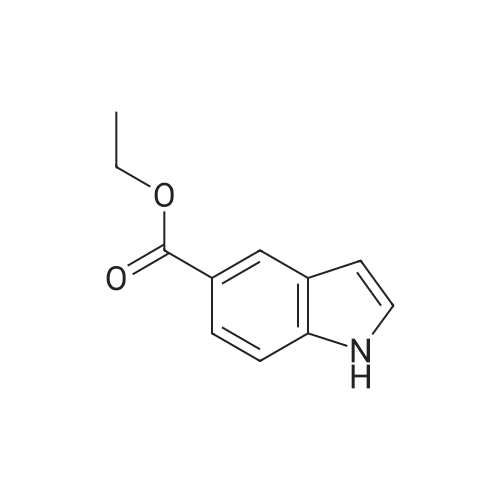
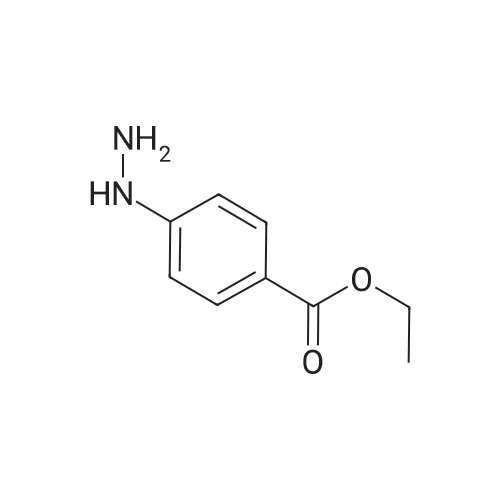
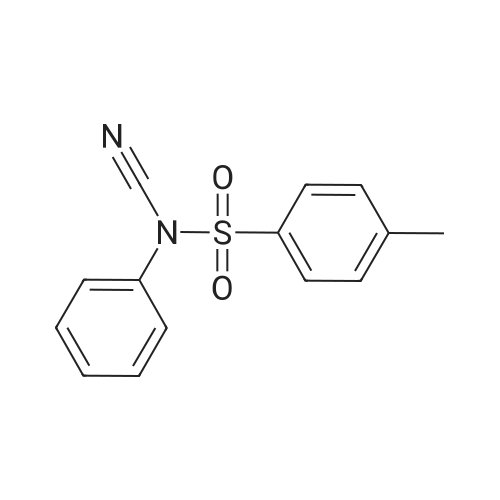
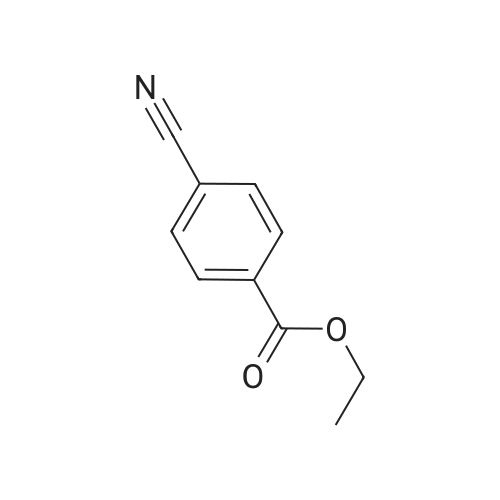
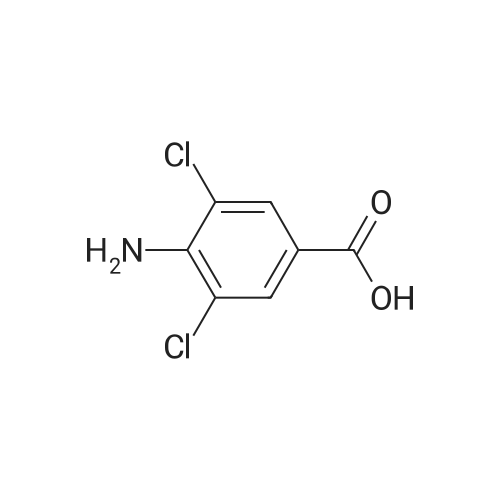
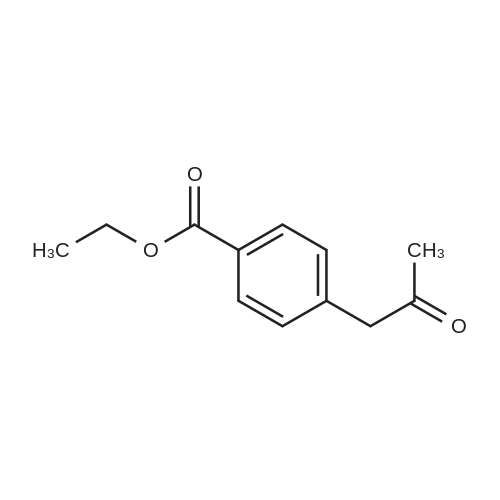
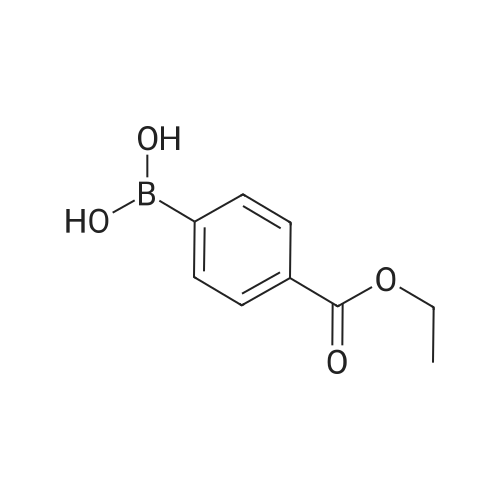
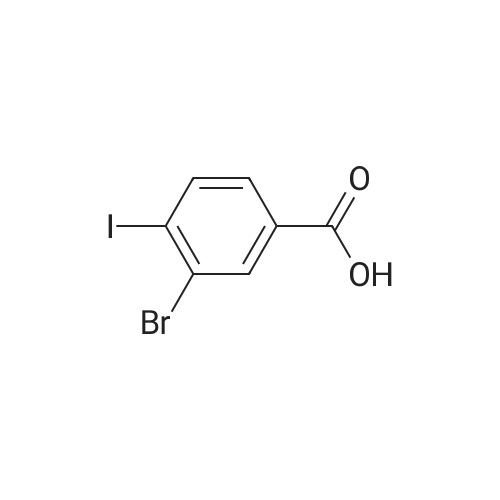
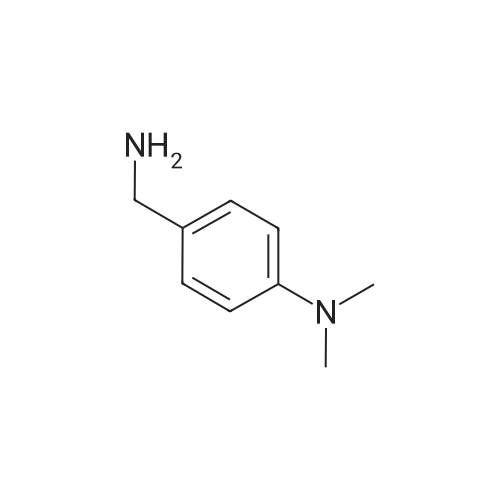
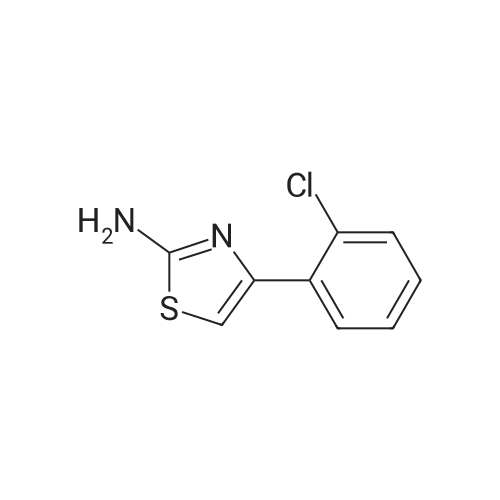
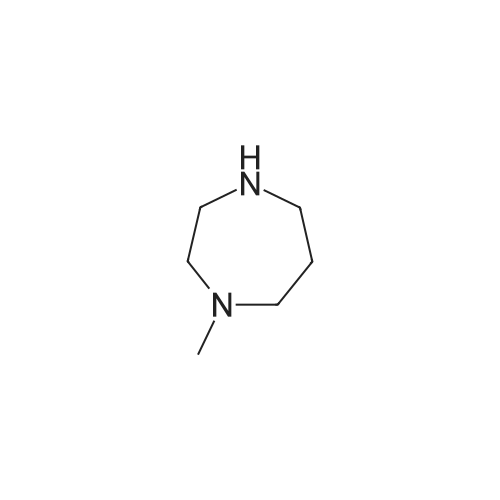
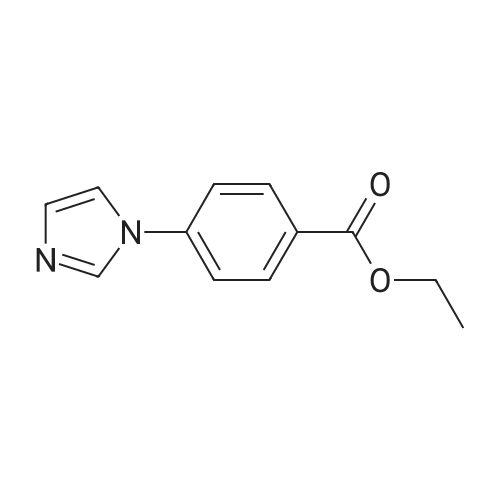
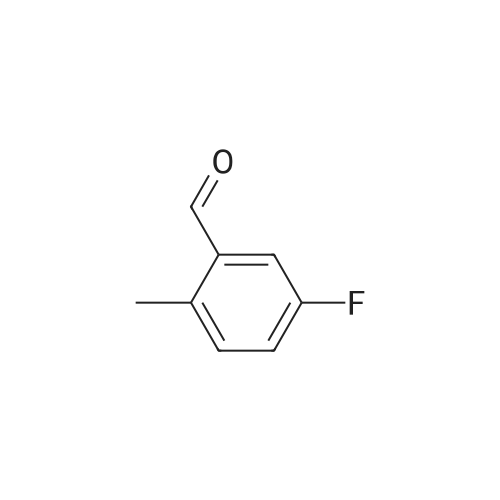
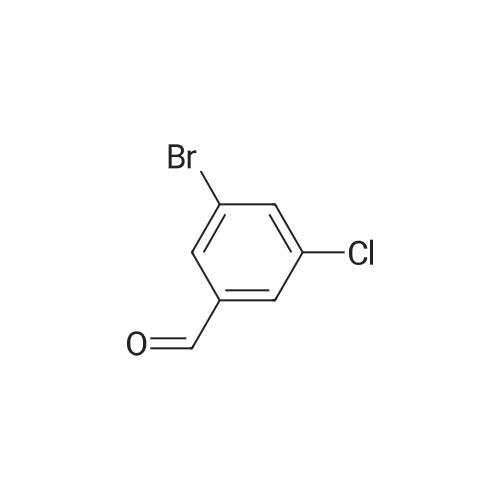
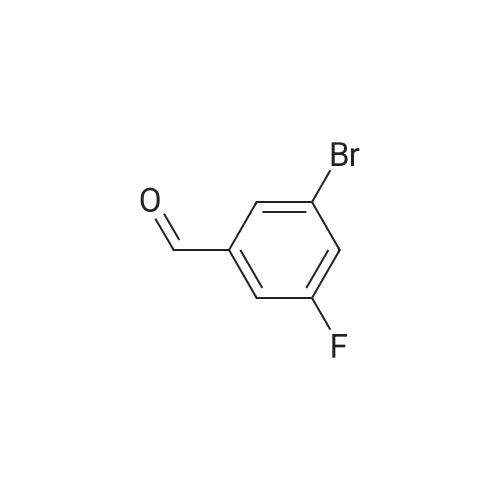
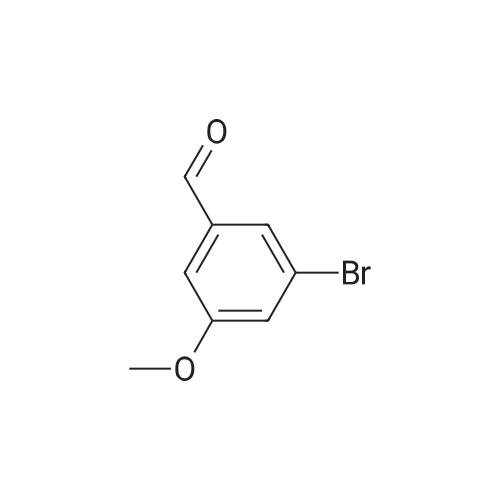
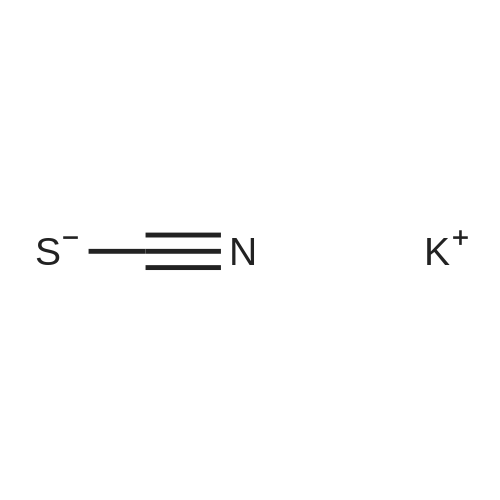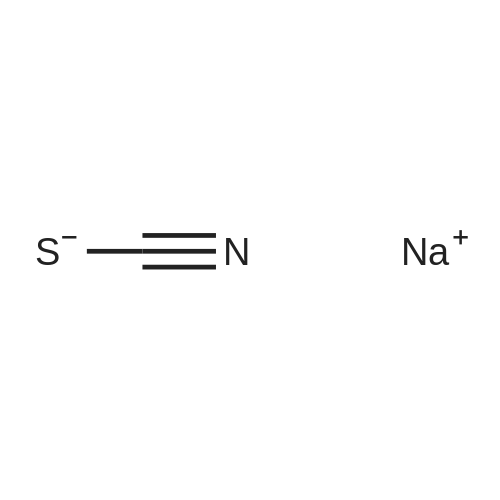* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
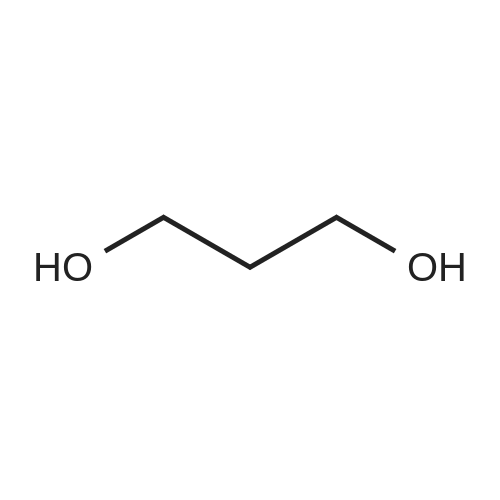
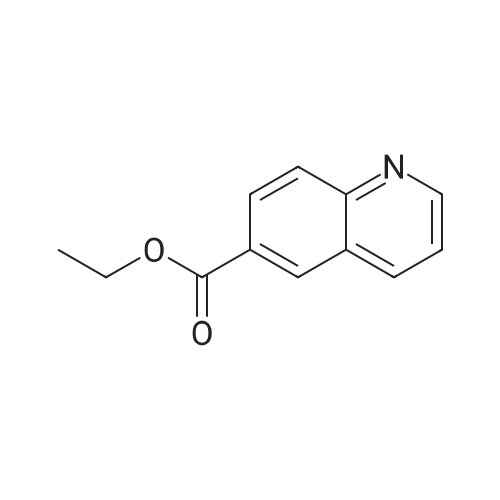
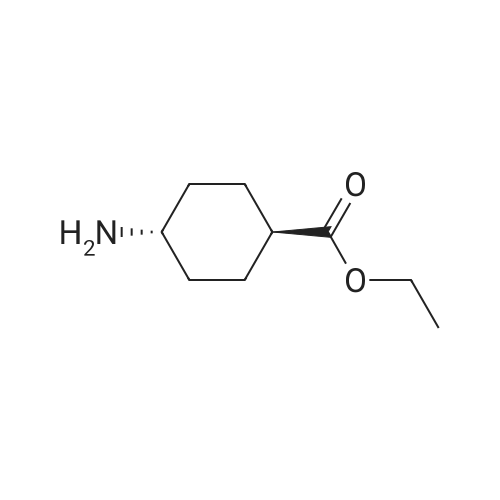
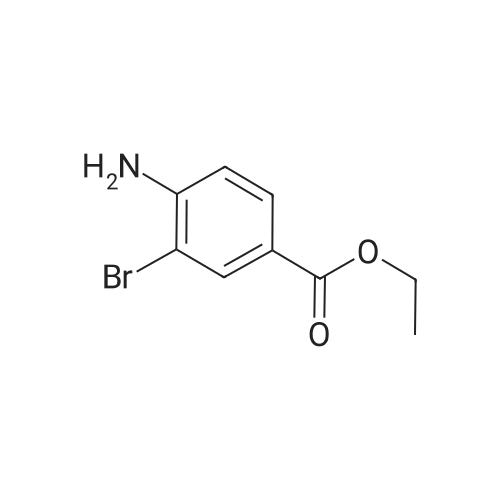
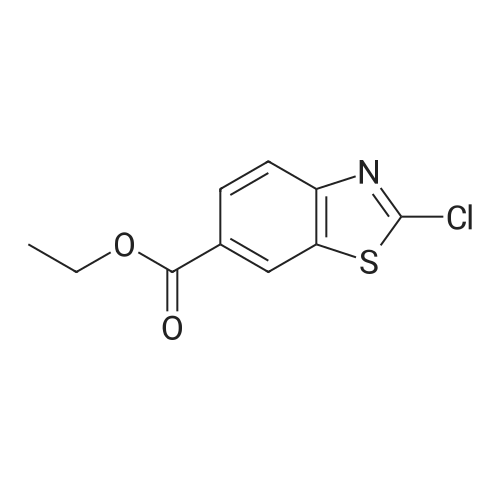
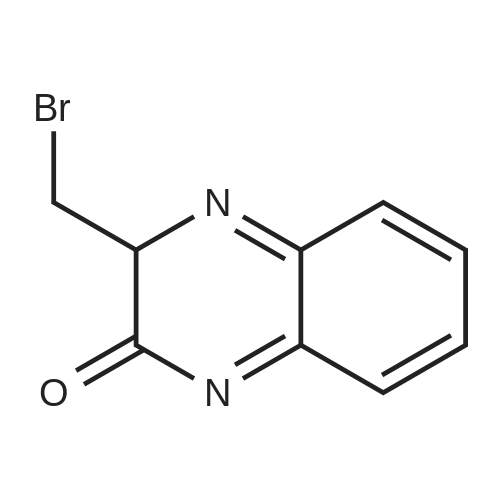
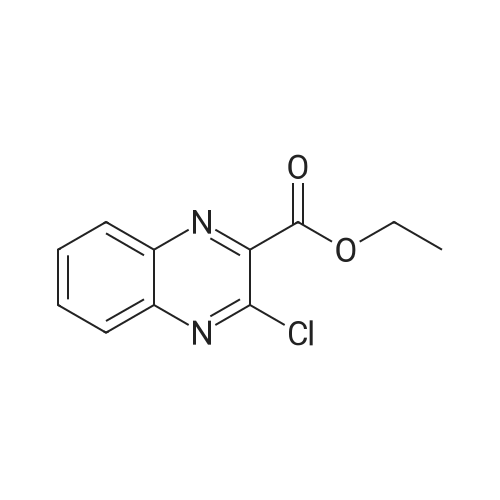
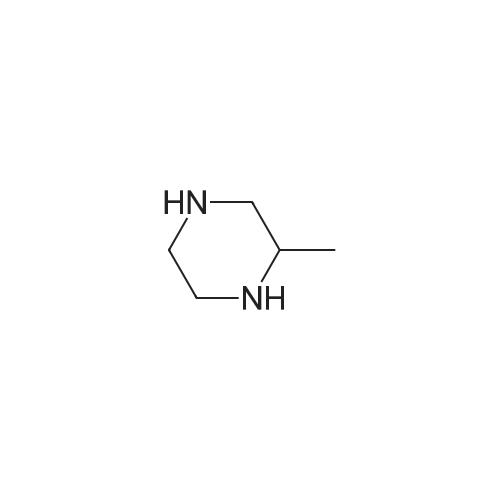
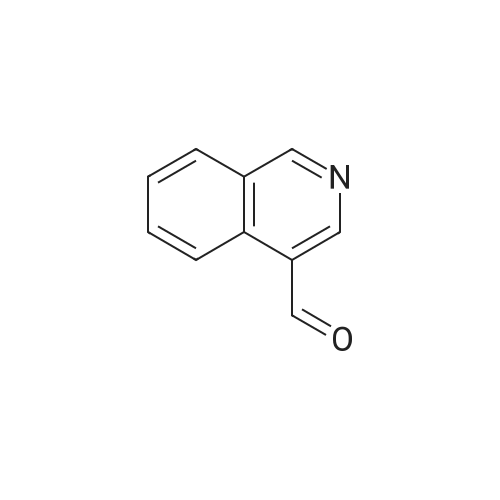
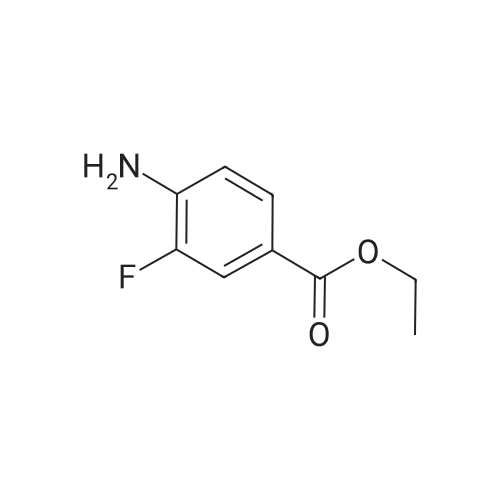
Accurately weigh 0.005mmol (l. Lmg) of the transition metal, add 10mL has been put into the magnetic stirrer Young's reaction tube, The oxygen reaction was carried out in the Young's reaction tube, and the reaction was carried out under oxygen conditions. The syringe was applied to the Young's reaction tubeA solution of 0.04 mmol of co-catalyst I, 0.08 mmol of co-catalyst II, 0.2 mmol of 4-carboxylate and 1 ml of l-3'-propylene glycol, the above-mentioned Young's reaction tube was placed on a magnetic stirrer and stirred at 150 ° C for 12 h. After the reaction was completed,The pH of the solution was neutral, and the reaction solution was post-treated to obtain pure product of ethyl 6-formate quinoline in a yield of 45percent.
Reference:
[1] Journal of Organic Chemistry, 2017, vol. 82, # 6, p. 3284 - 3290
[2] Patent: CN106543078, 2017, A, . Location in patent: Paragraph 0113; 0114
2
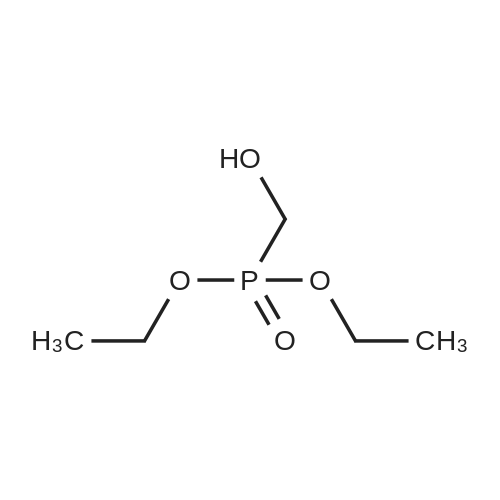
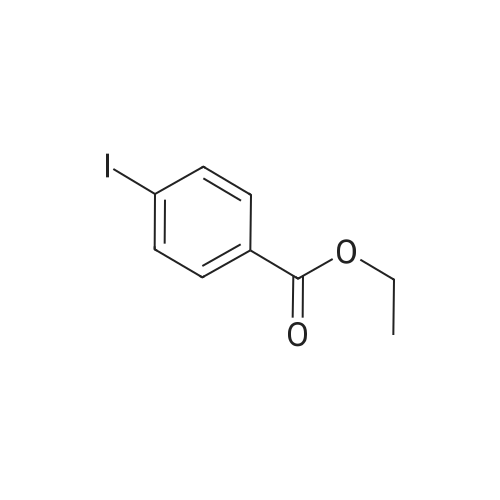
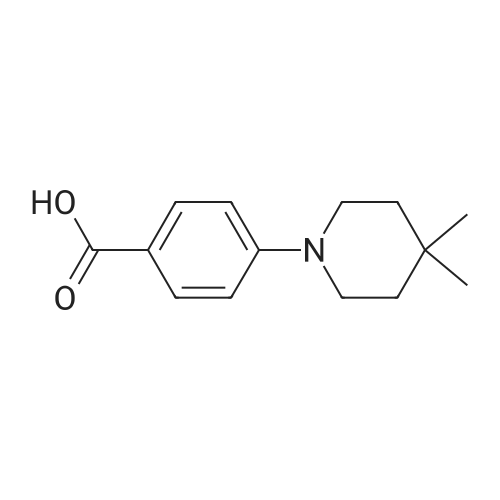
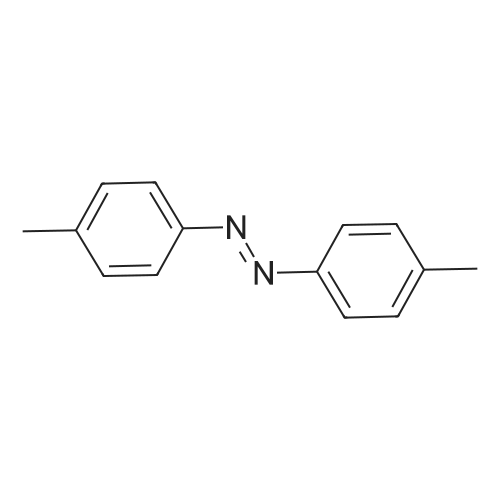
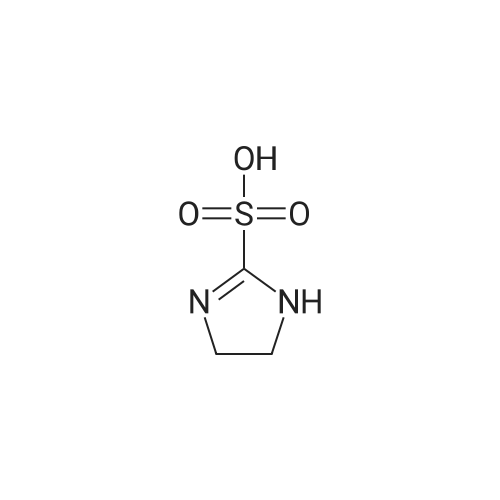
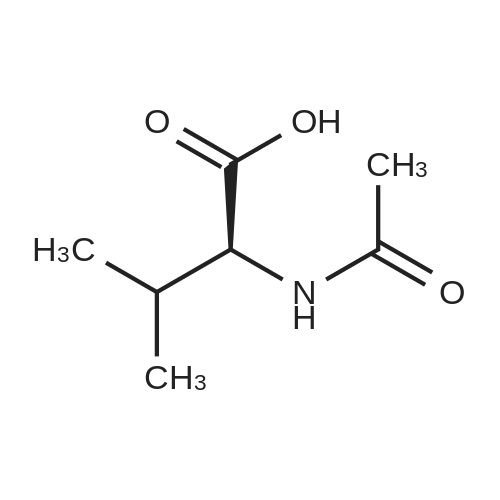
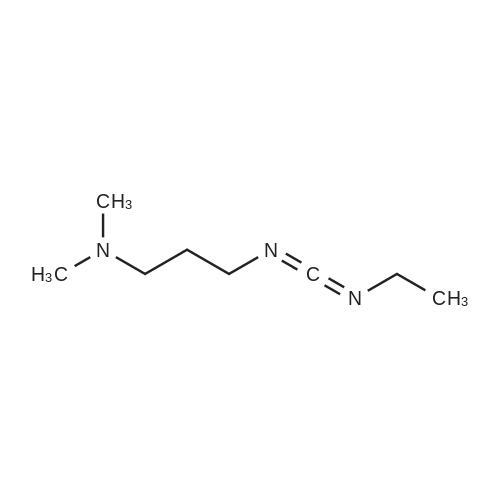
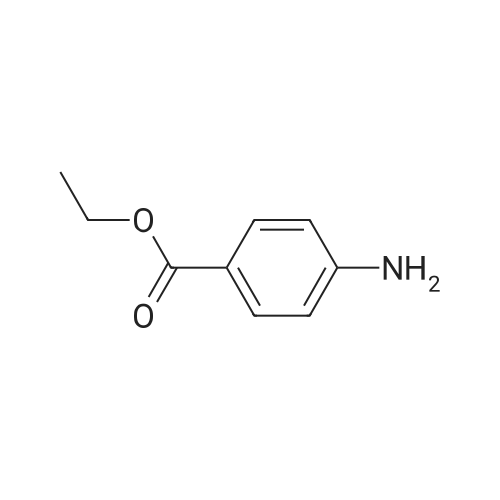
[ 107-18-6 ]
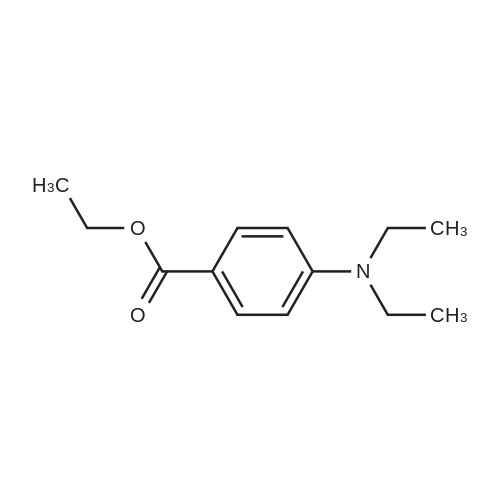
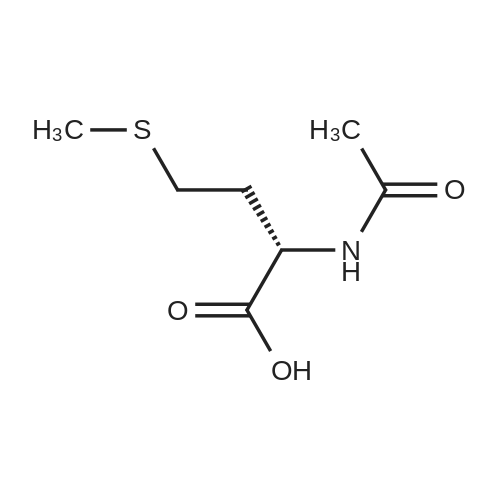
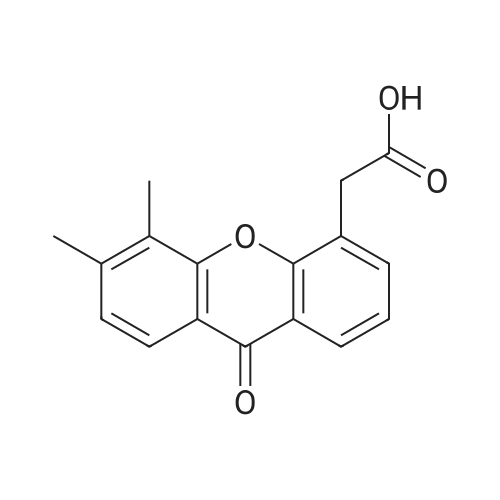
[ 94-09-7 ]
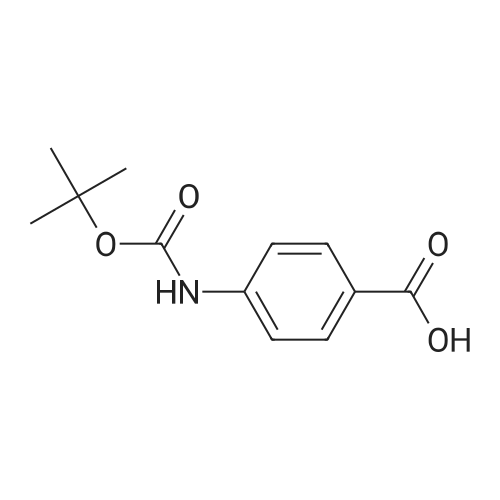
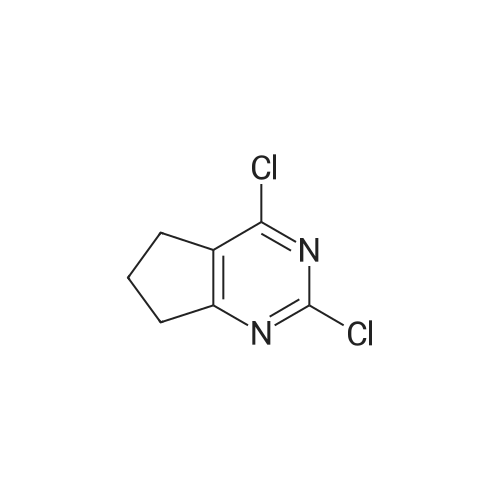
[ 73987-38-9 ]
Reference:
[1] Chemistry - A European Journal, 2017, vol. 23, # 63, p. 15874 - 15878
3
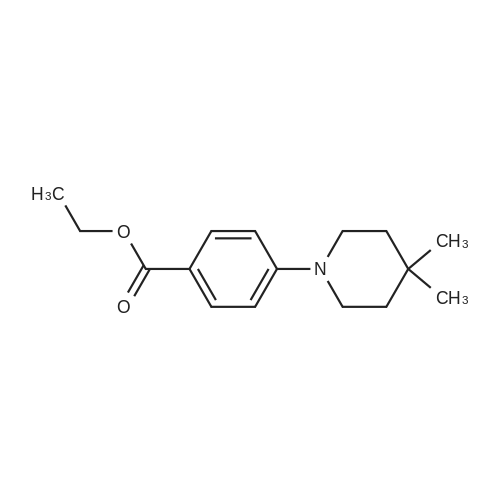
[ 64-17-5 ]
[ 94-09-7 ]
[ 855763-77-8 ]
Yield
Reaction Conditions
Operation in experiment
65%
at 150℃; for 36 h; Green chemistry
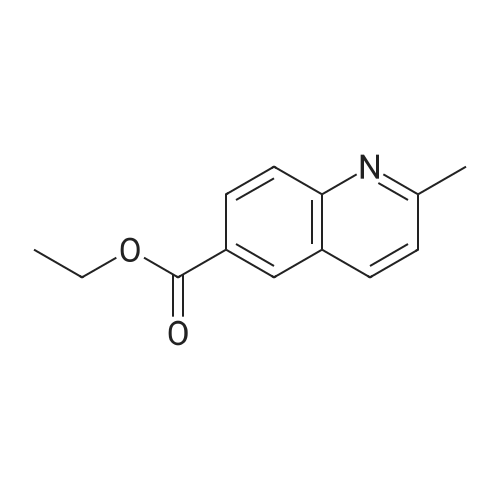
Accurately weigh 0.005mmol (l. Lmg) of the transition metal, add 10mL has been put into the magnetic stirrer Young's reaction tube, The oxygen reaction was carried out in the Young's reaction tube, and the reaction was carried out under oxygen conditions. The syringe was applied to the Young's reaction tubeAdd 0.04 mmol of co-catalyst I, 0.08 mmol of co-catalyst II, 0.2 mmol of ethyl 4-aminobenzoate and lmlWater, the Young's reaction tube was placed on a magnetic stirrer and stirred at 150 ° C for 36 hours. After completion of the reaction, the reaction solutionThe pH was neutral, and the reaction solution was post-treated to give pure 2-methyl-6-quinolinecarboxylic acid ethyl ester in 65percent yield.
Reference:
[1] Journal of Organic Chemistry, 2017, vol. 82, # 6, p. 3284 - 3290
[2] Patent: CN106543078, 2017, A, . Location in patent: Paragraph 0101; 0102
4
[ 94-09-7 ]
[ 93-85-6 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1962, vol. 27, p. 1533 - 1548
5
[ 333-20-0 ]
[ 94-09-7 ]
[ 50850-93-6 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 19 - 20℃;
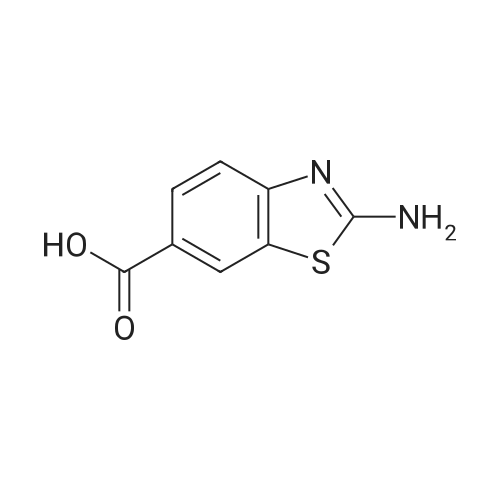
Ethyl 4-aminobenzoate (4.9 g, 30.0 mmol) and potassium thiocyanate (11.6 g,120.0 mmol) are dissolved in acetic acid(180 mL), followed by dripping acetic acid solution(20 mL) with bromine (1.5 mL,30.0 mmol) at 19 °C in 20 min, then the temperature is increased to room temperature and the mixture continues to react overnight. Then, the reaction solution is poured into water (100 mL), and stirred for 10 min, then ammonium hydroxide is dripped into the mixture to regulate pH to approximately 8, then the mixture is filtered, wherein filter cake is successively washed with water and petroleum ether, and finally the product is vacuumed to dryness, so as to obtain a yellow solid of 6.7 g, that is ethyl 2-aminobenzo[d]thiazole-6-carboxylate, with a yield of 100percent. Spectrum is: 1H NMR (400 MHz, DMSO) δ: 8.28(d, J = 1.6 Hz, 1H), 7.88(s ,2H), 7.82(dd, J = 1.6 Hz, 8.4 Hz, 1H), 7.37(d, J = 8.4Hz, 1H), 4.29(q, J = 7.2 Hz ,2H), 1.32(t, J = 7.2 Hz, 3H).
97%
With copper(ll) sulfate pentahydrate In methanol for 6 h; Reflux
To methanol (800 mL) were added ethyl 4-aminobenzoate 4 (49.5 g, 0.3 mol), KSCN (291 g, 3 mol) and CuSO4*5H2O (275 g,1.5 mol). The mixture was heated to reflux by mechanical stir for 6 h. After cooling to room temperature, the precipitate was filtered, about 2/3 of the filtrate was evaporated under reduced pressure. Water (600 mL) was added, the precipitate obtained was filtered and added to 2 N NaOH solution (500 mL), stirred for 10 min at room temperature. The precipitate was filtered, washed with waterand dried to get 5 (52.6 g, 79percent) as white powder, mp: 242-243 °C([45], mp, 241-242 °C).
69%
Stage #1: at 20℃; for 0.333333 h; Stage #2: at 10 - 20℃;
Ethyl 4-aminobenzoate (1 eq.) and KSCN (4 eq.) were dissolved in acetic acid (4 mL/mmol) and stirred at rt for 20mins. Then the reaction mixture was cooled to 10 °C and bromine (2 eq.) dissolved in small amount of acetic acid was added dropwise. Afterwards the reaction mixture was left to warm up to rt and stirred overnight. After the reaction was completed (monitored by TLC), reaction mixture was added dropwise into the sat. aq. NH3 solution (15 mL/mmol) while cooling in an ice bath. The product was extracted to EtOAc and the organic layer was washed with Na2S2O3, sat. aq. NaHCO3 and brine, dried using anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was recrystallized from diethyl ether to obtain ethyl 2-aminobenzo[d]thiazole-6-carboxylate in 69percent yield. 1H NMR (500 MHz, DMSO-d6): (ppm) 8.27 (d, J = 1.8Hz, 1H), 7.88 (s, 2H), 7.81 (dd, J = 8.4, 1.8 Hz, 1H), 7.36 (d,J = 8.4 Hz, 1H), 4.27 (q, J = 7.1 Hz, 2H), 1.30 (t, J = 7.1 Hz,3H).
67%
Stage #1: at 20℃; Stage #2: With ammonium hydroxide In water
Compound 28 (2.0 g, 12 mmol) was dissolved in 16 mL of acetic acid, and to the resulting solution was suspended potassium thiocyanate (4.67 g, 48 mmol). A solution of 0.61 mL of bromine in 8 ml of acetic acid was slowly added, and the reaction mixture was stirred at room temperature overnight. Water was added and the mixture was made neutral by addition of aqueous ammonium hydroxide. The precipitation was collected by filtration and then washed with water and subsequently dried to afford compound 29 (1.80 g, 67percent) as light yellow solid. The compound was used, without structure determination, directly for the next step.
50%
With copper(II) sulfate In methanol at 70℃; for 5 h;
Step: 1 Preparation of ethyl 2-amino-l,3-benzothiazole-6-carboxylate.A mixture of ethyl-4-amino benzoate (1.65g, lOmmol), KCNS (9.72g, 100 mmol) and CuSO4 (8g, 50mmol) in methanol (3OmL) was stirred for 5 hours at 7O0C. Subsequently, the suspension was cooled and filtered; the filtrate was diluted with water (4OmL) and heated to boiling. Ethanol was added to the boiling filtrate until a clear, although slightly yellowish solution was formed. Cooling of this solution resulted in crystallization of the product (l.lg, 50percent yield) as a pale yellow solid. NMR <n="17"/>o(DMSO-de) 1H δ (ppm): 8.34 (IH, d, Ar-H), 7.87 (IH, dd, Ar-H), 7.42 (IH, d, Ar-H), 4.30 (2H, q, CH2), and 1.32 (3H, t, CH3). MS m/z: 223 (M+l)
46%
at 10 - 20℃;
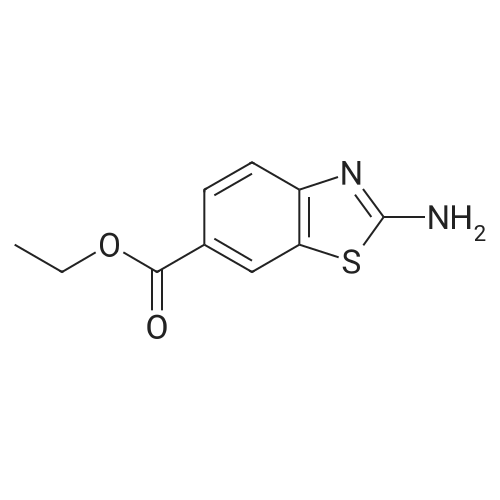
[1067] Step 1: ethyl 2-aminobenzo[d]thiazol-6-carboxylate[1068] To a solution of ethyl 4-aminobenzoate (16.5 g, 100.0 mmol) and potassium thiocyanate (10.7 g, 110.0 mmol) in acetic acid (150 mL) was added dropwise a solution of bromine (5.1 mL, 100.0 mmol) in acetic acid (50 mL) at below 10 ℃. The reaction mixture was stirred at room temperature overnight. The resulting solid was filtered and then dissolved in warm water (50 ℃). The aqueous layer was washed with chloroform and then basified to pH 11 with solid sodium carbonate. The resulting solid was filtered, washed with water, and then dried under reduced pressure to give 10.2 g of the titled compound as a white solid (Yield: 46percent).
46%
at 10 - 20℃;
Step 1: ethyl 4-aminobenzo[d]thiazol-6-carboxylate To a solution of ethyl 4-aminobenzoate (16.5 g, 100.0 mmol) and potassium thiocyanate (10.7 g, 110.0 mmol) in acetic acid (150 mL) was added dropwise a solution of bromine (5.1 mL, 100.0 mmol) in acetic acid (50 mL) at below 10° C. The reaction mixture was stirred at room temperature overnight. The resulting solid was filtered and then dissolved in warm water (50° C.). The aqueous layer was washed with chloroform and then basified to pH 11 with solid sodium carbonate. The resulting solid was filtered, washed with water, and then dried under reduced pressure to give 10.2 g of the titled compound as a white solid (Yield: 46percent).
Reference:
[1] Patent: EP3401315, 2018, A1, . Location in patent: Paragraph 0062; 0063
[2] Synlett, 2012, vol. 23, # 15, p. 2219 - 2222
[3] European Journal of Medicinal Chemistry, 2013, vol. 63, p. 702 - 712
[4] Medicinal Chemistry, 2017, vol. 13, # 4, p. 345 - 358
[5] European Journal of Medicinal Chemistry, 2013, vol. 61, p. 26 - 40
[6] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1991, vol. 30, # 5, p. 494 - 498
[7] Bioorganic and Medicinal Chemistry, 2005, vol. 13, # 7, p. 2509 - 2522
[8] Patent: WO2007/113644, 2007, A2, . Location in patent: Page/Page column 15-16
[9] Patent: WO2013/43001, 2013, A1, . Location in patent: Paragraph 1067; 1068
[10] Patent: US2015/11528, 2015, A1, . Location in patent: Paragraph 0600
[11] Journal of Medicinal Chemistry, 1999, vol. 42, # 15, p. 2828 - 2843
[12] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 23, p. 5561 - 5565
6
[ 94-09-7 ]
[ 50850-93-6 ]
Yield
Reaction Conditions
Operation in experiment
31%
With bromine In acetic acid
(195-1) A solution of ethyl p-aminobenzoate (23.2 g) and potassium thiocyanate (40.9 g) in acetic acid (280 mL) was cooled to 0° C., and thereto was added dropwise bromine (22.4 g), and the mixture was stirred at 0° C. for 20 minuts, and stirred at room temperature for 135 minutes, and stirred for 5 hours. The precipitated solid was collected by filtration to give ethyl 2-amino-1,3-benzothiazole-6-carboxylate (9.62 g, 31percent) 1H NMR (DMSO-d6, 400 MHz) δ 8.65 (brs, 2H), 8.38 (d, 1H, J=1.7 Hz), 7.89 (dd, 1H, J=8.5, 1.7 Hz), 7.44 (d, 1H, J=8.5 Hz), 4.30 (q, 2H, J=7.1 Hz), 1.32 (t, 3H, J=7.1 Hz).
31%
With bromine In acetic acid
(195-1) A solution of ethyl p-aminobenzoate (23.2 g) and potassium thiocyanate (40.9 g) in acetic acid (280 mL) was cooled to 0°C, and thereto was added dropwise bromine (22.4 g), and the mixture was stirred at 0°C for 20 minuts, and stirred at room temperature for 135 minutes, and stirred for 5 hours. The precipitated solid was collected by filtration to give ethyl 2-amino-1,3-benzothiazole-6-carboxylate (9.62 g, 31 percent). 1H NMR (DMSO-d6, 400MHz) δ 8.65 (brs, 2H), 8.38 (d, 1H, J=1.7Hz), 7.89 (dd, 1H, J=8.5, 1.7Hz), 7.44 (d, 1H, J=8.5Hz), 4.30 (q, 2H, J=7.1Hz), 1.32 (t, 3H, J=7.1Hz).
Reference:
[1] Patent: US2003/181496, 2003, A1,
[2] Patent: EP1479384, 2004, A1,
[3] Patent: US5496816, 1996, A,
[4] Archiv der Pharmazie (Weinheim, Germany), 1928, p. 215[5] Archiv der Pharmazie (Weinheim, Germany), 1929, p. 205
[6] Patent: US5498777, 1996, A,
[7] Patent: US5538964, 1996, A,
7
[ 540-72-7 ]
[ 94-09-7 ]
[ 50850-93-6 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1962, vol. 27, p. 1533 - 1548
[2] Journal of Medicinal Chemistry, 1997, vol. 40, # 1, p. 105 - 111
[3] Tetrahedron, 2005, vol. 61, # 38, p. 9075 - 9081
8
[ 1147550-11-5 ]
[ 94-09-7 ]
[ 50850-93-6 ]
Reference:
[1] Farmaco, 1994, vol. 49, # 3, p. 153 - 166
[2] Yakugaku Zasshi, 1950, vol. 70, p. 271,276[3] Chem.Abstr., 1951, p. 10212
9
[ 15192-76-4 ]
[ 94-09-7 ]
[ 50850-93-6 ]
Reference:
[1] Chemische Berichte, 1934, vol. 67, p. 944,947
[2] Yakugaku Zasshi, 1946, vol. 66, p. Ausg. B, S. 75[3] Chem.Abstr., 1952, p. 112
10
[ 94-09-7 ]
[ 582-80-9 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 1, p. 39 - 44
[2] Patent: CN106588914, 2017, A,
[3] Patent: CN107056789, 2017, A,
[4] Patent: CN106588913, 2017, A,
[5] Patent: CN106317057, 2017, A,
11
[ 94-09-7 ]
[ 32996-16-0 ]
Reference:
[1] Angewandte Chemie - International Edition, 2007, vol. 46, # 12, p. 2074 - 2077
12
[ 94-09-7 ]
[ 14685-90-6 ]
Reference:
[1] Berichte der Deutschen Pharmazeutischen Gesellschaft, vol. 31, p. 71[2] Chem. Zentralbl., 1921, vol. 92, # I, p. 584
[3] Journal and Proceedings of the Royal Society of New South Wales, 1938, vol. 71, p. 418[4] Chem. Zentralbl., 1938, vol. 109, # II, p. 4062
[5] Polish Journal of Chemistry, 2003, vol. 77, # 3, p. 329 - 332
[6] Patent: WO2011/47156, 2011, A1, . Location in patent: Page/Page column 77
13
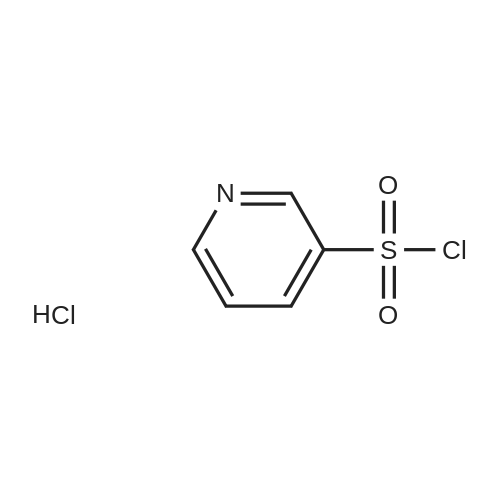
[ 94-09-7 ]
[ 100-37-8 ]
[ 59-46-1 ]
Reference:
[1] CIOS Rep. XXIII-23 <1945> 9,
[2] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
[3] Pharmaceutical Chemistry Journal, 2001, vol. 35, # 10, p. 556 - 559
Stage #1: With toluene-4-sulfonic acid In toluene at 50 - 60℃; for 2 h; Green chemistry Stage #2: for 5 h; Reflux; Green chemistry
165 g (1 mol) of ethyl p-aminobenzoate, 117.7 g (1.1 mol) of N-methylaniline, 10 g of p-toluenesulfonic acid,200mL with toluene toluene added to the 1000mL reaction flask, stirred and warmed to 50-60 ° C, 2h dropwise 59.53g (1.1mol) formic acid(Content of 85percent),Heating to reflux reaction, continue to separate the generated water, the reaction of about 5h to stop water when there is no water. The reaction solution was distilled off with atmospheric toluene recovery agent, toluene was recovered, the remaining vacuum distillation reaction solution,A pale yellow viscous liquid N- (4-ethoxycarbonylphenyl) -N'-methyl-N'-phenyl formamidine 262g, yield 93percent, purity 99.4percent.
Reference:
[1] Patent: CN107235859, 2017, A, . Location in patent: Paragraph 0012-0018
20
[ 122-51-0 ]
[ 94-09-7 ]
[ 100-61-8 ]
[ 57834-33-0 ]
Yield
Reaction Conditions
Operation in experiment
95.7%
at 50 - 60℃; for 2 h;
A mixture of 165 g (1 mol) of ethyl p-aminobenzoate, 112.35 g (1.05 mol) of N-methylaniline, 159 g (1.5 mol) of trimethyl orthoformate and 10.2 g (0.1 mol) of a specific surface area of 137 m2 / g Γ-type activated alumina into the 500ml reaction flask,Stirring and heating to 50 ~ 60 , incubation reaction 2h. Open the vacuum pump, and gradually increase the vacuum, vacuum distillation of the resulting methanol, to no methanol distillation.The reaction solution was filtered while hot, the recovered active alumina was repeatedly used and the mother liquor was distilled under reduced pressure to give a pale yellow viscous liquid N- (4-ethoxycarbonylphenyl) -N'-methyl-N'-phenyl Formamidine 270 g, yield 95.7percent, purity 99.5percent.
Reference:
[1] Patent: CN106431990, 2017, A, . Location in patent: Paragraph 0014; 0015; 0017; 0018; 0020; 0021
21
[ 94-09-7 ]
[ 1678-68-8 ]
[ 1678-68-8 ]
Reference:
[1] Patent: JP2005/350392, 2005, A, . Location in patent: Page/Page column 9-10
Reference:
[1] Yakugaku Zasshi, 1953, vol. 73, p. 294,296[2] Chem.Abstr., 1954, p. 2003
[3] Farmaco, Edizione Scientifica, 1954, vol. 9, p. 384
25
[ 94-09-7 ]
[ 94-24-6 ]
Reference:
[1] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
[2] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
[3] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
[4] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
26
[ 94-09-7 ]
[ 4740-24-3 ]
Reference:
[1] Zeitschrift fuer Chemie (Stuttgart, Germany), 1980, vol. 20, # 7, p. 259 - 260
[2] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
[3] Journal of the Society of Chemical Industry, London, 1945, vol. 64, p. 177
27
[ 94-09-7 ]
[ 56961-25-2 ]
Reference:
[1] Journal of the American Chemical Society, 1960, vol. 82, p. 3454 - 3456
28
[ 108-22-5 ]
[ 94-09-7 ]
[ 73013-51-1 ]
Reference:
[1] Green Chemistry, 2018, vol. 20, # 6, p. 1290 - 1296
[2] Journal of Organic Chemistry, 2012, vol. 77, # 22, p. 10347 - 10352
29
[ 94-09-7 ]
[ 4334-88-7 ]
Yield
Reaction Conditions
Operation in experiment
65%
Stage #1: With hydrogenchloride; sodium nitrite In methanol; water at 0 - 5℃; for 0.5 h; Stage #2: With tetrahydroxydiboron In methanol; water at 20℃; for 1 h;
General procedure: To a solution of arylamine (0.5 mmol, 1.0 equiv) in MeOH(1.0 mL) was added HCl (0.5 mL, 1.5 mmol, 3.0 equiv), followed by H2O (0.5 ml). This mixture was stirred 2 min,and the NaNO2 solution (0.25 mL) was then added. The NaNO2 solution was prepared by dissolving 35 mg ofNaNO2 in H2O (0.25 mL). This mixture was stirred 30 minat 0–5 °C, followed by HCl (135 mg, 1.5 mmol, 3.0 equivalents) in MeOH (1.0 mL). This mixture was stirred 60min. H2O (10 mL) was added to reaction mixture, then extracted with CH2Cl2 (50 mL, 3×). The combined organic layer was dried over Na2SO4, followed by evaporation to give the products.
Reference:
[1] Angewandte Chemie - International Edition, 2012, vol. 51, # 8, p. 1958 - 1961
31
[ 94-09-7 ]
[ 7149-03-3 ]
Yield
Reaction Conditions
Operation in experiment
91%
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0 - 20℃; for 18.5 h; Darkness
To a solution of ethyl 4-aminobenzoate (10 g, 61 mmol) in DMF (150 mL) cooled in ice-bath was added NBS (10.8 g, 61 mmol) portionwise and the mixture was stirred for 30 min at the same temperature. Then the ice-bath was removed and the stirring was continued at rt overnight (18 h). The mixture was poured into water (500 mL) and filtered, the orange solid was washed with water (50 mL × 2) and recrystallized from EtOH to afford pale-yellow crystals (13.3 g, 91percent) Rf = 0.38 (PE : EtOAc = 5 : 1); m.p. 86 - 88 °C; 1H-NMR (CDCl3, 300 MHz) δ: 8.07 (s, 1H), 7.79 (t, J = 11.55 Hz,1H), 6.70 (d, J = 8.10 Hz,1H), 4.58 (br, 2H), 4.37 (dd, J = 6.6 Hz, J = 6.6 Hz , 2H), 1.34 (t, J = 7.2 Hz, 3H).
88%
With bromine In dichloromethane at -10 - 20℃; for 18 h;
EXAMPLE 1 Preparation of Ethyl 4-amino-3-bromobenzoate A solution of bromine (7.0 mL, 137.3 mmol) in dichloromethane is added dropwise to a cold (-10° C.) solution of ethyl 4-aminobenzoate (22.0 g, 133.3 mmol) in dichloromethane. The reaction mixture is allowed to come to room temperature, stirred for 18 h and diluted with water. The organic phase is separated, washed twice with brine, dried over MgSO4 and concentrated in vacuo. The resultant residue is purified by flash chromatography (SiO2, 8/1 hexanes/EtOAc as eluent) to afford the title compound as a white solid, 28.6 g (88percent yield), identified by H-NMR and mass spectral analyses. MS m/e 242 (M-H)+; 1H NMR (400 MHz, DMSO-d6) δ 1.28 (t, J=7.01 Hz, 3 H), 4.22 (q, J=7.16 Hz, 2 H), 6.18 (brs, 2 H), 6.81 (d, J=7.91 Hz, 1H), 7.65 (dd, J=8.54, 1.98 Hz 1 H), 7.89 (d, J=1.83 Hz, 1H).
77%
at 90℃; for 8 h; Green chemistry
General procedure: To a round bottom flask was added 5 mmol of acetophenone, 0.5 mmol of 2-methylpyridine nitrate and 5.5 mmol of 40 wtpercent HBr, 60 ° C under the open flask stir reaction 6h, The yield of α-bromoacetophenone was 95percent The product was isolated by silica gel column chromatography (ethyl acetate / boiling point 60-90 ° C petroleum ether) at 200-300 mesh, The isolated yield was 88percent.
4.0 g
With N-Bromosuccinimide In chloroform at 0℃; for 3 h; Inert atmosphere
To a solution of ethyl 4-aminobenzoate (8.0 g, 0.048 mol) in chloroform (100 ml) was addedN-bromosuccinamide (8.62 g, 0.048 mol) at 0°C under inert atmosphere and reaction masswas stirred for 3 h at 0°C. Then the reaction mixture was diluted with EtOAc. The reaction mixture was quenched with water, extracted with EtOAc and organic layer was separated, dried over Na2504 and concentrated to afford 4.0 g of title product. ‘H NMR (300 MHz, DMSO-d6): 7.87 (s, 1H), 7.65-7.62 (d, J = 9.0 Hz, 1H), 6.80-6.78 (d, J = 8.7 Hz, 1H), 6.18(s, 2H), 4.28-4.18 (d, J= 6.9Hz, 2H), 1.29-1.24 (t, J= 6.9Hz, 3H); MS [M+Hf’: 244.
11.8 g
With bromine In methanol at 0 - 20℃; for 2 h; Inert atmosphere
Compound 11a (15.00 g, 90.81 mmol) was dissolved in anhydrous methanol (200 mL) at 0° C. and a solution of liquid bromine (13.06 g, 81.72 mmol) in methanol (100 mL) was added dropwise under nitrogen.The resulting reaction solution was reacted at 20°C for 2 hours. After the reaction is complete, directly concentrate, add water (100 mL), and extract with ethyl acetate (200 mL×3). Dry the organic phase over anhydrous sodium sulfate, filter, and concentrate.The mixture was purified by flash silica gel column chromatography (petroleum ether/ethyl acetate=100-0percent) to give compound 11b (11.8 g).
Reference:
[1] Journal of the American Chemical Society, 2002, vol. 124, # 19, p. 5350 - 5364
[2] European Journal of Organic Chemistry, 2010, # 24, p. 4662 - 4670
[3] Tetrahedron Letters, 2012, vol. 53, # 39, p. 5248 - 5252
[4] Patent: US2005/282825, 2005, A1, . Location in patent: Page/Page column 8
[5] Journal of Medicinal Chemistry, 2012, vol. 55, # 17, p. 7360 - 7377
[6] Patent: CN106866425, 2017, A, . Location in patent: Paragraph 0026-0029
[7] Synthesis, 2001, # 14, p. 2143 - 2155
[8] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 1, p. 153 - 158
[9] Journal de Pharmacie et de Chimie, 1928, vol. <8> 8, p. 58[10] Chem. Zentralbl., 1928, vol. 99, # II, p. 1325
[11] Tetrahedron Letters, 1996, vol. 37, # 52, p. 9325 - 9328
[12] Advanced Synthesis and Catalysis, 2008, vol. 350, # 13, p. 2052 - 2058
[13] Journal of the American Chemical Society, 2011, vol. 133, # 18, p. 6868 - 6870
[14] Chemistry - A European Journal, 2011, vol. 17, # 49, p. 13665 - 13669
[15] Angewandte Chemie - International Edition, 2012, vol. 51, # 8, p. 1958 - 1961
[16] Patent: US2002/19527, 2002, A1,
[17] Tetrahedron Letters, 2014, vol. 55, # 38, p. 5338 - 5341
[18] Patent: WO2016/55947, 2016, A1, . Location in patent: Page/Page column 76
[19] Journal of the American Chemical Society, 2016, vol. 138, # 40, p. 13147 - 13150
[20] Advanced Synthesis and Catalysis, 2017, vol. 359, # 7, p. 1144 - 1151
[21] Patent: CN107987072, 2018, A, . Location in patent: Paragraph 0217; 0218; 0219
[22] Patent: EP1726580, 2006, A1, . Location in patent: Page/Page column 120
32
[ 94-09-7 ]
[ 40566-85-6 ]
Yield
Reaction Conditions
Operation in experiment
2.2 g
With hydrogenchloride; sodium nitrite In water at -20 - -10℃; for 0.5 h;
Step 1 : Ethyl 4-hydrazinylbenzoate. hydrochloride: To a stirred and cooled (-20°C) solution of ethyl 4-aminobenzoate (2 g, 12.181 mmol) in cone. HC1 (22 mL) was added aqueous solution of sodium nitrite (925 mg, 13.40 mmol). This mixture was added very slowly to a precooled (-10 °C) mixture of tin chloride (13.8 g, 60.905 mmol) in cone. HC1 (15 mL) and stirred at the same temperature for 30 min. The precipitate obtained was filtered and washed with diethyl ether (2 x 20 mL) to yield 2.2 g of the title product as off white solid; 1H NMR (300 MHz, DMSO-i) δ 1.29 (t, J = 7.2 Hz, 3H), 4.26 (q, J = 7.5 Hz, 2H), 6.99 (d, = 7.8 Hz, 2H), 7.87 (d, = 8.4 Hz, 2H), 8.95 (br s, 1H), 10.48 (br s, 2H); APCI-MS (m/z) 181 (M+H)+.
Reference:
[1] Journal of Medicinal Chemistry, 1993, vol. 36, # 11, p. 1529 - 1538
[2] Patent: WO2015/87234, 2015, A1, . Location in patent: Page/Page column 39
Reference:
[1] Journal of the American Chemical Society, 2002, vol. 124, # 19, p. 5350 - 5364
[2] Tetrahedron Letters, 1996, vol. 37, # 52, p. 9325 - 9328
38
[ 94-09-7 ]
[ 82765-44-4 ]
Yield
Reaction Conditions
Operation in experiment
86%
With N-chloro-succinimide In acetonitrile for 5 h; Inert atmosphere; Reflux
To a solution of ethyl 4-aminobenzoate (2.0 mmol) inacetonitrile (2 mL mmol-1), NCS (2.05 mmol) was added.The mixture was refluxed for 5 h. Extraction was performedwith ethyl acetate and the organic layer was washed with 5percentNaOH, dried over anhydrous MgSO4, and the solvent wasremoved under reduced pressure. The product was purifiedby crystallization in hexane, which furnished 4-amino-3‑chlorobenzoate 27c as a purple crystal (mp 81-83 °C)in 86percent yield. 1H NMR (300 MHz, CDCl3) δ 1.33 (t, 3H,J 7.0 Hz, CH3), 4.29 (q, 2H, J 7.0 Hz, CH2), 6.70 (d, 1H,J 8.5 Hz, Ar‑H), 7.72 (dd, 1H, J 8.4, 1.9 Hz, Ar-H), 7.92 (d, 1H,J 1.9 Hz, Ar-H); 13C NMR (75 MHz, CDCl3) δ 14.30, 60.62,114.35, 118.06, 120.63, 129.52, 131.12, 146.97, 165.74.
Reference:
[1] Organic Letters, 2008, vol. 10, # 1, p. 113 - 116
[2] Journal of the Brazilian Chemical Society, 2018, vol. 29, # 1, p. 109 - 124
[3] Journal of Medicinal Chemistry, 2004, vol. 47, # 27, p. 6973 - 6982
[4] Patent: US5502073, 1996, A,
[5] Patent: US5516774, 1996, A,
[6] Patent: US5733905, 1998, A,
[7] Patent: US5736540, 1998, A,
39
[ 94-09-7 ]
[ 82765-44-4 ]
Yield
Reaction Conditions
Operation in experiment
76%
With N-chloro-succinimide In dichloromethane (CH2 Cl2); acetonitrile (CH3 CH)
Ethyl p-aminobenzoate, 49.50 g (0.30 moles) was dissolved in 500 ml of acetonitrile (CH3 CH) and heated to reflux. When the mixture became homogeneous, 42.0 g (0.315 moles) of N-chlorosuccinimide was added in several portions over one hour and the mixture was stirred at reflux overnight. By TLC (Hexane: EtOAc, 3:1), the mixture contained no starting material but only the desired product (Rf=0.55) and a minor impurity, which was probably dichlorinated material (Rf=0.65). The mixture was concentrated on a rotary evaporator and the residue was redissolved in 250 mL of dichloromethane (CH2 Cl2) and washed twice with 100 mL of 5percent sodium hydroxide (NaOH). The organic layer was dried over anhydrous potassium carbonate (K2 CO3) and concentrated on a rotary evaporator to yield 62.0 g of a reddish brown solid. The solid was recrystallized from 1.25 L of boiling hexane to give 52 g of a brown solid. The solid was recrystallized twice more from 1 L of boiling hexane to give 45.5 g (76percent) of tan solid ethyl 4-amino-3-chlorobenzoate (2), mp 82-83° C. and homogeneous by TLC. Additional material, 9.6 g (16percent) of like quality, was obtained by repeated recrystallization from hexane. The total yield of pure ethyl 4-amino-3-chlorobenzoate (2) was 92percent. 1H NMR (CDCl3) d 7.97-7.96 (t, 1H, J=0.7 Hz, ArH), 7.80-7.75 (dt, 1H, J=0.7 Hz, ArH), 6.77-6.73 (dd, 1H, J=0.5 Hz, ArH), 4.40-4.29 (q, 2H, J=2.8 Hz, OCH2), 1.41-1.34 (t, J=2.8 Hz, 2H, OCH2 CH3). IR (KBr) cm-1, 3500, 3370 (NH2, m), 1695, (C=O, s) 1630. MS (El) m/e 199 (M+), 201 (M+2), Anal. Calc'd for C9 H10 NO2 Cl: C, 54.15; H, 5.05: N, 7.02. Fd. C, 54.14; H, 5.17, N, 6.93. The formula is: STR2 4-Amino-3-chlorobenzoic acid (3)
Reference:
[1] Patent: US6028111, 2000, A,
40
[ 94-09-7 ]
[ 362527-61-5 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 1, p. 153 - 158
41
[ 94-09-7 ]
[ 406233-26-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 2006, vol. 49, # 3, p. 1165 - 1181
42
[ 94-09-7 ]
[ 406233-25-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 2006, vol. 49, # 3, p. 1165 - 1181
43
[ 94-09-7 ]
[ 1020149-73-8 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 6, p. 1631 - 1635
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 3, p. 701 - 713
With hydrogenchloride; sodium nitrite; In water; at -20 - -10℃; for 0.5h;
Step 1 : Ethyl 4-hydrazinylbenzoate. hydrochloride: To a stirred and cooled (-20C) solution of ethyl 4-aminobenzoate (2 g, 12.181 mmol) in cone. HC1 (22 mL) was added aqueous solution of sodium nitrite (925 mg, 13.40 mmol). This mixture was added very slowly to a precooled (-10 C) mixture of tin chloride (13.8 g, 60.905 mmol) in cone. HC1 (15 mL) and stirred at the same temperature for 30 min. The precipitate obtained was filtered and washed with diethyl ether (2 x 20 mL) to yield 2.2 g of the title product as off white solid; 1H NMR (300 MHz, DMSO-i) delta 1.29 (t, J = 7.2 Hz, 3H), 4.26 (q, J = 7.5 Hz, 2H), 6.99 (d, = 7.8 Hz, 2H), 7.87 (d, = 8.4 Hz, 2H), 8.95 (br s, 1H), 10.48 (br s, 2H); APCI-MS (m/z) 181 (M+H)+.
(a) 4-Methanesulfonylamino-benzoic acid. Ethyl 4-amino-benzoate (5 g, 30 mmol, 1.0 equiv) was mixed in 50 mL of CH2Cl2 with Et3N (6.1 g, 60 mmol, 2.0 equiv). The solution was cooled to 0 C. and methanesulfonyl chloride (3.8 g, 33 mmol. 1.1 equiv) was added as a solution in 15 mL of CH2Cl2 dropwise over a 15 min period. The reaction was stirred for 4 h at which time it was diluted with 50 mL of water. Layers were separated and the organic layer was washed with 50 mL of saturated sodium bicarbonate solution and concentrated. The resulting ester was dissolved in 50 mL of MeOH and treated with 50 mL of 5 N NaOH for 4 h. The reaction solution was extracted with Et2O and the aqueous layer was acidified to give a white precipitate that was collected by filtration. The solid was dried and used without further purification.
With pyridine; In dichloromethane;
Preparation 1 4-[(Methylsulfonyl)amino]benzoic acid To 215.2 g (2.72 mole) of pyridine in 1250 ml of dichloromethane is added 238 g (1.44 mole) of ethyl 4-aminobenzoate. After cooling to 0 C., a solution of 177.6 g (1.55 mole) of methanesulfonyl chloride in 250 ml of dichloromethane is added with stirring. Upon completion of the addition, the ice bath is removed and the reaction is stirred for one hour at room temperature. The reaction mixture is extracted with 3*500 ml of 4N sodium hydroxide and the aqueous extract is first washed with 3*500 ml of dichloromethane and then with 3*250 ml of ether. The aqueous extract is refluxed for two hours and allowed to cool overnight. The reaction is acidified with concentrated hydrochloric acid until a white precipitate forms. Collection by filtration followed by drying provides 310 g of the title compound. NMR (DMSO-d6): delta=3.2(s,3), 7.25-7.55(m,2) and 7.85-8.20 (m,2)ppm.
1. Ethyl-p-hydrazinobenzoate, Hydrochloride A solution of sodium nitrite (17.0 g, 0.24 mol) in water (90 ml) was added to a cooled solution of ethyl-p-amino benzoate (40 g, 0.24 mol) in concentrated hydrochloric acid (225 ml) at such a rate that the temperature did not exceed 0 C. The mixture was stirred at 0 C. for 0.1 h before adding to a stirred solution of tin (II) chloride dihydrate (202 g, 0.89 mol) in concentrated hydrochloric acid (135 ml) at such a rate that the temperature did not exceed -5 C. The resulting suspension was allowed to warm to room temperature over a 1 h period, filtered and washed with ether, mp 215-217 C., delta(360 MHz, D2 O) 1.38 (3H, t, J=7.1 Hz, Me), 4.37 (2H, q, J=7.1 Hz, CH2), 7.06 (1H, d, J=9 Hz, aromatic-H), 8.03 (1H, d, J=9 Hz, aromatic-H).
With sodium nitrite; In hydrogenchloride; water;
1. Ethyl-p-hydrazinobenzoate- Hydrochloride. A solution of sodium nitrite (17.0g, 0.24mol) in water (90ml) was added to a cooled solution of ethyl-p-amino benzoate (40g, 0.24mol) in concentrated hydrochloric acid (225ml) at such a rate that the temperature did not exceed 0C. The mixture was stirred at 0C for 0.1h before adding to a stirred solution of tin (II) chloride dihydrate (202g, 0.89mol) in concentrated hydrochloric acid (135ml) at such a rate that the temperature did not exceed -5C. The resulting suspension was allowed to warm to room temperature over a 1h period, filtered and washed with ether, mp 215-217C delta (360MHz, D2O) 1.38 (3H, t, J = 7.1Hz, Me), 4.37 (2H, q, J = 7.1Hz, CH2), 7.06 (1H, d, J = 9Hz, aromatic-H), 8.03 (1H, d, J = 9Hz, aromatic-H).
ethyl 4-[(4-isoquinolinylmethyl)amino]benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium borohydrid;toluene-4-sulfonic acid; In ethanol; dichloromethane; water; toluene;
EXAMPLE 2 A mixture of 8.0 g of <strong>[22960-16-3]4-isoquinolinecarboxaldehyde</strong> and 8.4 g of ethyl 4-aminobenzoate in 100 ml of toluene was refluxed in the presence of a catalytic amount of 4-toluenesulfonic acid. The water which formed was collected until no more was formed. The toluene was then removed under reduced pressure and the resulting residue was dissolved in 100 ml of ethanol. To the solution was then added 2 g of sodium borohydride and the mixture was refluxed for 30 minutes and then stirred at room temperature for 16 hours. The solution was concentrated to a volume of 50 ml and then poured into 1 liter of water. A thick pasty precipitate formed. The water was decanted and the precipitate was dissolved in methylene chloride and dried over sodium sulfate and the solvent was evaporated. The dark residue was triturated with 1:1 methylene chloride/hexane whereupon crystals formed. The solid was then separated by filtration and washed with additional solvent before further drying. Recrystallization of the solid from 50 ml of ethanol then gave ethyl 4-[(4-isoquinolinylmethyl)amino]benzoate melting at about 140-141.5 C.
Example 147 Ethyl N-[5-(5,6-dimethoxy-3-methyl-1,4-benzoquinon-2-yl)methyl-2-acetoxybenzoyl]-4-aminobenzoate Ethyl 4-aminobenzoate (-0.265 g, 1.604 mmol), triethylamine (0.162 g, 1.604 mmol) and <strong>[37091-73-9]2-chloro-1,3-dimethylimidazolinium chloride</strong> (0.271 g, 1.604 mmol) were added to a methylene chloride solution (50 ml) of 5-(5,6-dimethoxy-3-methyl-1,4-benzoquinon-2-yl)methyl-2-acetoxybenzoic acid (0.200 g, 0.535 mmol) and the resulting solution was stirred at room temperature for 12 hours. The reaction solution was poured into ice water and then extracted with methylene chloride. The extract was washed with water and then dried, and the solvent was removed by distillation. The obtained residue was purified by preparative thin-layer chromatography (hexane: ethyl acetate = 1:1) and then recrystallized from ether to obtain the titled compound (0.165 g, 0.316 mmol, 59percent).
ethyl 4-[(4,5-dihydro-1H-imidazole-2-yl)amino]benzoate trifluoroacetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In acetonitrile;
Step 2 Synthesis of ethyl 4-[(4,5-dihydro-1H-imidazole-2-yl)amino]benzoate trifluoroacetate: 991 mg (6.0 mmol) of ethyl 4-aminobenzoate, 1.0 g (6.66 mmol) of <strong>[64205-92-1]imidazoline-2-sulfonic acid</strong> and 1.35 g (13.3 mmol) of triethylamine were heated under reflux in 30 ml of acetonitrile overnight. The solvent was distilled off. After the extraction of the product from the residue with ethyl acetate, the extract was washed with 1 N aqueous sodium hydroxide solution and saturated aqueous common salt solution, and then dried over anhydrous magnesium sulfate.
In 1-methyl-pyrrolidin-2-one; at 130℃; for 1h;Product distribution / selectivity;
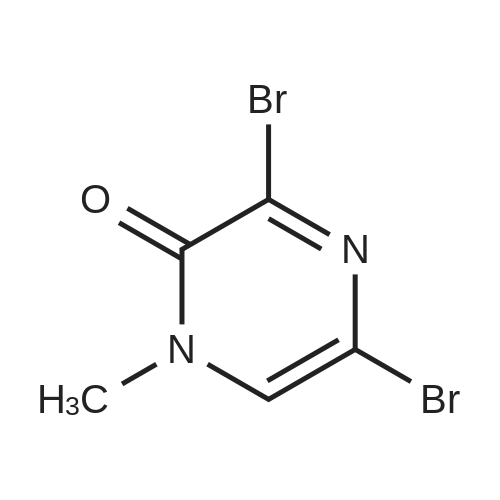
A mixture of 3,5-dibromo-l-methyl-2(lH)pyrazinone (1) (21.3g;79.5mmol), ethyl 4-aminobenzoate (13. Ig; 79.5mmol), and l-methyl-2-pyrollidinone (1OmL) was heated at 130 degrees for lhr. The mixture was cooled to room temperature, diluted with dichloromethane and filtered to give a dull brown solid. This was slurried with 0.5N NaOH, filtered, washed with water and diethyl ether to give 4-(6-bromo-4-methyl-3-oxo-3,4-dihydro-pyrazin-2-ylamino)-benzoic acid ethyl ester (2) as a light brown solid (21.3g); A mixture of 3,5-dibromo-l-methyl-2(lH)pyrazinone (21.3g;79.5mmol), ethyl 4-aminobenzoate (13. Ig; 79.5mmol), and l-methyl-2-pyrrolidinone (1OmL) was heated at 130 degrees for lhr. The mixture was cooled to room temperature, diluted with dichloromethane and filtered to give a dull brown solid. This was slurried with 0.5N NaOH, filtered, washed with water and diethyl ether to give 4-(6-bromo-4-methyl-3-oxo-3,4-dihydro-pyrazin-2-ylamino)-benzoic acid ethyl ester (1) as a light brown solid (21.3g)
With tert.-butylnitrite; dibenzoyl peroxide; In acetonitrile; at 20℃; for 4h;
EXAMPLE 4 Synthesis of ethyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate B2pin2 (1.0 mmol, 254 mg), benzoyl peroxide (0.02 mmol, 5 mg), ethyl 4-aminobenzoate (1 mmol, 165 mg) and acetonitrile (3 mL) were added in a 25 mL tube-type reactor, followed by the addition of tert-butyl nitrite (1.5 mmol, 154 mg). The reaction was conducted at room temperature for 4 h. The solution was concentrated after the reaction and the resultant was purified by column chromatography (eluted by petroleum ether:ethyl acetate=20:1, V:V) to give ethyl 4-(4,4,5,5 -tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate having the following structure: This compound is pale yellow liquid and obtained in 79% yield. Its NMR data are as follows: 1H NMR (400 MHz, CDCl3) delta 8.02 (d, 2H, J=8.4 Hz), 7.87 (d, 2H, J=8.4 Hz), 4.38 (q, 1H, J=7.1 Hz), 1.42~1.35(m, 15H); 13C NMR (100 MHz, CDCl3) delta 166.6, 134.5, 132.6, 128.5, 84.1, 61.0, 24.8, 14.2.
79%
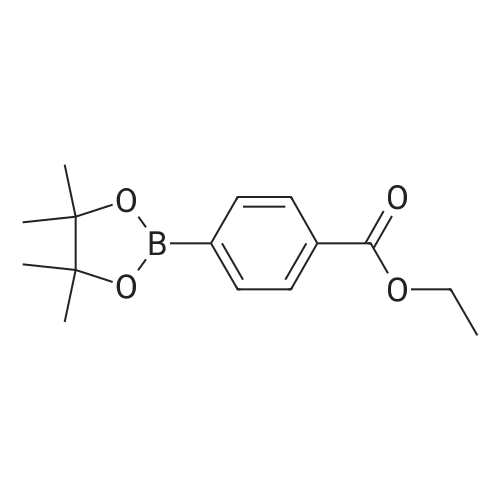
With tert.-butylnitrite; dibenzoyl peroxide; In acetonitrile; at 20℃; for 4h;
B2pin2 (1.0 mmol, 254 mg), benzoyl peroxide (0.02 mmol, 5 mg), ethyl 4-aminobenzoate (1 mmol, 165 mg) and acetonitrile (3 mL) were added in a 25 mL tube-type reactor, followed by the addition of tert-butyl nitrite (1.5 mmol, 154 mg). The reaction was conducted at room temperature for 4 h. The solution was concentrated after the reaction and the resultant was purified by column chromatography (eluted by petroleum ether : ethyl acetate = 20:1, V:V) to give ethyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate having the following structure: This compound is pale yellow liquid and obtained in 79% yield. Its NMR data are as follows: 1H NMR (400MHz, CDCl3) delta 8.02 (d, 2H, J=8.4Hz), 7.87 (d, 2H, J=8.4Hz), 4.38 (q, 1H, J=7.1Hz), 1.42~1.35(m, 15H); 13C NMR (100MHz, CDCl3) delta 166.6, 134.5, 132.6, 128.5, 84.1, 61.0, 24.8, 14.2.
77%
General procedure: An arylamine (50 mmol) was dissolved in 50% hydrofluoroboric acid(17 mL) and water (20 mL). After cooling the reaction mixture to 0 C, asolution of sodium nitrite (3.4 g in 7.5 mL water) was added dropwise tothe reaction system (over 5 min). The resulting mixture was stirred for 1h and the precipitate was collected by filtration. It was redissolved in theminimum amount of acetone and then diethyl ether was added to precipitatethe aryl diazonium tetrafluoroborate. The product was filtered, washedwith diethyl ether and dried under reduced pressure.Borylation of aryldiazonium salts; general procedureThe aryldiazonium salt (0.5 mmol) and (Bpin)2 (0.75 mmol) were added toan oven-dried Schlenk tube. The tube was evacuated and backfilled with argon (three times). CH3OH (0.8 mL) was added to this Schlenk tube.The tube was sealed and the mixture was stirred at room temperature(22-25 C) for 36 h. After evaporation of the solvent, the residue waspurified by column chromatography to afford the product.The arylboronates were purified by chromatography on a silica column eluting with petroleum ether (boiling range 60-90 C) or a petroleumether/ethyl acetate mixture (ca. 60:1) by volume giving Rf values for theboronates of ca. 0.2-0.3.
75%
General procedure: To a solution of arylamine (0.5 mmol, 1.0 equiv) in MeOH(1.0 mL) was added HCl (0.5 mL, 1.5 mmol, 3.0 equiv) followed by H2O (0.5 ml). This mixture was stirred 2 min, and the NaNO2 solution (0.25 mL) was then added. The NaNO2 solution was prepared by dissolving 35 mg of NaNO2 in H2O (0.25 mL). This mixture was stirred 30 minat 0-5 C followed by B2pin2 (2, 381 mg, 1.5 mmol, 3.0equiv) in MeOH (1.0 mL). This mixture was stirred 60 min.H2O (10 mL) was added to the reaction mixture, then extracted with CH2Cl2 (50 mL, 3×). The combined organic layers were washed with sat. NaHCO3, dried over Na2SO4, followed by evaporation, and the crude residue was purified by flash chromatography.
pyridine; In dichloromethane; at 0 - 20℃;Inert atmosphere;
Procedure for the synthesis of compound 20: To a solution of 2-methoxy-5-chlorobenzenesulfonyl chloride (878 mg, 0.00362 moles, 1.5 eq) in dichloromethane was added a solution of ethyl 4-aminobenzoate (400 mg, 0.00242 moles, 1 eq) in 1:1 ratio of pyridine and DCM (5 mL/5 mL) under nitrogen atmosphere at 0 C. The reaction mixture was stirred at room temperature overnight. The progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was diluted with DCM, washed twice with water, and dried with anhydrous sodium sulphate. The organic layer was concentrated, dried, washed with ether followed by n-hexane and dried to provide 20 as an off-white solid (0.8 g, 89% yield).1H NMR (500 MHz, CDCl3) 1.34 (t, 3H), 4.0 (s, 3H), 4.22-4.32 (q, 2H), 6.90 (d, 1H), 7.16 (d, 2H), 7.20 (s, 1H), 7.84 (s, 1H), 7.92 (d, 2H).
Example 60Cyclopropanesulfonic acid [2-(5-fluoro-2-methyl-phenyl)-3,3-dimethyl-l,2,3,4- tetrahydro-quinoline-6-carbonyl] -amideA mixture of 4-amino-benzoic acid ethyl ester (1.65 g, 10 mmol) and 5-fiuoro-2-methyl- benzaldehyde (1.38 g, 10 mmol) in ethanol (10 mL) was heated to reflux for 2 hours. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[l-(5-fiuoro-2-methyl-phenyl)- methylidene]-amino}-benzoic acid ethyl ester (2.0 g, 70%) as a white solid: LC/MS m/e calcd for Ci7Hi6FN02 (M+H)+: 285.3, observed: 286.3.
70%
In ethanol; for 2h;Reflux;
A mixture of 4-amino-benzoic acid ethyl ester (1.65 g, 10 mmol) and <strong>[22062-53-9]5-fluoro-2-methyl-benzaldehyde</strong> (1.38 g, 10 mmol) in ethanol (10 mL) was heated to reflux for 2 hours. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[1-(5-fluoro-2-methyl-phenyl)-methylidene]-amino}-benzoic acid ethyl ester (2.0 g, 70%) as a white solid: LC/MS m/e calcd for C17H16FNO2 (M+H)+: 285.3, observed: 286.3
Example 55 2- (3 -Chloro-5-morpholin-4-yl-phenyl)-3 ,3 -dimethyl- 1 ,2,3 ,4-tetrahydro-quinoline-6- carboxylic acidA mixture of 4-amino-benzoic acid ethyl ester (8.3 g, 50.3 mmol) and 3-bromo-5-chloro- benzaldehyde (11.0 g, 50.3 mmol) in ethanol (100 mL) was heated to reflux for 2 hours. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-{ [l-(3-bromo-5-chloro-phenyl)- methylidene] -amino}-benzoic acid ethyl ester (6.4 g, 35%) as a white solid: LC/MS m/e calcd for Ci6Hi3BrClN02 M+: 366.6, observed: 366.6.
35%
In ethanol; for 2h;Reflux;
A mixture of 4-amino-benzoic acid ethyl ester (8.3 g, 50.3 mmol) and <strong>[188813-05-0]3-bromo-5-chloro-benzaldehyde</strong> (11.0 g, 50.3 mmol) in ethanol (100 mL) was heated to reflux for 2 hours. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[1-(3-bromo-5-chloro-phenyl)-methylidene]-amino}-benzoic acid ethyl ester (6.4 g, 35%) as a white solid: LC/MS m/e calcd for C16H13BrClNO2 M+: 366.6, observed: 366.6.
Example 49N- [2- (4'-tert-Butyl-5-fluoro-biphenyl-3 -yl) -3 ,3 -dimethyl- 1 ,2,3 ,4-tetrahydro-quinoline- 6-carbonyl] -methanesulf onamide A mixture of 4-amino-benzoic acid ethyl ester (1.65 g, 10 mmol) and 3-bromo-5-fiuoro- benzaldehyde (2.03 g, 10 mmol) in ethanol (20 mL) was heated to reflux for 2 h. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[l-(3-bromo-5-fiuoro-phenyl)-methylidene]- aminoj-benzoic acid ethyl ester (2.25 g, 64%) as a white solid: LC/MS m/e calcd for Ci6Hi3BrFN02 M+: 350.2, observed: 350.2, 352.2.
64%
In ethanol; for 2h;Reflux;
A mixture of 4-amino-benzoic acid ethyl ester (1.65 g, 10 mmol) and <strong>[188813-02-7]3-bromo-5-fluoro-benzaldehyde</strong> (2.03 g, 10 mmol) in ethanol (20 mL) was heated to reflux for 2 h. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[1-(3-bromo-5-fluoro-phenyl)-methylidene]-amino}-benzoic acid ethyl ester (2.25 g, 64%) as a white solid: LC/MS m/e calcd for C16H13BrFNO2 M+: 350.2, observed: 350.2, 352.2.
Example 662- (3 -Methoxy-5-morpholin-4-yl-phenyl)-3 ,3 -dimethyl- 1 ,2,3 ,4-tetrahydro-quinoline-6- carboxylic acidA mixture of 4-amino-benzoic acid ethyl ester (6.1 g, 36.9 mmol) and 3-bromo-5- methoxy-benzaldehyde (8.0 g, 36.9 mmol) in ethanol (100 mL) was prepared. The reaction mixture was heated to reflux for 2 hours. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[l-(3-bromo-5-methoxy-phenyl)-methylidene]-amino}-benzoic acid ethyl ester (4.5 g, 34%) as a white solid: LC/MS m/e calcd for Ci7Hi6BrN03 M+: 362.2, observed: 362.1.
34%
In ethanol; for 2h;Reflux;
A mixture of 4-amino-benzoic acid ethyl ester (6.1 g, 36.9 mmol) and <strong>[262450-65-7]3-bromo-5-methoxy-benzaldehyde</strong> (8.0 g, 36.9 mmol) in ethanol (100 mL) was prepared. The reaction mixture was heated to reflux for 2 hours. Then the reaction mixture cooled to room temperature. The solvent was removed in vacuo and the residue was washed with ether to afford 4-[1-(3-bromo-5-methoxy-phenyl)-methylidene]-amino}-benzoic acid ethyl ester (4.5 g, 34%) as a white solid: LC/MS m/e calcd for C17H16BrNO3 M+: 362.2, observed: 362.1
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 3h;Inert atmosphere;
General procedure: A suspension of 8A or 8B, Ethyl 4-aminobenzoate (1 equiv), DMAP (0.5equiv) and EDCI (1-2 equiv) in CH2Cl2 were stirred for 3 h. They were performed under an atmosphere of Nitrogen gas. The reaction mixture was diluted with 10 volumes CH2Cl2. The organic phase was washed sequentially with 1M HCl , saturated aqueous NaHCO3, and brine, dried over MgSO4 , filtered and concentrated . It is used in the next step without further purication.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 3h;Inert atmosphere;
General procedure: A suspension of 8A or 8B, Ethyl 4-aminobenzoate (1 equiv), DMAP (0.5equiv) and EDCI (1-2 equiv) in CH2Cl2 were stirred for 3 h. They were performed under an atmosphere of Nitrogen gas. The reaction mixture was diluted with 10 volumes CH2Cl2. The organic phase was washed sequentially with 1M HCl , saturated aqueous NaHCO3, and brine, dried over MgSO4 , filtered and concentrated . It is used in the next step without further purication.
With ytterbium(III) trifluoromethanesulfonate hydrate; In acetonitrile; at 90℃; for 12h;sealed reaction bottle;
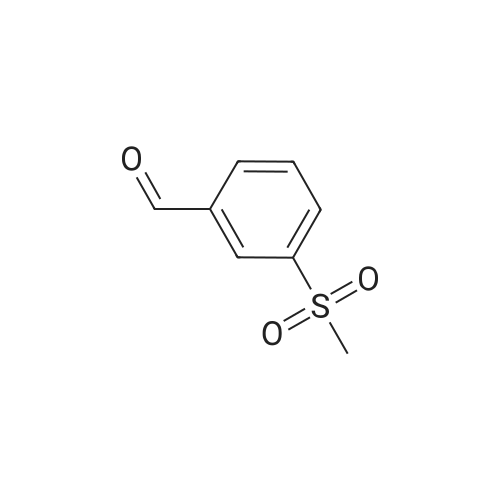
A mixture of 4-aminobenzoic acid ethyl ester (3.3 g, 20 mmol), <strong>[43114-43-8]3-methanesulfonyl benzaldehyde</strong> (3.68 g, 20 mmol) and ytterbium(III) triflate hydrate (1.86 g, 3 mmol) in acetonitrile (150 mL) was cooled to 0 C. in a sealed reaction bottle. Then a cooled solution of isobutene (5.6 g, 100 mmol) was added into. The reaction mixture was heated to 90 C. and stirred for 12 h. The solvent was removed in vacuo and the residue was purified on flash silica gel chromatography (silica gel from QingDao, 200-300 mesh, glass column from Shanghai SD company) (10% ethyl acetate/hexanes) to afford 2-(3-methanesulfonyl-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinoline-6-carboxylic acid ethyl ester (3.3 g, 43%) as a white solid: MS (ESI) M+1=388.3.
43%
ytterbium(III) trifluoromethanesulfonate hydrate; In acetonitrile; at 0 - 90℃; for 12h;Sealed tube;
Example 37 N- [2- (3-methanesulf onyl-phenyl) -4,4-dimethyl- 1 ,2,3,4- tetrahydro- quinoline-6- carbonyl] -methanesulfonamideA mixture of 4-aminobenzoic acid ethyl ester (3.3 g, 20 mmol), <strong>[43114-43-8]3-methanesulfonyl benzaldehyde</strong> (3.68 g, 20 mmol) and ytterbium(III) triflate hydrate (1.86g, 3 mmol) in acetonitrile (150 mL) was cooled to 0 C in a sealed reaction bottle. Then a cooled solution of isobutene (5.6 g, 100 mmol) was added into. The reaction mixture was heated to 90 C and stirred for 12 h. The solvent was removed in vacuo and the residue was purified on flash silica gel chromatography (silica gel from QingDao, 200-300 mesh, glass column from Shanghai SD company)(10% ethyl acetate/hexanes) to afford 2-(3-methanesulfonyl- phenyl)-4,4-dimethyl-l,2,3,4-tetrahydro-quinoline-6-carboxylic acid ethyl ester (3.3 g, 43%) as a white solid: MS(ESI) M+l = 388.3.A mixture of 2-(3-methanesulfonyl-phenyl)-4,4-dimethyl-l,2,3,4-tetrahydro-quinoline-6- carboxylic acid ethyl ester (3.3 g, 8.53 mmol), sodium hydroxide (3.41 g, 85.3 mmol), water (10 mL) in acetonitrile (30 mL) was stirred at 60C for 12 h. The mixture was neutralized with a 3 mol/L aqueous hydrochloric acid solution and extracted with ethyl acetate (2 x 50 mL), washed with water, dried over anhydrous sodium sulfate and then concentrated in vacuo to afford 2-(3-methanesulfonyl-phenyl)-4,4-dimethyl-l,2,3,4- tetrahydro-quinoline-6-carboxylic acid (2.76 g, 90%) as a white solid which was used for next step without further purification: MS(ESI) M+l = 360.3.To a suspension of methanesulfonamide (480 mg, 5.0 mmol) in N,N-dimethylformamide (5 mL) was added sodium hydride (200 mg, 5.0 mmol). The resulting mixture was stirred at 25C for 1 h to afford Solution A37. A solution of 2-(3-methanesulfonyl-phenyl)-4,4- dimethyl- 1, 2,3, 4-tetrahydro-quinoline-6-carboxylic acid (359 mg, 1.0 mmol) and 1,1'- carbonyldiimidazole (320 mg, 2.0 mmol) in N,N-dimethylformamide (5 mL) was stirred at 70C for 1 h and cooled to room temperature to afford Solution B37. Solution B37 was added to Solution A37 and the resulting mixture was stirred at 25C for 1 h. To the reaction mixture was added water (0.5 mL). The mixture was filtered to remove the insoluble solid, and the filtrate was purified by Waters automated flash system (column: Xterra 30 mm x 100 mm, sample manager 2767, pump 2525, detector: ZQ mass and UV 2487, solvent system: acetonitrile and 0.1% ammonium hydroxide in water) afforded N-[2- (3-methanesulfonyl-phenyl)-4,4-dimethyl-l,2,3,4-tetrahydro-quinoline-6-carbonyl]- methanesulfonamide (100 mg, 23%) as a white solid: MS(ESI) M+l = 437.3.
2-(5-bromo-2-chloro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinoline-6-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
33%
With ytterbium(III) triflate; In acetonitrile; at 85℃; for 16h;sealed tube;
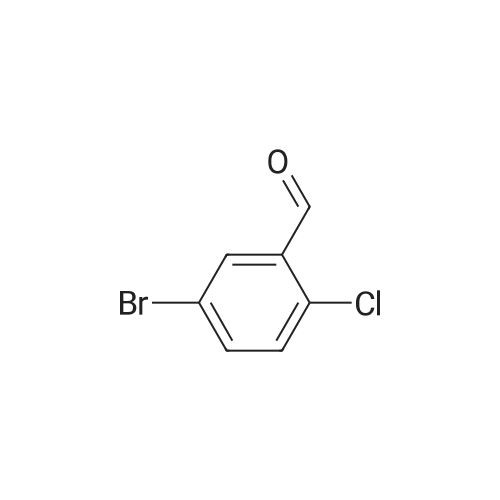
To a stirred solution of 4-amino-benzoic acid methyl ester (8.25 g, 50 mmol) and <strong>[189628-37-3]5-bromo-2-chloro-benzaldehyde</strong> (13.2 g, 60 mmol) in acetonitrile (200 mL) were added isobutene (14 mL, 0.2 mmol) and ytterbium(III) trifluoromethanesulfonate (Yb(OTf)3) (4.65 g, 7.5 mmol). The resulting mixture was stirred at 85 C. for 16 h in sealed tube. The mixture was diluted with ethyl acetate (300 mL) and washed with water (100 mL×2) and brine (100 mL×2) and then dried over anhydrous sodium sulfate. The solvent was removed in vacuo and the residue was purified by ISCO combi-flash chromatography (gradient elution, 10-50% ethyl acetate in petroleum ether) to afford 2-(5-bromo-2-chloro-phenyl)-4,4-dimethyl-1,2,3,4-tetrahydro-quinoline-6-carboxylic acid methyl ester (6.99 g, 33%) as a light yellow solid: MS (ESI) M+1=422.0.
33%
ytterbium(III) triflate; In acetonitrile; at 85℃; for 16h;Sealed tube;
Example 23N-[2-(2-chloro-5-morpholin-4-yl-phenyl)-4,4-dimethyl-l,2,3,4-tetrahydro-quinoline-6- carbonyl] -methanesulfonamideTo a stirred solution of 4-amino-benzoic acid methyl ester (8.25 g, 50 mmol) and 5-bromo- 2-chloro-benzaldehyde (13.2 g, 60 mmol) in acetonitrile (200 mL) were added isobutene (14 mL, 0.2 mmol) and ytterbium(III) trifluoromethanesulfonate (Yb(OTf)3) (4.65 g, 7.5 mmol). The resulting mixture was stirred at 85 C for 16 h in sealed tube. The mixture was diluted with ethyl acetate (300 mL) and washed with water (100 mL x 2) and brine (100 mL x 2) and then dried over anhydrous sodium sulfate. The solvent was removed in vacuo and the residue was purified by ISCO combi-flash chromatography (gradient elution, 10 - 50% ethyl acetate in petroleum ether) to afford 2-(5-bromo-2-chloro-phenyl)-4,4-dimethyl- l,2,3,4-tetrahydro-quinoline-6-carboxylic acid methyl ester (6.99 g, 33%) as a light yellow solid: MS(ESI) M+l = 422.0.A mixture solution of 2-(5-bromo-2-chloro-phenyl)-4,4-dimethyl- 1,2,3, 4-tetrahydro- quinoline-6-carboxylic acid methyl ester (3 g, 7 mmol), morpholine (6.2 mL, 71 mmol), copper(I) iodide (0.8 g, 4.2 mmol), N,N-dimethylglycine hydrochloride (0.78 g, 5.6 mmol), and potassium carbonate (2.9 g, 21 mmol) in dimethyl sulfoxide (25 mL) was stirred at 120C for 16 h. Then the reaction mixture was cooled to room temperature. The reaction mixture was extracted with ethyl acetate (200 mL x 2), washed with saturated aqueous ammonium chloride solution (50 mL x 3) and brine (50 mL x 2), dried over anhydrous sodium sulfate and then concentrated in vacuo and the residue was purified by ISCO combi-flash chromatography (gradient elution, 30 - 50% ethyl acetate in petroleum ether) to afford 2-(2-chloro-5-morpholin-4-yl-phenyl)-4,4-dimethyl- 1,2,3, 4-tetrahydro-quinoline- 6-carboxylic acid ethyl ester (2.8 g, 93%) as a white solid: MS(ESI) M+l = 429.1.To a stirred mixture solution of 2-(2-chloro-5-morpholin-4-yl-phenyl)-4,4-dimethyl- l,2,3,4-tetrahydro-quinoline-6-carboxylic acid ethyl ester (2.8 g, 6.5 mmol) in methanol (50 mL) and tetrahydrofuran (50 mL) was added 30% sodium hydroxide in water (10 mL). The reaction mixture was stirred at 60C for 16 h. The mixture was neutralized with a 3 N aqueous hydrochloric acid solution and extracted with ethyl acetate (100 mL x 2), washed with water, dried over anhydrous sodium sulfate and then concentrated in vacuo to afford 2-(2-chloro-5-morpholin-4-yl-phenyl)-4,4-dimethyl- 1 ,2,3, 4-tetrahydro-quinoline-6- carboxylic acid (2.4 g, 92%) as a light white solid which was used for next step without further purification: MS(ESI) M+l = 401.1.To a suspension of 60% sodium hydride (650 mg, 16 mmol) in N,N-dimethylformamide (5 mL) was added methanesulfonamide (1.53 g, 16 mmol) at room temperature. The resulting mixture was stirred at 25C for 1 h to afford Solution A23. A solution of 2-(2-chloro-5- morpholin-4-yl-phenyl)-4,4-dimethyl-l,2,3,4-tetrahydro-quinoline-6-carboxylic acid (900 mg, 2.3 mmol) and Iota, Gamma-carbonyldiimidazole (730 mg, 4.5 mmol) in N,N- dimethylformamide (3 mL) was stirred at 70C for 1 h and cooled to room temperature to afford Solution B23. Solution B23 was added to Solution A23 and the resulting mixture was stirred at 25C for 1 h. To the reaction mixture was added water (0.5 mL). The mixture was filtered to remove the insoluble solid, and the filtrate was purified by Waters automated flash system (column: Xterra 30 mm x 100 mm, sample manager 2767, pump 2525, detector: ZQ mass and UV 2487, solvent system: acetonitrile and 0.1% ammonium hydroxide in water) afforded N-[2-(2-chloro-5-morpholin-4-yl-phenyl)-4,4-dimethyl- 1, 2,3, 4-tetrahydro-quinoline-6-carbonyl] -methanesulfonamide (190 mg, 17%) as a white solid: MS(ESI) M+l = 478.0.
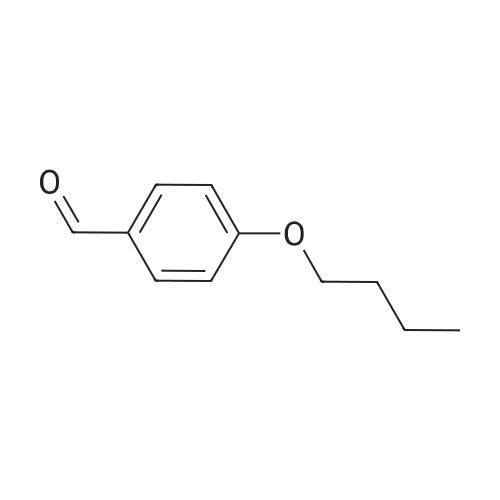
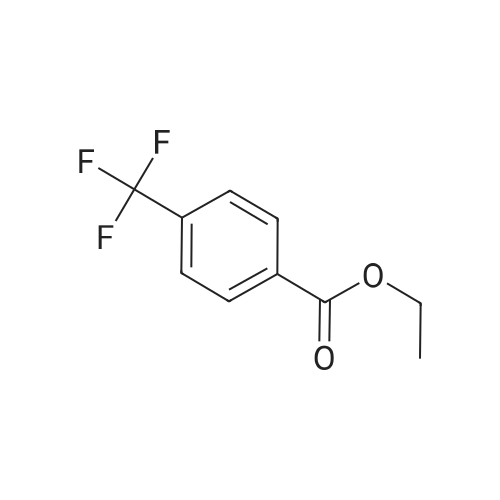
General procedure: An oven-dried Schlenk tube (A) equipped with a magnetic stir bar was charged with AgF (132.2 mg, 1.05 mmol, 3.5 equiv), sealed with a septum, and degassed by alternating vacuum evacuation and nitrogen backfill (three times) before freshly distilled EtCN (3 mL)was added. To the resulting suspension, which was precooled to -78 C (dry ice-acetone bath), was added TMSCF3 (149.3 mg, 1.05 mmol, 3.5 equiv) by microsyringe. The mixture was allowed towarm to r.t. and stirring was continued for an additional 15 min. In due course, AgF solid dissolved and a gray, dark solution of [Ag-CF3] formed. Another Schlenk tube (B) equipped with a magnetic stir bar was charged with the aniline (ArNH2; 0.30 mmol, 1.0 equiv) in freshly distilled EtCN (1.5 mL). To the resulting solution, which was precooled to 0 C (ice bath), aq HCl (12 M; 50.0 muL, 0.60mmol, 2.0 equiv) was added; precipitate formed immediately. After 5 min stirring, t-BuONO (37.7 mg, 0.33 mmol, 1.1 equiv) was added by microsyringe, and the mixture was allowed to stir at 0 C for 15 min. The resulting suspension in Schlenk tube (B) was degassed by alternating vacuum evacuation at -196 C (liquid nitrogen), then the solution was allowed to warm to r.t. under a nitrogen atmosphere (three times), and finally cooled to -78 C (dry ice-acetone bath). The gray, dark solution of [AgCF3] in Schlenk tube (A), which was precooled to -78 C (dry ice-acetone bath), was added to Schlenk tube (B) (ArN2+Cl-) by syringe at -78 C (dry ice-acetone bath) over a period of 1 h. After the addition was complete, the reaction mixture was stirred for 3 h at -78 C (dry ice-acetone bath), allowed to warm to r.t., and stirring was continued for an additional 1 h. An off-white precipitate was observed, and the reaction mixture was diluted with EtOAc (3 mL) and filtered through a short silica gel column. The solvent was removed under reduced pressure with a rotatory evaporator, and the crude residue was purified by silica gel column chromatography to give the desired trifluoromethylation product 3. The yields of products 3a, 3f, 3g, 3l, 3o, 3r, 3x, and 3zb are based on the 19F NMR spectra with 4-F3COC6H4OMe as internal standard. Analytical data for the representative product ethyl 4-(trifluoromethyl)benzoate (3i) are provided below. Data for other products can be found in the literature.
Step 1 To a stirred mixture of 8-bromo-2,2-dimethyl-4-(p-tolyl)chromene-6-carboxylic acid (56 mg, 0.15 mmol) in dichloromethane (4 mL) was added 4-(dimethylamino)-pyridine (44 mg, 0.36 mmol) and ethyl-4-aminobenzoate (37 mg, 0.225 mmol). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (43 mg, 0.225 mmol) was then added and the reaction mixture was stirred at room temperature for 24 hours. The resulting solution was directly submitted to column chromatography on silica gel using dichloromethane as the mobile phase. Ethyl 4-[[8-bromo-2,2-dimethyl-4-(p-tolyl)chromene-6-carbonyl]amino]benzoate (69 mg, 0.133 mmol) was obtained as a white solid. 1H NMR (300 MHz, CDCl3) ? 8.03 (2H, d, J=8.7 Hz), 7.91 (1H, d, J=2.1 Hz), 7.70 (1H, bs), 7.65 (2H, d, J=8.7 Hz), 7.50 (1H, d, 2.1 Hz), 7.25 (4H, s), 5.70 (1H, 1s), 4.359 (2H, q, J=7.1 Hz), 2.39 (3H, s), 1.56 (6H, s), 1.39 (3H, t, J=7.1 Hz).
General procedure: To a solution of arylamine (0.5 mmol, 1.0 equiv) in MeOH(1.0 mL) was added HCl (0.5 mL, 1.5 mmol, 3.0 equiv), followed by H2O (0.5 ml). This mixture was stirred 2 min,and the NaNO2 solution (0.25 mL) was then added. The NaNO2 solution was prepared by dissolving 35 mg ofNaNO2 in H2O (0.25 mL). This mixture was stirred 30 minat 0-5 C, followed by HCl (135 mg, 1.5 mmol, 3.0 equivalents) in MeOH (1.0 mL). This mixture was stirred 60min. H2O (10 mL) was added to reaction mixture, then extracted with CH2Cl2 (50 mL, 3×). The combined organic layer was dried over Na2SO4, followed by evaporation to give the products.
With palladium diacetate; caesium carbonate; 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In 1,4-dioxane; at 100℃; for 2h;Inert atmosphere;
Caesium carbonate (6.20 g, 19.04 mmol), B1NAP (592 mg, 0.95 mmol) and palladium(II) acetate (178 mg, 0.79 mmol) were added under a protective gas atmosphere to a solution of 2,4-dichloro-6,7-dihydro- 5H-cyclopenta[d]pyrimidine (3.00 g, 15.87 mmol) and 4-aminobenzoic acid ethyl ester (2.62 g, 15.87 mmol) in anhydrous 1 ,4-dioxane (60 ml) and the mixture was stirred at 100C for 2 h. Then the reaction mixture was poured onto a silica gel column and eluted with dichloromethane. White solid. Yield: 978 mg (19% of theoretical) LC-MS (method 2): R, = 3.85 min, m/z: [M+H]+ = 318.2 13C-NMR (101 MHz, DMSO-d6, 5 ppm): 14.2, 21.1, 27.8, 34.3, 60.2, 1 17.7, 122.3, 123.8, 130.1, 144.6, 155.1 , 158.7, 165.4, 177.7
A mixture of 165 g (1 mol) of ethyl p-aminobenzoate, 112.35 g (1.05 mol) of N-methylaniline, 159 g (1.5 mol) of trimethyl orthoformate and 10.2 g (0.1 mol) of a specific surface area of 137 m2 / g Gamma-type activated alumina into the 500ml reaction flask,Stirring and heating to 50 ~ 60 , incubation reaction 2h. Open the vacuum pump, and gradually increase the vacuum, vacuum distillation of the resulting methanol, to no methanol distillation.The reaction solution was filtered while hot, the recovered active alumina was repeatedly used and the mother liquor was distilled under reduced pressure to give a pale yellow viscous liquid N- (4-ethoxycarbonylphenyl) -N'-methyl-N'-phenyl Formamidine 270 g, yield 95.7%, purity 99.5%.
With transition metal catalyst; pyridine derviative; acid derviative; at 150℃; for 12.0h;Green chemistry;
Accurately weigh 0.005mmol (l. Lmg) of the transition metal, add 10mL has been put into the magnetic stirrer Young's reaction tube, The oxygen reaction was carried out in the Young's reaction tube, and the reaction was carried out under oxygen conditions. The syringe was applied to the Young's reaction tubeA solution of 0.04 mmol of co-catalyst I, 0.08 mmol of co-catalyst II, 0.2 mmol of 4-carboxylate and 1 ml of l-3'-propylene glycol, the above-mentioned Young's reaction tube was placed on a magnetic stirrer and stirred at 150 C for 12 h. After the reaction was completed,The pH of the solution was neutral, and the reaction solution was post-treated to obtain pure product of ethyl 6-formate quinoline in a yield of 45%.
General procedure: NMM (2 equiv.) was added to a solution of Boc-Lys(Boc)-OH (1 equiv.) in anhydrous DCM at 0C, followed by dropwise addition of IBCF (1.5 equiv.). The mixture was stirred for 30min, then benzocaine or compound 6 (1.5 equiv.) was added, and the resulting mixture was stirred at 0C for 1h and then at room temperature for 12h. The precipitate was filtered off and the filtrate was washed with H2O (2×40mL), brine (40mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EA/DCM=1/2, v/v) to yield pure compound 10 (37%) or 11 (25%).
165 g (1 mol) of ethyl p-aminobenzoate, 117.7 g (1.1 mol) of N-methylaniline, 10 g of p-toluenesulfonic acid,200mL with toluene toluene added to the 1000mL reaction flask, stirred and warmed to 50-60 C, 2h dropwise 59.53g (1.1mol) formic acid(Content of 85%),Heating to reflux reaction, continue to separate the generated water, the reaction of about 5h to stop water when there is no water. The reaction solution was distilled off with atmospheric toluene recovery agent, toluene was recovered, the remaining vacuum distillation reaction solution,A pale yellow viscous liquid N- (4-ethoxycarbonylphenyl) -N'-methyl-N'-phenyl formamidine 262g, yield 93%, purity 99.4%.
With acetic acid; In Petroleum ether; at 50 - 65℃; for 2.25h;Green chemistry;Catalytic behavior;
1) Accurately weigh 16.5 g of ethyl p-aminobenzoate, 16.5 g of trimethyl orthoformate, 16.8 g of N-methylaniline into a 150 mL three-necked flask, and add 60 mL of petroleum ether to a three-necked flask and place it in magnetic stirring. A dropping funnel, a water separator and a thermometer were placed on the three-necked flask, and the outer wall of the water separator was pre-passed with a freezing liquid of -5 C. A three-necked flask was placed in a magnetic stirrer. 2) Stir and raise the temperature to a temperature of 50 C in a three-necked flask .9 g of glacial acetic acid was added dropwise to the reaction vessel within 15 min, and the reaction was kept for 0.6 h; The temperature was raised to 50 C, the reaction was 0.8 h; the temperature was further raised to 65 C, and the reaction was 0.6 h; The liquid in the lower layer of the water separator is continuously separated, and the reflux is stopped at the upper end of the water separator. 3) The reaction mixture was desolvated under a vacuum, then added 60 mL of toluene, cooled to 40 C; 4) Washing 3 times with hydrochloric acid aqueous solution of pH=2, using 50 mL of hydrochloric acid aqueous solution each time, washing for 8 min each time; then washing with 0.6 mol/L sodium bicarbonate aqueous solution 30 mL, adjusting pH to 8; finally washing 3 times with distilled water Every time you use40 mL of distilled water. 5) The organic layer after the water washing treatment was transferred to a rotary steaming flask for vacuum distillation, and the temperature of the vacuum distillation was controlled at about 125 C, and the product was purified by distillation under reduced pressure, the yield was 98%, and the chroma value was 26 Hazen.
General procedure: Xantphos (28.9 mg, 0.05 mmol), Cs2CO3 (32.6 mg, 0.10 mmol) and [Ru(p-cymene)Cl2]2 (15.3 mg, 0.025 mmol) were added to a Schlenk tube. Then the tube was evacuated and refilled with nitrogen. Dry toluene as solvent (2.0 mL)were added and the solution was stirred at room temperature under nitrogen for 30 min. Activated molecular sieves 4A (0.10 g) and triol 6a (106.1 mg, 1.00 mmol) were then added. And the solution was stirred at room temperature under nitrogen for 40 min. Subsequently, aniline 5a (46.6 mg, 0.50 mmol) was added and the solution was stirred at room temperature under nitrogen for 20 min. Then the resulting solution was heated to reflux and stirred for 48 h. The reaction was monitored by TLC. After completion, the solution was filtered through a plug of Celite and washed with EtOAc (3 mL x 3). The filtrate was concentrated under vacuum. Purification via flash column chromatography with silica gel (eluting with PE/EA = 5/1 (v/v)) yielded 4a (43.0 mg, 81% yield) as light yellow oil.
With p-toluenesulfonic acid monohydrate; sodium sulfate; In chloroform; for 48h;Reflux;
General procedure: A solution of 4-substituted aniline (0.616 mmol), benzoylacetate(0.71 mmol), TsOHH2O (0.03 mmol) and Na2SO4 (6.16 mmol) inCHCl3 (3 ml) was stirred and refluxed for 48 h. The reaction mixture was concentrated in vacuo and washed with hexane or Et2O. Next,collected filtrate and evaporated to get imine. The imine was dissolvedin diphenylether (1 ml) and heated to 250 C for 10 min. Thereaction mixture was cooled to room temperature. After triturationwith Et2O, the product was collected by filtration.
With thionyl chloride; In toluene; at 20℃; for 19.5h;
General procedure: 33.8 g (0.25 mol) of N-methylformylaniline, 12.5 g of toluene, and 1.65 g (0.01 mol) of ethyl paraaminobenzoate were added to the reaction device. After stirring and dissolving, 29.8 g of sulfoxide was added dropwise. (0.25 mol), after stirring (about 70 min), continue stirring for 20 min.Then add 225g of toluene, and then add 39.6g (0.24mol) of ethyl p-aminobenzoate step by step. After the addition (about 90min), stir at room temperature for 18h.The material was orange-yellow, and the purity of UV-1 was 93.0% by sampling (HPLC, the same below). The material liquid was heated to 55 ± 1 C and kept for 7 hours. The material liquid was yellow. The purity of UV-1 was 97.3%. Continue heating the material liquid to 90 ± 1 , stop heating, and recover sulfoxide. Cool the recovered sulfoxide to 35 ± 1 , neutralize it with 10wt% sodium carbonate aqueous solution to pH = 6, and control the temperature below 35 . After neutralization, let it stand and separate layers, and layer Desolvation of the organic layer,68.3 g of ultraviolet absorber UV-1 is obtained,The yield was 96.7% and the purity was 99.6%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping