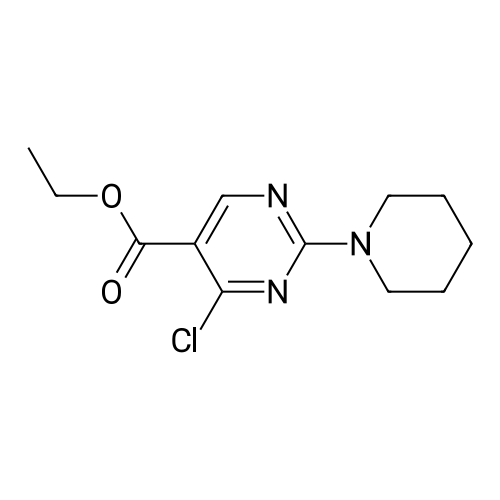
|
|
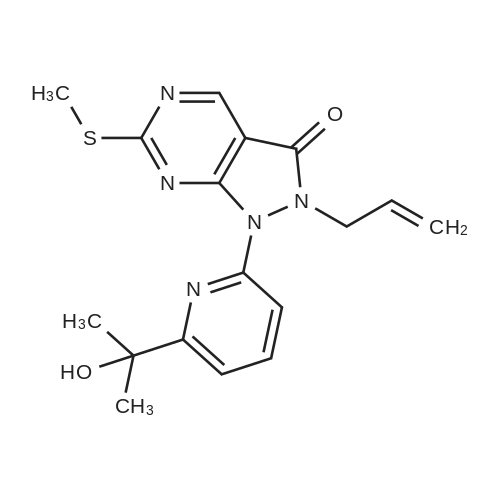
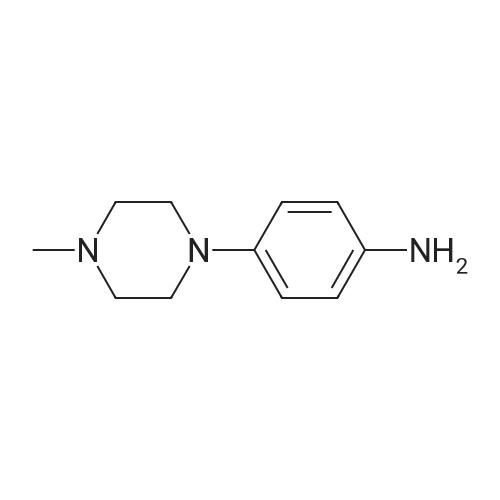
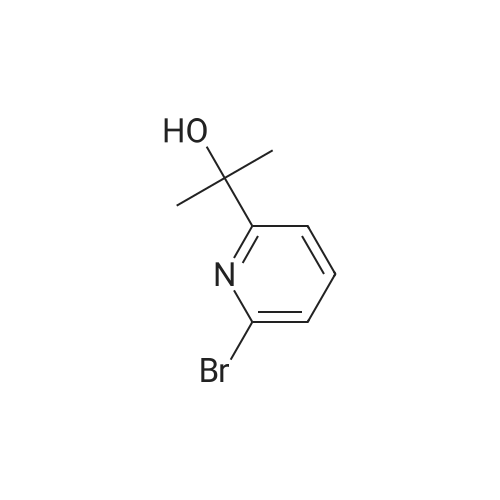
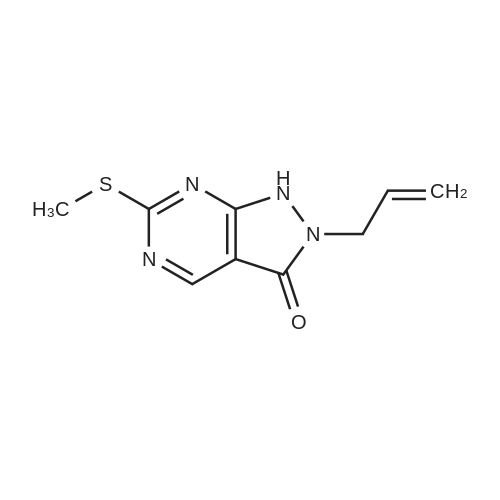
Reference Example 4:Production of Form D of Compound A20.0 g of m-chloroperbenzoic acid (> 65 %) was added to toluene (500 mL) solution of 24.3 g of 2-allyl-l -[6-(I -hydroxy- l-methylethyl)-2-pyridinyl]-6-(methylthio)- 1,2- dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one, and stirred for 40 minutes. 35.5 mL of N,N- diisopropylethylamine and 14.3 g of 4-(4-methylpiperazin-l-yl)aniline were added to the reaction liquid, and stirred overnight. Tetrahydrofuran (500 mL) and aqueous saturated sodium hydrogen carbonate solution were added to the reaction liquid, extracted with ethyl acetate, washed with brine, and dried with anhydrous magnesium sulfate. After the solvent was evaporated away, the solid was collected by filtration and washed with ethyl acetate to give 11.Og of the crude entitle compound. The filtrate was concentrated and the residue was purified through silica gel column chromatography (hexane/ethyl acetate = 1/1 to 0/1 and chloroform/methanol = 1/0 to 7/1). After the solvent was evaporated away, the solid was collected by filtration and washed with ethyl acetate to give 16.9g of the entitle compound. The filtrate was concentrated and the residue was purified through silica gel basic column chromatography (hexane/ethyl acetate = 1/1 to 0/1, ethyl acetate/ethanol = 98/2). After the solvent was evaporated away, the solid was collected by filtration and washed with ethyl acetate to give 2.5Og of the entitle compound. The combined crude title compounds (30.4g) was recrystallized from isopropanol (300 mL) to obtain 32.2g of the entitled compound as 1 isopropanol adduct. The entitled compound 1 isopropanol adduct (32.2g) was dissolved in ethyl acetate (300 mL) under reflux and the solution was stirred over night in room temperature. The solid was collected by filtration and dried in vacuo to obtain 21.2g of the entitled compound as a yellow solid. XRPD patterns:(2 theta(degrees), Intensity(cps)): (6.5, 71.0), (10.3, 61.5), (9.3, 40.8), (14.6, 22.5), (18.7, 22.0), (19.5, 55.9), (22.2, 32.2). DSC:When DSC of Form D was measured using a TA Instruments DSC Ql 000 instrument, the extrapolated melting temperature onset of Form D was 173C with an enthalpy of fusion of 66.2 J/g at 5 C/min under nitrogen in crimped aluminum pan. The peak melting temperature was 174C. Before melting, a small endotherm peak with form conversion was detected at 135~150C. |
| 1.2 g |
|
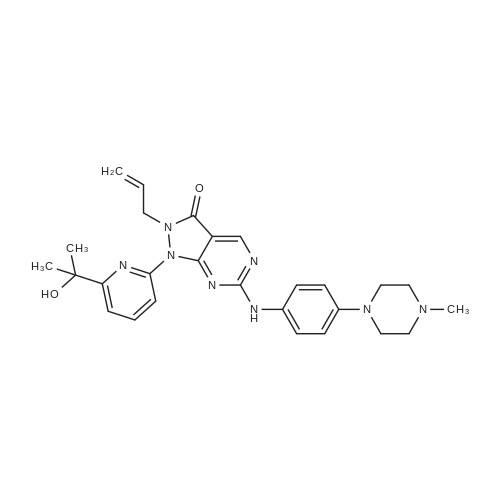
817 mg of m-chloroperbenzoic acid (> 65%) was added to toluene (20 mL) solution of 1.10 g of the above produce, and stirred for 20 minutes. 1.61 mL of N,N- diisopropylethylamine and 706 mg of 4-(4-methylpiperazin-l-yl)aniline were added to the reaction liquid, and stirred overnight. Aqueous saturated sodium hydrogencarbonate solution was added to the reaction liquid, extracted with ethyl acetate, washed with saturated saline water, and dried with anhydrous magnesium sulfate. The solvent was evaporated away, and the residue was purified through basic silica gel column chromatography (hexane/ethyl acetate = 1/1 to 0/1, ethyl acetate/ethanol = 98/2). After concentrated, this was recrystallized from ethyl acetate to obtain 1.20 g of the entitled compound as a yellow solid. iH-NMR (400 MHz, CDCI3) delta: 8.83 (IH, s), 7.86 (IH, dd, J=8.0, 7.8 Hz), 7.75 (IH, d, J=7.3 Hz), 7.49 (IH, brs), 7.48 (2H, d, J=9.0 Hz), 7.34 (IH, d, J=7.4 Hz), 6.93 (2H, d, J=9.0 Hz), 5.70 (IH, ddt, J=17.2, 10.0, 6.5 Hz), 5.04 (IH, d, J=10.0 Hz), 4.94 (IH, d, J=17.2 Hz), 4.74 (2H, d, J=6.5 Hz), 3.26 (4H, t, J=4.8 Hz), 2.73 (4H, brs), 2.44 (3H, s), 1.59 (6H, s). ESI-MS Found: m/z[M+H]+ 501 |
| 1.2 g |
|
Step 3) Production of 2-allyl-l-[6-(l -hydroxy- l-methylethyl)pyridin-2-yl]-6- [4-(4- methylpiperazin- 1 -yl)phenyl] amino} - 1 ,2-dihydro-3H-pyrazolo[3 ,4-d]pyrimidin- 3 -one: 817 mg of m-chloroperbenzoic acid (> 65%) was added to toluene (20 mL) solution of 1.10 g of the above produce, and stirred fro 20 minutes. 1.61 mL of N,N- diisopropylethylamine and 706 mg of 4-(4-methylpiperazin-l-yl)aniline were added to the reaction liquid, and stirred overnight. Aqueous saturated sodium hydrogencarbonate solution was added to the reaction liquid, extracted with ethyl acetate, washed with saturated saline water, and dried with anhydrous magnesium sulfate. The solvent was evaporated away, and the residue was purified through basic silica gel column chromatography (hexane/ethyl acetate = 1/1 to 0/1, ethyl acetate/ethanol = 98/2). After concentrated, this was recrystallized from ethyl acetate to obtain 1.20 g of the entitled compound as a yellow solid. iH-NMR (400 MHz, CDCI3) delta: 8.83 (IH, s), 7.86 (IH, dd, J=8.0, 7.8 Hz), 7.75 (IH, d, J=7.3 Hz), 7.49 (IH, brs), 7.48 (2H, d, J=9.0 Hz), 7.34 (IH, d, J=7.4 Hz), 6.93 (2H, d, J=9.0 Hz), 5.70 (IH, ddt, J=17.2, 10.0, 6.5 Hz), 5.04 (IH, d, J=10.0 Hz), 4.94 (IH, d, J=17.2 Hz), 4.74 (2H, d, J=6.5 Hz), 3.26 (4H, t, J=4.8 Hz), 2.73 (4H, brs), 2.44 (3H, s), 1.59 (6H, s). ESI-MS Found: m/z[M+H]+ 501. |
|
|
817 mg of m-chloroperbenzoic acid (>65%) was added to toluene (20 mL) solution of 1.10 g of the above produce, and stirred for 20 minutes. 1.61 mL of N,N-diisopropylethylamine and 706 mg of 4-(4-methylpiperazin-1-yl)aniline were added to the reaction liquid, and stirred overnight. Aqueous saturated sodium hydrogencarbonate solution was added to the reaction liquid, extracted with ethyl acetate, washed with saturated saline water, and dried with anhydrous magnesium sulfate. The solvent was evaporated away, and the residue was purified through basic silica gel column chromatography (hexane/ethyl acetate=1/1 to 0/1, ethyl acetate/ethanol=98/2). After concentrated, this was recrystallized from ethyl acetate to obtain the entitled compound as a yellow solid. 1H-NMR (400 MHz, CDCl3) delta: 8.83 (1H, s), 7.86 (1H, dd, J=8.0, 7.8 Hz), 7.75 (1H, d, J=7.3 Hz), 7.49 (1H, brs), 7.48 (2H, d, J=9.0 Hz), 7.34 (1H, d, J=7.4 Hz), 6.93 (2H, d, J=9.0 Hz), 5.70 (1H, ddt, J=17.2, 10.0, 6.5 Hz), 5.04 (1H, d, J=10.0 Hz), 4.94 (1H, d, J=17.2 Hz), 4.74 (2H, d, J=6.5 Hz), 3.26 (4H, t, J=4.8 Hz), 2.73 (4H, brs), 2.44 (3H, s), 1.59 (6H, s). ESI-MS Found: m/z[M+H]+ 501. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping