
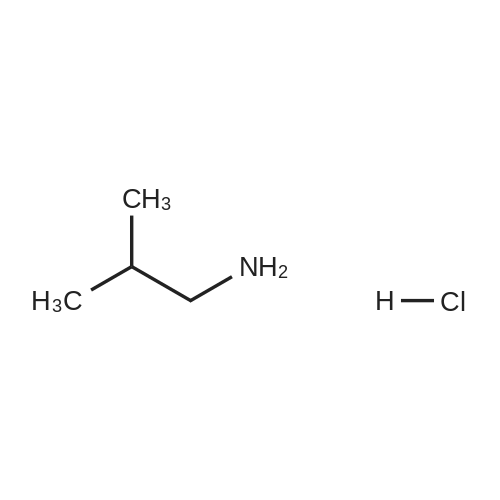
[1] Patent: US6268363, 2001, B1,
8
[ 96-26-4 ]
[ 42365-50-4 ]
[ 38993-84-9 ]
[ 226931-24-4 ]
Reference:
[1] Patent: US6268363, 2001, B1,
9
[ 96-26-4 ]
[ 42365-42-4 ]
[ 38993-84-9 ]
[ 226931-17-5 ]
Reference:
[1] Patent: US6268363, 2001, B1,
10
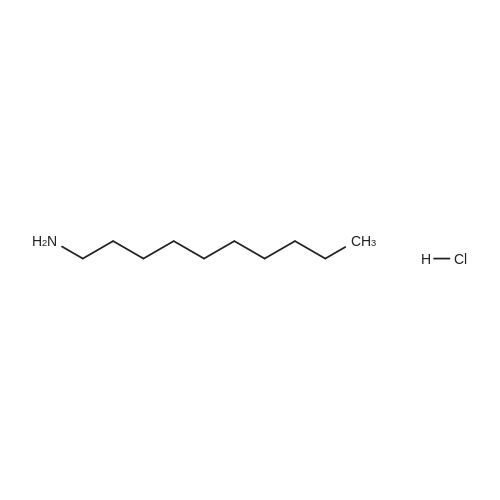
[ 96-26-4 ]
[ 22680-44-0 ]
[ 38993-84-9 ]
[ 226931-19-7 ]
Reference:
[1] Patent: US6268363, 2001, B1,
11
[ 96-26-4 ]
[ 18600-41-4 ]
[ 38993-84-9 ]
[ 226931-04-0 ]
Reference:
[1] Patent: US6268363, 2001, B1,
12
[ 226931-60-8 ]
[ 96-26-4 ]
[ 38993-84-9 ]
[ 226931-08-4 ]
Reference:
[1] Patent: US6268363, 2001, B1,
13
[ 96-26-4 ]
[ 50877-01-5 ]
[ 38993-84-9 ]
[ 226930-90-1 ]
Reference:
[1] Patent: US6268363, 2001, B1,
14
[ 96-26-4 ]
[ 541-23-1 ]
[ 38993-84-9 ]
[ 226930-98-9 ]
Reference:
[1] Patent: US6268363, 2001, B1,
15
[ 96-26-4 ]
[ 142-95-0 ]
[ 38993-84-9 ]
[ 226930-94-5 ]
Reference:
[1] Patent: US6268363, 2001, B1,
16
[ 96-26-4 ]
[ 143-09-9 ]
[ 38993-84-9 ]
[ 226930-96-7 ]
Reference:
[1] Patent: US6268363, 2001, B1,
17
[ 96-26-4 ]
[ 142-65-4 ]
[ 38993-84-9 ]
[ 226930-92-3 ]
Reference:
[1] Patent: US6268363, 2001, B1,
18
[ 96-26-4 ]
[ 5041-09-8 ]
[ 226930-88-7 ]
[ 38993-84-9 ]
Reference:
[1] Patent: US6268363, 2001, B1,
19
[ 96-26-4 ]
[ 17061-61-9 ]
[ 38993-84-9 ]
[ 226931-15-3 ]
Reference:
[1] Patent: US6268363, 2001, B1,
20
[ 96-26-4 ]
[ 61892-95-3 ]
Reference:
[1] Green Chemistry, 2017, vol. 19, # 15, p. 3515 - 3519
21
[ 96-26-4 ]
[ 5392-28-9 ]
[ 945-24-4 ]
Yield
Reaction Conditions
Operation in experiment
54%
Stage #1: With barium(II) chloride In water at 100℃; for 0.166667 h; Stage #2: With sodium acetate; DL-cysteine hydrochloride In water at 20℃; for 24 h;
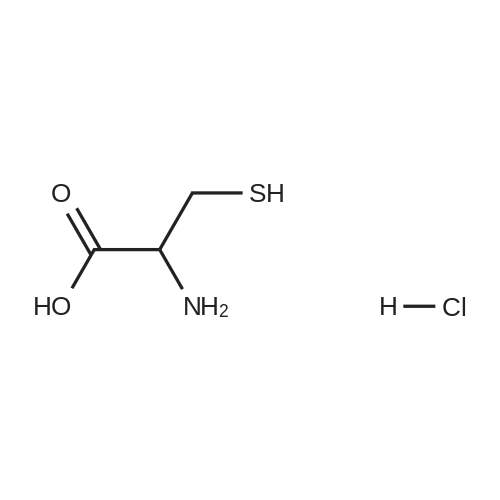
To a suspension of tetraaminopyrimidine sulfate (7.14 g, 30 mmol) in water is added barium chloride (7.32 g, 30 mmol) at once. The mixture is heated at 100 0C for 10 min and cooled to RT. The solid barium sulfate is removed by filtration. The filtrate is added to a solution of 450 mL of 4 M aqueous sodium acetate solution containing dihydroxyacetone (8 g, 90 mmol) and cysteine hydrochloride monohydrate (3.63 g, 30 mmol) in a 1 liter 3-neck round bottom flask attached with a mechanical stirrer and stirred for 24 h at RT open to air. The precipitated yellow solid is filtered, washed with water, and ethanol and dried overnight in a heated vacuum oven to give 3.4 g (66percent) of product. This product is further purified as per the following procedure. The yellow solid is dissolved in 10percent acetic acid with aid of few drops of cone. HCl at 75 0C. The hot solution is treated with activated charcoal and filtered. The filtrate is neutralized with cone. NH4OH. The bright yellow solid is collected, washed with water, water-ethanol and finally ethanol and dried overnight in a heated vacuum oven to provide 2.8 g of the title compound (54percent).
1. 2,4-DIAMINO-6-HYDROXYMETHYLPTERIDINE SPC2 A solution of 6.4 grams of barium chloride in a minimum amount of hot water was added with stirring at a temperature of 70°-80° C to a suspension of 7.6 grams of 2,4,5,6-tetraaminopyrimidine sulphate in 104 ml water. The resulting suspension was stirred for 30 minutes and the formed barium sulphate was removed by filtration and washed on the funnel with 26 ml water at a temperature of 70° C. The solution containing the 2,4,5,6-tetraaminopyrimidine dihydrochloride is diluted with water to a final volume of 400 ml. A solution of 128 grams of sodium acetate, 136 grams of bisulphite addition product of 1,3-dihydroxyacetone (free of methyl glyoxal) and 46 grams of cysteine hydrochloride in 390 ml water was prepared at room temperature in a 2 liter three-necked flask fitted with a stirrer, an air-bubbling system and a dropping funnel. To this solution, the 400 ml of the previously prepared solution of 2,4,5,6-tetraaminopyrimidine dihydrochloride were added with energic stirring and air-bubbling. A solution of 8 g of selenium dioxide dissolved in the minimum amount of water was made. Half of this solution was added to the reaction mixture immediately after the addition of the tetraaminopyrimidine solution and the other half 4-7 hours later. The reaction was allowed to proceed for 24 hours at room temperature. The reaction can be carried out in a similar manner in a range of temperatures from 20° to 100° C, but the yield is lower. After the end of the reaction, the solution is kept 1 hour at 4° C. The precipitate was filtered off, washed on the funnel with cold alcohol, alcohol:ethyl ether (1:1) and ethyl ether, then dried under vacuum for 24 hours at 50°. The yield is 3,8 g (72 percent) of 2,4-diamino-6-hydroxymethylpteridine.
Reference:
[1] Patent: US3989703, 1976, A,
25
[ 96-26-4 ]
[ 534-03-2 ]
[ 78531-52-9 ]
Yield
Reaction Conditions
Operation in experiment
71%
With ammonium hydroxide; hydrogen In methanol; water at 65℃; for 6 h; Autoclave
In an autoclave, a mixture of Raney nickel (4 g, wet) and ammonium hydroxide solution (257 g, 28percent NH3 in water, 4.2 mol) was heated to 65° C. under 250 psi of hydrogen atmosphere. A solution of 1,3-dihydroxyacetone (38 g, 0.42 mol) in methanol (76 ml) was pumped into the autoclave over a period of 3 h. After the addition was complete, the reaction mixture was maintained at 65° C. and 250 psi hydrogen pressure for additional 3 h. The autoclave was then cooled to ambient temperature and depressurized. The resulting reaction mixture was filtered to remove the catalyst. Ammonia and most of the water were removed by distillation under reduced pressure. GC analysis of the crude product showed a mixture of 91percent serinol (2-amino-1,3-propanediol) and 7percent bis-adduct (2,2′-iminobis-1,3-propanediol). (0025) An aqueous solution of the crude product mixture was loaded to a column of Amberlyst® 15 and eluted with water until neutral pH. GC analysis of the water eluate showed mainly the bis-adduct and other unidentified impurities. The column was then eluted with 300 ml of 4M ammonium hydroxide solution to recover the serinol. The eluate was then stripped under vacuum to remove ammonia and water. The residue was triturated with a 1:1 mixture of 2-butanol and 2-methyltetrahydrofuran to yield crystalline serinol (27 g, 71percent yield). Assay by GC: 97percent; Assay by titration: 101percent
61%
With ammonia; hydrogen In methanol; water at 65℃; for 5 h;
In an autoclave, ammonia gas (13 g, 0.77 mol) was charged to a suspension of Raney nickel (4 g, wet) in methanol (100 ml) and then heated to 65° C. under 250 psi of hydrogen atmosphere. A solution of 1,3-dihydroxyacetone (58percent solution in water, 200 g, 1.29 mol) was pumped into the autoclave over a period of 2 h. After the addition was complete, the reaction mixture was continued to hydrogenate at 65° C. temperature and 250 psi hydrogen pressure for an additional 3 h. The autoclave was cooled to ambient temperature and depressurized. The reaction mixture was filtered to remove the catalyst and evaporated under vacuum to remove ammonia and most of the water. The crude product was shown to contain 2.4percent serinol and 90percent 2,2′-iminobis-1,3-propanediol by GC analysis. (0029) An aqueous solution of the crude product was loaded to a column of Amberlyst® 15 and eluted with water until neutral pH. GC analysis of the water eluate showed complete removal of serinol (FIG. 5). The water eluate was then evaporated under vacuum, the residue was dissolved in 1:1 mixture of 2-butanol and tetrahydrofuran, treated with activated carbon, and concentrated to yield the bis-adduct 2,2′-iminobis-1,3-propanediol as a viscous oil (65 g, 61percent). Assay by GC: 99percent
Reference:
[1] Journal of Molecular Catalysis B: Enzymatic, 2015, vol. 120, p. 141 - 150
27
[ 96-26-4 ]
[ 142-84-7 ]
[ 6282-00-4 ]
Yield
Reaction Conditions
Operation in experiment
81%
With Cu/Al2O3; dihydrogen peroxide In water at 25℃; for 24 h; Green chemistry
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35percent hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 ° C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33μm capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
Reference:
[1] Patent: CN108658715, 2018, A, . Location in patent: Paragraph 0040; 0048; 0049
Reference:
[1] Chemical Communications, 2005, # 21, p. 2716 - 2718
30
[ 67-56-1 ]
[ 96-26-4 ]
[ 6342-56-9 ]
[ 547-64-8 ]
Reference:
[1] ChemCatChem, 2015, vol. 7, # 7, p. 1152 - 1160
[2] ChemSusChem, 2013, vol. 6, # 8, p. 1352 - 1356
[3] Chemical Communications, 2005, # 21, p. 2716 - 2718
31
[ 57-48-7 ]
[ 849585-22-4 ]
[ 4573-78-8 ]
[ 87-79-6 ]
[ 87-81-0 ]
[ 551-68-8 ]
[ 96-26-4 ]
[ 56-82-6 ]
Yield
Reaction Conditions
Operation in experiment
26.7 %Spectr.
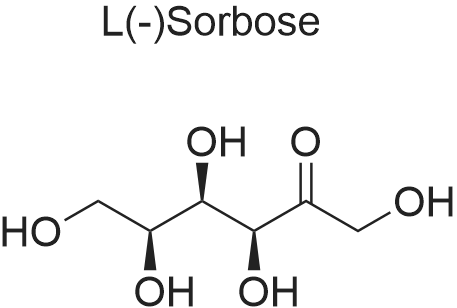
With molybdenum(VI) oxide In water at 100℃; for 4 h;
FIG. 8 shows 1H NMR spectra of D-fructose standard solution and of the fructose-containing fraction isolated after reaction of D-fructose with MoO3 in water at 100° C. for 4 h. Sorbose is present in the collected fraction.
With hydrogenchloride; ammonium cerium (IV) nitrate; sodium ethanolate In ethanol; acetonitrile at 60℃; for 8 h;
A mixture of acetonitrile and ethanol in a volume ratio of 2: 1 was added to the reactor,100 mmol of 1,3-dihydroxyacetone and 200 mmol of formaldehyde were dissolved in an organic solvent,100 mmol of cerium ammonium nitrate was added,Under normal pressure 60 conditions,Adding sodium ethoxide to adjust the pH to 10,15 mmol of catalyst was added,Stirring reaction 8h, obtained 4-hydroxymethyl imidazole,After completion of the reaction,Dropping concentrated HCl,Adjust pH = 2,filter,The filtered solid,Washed with saturated brine,Recrystallization from ethanol,Dried in vacuo to give the product 4-hydroxymethylimidazole hydrochloride.The purity of the prepared 4-hydroxymethylimidazolidine hydrochloride was 98.2percentThe yield was 87.5percent.
Reference:
[1] Patent: CN106349165, 2017, A, . Location in patent: Paragraph 0016-0030
35
[ 96-26-4 ]
[ 3473-63-0 ]
[ 32673-41-9 ]
Reference:
[1] Synthesis, 1983, # 7, p. 576
36
[ 96-26-4 ]
[ 83465-22-9 ]
[ 83480-29-9 ]
Yield
Reaction Conditions
Operation in experiment
85%
With hydrogenchloride; sodium cyanoborohydride In N,N-dimethyl-formamide at 60℃; for 24 h;
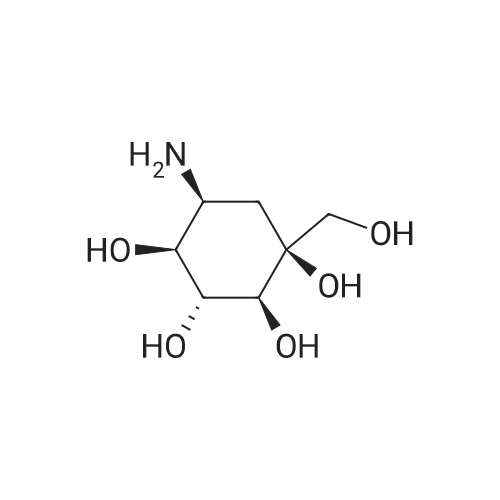
III (0.2 g, 0.001 mmol) was added to DMF (8 mL), stirred and dissolved, followed by the addition of 1,3-dihydroxyacetone (0.3 g, 0.03 mmol), 2N HCl (0.15 mL) and NaBH3CN (0.22 g, 0.004 mol) at 60 °C for 24 h cooled to room temperature, add water (3mL), adjusted to pH=4.0 with 2 Ν HCl, stirred for 30min, 1N NaOH was added to adjust PH = 7.0, the solvent was distilled off under reduced pressure through the UBK resin, followed by elution with water, 2N ammonia, and the eluate was collected and concentrated. After passing through CG50, water: ethanol (6: 4) was eluted and the eluate was collected, and then decolorized by Dowex1 x 2 to give 0.24 g of white powder voglibose, 85percent.
82%
With hydrogenchloride; water; sodium cyanoborohydride In water; N,N-dimethyl-formamide at 70℃; for 16 h;
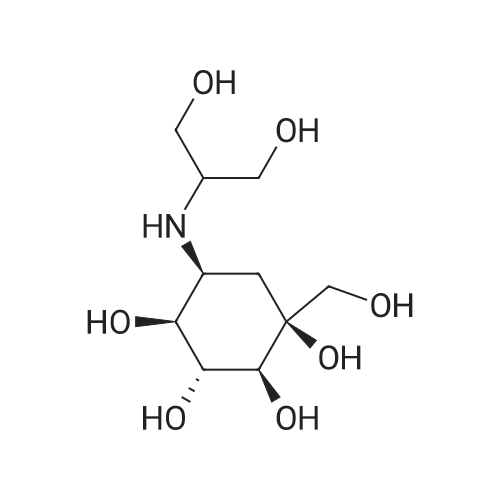
To a solution of 6 (483mg, 2.5mmol) in DMF (30mL), 1,3-dihydroxyacetone (900mg, 10.0mmol) and 2.0M hydrochloric acid (0.50mL) and NaBH3CN (628mg, 10.0mmol) were added successively. The mixture was heated for 16h at 70°C with stirring and cooled to rt and then evaporated. The residual product was dissolved in H2O (25mL) and adjusted to pH=4.0 with 2.0M hydrochloric acid under ice bath. The mixture was stirred at rt and adjusted to pH=4.5 with 1.0M NaOH and then concentrated. The residual product was eluted from a column of Amberlite CG-50 (NH4+) resin (100mL) with deionized water (100mL) and 1.0M aqueous ammonia (200mL) successively to give white product (550mg, 82percent). (0031) Amorphous white solid, mp 161.5–162.1°C (lit.11 mp 162–163°C). [α]D25 +25.9(c 1.02, H2O) (lit.11 [α]D25 +26.2(c 1.00, H2O)). FT-IR (Neat): νmax=3453, 3295, 2928, 1417, 1115, 1086, 1055, 1031, 992, 848cm−1; 1H NMR (400MHz, D2O): δ=3.77 (t, J=9.7Hz, 1H), 3.66 (dd, J=8.4, 3.8Hz, 1H), 3.63 (dd, J=6.9, 4.8Hz, 2H), 3.58 (dd, J=12.1, 3.9Hz, 2H), 3.52 (dd, J=11.5, 6.7Hz, 1H), 3.46 (d, J=11.3Hz, 1H, ABq), 3.42 (d, J=11.3Hz, 1H, ABq), 3.35 (d, J=9.5Hz, 1H), 3.32 (d, J=3.5Hz, 1H) ppm. 13C NMR (101MHz, D2O): δ=76.67, 74.50, 73.55, 72.47, 65.60, 62.65, 58.93, 56.93, 54.87, 29.75ppm.
Reference:
[1] Patent: CN107129474, 2017, A, . Location in patent: Paragraph 0060; 0068; 0069
[2] Tetrahedron, 2013, vol. 69, # 34, p. 7031 - 7037
[3] Journal of Medicinal Chemistry, 1986, vol. 29, # 6, p. 1038 - 1046
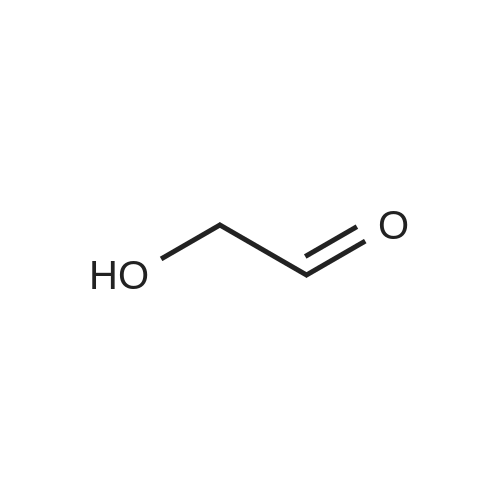
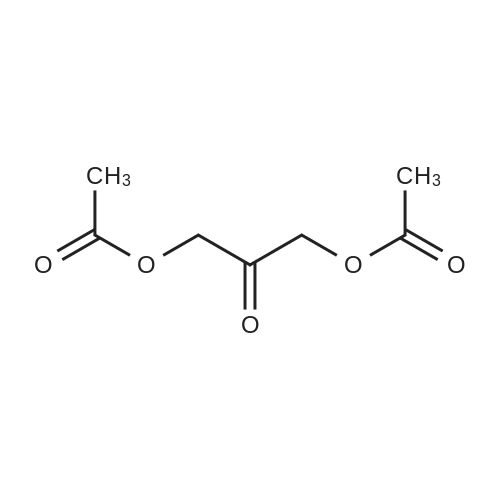
A 16 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a reflux condenser and a nitrogen inlet was charged with 1.00 kg (10.9 mol) 1,3-<strong>[96-26-4]dihydroxyacetone</strong> and 3.25 L (4.03 mol) pyridine. To this suspension 3.31 L (34.8 mol) acetic acid anhydride was added during 35 min, maintaining the temperature between 15 and 22 C. with a cooling bath. During the addition the suspension turned into a clear, slightly reddish solution. The mixture was stirred for 2.5 h at RT before it was concentrated on a rotatory evaporator at 50-55 C./10 mbar. The oily residue was dissolved in 10.0 L dichloromethane and washed two times with 5.0 L 2N hydrochloric acid, then with 5.0 L water. The organic layer was concentrated on a rotatory evaporator at 40 C./10 mbar and the oily residue was further dried under these conditions for 1.5 h. The dark red crude product (2 kg) was dissolved in 5.7 L toluene and the solution was warmed to 30 C. 5 L heptane were added during 10 min and the resulting turbid solution was seeded with product crystals, whereas fast crystallization occurred. 5 L heptane was added to the suspension to improve stirability. After stirring overnight at RT the suspension was cooled to 0 C. and stirred for 2 h at that temperature. The crystals then were filtered off and washed portionwise with totally 7 L of pre-cooled heptane. The crystals were dried at 30-35 C. at <=10 mbar over the weekend, to give 1.47 kg 1,3-diacetoxyacetone (78% yield; assay: 100%).
With pyridine; dmap; for 2.5h;Inert atmosphere;
To a stirred solution of <strong>[96-26-4]dihydroxyacetone</strong> (20.0 g, 0.22 mol) in pyridine (300 mL) was added DMAP (1.4 g, 11.8 mmol, 5 mol%) and acetic anhydride (63 mL, 0.66 mmol, 3 eq.) under argon. After 2 h 30 the solvent was removed under reduced pressure and the mixture was diluted with CH2Cl2 (300 mL). The organic layer was washed with HCl (3N) until pH = 5, with NaHCO3 until pH = 7 and then with water (50 mL). The organic layer was dried over MgSO4, filtered and evaporated under reduced pressure. The crude product was used without further purification. Yield: 90 % as a pale yellow oil
With pyridine; at 0 - 20℃; under 760.051 Torr; for 16h;
To a solution of l,3-dihydroxypropan-2-one (90 g, 999 mmol) in pyridine (400 ml) was added acetic anhydride (408 g, 3997 mmol) at 0 C. After the resulting mixture was stirred at 20 C for 16 hours, it was concentrated under reduced pressure. The residue was diluted with DCM (1000 mL), washed with 2N HC1 (2 x 1000 mL), NaHC03 (3 x 1000 mL). The combined organic layers was dried with anhydrous Na2S04, filtered, and concentrated under reduced pressure. The residue was poured into stirring petroleum ether (1500 mL), the product was precipitated out and filtered to give 2-oxopropane-l,3-diyl diacetate. H- MR: (300 MHz, CDC13, ppm): delta 4.76 (s, 4H), 2.18 (s, 6H).
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; In water; acetonitrile; at 60℃; under 760.051 Torr; for 12h;
Glycerol (1.0 mmol) and TEMPO (2.0 mmol) were placed in a flask under an air atmosphere (1 atm) and dissolved by adding a mixed solvent of 1 mL of water and 3 mL of acetonitrile. Here, 50 mg (containing Cu 2 +: 4.5 mg) of "Cu (II) -AlO (OH)" obtained in Preparation Example 1 was added and the reaction was carried out at 60 C. for 12 hours. The reaction progresses to TLC (I2 coloration) I monitored. After completion of the reaction, the reaction solution was centrifuged to remove "Cu (II) -AlO (OH)" and the supernatant was passed through a silica gel column (acetone eluent) to remove TEMPO, And concentrated to isolate a pale yellow oily reaction product. HPLC of the reaction product, 1 H-NMR, and As a result of 13 C-NMR analysis, It was confirmed to be dihydroxyacetone, The raw material conversion was 100%, and the yield of dihydroxyacetone was 95%.
55%
With Pd-Ce nanoparticles supported on functional Fe-MIL-101-NH2; In water; acetonitrile; at 60℃; under 1034.32 Torr; for 6h;
A 70.0 mL solution of acetonitrile/water (6:1, v/v) was used todissolve glycerol (1610.0 mg, 17.5 mmol). A 7.0 mL of this glyc-erol solution was next mixed with 25.0 mg of each catalyst in abatch reactor. A magnetic stirring bar was also placed inside eachone of the batch reactor. The containers were then placed in apressure chamber, and the setup was flushed with oxygen. A reac-tion condition of 60C and pressure at 20 psi was maintainedfor 6 h. After reaction, the reactor was cooled to room tempera-ture and the vessels were taken out for filtration. The mixtureswere filtered through syringe filters to remove the solid catalysts.The filtrates were collected in separate round bottom flasks. Thesolvent was removed under reduced pressure by using a rotaryevaporator, and the resulting product mixtures were analyzedusing gas chromatography (Agilent Gas Chromatograph (GC-FID)6850, column: Agilent 19091Z-433, flow rate: 1.6 mL/min) andthe high-performance liquid chromatography (Agilent, column:Aminex HPX-87H Column, wash solution: 0.02 N H2SO4, flow rate1.0 mL/min, column temperature 60C, UV detector 210 nm). Theyield was calculated according to the amount of glycerol added. Inaddition, the products were analyzed and identified by1H and13CNMR. Dihydroxyacetone, HOCH2C( O)CH2OH, yield 55% (withdimer).1H NMR (D2O, ppm), = 4.29 (s, 4H, 2CH2).13C NMR (D2O,ppm), = 211.93 (CO), 64.77 (-CH2-).
52%
With trichloroisocyanuric acid; In acetonitrile; at 20℃; for 24h;Inert atmosphere;
A 25mL round-bottom flask was equipped with magnetic stirring bar in which 0.10g of glycerol (1.34mmol), 0.05g (0.02mmolPd) of 3 and 0.33g (1.43mmol) of trichloroisocyanuric acid were added and then made the suspension of the mixture in 6mL of acetonitrile. This reaction mixture was stirred at room temperature for 24h and then evaporated to dryness. The obtained crude product was purified by flash chromatography (SiO2) eluting with Et2O/acetone (v/v=3/2) to give dihydroxyacetone in 52% yield as the known product as identified by 1H and 13C NMR spectra that are consistent with the literature data. A same procedure was used to test Pd NPs/polystyrene to produce DHA in 27% yield.
8%
With oxygen; alpha,alpha'-azodiizobutyramidine-dihydrochloride; In water; at 60℃; under 7500.75 Torr; for 20h;Autoclave;
General procedure: Glycerol oxidation was performed in a high-pressure stainless-steelautoclave with a polytetrafluoroethylene insert (10 mL) at a constanttemperature of 60 C. The autoclave was connected to the O2 supplysystem, which kept the pressure constant. The solution was stirredmagnetically. Typically, 5.0 mL of 1.0M glycerol in water was oxidizedin the presence of 4.0mM catalyst at 60 C at aconstant pressure of10 bars of O2. After desired time, the reactor was quickly cooled down,depressurized and the catalyst was removed by extraction with diethylether. The stability of the catalysts after reaction was tested by FTIR andXRD, which did not change compared with the fresh ones [Fig. S3]. Theremaining solution was diluted 10 times with distilled water and analyzedby high performance liquid chromatography (HPLC) using aShimadzu LC10A-VP chromatograph equipped with SPB-10 A UV andRID-10 A R.I. detectors, and a Prevail TM C18 (4.6mm×250 mm) column. A solution of H2SO4 (0.1% w/w) in H2O/acetonitrile (1/2 v/v)was used as the eluent at a flow rateof 1.0 mL min-1 at 50 C. Theglycerol conversion, alpha, and the selectivity for lactic acid (LA), SLA, werecalculated using Eqs. (3) and (4):
With reconbinant Escherichia coli JM109 glycerol dehydrogenase; nicotinamide adenine dinucleotide; ammonium chloride; sodium hydroxide; In aq. buffer; at 30℃;pH 10.5;Enzymatic reaction;Kinetics;
General procedure: GroDHase activity was assayed as described previously [5], with minor modifications. In the direction of Gro oxidation, the standard medium contained 100mM CAPS-NaOH pH 10.5, 5mM NAD+, and 500mM Gro. In the direction of DHA reduction, the standard medium was composed of 100mM MES-NaOH pH 6.5, 0.2mM NADH, and 200mM DHA. Alternatively, 50mM phosphate/carbonate pH 10.3 (Gro oxidation) and 6.5 (DHA reduction) were used as buffers. Both assays were initiated by the addition of an appropriate amount of enzyme in a final volume of 50mul. Reactions were performed at 30C and the reduction/oxidation of NAD+/NADH was followed at 340nm using a Multiskan Ascent 384-microplate reader (Thermo Electron Corporation, Waltham, MA, USA). One unit of enzyme activity (U) is defined as the amount of enzyme catalyzing the formation of 1mumol of product (NADH or NAD+) in 1min under the specified assay conditions.
With PdSn; potassium hydroxide; at 25℃;pH 13.1;
General procedure: Electrochemical evaluations were made with total metal loadings (Pt+Sn) of 0.2 mg cm-2. The electrochemical analyses were performed using a Pine Instrument Company electrochemicalanalysis system with an electrode rotation rate of 1300 rpm (revolutionsper minute), at roomtemperature in N2-saturated 1.0MKOH electrolytecontaining 1.0 M alcohol. Platinum wire and Hg/HgO electrode were used as counter and reference electrodes, respectively, althoughall potentials are quoted versus the reversible hydrogen electrode(RHE).
With oxygen; In water; at 20 - 80℃; under 7500.75 Torr; for 2h;Autoclave;
In a molar ratio of glycerin and gold 100: 1, 24mL0.1mol / L aqueous glycerol solution, 0.1576g 3% Au / ZnO 50mL was added to the autoclave, completely sealed after evacuation at room temperature three times with high purity oxygen, then charged 1.0MPa high purity oxygen, stirred and heated to 80 deg.] C, a constant temperature for 2 hours. after completion of the reaction, was cooled with an ice bath to room temperature, centrifugation, filtration, the reaction liquid was detected by HPLC, and measuring the conversion rate of glycerol the yield of 1,3-dihydroxyacetone.Analysis results, the glycerol conversion was 87.1%, selectivity of 1,3-dihydroxyacetone of 74.8%.
With hydrogenchloride; sodium cyanoborohydride; In N,N-dimethyl-formamide; at 60℃; for 24h;
III (0.2 g, 0.001 mmol) was added to DMF (8 mL), stirred and dissolved, followed by the addition of 1,3-dihydroxyacetone (0.3 g, 0.03 mmol), 2N HCl (0.15 mL) and NaBH3CN (0.22 g, 0.004 mol) at 60 C for 24 h cooled to room temperature, add water (3mL), adjusted to pH=4.0 with 2 Nu HCl, stirred for 30min, 1N NaOH was added to adjust PH = 7.0, the solvent was distilled off under reduced pressure through the UBK resin, followed by elution with water, 2N ammonia, and the eluate was collected and concentrated. After passing through CG50, water: ethanol (6: 4) was eluted and the eluate was collected, and then decolorized by Dowex1 x 2 to give 0.24 g of white powder voglibose, 85%.
82%
With hydrogenchloride; water; sodium cyanoborohydride; In water; N,N-dimethyl-formamide; at 70℃; for 16h;
To a solution of 6 (483mg, 2.5mmol) in DMF (30mL), 1,3-dihydroxyacetone (900mg, 10.0mmol) and 2.0M hydrochloric acid (0.50mL) and NaBH3CN (628mg, 10.0mmol) were added successively. The mixture was heated for 16h at 70C with stirring and cooled to rt and then evaporated. The residual product was dissolved in H2O (25mL) and adjusted to pH=4.0 with 2.0M hydrochloric acid under ice bath. The mixture was stirred at rt and adjusted to pH=4.5 with 1.0M NaOH and then concentrated. The residual product was eluted from a column of Amberlite CG-50 (NH4+) resin (100mL) with deionized water (100mL) and 1.0M aqueous ammonia (200mL) successively to give white product (550mg, 82%). (0031) Amorphous white solid, mp 161.5-162.1C (lit.11 mp 162-163C). [alpha]D25 +25.9(c 1.02, H2O) (lit.11 [alpha]D25 +26.2(c 1.00, H2O)). FT-IR (Neat): numax=3453, 3295, 2928, 1417, 1115, 1086, 1055, 1031, 992, 848cm-1; 1H NMR (400MHz, D2O): delta=3.77 (t, J=9.7Hz, 1H), 3.66 (dd, J=8.4, 3.8Hz, 1H), 3.63 (dd, J=6.9, 4.8Hz, 2H), 3.58 (dd, J=12.1, 3.9Hz, 2H), 3.52 (dd, J=11.5, 6.7Hz, 1H), 3.46 (d, J=11.3Hz, 1H, ABq), 3.42 (d, J=11.3Hz, 1H, ABq), 3.35 (d, J=9.5Hz, 1H), 3.32 (d, J=3.5Hz, 1H) ppm. 13C NMR (101MHz, D2O): delta=76.67, 74.50, 73.55, 72.47, 65.60, 62.65, 58.93, 56.93, 54.87, 29.75ppm.
3 g
The valionamine (3.0 g) prepared in Example 4 was dissolved in 75 ml of N,N-dimethylformamide, and 1,3-dihydroxyacetone (5.1 g) was added thereto, followed by addition of 2.3 ml of 2M hydrochloric acid at room temperature. After stirring for 1 hour, sodium cyanoborohydride (4.0 g) was added, roomStir for 16 hours. The reaction solution was concentrated, and the residue was poured into water (150 ml), and the pH was acidified, and the mixture was stirred for 1 hour in an ice bath, and passed through a column of DOWEX 50WX8 (H+, 100 ml). After washing, the product was washed with 2 mol/L ammonia water and concentrated. About 30ml, decolorized by activated carbon, the filtrate was concentrated to obtain an oil, and dried in ethanol to obtain 3.5g of white solid, ethanol: water = 7.5:1 mixed solvent.Crystals gave 3.0 g of white crystals.
With 1H-imidazole; xylose isomerase; sodium chloride; magnesium chloride; In water; at 25℃;pH 8;Enzymatic reaction;
Conversion of DHA to D-glyceraldehyde (DGA) [00152] Xylose isomerase used for the production DGA from DHA is bought from commercial sources and used without further purification. The xylose isomerase catalyzed reactions of DGA and DHA in H20 at pH 7.0 or 8.0 in the presence of 24 mM imidazole buffer and 10 mM MgC12 at 25 C and I = 0.1 (NaCl) is monitored by HPLC. The reactions (10 mL volume) are initiated by making a 5-fold dilution of a 50 mM stock solution of DHA (I = 0.1, NaCl) into 30 mM imidazole buffer (I = 0.1, NaCl) containing 12.5 mM MgC12 to give final concentrations of 11 mM substrate, 24 mM imidazole, 10 mM MgC12 and 0.30 mM xylose isomerase for the reaction at pH 7.0 and 0.20 mM xylose isomerase for the reaction at pH 8.0. At timed intervals an aliquot (1 mL) is withdrawn and the solution is adjusted to pH ~ 5 with 10 mu. or 25 mu. of 1 M CH3COOH for reactions at pH 7.0 or 8.0, respectively. Control experiments shows that the drop in pH effectively quenches the slow xylose isomerase catalyzed reaction. The enzyme is removed by ultrafiltration at 5C using a Microcon YM-10 centrifugal filtration device. The filtrate is then flash frozen over solid C02 and stored at -15C. HPLC analyses of the mixture are performed within one week of freezing. HPLC analyses is performed on a XBridge C18, 5muiotaeta, 4.6 x 250 mm column from Waters (Milford, USA). Samples (10 mu,) are injected and eluted with the following conditions. The solvent system used is: Solvent (A): 0.1% (v/v) aqueous trifluoroacetic acid (TFA) and Solvent (B): 0.095% (v/v)TFA in CH3CN/H20 4/1; gradient elution from 10% to 70% B in 30 min, flow rate 1 mL min i; detection at 215 nm. Analysis of the reaction mixture after 24h shows a 1:1 mixture of DHA and enantiomerically pure DGA.
With pyridine; In dichloromethane; at 20℃; for 16h;Inert atmosphere;
To a mixture of 1,3-dihydroxypropan-2-one (77 g, 0.85 5 mol) and anhydrous pyridine (140 g, 1.76 mol) in anhydrous dichloromethane (2500 mL) under nitrogen at room temperature, was added with palmitic chloride (453 g, 1.64 mol). The mixture was stirred at room temperature for 16 h. It was diluted with MeOH (1000 mL) and water (2000 mL) and stirred for 30 mm. The precipitate was collected by filter and dried to afford mt-i (462 g, 0.8 15 mmol, 95% yield) as a white solid.
95%
With pyridine; In dichloromethane; at 20℃; for 16h;Inert atmosphere;
To a mixture of 1,3-dihydroxypropan-2-one (77 g, 0.855 mol) and anhydrous pyridine (140 g, 1.76 mol) in anhydrous dichloromethane (2500 mL) under nitrogen at room temperature, was added with palmitic chloride (453 g, 1.64 mol). The mixture was stirred at room temperature for 16 h. It was diluted with MeOH (1000 mL) and water (2000 mL) and stirred for 30 min. The precipitate was collected by filter and dried to afford Int-1 (462 g, 0.815 mmol, 95% yield) as a white solid.
With pyridine; In chloroform; at 20 - 25℃; for 3h;
DHA (3.5 gm, 39 mmole) was stirred in 150 ml of chloroform <n="59"/>under flow of N2 at room temperature, followed by sequential dropwise addition of hexadecanoyl chloride (22.1 ml, 80 mmol) and anhydrous pyridine (7.5 ml), in that order. The mixture was stirred for 3 hrs at room temperature followed by extraction (2x) of the chloroform layer with 150 ml water portions. Chloroform was evaporated by rotary evaporation and the remaining solid was re-crystallized using methylene chloride-diethyl ether (1 : 1), Figure 1. 1H-NMR spectra were recorded at room temperature with a Brucker AF-300 spectrometer operating at 300.13 MHz.
With triethylamine; In dichloromethane; at 20℃;Cooling with ice; Inert atmosphere;
10.25 g (40 mmol) of palmitic acid was dissolved in 100 ml of anhydrous dioxane, and 3 drops of N,N-dimethylformamide were added.4.9 g (41 mmol) of thionyl chloride was added dropwise, and the mixture was evaporated to dryness vacuo.1.75 g (20 mmol) of 1,3-<strong>[96-26-4]dihydroxyacetone</strong> was dissolved in 100 ml of anhydrous dichloromethane.5 ml (40 mmol) of triethylamine was added, and the above oil was quickly added dropwise under ice-cooling, and the mixture was filtered under nitrogen, and allowed to react overnight at room temperature. The reaction mixture was concentrated, and the mixture was washed twice with water, and the aqueous layer was washed with chloroform.The crude product was recrystallized from dichloromethane: diethyl ether 1:1.A white solid was obtained.2.9 g (5 mmol) of the above white solid was dissolved in 100 ml of a mixed solvent (THF: benzene: water 10:2:1).Add 0.3g (7.8mmol) of sodium borohydride, react at 5 C for 30min, add 0.9ml glacial acetic acid to stop the reaction, add 80ml chloroform and mix with the reaction solution, wash once with distilled water, wash with 4% sodium bicarbonate solution Once, wash once with saturated brine and dry over anhydrous sodium sulfate.The solvent was distilled off. The crude product was recrystallized from n-hexane:ethyl acetate 97:3. Pure 1,3-dipalmitin was obtained.
1,3-bis-O-(tert-butyldiphenylsilyl)-1,3-dihydroxy-2-propanone (1c). tert-Butylchlorodiphenyl-silane (50 g, 181.9 mmol) was added over a period of 30 min to a chilled (0 C.) solution of 1,3-<strong>[96-26-4]dihydroxyacetone</strong> (1a, 7.28 g, 0.42 mmol) and DMAP (2.47 g, 20.2 mmol) in dry pyridine (100 mL). The reaction mixture was allowed to reach room temperature, and stirring was continued for a total of 4 days. The reaction mixture was poured over crushed ice (500 g) and left overnight. The precipitated solid was filtered and washed with water (ca. 500 mL). The filtrate was extracted with EtOAc (3*150 mL) and the combined organic extract was washed with 1N HCl (3*100 mL), water (2*100 mL), dried (Na2 SO4), and evaporated under reduced pressure to give a white solid. The solid was recrystallized from EtOAc/hexane to give 1c (41.7 g, 91%) as a white solid, mp 100-101 C.; IR (KBr) 1749.7 cm-1; 1 H NMR (CDCl3) delta 1.05 (s, 18 H, C(CH3)3), 4.40 (s, 4 H, CH2 OSi), 7.30-7.60 (m, 20 H, Ph); 13 C NMR delta 19.17, 26.68, 68.53, 127.81, 129.91, 132.58, 135.46. Anal. Calcd for C35 H42 O3 Si2: C,74.15; H, 7.47. Found: C,74.12; H, 7.50.
With 1H-imidazole; In dichloromethane; at 20℃;
Step i:; 1 ,3-<strong>[96-26-4]dihydroxyacetone</strong> 11a1 (964 mg, 10.7 mmol) is dissolved in DCM (25 mL) and imidazole (2.19 g, 32.1 mmol) followed by fert-butyldiphenylchlorosilane (5.8 mL, 22.5 mmol) are added. The mixture is stirred at RT until the reaction is complete, then water is added. The layers are separated; the organics are dried over MgSO4, filtered and concentrated under reduced pressure to afford 11a2 which is used without further purification.
With toluene-4-sulfonic acid; In methanol; at 20℃; for 24h;
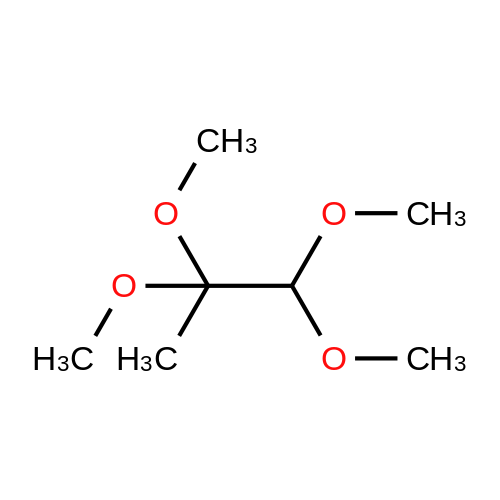
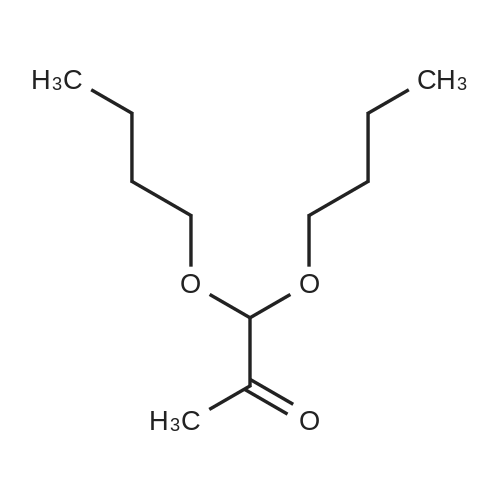
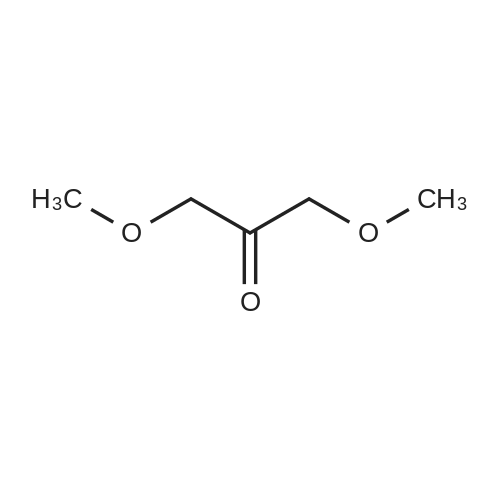
Dihydroxypropanone (DHA) (25 g, 0.139 mol), trimethylorthoformate (30.4 mL, 0.278 mol) andp-toluenesulfonic acid (TsOH) (100 mg) was dissolved in 300 mL of methano in a round-bottom flask, andallowed to stir for 24 h. After that, 300mg Na2CO3 was added, and the reaction mixture was keep stirred foranother 12 h, till most of the salt was dissolved. The solvent and unreacted trimethyl orthoformate wereremoved by evaporation under reduced pressure, the resulting solid was recrystallized from ethyl ether togive 17.5 g of 2,2-dimethoxy-propane-1,3-diol (51%).
A mixture of 1, 3-dihydroxypropan-2-one (24.45 g), trimethyl orthoformate (30 mL), and para-toluenesulfonic acid monohydrate (100 mg) in methanol (300 mL) was stirred overnight at room temperature. Sodium carbonate (300 mg) was added, and the mixture was concentrated. The concentrate was purified by flash chromatography (Analogix, SF65-330, 7% methanol/dichloromethane). MS (ESI) m/z 90.0 (M-OC2H6).
With 1H-imidazole; In N,N-dimethyl-formamide; at 0 - 30℃;
The following example describes a procedure for the TBS -protection of <strong>[96-26-4]dihydroxyacetone</strong> (Preparation of Compound 2), as described in Sodeoka et al., J. Am. Chem. Soc. 1990, 112, 4906, the contents of which are incorporated herein by reference. To a solution of <strong>[96-26-4]dihydroxyacetone</strong> 1 (250. OOg, 2.78mol) and imidazole (472.34g, 6.94mol; 2.5equiv) in DMF (1250mL) TBS-C1 (1045.70g, 6.94mol; 2.5 equiv) was added over about 40min between 0 to 30 C. Then, the mixture was stirred at room temperature until <strong>[96-26-4]dihydroxyacetone</strong> was disappeared (typical reaction time is 4 to 5 hours. Reaction progress was monitored by TLC). After completion of the reaction, ice- water (1500mL) was added to the reaction mixture and extracted with ethyl acetate (1500mL). The aqueous layer was separated and organic layer was washed with water (1500mL), then 20% NaCl aq. (1500mL). The organic layer was evaporated to dryness and the residue was purified by distillation (bp. 135-138 C at 3mmHg) to give 2 as colorless oil (849.83g, 96% yield).
84.5%
With 1H-imidazole; In N,N-dimethyl-formamide; at 20℃; for 12h;
Dihydroxyacetone (2 g, 11.10 mmol) is dissolved in 50 ml of dimethyl formamide (DMF), tert-butyl dimethylsilyl chloride (tBuDMSCl) (4.8 eq, 8.03 g) and imidazole (10 eq, 7.56 g) are added and the mixture is agitated 12 h at 20 C. The mixture is evaporated to dryness, taken up in 150 ml AcOEt. The organic phase is washed with water H2O (2×50 ml), 10% HCl (2×50 ml), saturated NaCl, then dried on Na2SO4 and evaporated under reduced pressure to give 20.1 g of crude product. The mixture is chromatographed on silica with CHex/AcOEt 8/2 as eluent to give 5.96 g of product. Yield: 84.5%. Rf (CH2Cl2/MeOH/AcOH:9/1/0.5): 0.24.
56%
To a stirred solution of <strong>[96-26-4]dihydroxyacetone</strong> (dimer form, 1.8 g, 10 mmol) in dry THF (15 mL) was added N-methylimidazole (9.52 mL, 120 mmol), followed after 15 min by tert-butyldimethylsilyl chloride (6.63 g, 44 mmol) and iodine (0.8 g, 120 mmol). After 16 h, the solvent was evaporated. A solution of the residue in CH2Cl2 was washed with saturated aq. Na2S2O3, brine, and water and evaporated under reduced pressure. Purification of the residue by column chromatography (Hexane: EtOAc, 9.0: 1, Rf 0.7) gave the ketone 9 (7.0 g, 56%) as an oil. [Found: M+Na+ (+ESI), 341.1929, C15H34O3Si2 requires M+Na, 341.1939]; deltaH (400 MHz; CDCl3) 0.07 (12H, s, 2 x Me2Si), 0.90 (18H, s, 2 x tBu), 4.39 (4H, s, 2 x OCH2); deltaC (100 MHz, CDCl3) -5.55 (Me2Si), 18.3 (Me3C), 25.7 (Me3C), 67.9 (OCH2), 208.6 (C=O); IR, numax/cm-1 2931 (C-H), 1731 (C=O), 1472 (C-H), 1253 (C-O).
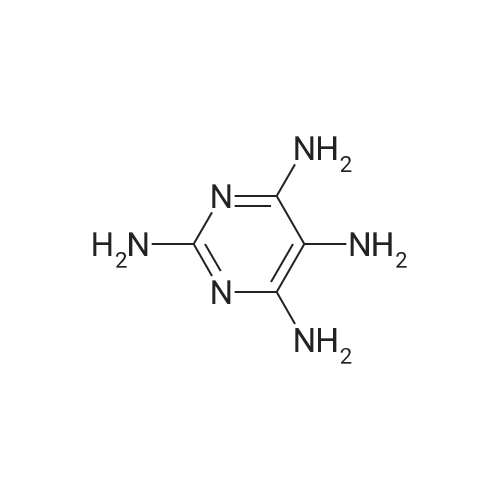
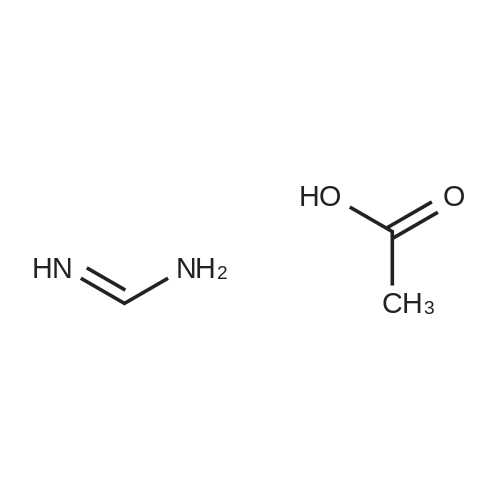
790mg (3.3mmol) of 2,4,5,6 four amino pyrimidine sulfate (Compound 10) was dissolved in 15ml of water, to which was dissolved in 4ml of water was added 690mg (3.3mmol) of barium chloride, the reaction solution was set to open bath for 10 minutes. After spontaneous cooling to room temperature, filtered and precipitated barium sulfate precipitate was washed with water to about 4ml. The combined filtrate and washings were diluted with water to about 50ml, and then added to a previously added 900mg (10mmol) 50ml4M sodium acetate solution of 1,3-dihydroxyacetone, the reaction mixture was stirred at room temperature 24h. After which the reaction flask was transferred to a 4 C refrigerator stored 12 h, then the precipitate was filtered off and washed with ice-water. The precipitate was suspended in 40ml of water, heated to boiling, a small amount of added dropwise 1N aqueous sodium hydroxide solution was suspendedIt was completely clarified. Slowly add 0.2g of activated carbon, stir hot filtration (also preheated funnel). After cooling to room temperature with 1N aqueous hydrochloric acid solution adjusting pH to about 6.0, and the reaction solution was cooled at 4 degree refrigerator 12h. The precipitate was filtered off, washed with ice water, ethanol, ethanol - ether (50:50), washed with ether, and dried in vacuo to give 350mg after compound (11), white solid, a yield of 55.2%
Compound BB-3A-1 (150 g, 1.57 mol), 1,3-<strong>[96-26-4]dihydroxyacetone</strong> (110.28 g, 1.22 mol) and potassium thiocyanate (178.46 g, 1.84 mol) were dissolved in n-butanol (2 L), followed by addition of acetic acid (133.74 mL, 2.34 mol) at room temperature, and stirred for 16 hours at room temperature. 60 mL of water was added to the reaction solution, and stirred for 30 minutes at room temperature, followed by filtration. The filter cake was washed with water and dried to obtain a white powdery compound BB-3A-2 (210 g, yield 77.7%). MS m/z : 173.0 [M+H]+.
To an ice cold solution of 1,3- dihydroxypropan-2-one (1, 25.0 g, 0.277 mol) in dichloromethane (500 mL) was added 4- dimethylaminopyridine (10.17 g, 0.083 mol) and pyridine (49.2 mL, 0.610 mol) and stirred for next 5 min. To the above mixture octanoyl chloride (2, 105.4 mL, 0.610 mol) was added dropwise at 0 C and the reaction mixture was stirred at room temperature for 16h. After completion, reaction mixture was filtered; the solid was washed with dichloromethane (100 mL), filtrate was washed with brine (200 mL), saturated solution of sodium bicarbonate (200 mL) and 0.1 N HC1 solution (100 mL). Organic layer was separated and dried over anhydrous sodium sulfate and solvent was removed under reduced pressure to get crude. The crude was purified by silica gel (100-200 mesh) column chromatography eluting with 10% ethyl acetate in hexanes to afford the desired product as white solid. Yield: 70.0 g, 73%. MS (ESI) m/z 343.19[M+1]+; 1H NMR (400 MHz, DMSO-d6); delta 4.84 (s, 4H), 2.37 (t, = 7.2 Hz, 4H), 1.45-1.62 (m, 4H), 1.15-1.35 (m, 16H), 0.78-0.92 (m, 6H).
73%
To an ice cold solution of 1,3-dihydroxypropan-2-one (1, 25.0 g, 0.277 mol) in dichloromethane (500 mL) was added 4- dimethylaminopyridine (10.17 g, 0.083 mol) and pyridine (49.2 mL, 0.610 mol) and stined for next 5 mm. To the above mixture octanoyl chloride (2, 105.4 mL, 0.610 mol) was added dropwise at 0 C and the reaction mixture was stined at room temperature for 16h. After completion, reaction mixture was filtered; the solid was washed with dichloromethane (100 mL), filtrate was washed with brine (200 mL), saturated solution of sodium bicarbonate (200 mL) and 0.1 N HC1 solution (100 mL). Organic layer was separated and dried over anhydrous sodium sulfate and solvent was removed under reduced pressure to get crude. The crude was purified by silica gel (100-200 mesh) column chromatography eluting with 10% ethyl acetate in hexanes to afford the desired product as white solid. Yield: 70.0 g, 73%;MS (ESI) mlz 343.19[M+1]?H NMR (400 MHz, DMSO-d6); oe 4.84 (s, 4H), 2.37 (t, J = 7.2 Hz, 4H), 1.45-1.62 (m, 4H), 1.15-1.35 (m, 16H), 0.78-0.92 (m, 6H).
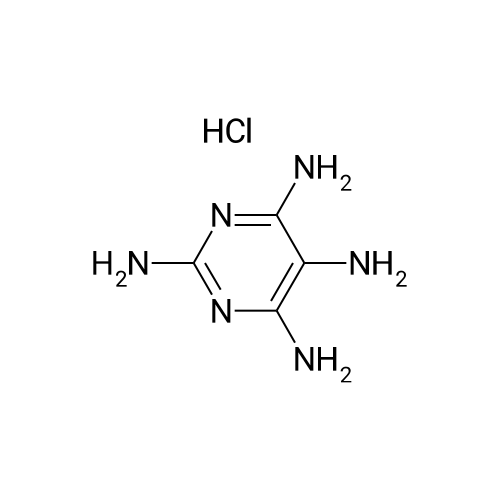
2,4,5,6-Tetraminopyrimidine.H2SO4.H2O (75.0 g, 0.293 mole) was added to a stirred solution of BaCl2.2H2O (71.5 g, 0.293 mole) in H2O (1.45 l.) at 85-90 C. The mixture was stirred rapidly at about 90 C. for 15 min, cooled to 40 C., and filtered from BaSO4, which was washed thoroughly on a funnel with H2O. The clear, yellow filtrate was then diluted further with H2O to give a volume of 4.35 l. This solution of the tetraminopyrimidine.2HCl was then added to a solution of NaOAc (4.35 l. of 4 N) in which dihydroxyacetone (79.3 g, 0.88 mole) and cysteine.HCl.H2O (51.5 g, 0.293 mole) had just been dissolved. The resulting solution was stirred mechanically at room temperature while a slow stream of air was continuously passed through it for 26 hr. (Yellow-orange solid began separating after 2 hr.) The mixture was then kept in a refrigerator for 16 hr before the solid was collected, washed successively with cold H2O, EtOH, and Et2O before it was dried to constant weight in vacuo over P2O5 at 25 C. [The crude product mixture (47 g) was weighed in order to obtain an estimate of the volume of 48% HBr required to form hydrobromide salts.] A mechanically stirred mixture of the dried solid and EtOH (6.05 l.) was heated to 70 C., and a solution of 48% HBr (28 ml) in EtOH (490 ml) was added in a thin stream while the mixture was maintained at 70-75 C. The mixture was then refluxed for about 5 min with rapid stirring while nearly all of the solid dissolved. The hot solution was treated with Norit and filtered through a Celite mat. The clear yellow filtrate was kept in a refrigerator overnight while a first crop of orange-colored solid separated. The collected solid was washed with EtOH, then dried in vacuo (56 C. over P2O5) to give 17.2 g of product. The filtrate was concentrated by evaporation (rotary evaporator, H2O aspirator, bath to 35 C.) to about 2 l. and then refrigerated to give a second crop, which was dried as before, of 10.2 g; total yield 27.4 g (34%). The 1H NMR spectrum of this material in CF3CO2D showed it to contain a barely detectable amount of methyl substituted 2,4-diaminopteridine.HBr as evidenced by very weak signals at delta2.83 (CH3) and delta8.85 (pteridine ring H). Strong signals produced by the desired product occur at delta5.28 (6-CH2O) and delta9.08 (C7-H). The proportion of desired product to the methyl-substituted contaminant was estimated from the 1H NMR integrals to be 20:1. The 1H NMR spectrum also revealed retention of a small amount of EtOH in the product dried as described but not enough to interfere with the conversion of it to 2.
With sodium acetate; DL-cysteine hydrochloride; In water; at 20℃; for 26h;
2,4,5,6-Tetraminopyrimidine.H2SO4.H2O (75.0 g, 0.293 mole) was added to a stirred solution of BaCl2.2H2O (71.5 g, 0.293 mole) in H2O (1.45 l.) at 85-90 C. The mixture was stirred rapidly at about 90 C. for 15 min, cooled to 40 C., and filtered from BaSO4, which was washed thoroughly on a funnel with H2O. The clear, yellow filtrate was then diluted further with H2O to give a volume of 4.35 l. This solution of the tetraminopyrimidine.2HCl was then added to a solution of NaOAc (4.35 l. of 4 N) in which dihydroxyacetone (79.3 g, 0.88 mole) and cysteine.HCl.H2O (51.5 g, 0.293 mole) had just been dissolved. The resulting solution was stirred mechanically at room temperature while a slow stream of air was continuously passed through it for 26 hr. (Yellow-orange solid began separating after 2 hr.) The mixture was then kept in a refrigerator for 16 hr before the solid was collected, washed successively with cold H2O, EtOH, and Et2O before it was dried to constant weight in vacuo over P2O5 at 25 C. [The crude product mixture (47 g) was weighed in order to obtain an estimate of the volume of 48% HBr required to form hydrobromide salts.] A mechanically stirred mixture of the dried solid and EtOH (6.05 l.) was heated to 70 C., and a solution of 48% HBr (28 ml) in EtOH (490 ml) was added in a thin stream while the mixture was maintained at 70-75 C. The mixture was then refluxed for about 5 min with rapid stirring while nearly all of the solid dissolved. The hot solution was treated with Norit and filtered through a Celite mat. The clear yellow filtrate was kept in a refrigerator overnight while a first crop of orange-colored solid separated. The collected solid was washed with EtOH, then dried in vacuo (56 C. over P2O5) to give 17.2 g of product. The filtrate was concentrated by evaporation (rotary evaporator, H2O aspirator, bath to 35 C.) to about 2 l. and then refrigerated to give a second crop, which was dried as before, of 10.2 g; total yield 27.4 g (34%). The 1H NMR spectrum of this material in CF3CO2D showed it to contain a barely detectable amount of methyl substituted 2,4-diaminopteridine.HBr as evidenced by very weak signals at 62.83 (CH3) and 68.85 (pteridine ring H). Strong signals produced by the desired product occur at delta5.28 (6-CH2O) and 69.08 (C7-H). The proportion of desired product to the methyl-substituted contaminant was estimated from the 1H NMR integrals to be 20:1. The 1H NMR spectrum also revealed retention of a small amount of EtOH in the product dried as described but not enough to interfere with the conversion of it to 2.
To an ice cold solution of 1,3- dihydroxypropan-2-one (1, 30.0 g, 0.33 mol) in dichloro methane (500 mL) was added 4- dimethylaminopyridine (20.30 g, 0.167 mol) and pyridine (107 mL, 0.1.332 mol) and stirred for next 5 min. To the above mixture dodecanoyl chloride 2A (218.50 g, 1.167 mol) was added dropwise at 0 C and the reaction mixture was stirred at room temperature for 16h. After completion, reaction mixture was filtered; the solid was washed with dichloro methane (100 mL), filtrate was washed with brine (200 mL), saturated solution of sodium bicarbonate (200 mL) and 0.1 N HC1 solution (100 mL). Organic layer was separated and dried over anhydrous sodium sulfate and solvent was removed under reduced pressure to get crude. The crude was triturated with diethyl ether to afford the desired product 3A as white solid. Yield: 78 g, 51%. MS (ESI) m/z 455.37[M+1]+;
With selenium(IV) oxide; sodium acetate; In water;
1. 2,4-DIAMINO-6-HYDROXYMETHYLPTERIDINE SPC2 A solution of 6.4 grams of barium chloride in a minimum amount of hot water was added with stirring at a temperature of 70-80 C to a suspension of 7.6 grams of 2,4,5,6-tetraaminopyrimidine sulphate in 104 ml water. The resulting suspension was stirred for 30 minutes and the formed barium sulphate was removed by filtration and washed on the funnel with 26 ml water at a temperature of 70 C. The solution containing the 2,4,5,6-tetraaminopyrimidine dihydrochloride is diluted with water to a final volume of 400 ml. A solution of 128 grams of sodium acetate, 136 grams of bisulphite addition product of 1,3-dihydroxyacetone (free of methyl glyoxal) and 46 grams of cysteine hydrochloride in 390 ml water was prepared at room temperature in a 2 liter three-necked flask fitted with a stirrer, an air-bubbling system and a dropping funnel. To this solution, the 400 ml of the previously prepared solution of 2,4,5,6-tetraaminopyrimidine dihydrochloride were added with energic stirring and air-bubbling. A solution of 8 g of selenium dioxide dissolved in the minimum amount of water was made. Half of this solution was added to the reaction mixture immediately after the addition of the tetraaminopyrimidine solution and the other half 4-7 hours later. The reaction was allowed to proceed for 24 hours at room temperature. The reaction can be carried out in a similar manner in a range of temperatures from 20 to 100 C, but the yield is lower. After the end of the reaction, the solution is kept 1 hour at 4 C. The precipitate was filtered off, washed on the funnel with cold alcohol, alcohol:ethyl ether (1:1) and ethyl ether, then dried under vacuum for 24 hours at 50. The yield is 3,8 g (72 %) of 2,4-diamino-6-hydroxymethylpteridine.
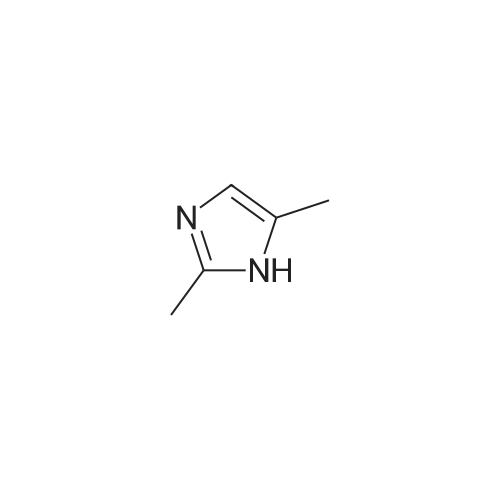
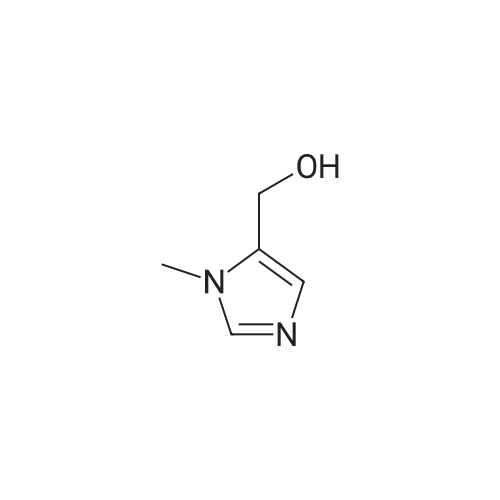
31-1) 5-Hydroxymethyl-1-methylimidazole The title compound was obtained in a yield of 32percent according to the procedure described in J. M. Dener, L-H Zhang, H. Rapoport, J. Org. Chem., 1993, 58, 1159 using dihydroxyacetone and methylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta3.67(s, 3H), 4.58(s, 2H), 5.37(brs, 1H), 6.76(s, 1H), 7.32(s, 1H)
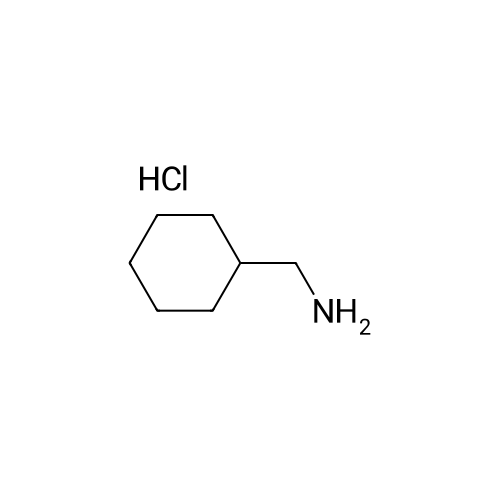
34-1) 5-Hydroxymethyl-1-cyclohexylmethylimidazole The title compound was obtained in a yield of 45percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and cyclohexylmethylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta0.94(m, 2H), 1.16(m, 3H), 1.50-1.70(m, 6H), 3.65(d, 2H), 4.24(brs, 1H), 4.60(s, 2H), 6.85(s, 1H), 7.45(s, 1H)
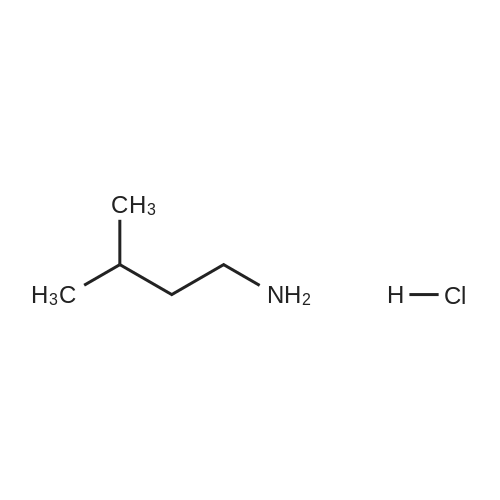
38-1) 5-Hydroxymethyl-1-(3-methyl)butylimidazole The title compound was obtained in a yield of 52percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and isoamylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta0.90(d, 6H), 1.32(m, 2H), 1.65(m, 1H), 3.67(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
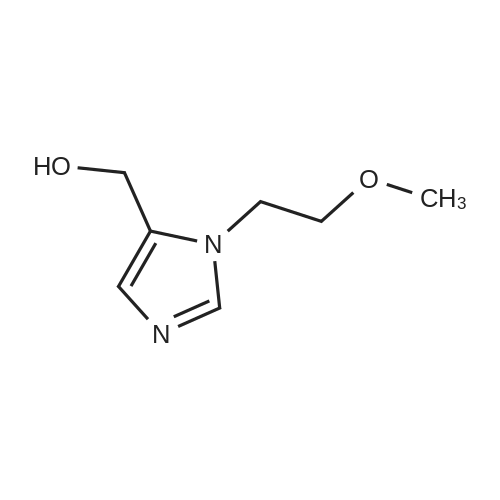
39-1) 5-Hydroxymethyl-1-(2-methoxy)ethylimidazole The title compound was obtained in a yield of 60percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 2-methoxyethylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta3.38(s, 3H), 3.42(t, 2H), 3.65(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
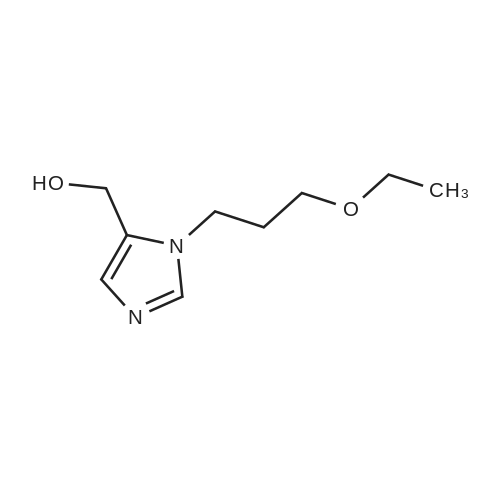
41-1) 5-Hydroxymethyl-1-(3-ethoxy)propylimidazole The title compound was obtained in a yield of 61percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 3-ethoxypropylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta1.20(t, 3H), 1.72(m, 2H), 3.50(s, 4H), 3.63(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
47-1) 5-Hydroxymethyl-1(4-methylbenzyl)imidazole The title compound was obtained in a yield of 65percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 4-methylbenzylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta2.32(s, 3H), 4.50(s, 2H), 5.19(s, 2H), 6.95(s, 1H), 7.05(d, 2H), 7.15(d, 2H), 7.59(s, 1H)
43-1) 5-Hydroxymethyl-1-(4-methoxybenzyl)imidazole The title compound was obtained in a yield of 30percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 4-methoxybenzylamine hydrochloride as starting materials. 1H NMR(CDCl3+CD3OD) delta3.75(s, 3H), 4.50(s, 2H), 5.15(s, 2H), 6.86(m, 3H), 7.08(d, 2H), 7.42(s, 1H)
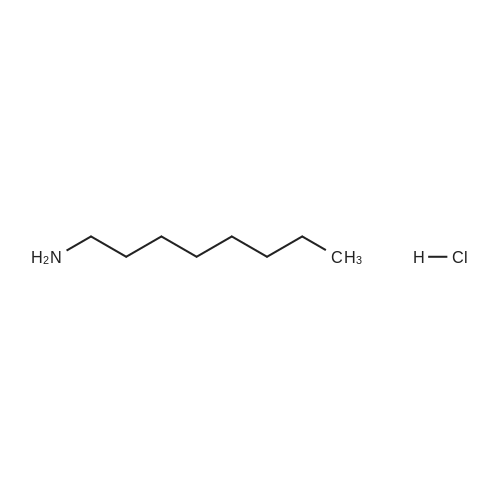
36-1) 5-Hydroxymethyl-1-octylimidazole The title compound was obtained in a yield of 52percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and octylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta0.88(t, 3H), 1.18(brs, 2H), 1.30(brs, 10H), 1.42(m, 2H), 3.67(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
48-1) 5-Hydroxymethyl-1-(3-methylbenzyl)imidazole The title compound was obtained in a yield of 60percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 3-methylbenzylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta2.27(s, 3H), 4.45(s, 2H), 4.52(br, 1H), 5.13(s, 2H), 6.80(d, 1H), 6.90(m, 2H), 7.08(m, 1H), 7.17(m, 1H), 7.34(s, 1H)
44-1) 5-Hydroxymethyl-1-(3-chlorobenzyl)imidazole The title compound was obtained in a yield of 60percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 3-chlorobenzylamine hydrochloride as starting materials. 1H NMR(CDCl3+CD3OD) delta3.81(s, 3H), 4.47(s, 2H), 5.25(s, 2H), 6.99(s, 1H), 7.05(m, 1H), 7.14(s, 1H), 7.30(d, 2H), 7.61(s, 1H)
37-1) 5-Hydroxymethyl-1-decylimidazole The title compound was obtained in a yield of 52percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and decylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta0.88(t, 3H), 1.04(brs, 2H), 1.30(brs, 14H), 1.42(m, 2H), 3.68(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
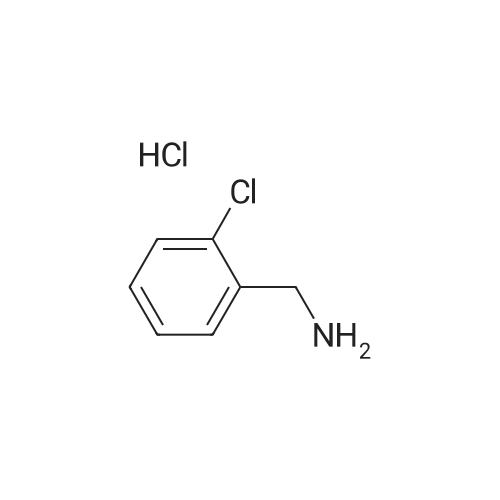
45-1) 5-Hydroxymethyl-1-(2-chlorobenzyl)imidazole The title compound was obtained in a yield of 60percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and <strong>[22680-44-0]2-chlorobenzylamine hydrochloride</strong> as sting materials. 1H NMR(CDCl3) delta3.24(s, 2H), 4.44(s, 2H), 5.26(s, 2H), 6.78(d, 1H), 6.90(s, 1H), 7.15(m, 1H), 7.21(m, 1H), 7.34(d, 1H), 7.38(s, 1H)
35-1) 5-Hydroxymethyl-1-pentylimidazole The title compound was obtained in a yield of 50percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and pentylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta0.90(t, 3H), 1.08(brs, 2H), 1.30(m, 4H), 1.45(m, 2H), 3.64(t, 2H), 4.24(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
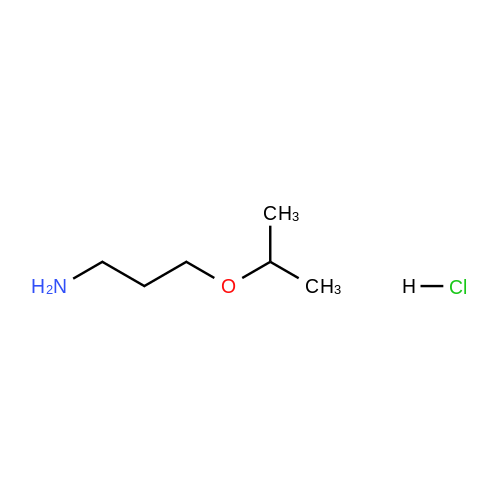
40-1) 5-Hydroxymethyl-1-(3-methoxy)propylimidazole The title compound was obtained in a yield of 61percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 3-methoxypropylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta1.72(m, 2H), 3.32(s, 3H), 3.46(t, 2H), 3.63(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
42-1) 5-Hydroxymethyl-1-(3-isopropoxy)propylimidazole The tide compound was obtained in a yield of 61percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 3-isopropoxypropylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta1.15(d, 6H), 1.71(m, 2H), 3.45-3.55(m, 3H), 3.63(t, 2H), 4.23(brs, 1H), 4.60(s, 2H), 6.84(s, 1H), 7.44(s, 1H)
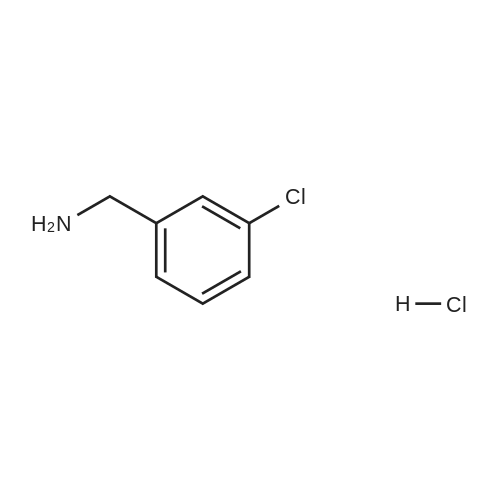
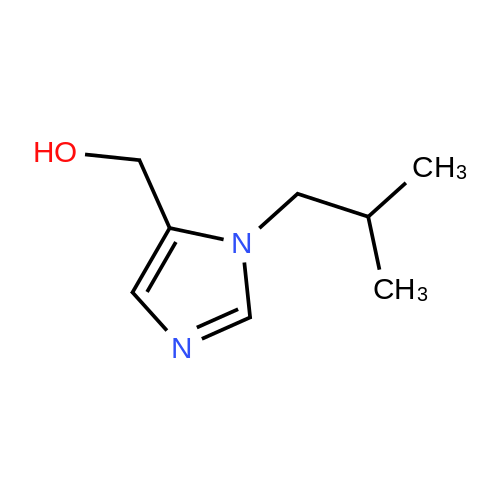
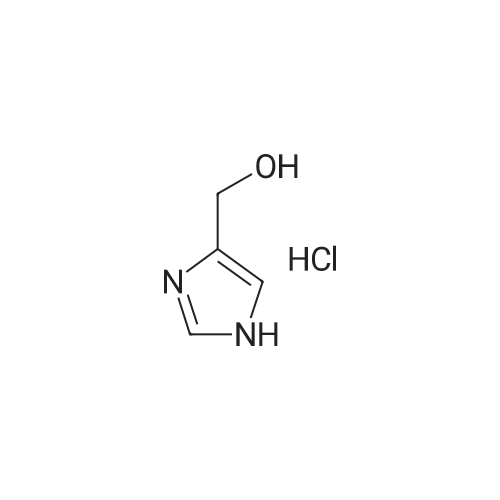
(2-fluorophenyl)methanamine hydrochloride[ No CAS ]
[ 38993-84-9 ]
[ 66973-95-3 ]
Yield
Reaction Conditions
Operation in experiment
71%
46-1) 5-Hydroxymethyl-1-(2-fluorobenzyl)imidazole The title compound was obtained in a yield of 71percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and 2-fluorobenzylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta3.25(s, 2H), 4.45(s, 2H), 5.27(s, 2H), 6.79(d, 1H), 7.17(m, 1H), 7.26(m, 1H), 7.35(d, 1H), 7.38(s, 1H)
33-1) 5-Hydroxymethyl-1-isobutylimidazole The title compound was obtained in a yield of 45percent according to the same procedure as Preparation 31-1) using dihydroxyacetone and isobutylamine hydrochloride as starting materials. 1H NMR(CDCl3) delta0.90(d, 6H), 1.76(m, 1H), 3.62(d, 2H), 4.24(brs, 1H), 4.60(s, 2H), 6.85(s, 1H), 7.45(s, 1H)
With ammonia; In water; isopropyl alcohol; at 0 - 70℃; under 9562.44 - 11769.2 Torr; for 11.5h;
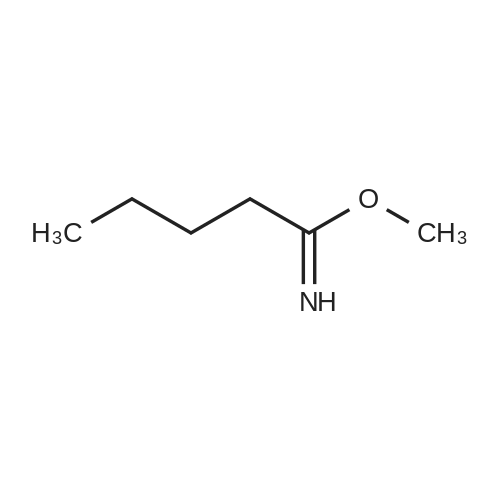
Comparative Example 1; a. Preparation of 2-butyl-4-hydroxymethylimidazole (BHI) 140 g (1.15 mol) methyl pentanimidate prepared according to the method of example 1, 95% purity and 84 g (0.93 mol) 1,3-<strong>[96-26-4]dihydroxyacetone</strong> were charged in 47 ml of isopropyl alcohol and cooled to 0-5 C. Ammonia gas was passed through at a pressure of 13-16 kg/cm2 at a temperature of 65-70 C. while stirring for 6 hrs. The reaction mixture was cooled to room temperature and the ammonia pressure was released. The reaction mixture was then subsequently heated to 55-60 C., transferred to 350 ml of deionised water, stirred for 30 min, and cooled to 0-5 C. The cold mixture was stirred for another 5 hrs at 0-5 C., filtered and washed with 25 ml of deionised water. Crude material was recrystallized by dissolution into 100 ml of acetonitrile at 65-70 C. The clear solution was then cooled to 0-5 C. and stirred for another 2 hrs. Finally, the solid material was filtered off, washed with 12.5 ml of acetonitrile and dried under vacuum at 30-40 C. for 6-8 hrs. The yield of 2-butyl-4-hydroxymethylimidazole obtained was 87 g (0.55 mol; 48%) with a purity of 97% by HPLC.
(3R,4S)-1,3,4-trihydroxy-4-(4-nitrophenyl)butan-2-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96%
With (2S,3R)-O-(n-octanoyl)-L-threonine; In 1-methyl-pyrrolidin-2-one; water; at 20℃; for 30h;
General procedure: To a solution of the aldehyde (1 mmol, 1 equiv) and catalyst 1c (0.05 mmol) in NMP (2 equiv water) was added hydroxyacetone or <strong>[96-26-4]dihydroxyacetone</strong> (3 mmol, 3 equiv). The resulting reaction mixture was stirred at room temperature until the aldehyde was consumed as monitored by TLC (30-96 h). The reaction mixture was diluted with ethyl acetate (5 mL) and poured into a half saturated NH4Cl solution. The mixture was extracted with ethyl acetate and the organic layers were combined and washed with brine, dried (Na2SO4), concentrated, and purified by flash column chromatography (mixtures of hexanes/ethyl acetate) to afford the desired aldol products 2a-p.
To a suspension of tetraaminopyrimidine sulfate (7.14 g, 30 mmol) in water is added barium chloride (7.32 g, 30 mmol) at once. The mixture is heated at 100 0C for 10 min and cooled to RT. The solid barium sulfate is removed by filtration. The filtrate is added to a solution of 450 mL of 4 M aqueous sodium acetate solution containing dihydroxyacetone (8 g, 90 mmol) and cysteine hydrochloride monohydrate (3.63 g, 30 mmol) in a 1 liter 3-neck round bottom flask attached with a mechanical stirrer and stirred for 24 h at RT open to air. The precipitated yellow solid is filtered, washed with water, and ethanol and dried overnight in a heated vacuum oven to give 3.4 g (66%) of product. This product is further purified as per the following procedure. The yellow solid is dissolved in 10% acetic acid with aid of few drops of cone. HCl at 75 0C. The hot solution is treated with activated charcoal and filtered. The filtrate is neutralized with cone. NH4OH. The bright yellow solid is collected, washed with water, water-ethanol and finally ethanol and dried overnight in a heated vacuum oven to provide 2.8 g of the title compound (54%).
In water; at 300℃; for 0.3h;Stainless steel flow reactor;
Example 4; Continuous reactions using the Materials of Example 2 were completed. The yields of products obtained from a continuous run are presented in Table 3. A reaction mixture under high pressure (from approximately 1500 psi to 3500 psi) was pumped sequentially through a high temperature zone (from approximately 200 C. to 325 C.), a heat exchanger, a backpressure regulator, and then into a collection vessel. The reaction mixture was generated by pumping an aqueous solution of glucose at room temperature into a heated source of 2-propanol (temperature from approximately 275 C. to 325 C.) which was being pumped into the high temperature zone. aContinuous reaction pumped through a stainless steel tube having about a 5 min residence time and a length to diameter ratio of about 20. Temperature was 300 C., and the sample was collected over a period of 18 min. Glucose input during this time was 0.64 g and solvent included 32 mL of 2-propanol/5% water. ball yields given in grams per gram starting material cDetected by NMR in the D2O solution as the hydrate but reported as the aldehyde. disopropyl ester tentatively assigned as isopropyl glycolate eisopropyl ester tentatively assigned as isopropyl acetate funidentified products not soluble in alcohol gunidentified products soluble in alcohol htotal identified products derived from carbohydrate material iproduced by the oxidation of 2-propanol
With carbon nanotube supported gold nanoparticles (0.5 wt%); oxygen; sodium hydroxide; at 60℃; under 2250.23 Torr; for 2h;
General procedure: The standard oxidation experiments were carried out with oxygen under 3 bar, at 60 C. NaOH solution (NaOH/glycerol molar ratio = 2) and catalyst (700 mg) were added to a 0.3 M aqueous solution of glycerol (V = 150 mL) under stirring at 1000 rpm. Additionally, a limited number of runs were carried out at different oxygen pressures (6, 10 bar) or temperatures (40, 80 C). After heating under nitrogen to the selected temperature, the reaction was initiated by switching from inert gas to oxygen.
With carbon nanotube supported gold nanoparticles (0.5 wt%); oxygen; sodium hydroxide; at 80℃; under 2250.23 Torr; for 2h;
General procedure: The standard oxidation experiments were carried out with oxygen under 3 bar, at 60 C. NaOH solution (NaOH/glycerol molar ratio = 2) and catalyst (700 mg) were added to a 0.3 M aqueous solution of glycerol (V = 150 mL) under stirring at 1000 rpm. Additionally, a limited number of runs were carried out at different oxygen pressures (6, 10 bar) or temperatures (40, 80 C). After heating under nitrogen to the selected temperature, the reaction was initiated by switching from inert gas to oxygen.
With Escherichia coli fructose-6-phosphate aldolase B; NADH;pH 8.5;Enzymatic reaction;Kinetics;
One unit of FSA activity was defined as the amount that cleaves 1 mumol of d-fructose-6-phosphate per min to afford d-G3P and DHA at pH 8.5 (glycylglycine buffer 50 mM) and 25 C. (0013) Kinetics of FSAB for general donor and acceptor substrates were measured following the corresponding previously described protocol [3c]. Kinetics of FSAA and B for retroaldol reaction from d-fructose and l-<strong>[87-79-6]sorbose</strong> were calculated by measuring the amount of DHA formed using a glycerol dehydrogenase from Cellulomonas sp. (GDHcell). One mL of each kinetic assay contained glycylglycine buffer 50 mM pH 8.5, 1.5 U FSA, substrate concentrations (d-fructose or l-<strong>[87-79-6]sorbose</strong>) from 20 mM to 800 mM, 10 U GDHcell and 0.5 mM NADH. Decrease of A340 nm was proportional to the amount of DHA produced in a reaction (?NADH = 6220 cm-1 M-1) where 1 mmol of NADH oxidized was equivalent to 1 mmol of substrate cleaved.
With ammonium hydroxide; hydrogen; In methanol; water; at 65℃; under 12929.0 Torr; for 6h;Autoclave;
In an autoclave, a mixture of Raney nickel (4 g, wet) and ammonium hydroxide solution (257 g, 28% NH3 in water, 4.2 mol) was heated to 65 C. under 250 psi of hydrogen atmosphere. A solution of 1,3-dihydroxyacetone (38 g, 0.42 mol) in methanol (76 ml) was pumped into the autoclave over a period of 3 h. After the addition was complete, the reaction mixture was maintained at 65 C. and 250 psi hydrogen pressure for additional 3 h. The autoclave was then cooled to ambient temperature and depressurized. The resulting reaction mixture was filtered to remove the catalyst. Ammonia and most of the water were removed by distillation under reduced pressure. GC analysis of the crude product showed a mixture of 91% serinol (2-amino-1,3-propanediol) and 7% bis-adduct (2,2?-iminobis-1,3-propanediol). (0025) An aqueous solution of the crude product mixture was loaded to a column of Amberlyst 15 and eluted with water until neutral pH. GC analysis of the water eluate showed mainly the bis-adduct and other unidentified impurities. The column was then eluted with 300 ml of 4M ammonium hydroxide solution to recover the serinol. The eluate was then stripped under vacuum to remove ammonia and water. The residue was triturated with a 1:1 mixture of 2-butanol and 2-methyltetrahydrofuran to yield crystalline serinol (27 g, 71% yield). Assay by GC: 97%; Assay by titration: 101%
61%
With ammonia; hydrogen; In methanol; water; at 65℃; under 12929.0 Torr; for 5h;
In an autoclave, ammonia gas (13 g, 0.77 mol) was charged to a suspension of Raney nickel (4 g, wet) in methanol (100 ml) and then heated to 65 C. under 250 psi of hydrogen atmosphere. A solution of 1,3-dihydroxyacetone (58% solution in water, 200 g, 1.29 mol) was pumped into the autoclave over a period of 2 h. After the addition was complete, the reaction mixture was continued to hydrogenate at 65 C. temperature and 250 psi hydrogen pressure for an additional 3 h. The autoclave was cooled to ambient temperature and depressurized. The reaction mixture was filtered to remove the catalyst and evaporated under vacuum to remove ammonia and most of the water. The crude product was shown to contain 2.4% serinol and 90% 2,2?-iminobis-1,3-propanediol by GC analysis. (0029) An aqueous solution of the crude product was loaded to a column of Amberlyst 15 and eluted with water until neutral pH. GC analysis of the water eluate showed complete removal of serinol (FIG. 5). The water eluate was then evaporated under vacuum, the residue was dissolved in 1:1 mixture of 2-butanol and tetrahydrofuran, treated with activated carbon, and concentrated to yield the bis-adduct 2,2?-iminobis-1,3-propanediol as a viscous oil (65 g, 61%). Assay by GC: 99%
With sodium hydroxide; In aq. phosphate buffer; at 85℃; for 24h;pH 7.4;
Pyrazine derivatives were synthesized from a one-pot reaction, in which 1,3-dihydroxyacetone, glycolaldehyde, and 2-aminoacetamidine-dihydrobromide (0.10 M 1:1:1 ratio) were mixed in aqueous solution with sodium phosphate (0.25 M) atpH 7.4 by addition of NaOH, then heated to 85 C for 24 h. In a2-mL microvial, reaction contents were de-aerated and purged with nitrogen before being sealed under vacuum. A 1.0 mL aliquot of the product mixture was diluted with 8 mL of water,and a 5 mL portion was then desalted on a 20 mL Discovery DSC-18 column (Supelco, Bellefonte, PA, USA). Material was eluted with 36 mL water and 44 mL 50% methanol/water, collected in 20 separate fractions. Salt-free fractions 10-20(from 37-80 mL eluent) were combined and concentrated to 0.5 mL syrup using a CentriVap centrifuge. After storage at -20 C, the mixture was suspended in 200 muL water, and 50 muL aliquots were further diluted in 300 muL water for adequate ampoule sampling volume.
With molybdenum(VI) oxide; In water; at 100℃; for 4h;
FIG. 8 shows 1H NMR spectra of D-fructose standard solution and of the fructose-containing fraction isolated after reaction of D-fructose with MoO3 in water at 100 C. for 4 h. Sorbose is present in the collected fraction.
With hydrogenchloride; ammonium cerium (IV) nitrate; sodium ethanolate; In ethanol; acetonitrile; at 60℃; for 8.0h;pH 10.0;
A mixture of acetonitrile and ethanol in a volume ratio of 2: 1 was added to the reactor,100 mmol of 1,3-dihydroxyacetone and 200 mmol of formaldehyde were dissolved in an organic solvent,100 mmol of cerium ammonium nitrate was added,Under normal pressure 60 conditions,Adding sodium ethoxide to adjust the pH to 10,15 mmol of catalyst was added,Stirring reaction 8h, obtained 4-hydroxymethyl imidazole,After completion of the reaction,Dropping concentrated HCl,Adjust pH = 2,filter,The filtered solid,Washed with saturated brine,Recrystallization from ethanol,Dried in vacuo to give the product 4-hydroxymethylimidazole hydrochloride.The purity of the prepared 4-hydroxymethylimidazolidine hydrochloride was 98.2%The yield was 87.5%.
A suspension of 346 2-fluoro-benzamidine hydrochloride (3 g, 17.18 mmol), 347 1,3-dihydroxy-propan-2-one (3.17 g, 35.22 mmol), and NH4Cl (4.2 g) in 348 NH4Cl (30 mL) was heated at 80 C. for 1 hr. The reaction mixture was extracted with EtOAc (50 mL×3), washed with brine, dried (Na2SO4) and evaporated in vacuo to give a solid that was purified by column chromatography on silica gel eluting with 2% methanol in 16 DCM to afford the 331 title compound as an off-white solid (2.2 g, 66.6%). 1HNMR (400 MHz, DMSO-d6, 100 C.) 4.50 (s, 2H), 7.01 (s, 1H), 7.25 (m, 2H), 7.38 (m, 1H), 7.99 (m, 1H). LCMS m/z=175 [M-Cl]+
(3R,4S)-1,3,4-trihydroxy-5-(4′-chlorophenyl)-pentan-2-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With Escherichia coli strain BL21-AI D-fructose-6-phosphate aldolase R134V/S166G variant; In acetonitrile; at 30℃;pH 8.0;Darkness; Enzymatic reaction;
Reactions were performed, in triplicate, in50 mM TEA buffer at pH 8.0, 30 C, and 25 rpm. Increasingconcentrations (0.5-5 mM) of 2a-c were set up with thechosen ketone (1a or 1b, 50 mM) and with 1 muM enzyme.Aliquots were removed after 5, 10, 15, and 20 min, and thereactions were terminated by addition of methanol (fortifiedwith the internal standard 3-phenyl-1-propanol at 2 mM) up toa final concentration of 50% (v/v). Samples were centrifugedfor 10 min at 16000g and stored at -20 C. Analysis was performedby reversed phase high-performance liquid chromatography(HPLC) as described above. The peak areas of theproducts were normalized by the average area of the internalstandard. For determination of the product concentration,calibration was performed with increasing concentrations ofreference aldol products. Steady state parameters weredetermined by fitting the Michaelis-Menten equation to theraw data using MMFIT and RFFIT.
With Cu/Al2O3; dihydrogen peroxide; In water; at 25℃; for 24h;Green chemistry;
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35% hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33mum capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
With Cu/Al2O3; dihydrogen peroxide; In water; at 25℃; for 24h;Green chemistry;
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35% hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33mum capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
With Cu/Al2O3; dihydrogen peroxide; In water; at 25℃; for 24h;Green chemistry;
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35% hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33mum capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
With Cu/Al2O3; dihydrogen peroxide; In water; at 25℃; for 24h;Green chemistry;
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35% hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33mum capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
With Cu/Al2O3; dihydrogen peroxide; In water; at 25℃; for 24h;Green chemistry;
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35% hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33mum capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
With Cu/Al2O3; dihydrogen peroxide; In water; at 50℃; for 12h;Green chemistry;
General procedure: The catalyst A 25 mg prepared in Example 1 was weighed, added to a 38 mL reaction tube with magnetic stirring, and then 465 mg (5 mmol) of aniline, 90 mg (1 mmol) of 1,3-dihydroxyacetone, 0.5 mL (6 mmol) 35% hydrogen peroxide and 5 mL chloroform. Thereafter, the temperature was raised to 50 C using an electric heating furnace and maintained for 12 hours. Then, the reaction tube was cooled to room temperature by water cooling, centrifuged at 8000 rpm for 5 minutes using a centrifuge (Shanghai Anting Scientific Instrument Factory), and separated to recover Catalyst A from the reaction mixture. Using N-formylated aniline standard product as a comparison, qualitative and quantitative analysis was performed using HP 6890/5973 GC-MS gas chromatography mass spectrometer and Agilent 7890A (30m×0.25mm×0.33mum capillary column, hydrogen flame ionization detector). A well-known method in the art, such as an industrial rectification process, obtains the target product N-formylated aniline, and the yield results are shown in Table 1 below.
With hydrogenchloride; In water; at 80℃; for 2h;Inert atmosphere;
(3): In a 1-liter three-necked flask,PDO (54 g, 0.3 mol) was added in sequence,Deionized water (500 ml), dissolved in an oil bath at 80 C,After passing nitrogen, hydrochloric acid (2 mol / liter, 3 ml) was added.The hydrolysis reaction was carried out for 2 hours. After the reaction, Add sodium hydroxide solution (2 mol / liter, 3 ml),After mixing well, the oil phase (benzaldehyde) was separated.The aqueous phase was added to 100 ml of dichloromethane to extract the benzaldehyde produced by the reaction, and the layers were separated.The aqueous phase was added to 500 ml of anhydrous ether.Azeotropic distillation to remove unreacted water,Obtain 29 grams of viscous liquid and add 30 ml of absolute ethanol.After mixing uniformly, crystallization is carried out at a low temperature of -10 C.After the obtained crystals were washed with ice-free ether,Dry at 40 C under vacuum,Get 26 grams of white crystals,Is DHA dimer, the yield is 96%,
With 3H(1+)*MoO40W12(3-); oxygen; In water; at 60℃; under 7500.75 Torr; for 20h;Autoclave;
General procedure: Glycerol oxidation was performed in a high-pressure stainless-steelautoclave with a polytetrafluoroethylene insert (10 mL) at a constanttemperature of 60 C. The autoclave was connected to the O2 supplysystem, which kept the pressure constant. The solution was stirredmagnetically. Typically, 5.0 mL of 1.0M glycerol in water was oxidizedin the presence of 4.0mM catalyst at 60 C at aconstant pressure of10 bars of O2. After desired time, the reactor was quickly cooled down,depressurized and the catalyst was removed by extraction with diethylether. The stability of the catalysts after reaction was tested by FTIR andXRD, which did not change compared with the fresh ones [Fig. S3]. Theremaining solution was diluted 10 times with distilled water and analyzedby high performance liquid chromatography (HPLC) using aShimadzu LC10A-VP chromatograph equipped with SPB-10 A UV andRID-10 A R.I. detectors, and a Prevail TM C18 (4.6mm×250 mm) column. A solution of H2SO4 (0.1% w/w) in H2O/acetonitrile (1/2 v/v)was used as the eluent at a flow rateof 1.0 mL min-1 at 50 C. Theglycerol conversion, alpha, and the selectivity for lactic acid (LA), SLA, werecalculated using Eqs. (3) and (4):
With MoO40W12(3-)*Fe(3+); oxygen; In water; at 60℃; under 7500.75 Torr; for 20h;Autoclave;
General procedure: Glycerol oxidation was performed in a high-pressure stainless-steelautoclave with a polytetrafluoroethylene insert (10 mL) at a constanttemperature of 60 C. The autoclave was connected to the O2 supplysystem, which kept the pressure constant. The solution was stirredmagnetically. Typically, 5.0 mL of 1.0M glycerol in water was oxidizedin the presence of 4.0mM catalyst at 60 C at aconstant pressure of10 bars of O2. After desired time, the reactor was quickly cooled down,depressurized and the catalyst was removed by extraction with diethylether. The stability of the catalysts after reaction was tested by FTIR andXRD, which did not change compared with the fresh ones [Fig. S3]. Theremaining solution was diluted 10 times with distilled water and analyzedby high performance liquid chromatography (HPLC) using aShimadzu LC10A-VP chromatograph equipped with SPB-10 A UV andRID-10 A R.I. detectors, and a Prevail TM C18 (4.6mm×250 mm) column. A solution of H2SO4 (0.1% w/w) in H2O/acetonitrile (1/2 v/v)was used as the eluent at a flow rateof 1.0 mL min-1 at 50 C. Theglycerol conversion, alpha, and the selectivity for lactic acid (LA), SLA, werecalculated using Eqs. (3) and (4):
With MoO40W12(3-)*Al(3+); In water; at 60℃; under 7500.75 Torr; for 20h;Autoclave; Inert atmosphere;
General procedure: Glycerol oxidation was performed in a high-pressure stainless-steelautoclave with a polytetrafluoroethylene insert (10 mL) at a constanttemperature of 60 C. The autoclave was connected to the O2 supplysystem, which kept the pressure constant. The solution was stirredmagnetically. Typically, 5.0 mL of 1.0M glycerol in water was oxidizedin the presence of 4.0mM catalyst at 60 C at aconstant pressure of10 bars of O2. After desired time, the reactor was quickly cooled down,depressurized and the catalyst was removed by extraction with diethylether. The stability of the catalysts after reaction was tested by FTIR andXRD, which did not change compared with the fresh ones [Fig. S3]. Theremaining solution was diluted 10 times with distilled water and analyzedby high performance liquid chromatography (HPLC) using aShimadzu LC10A-VP chromatograph equipped with SPB-10 A UV andRID-10 A R.I. detectors, and a Prevail TM C18 (4.6mm×250 mm) column. A solution of H2SO4 (0.1% w/w) in H2O/acetonitrile (1/2 v/v)was used as the eluent at a flow rateof 1.0 mL min-1 at 50 C. Theglycerol conversion, alpha, and the selectivity for lactic acid (LA), SLA, werecalculated using Eqs. (3) and (4):






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






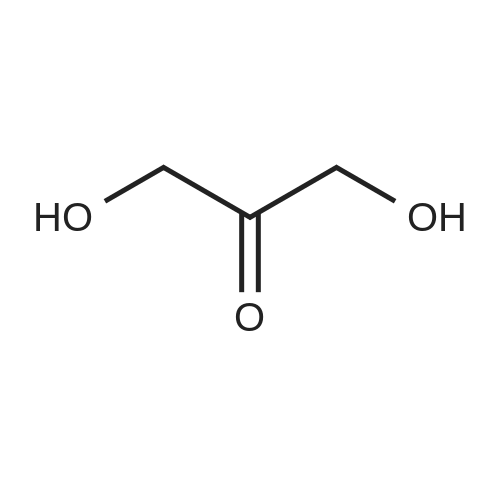

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping