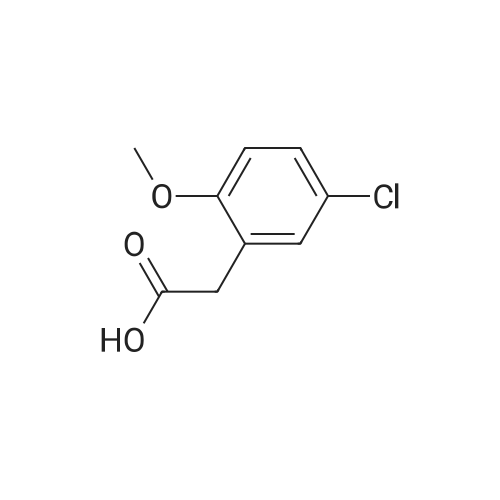
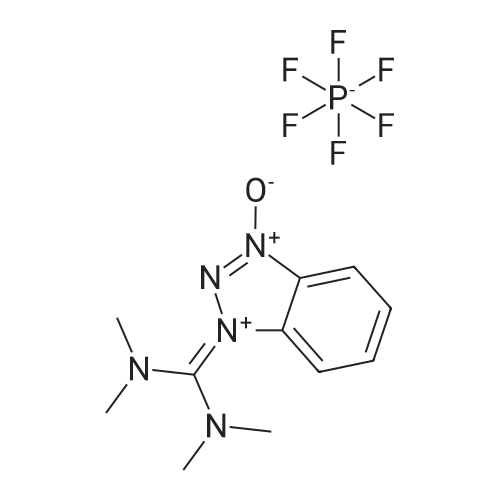
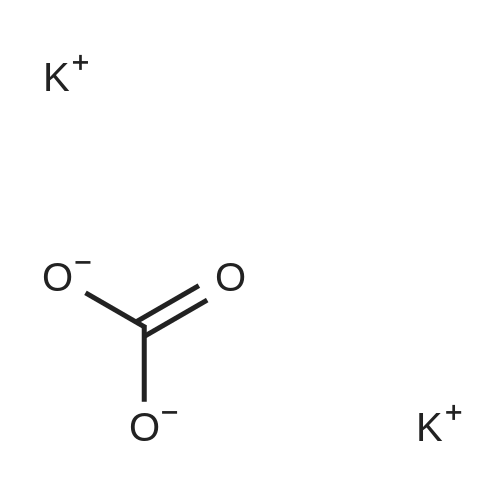
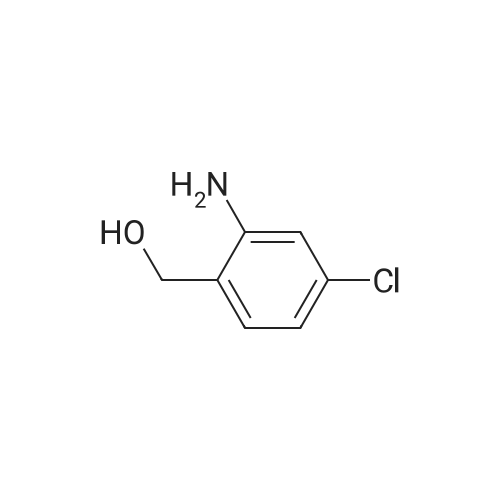
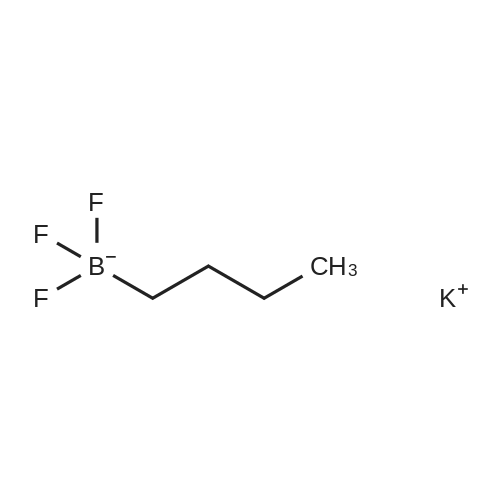
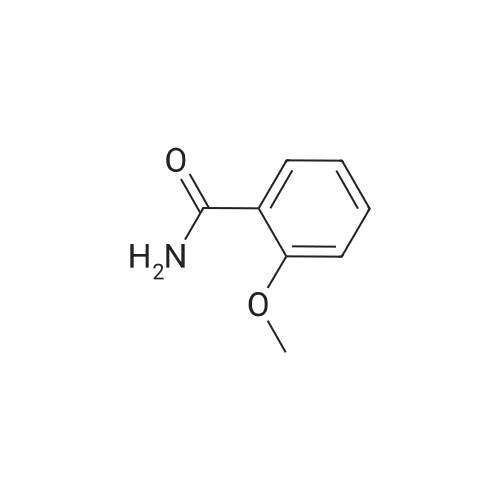
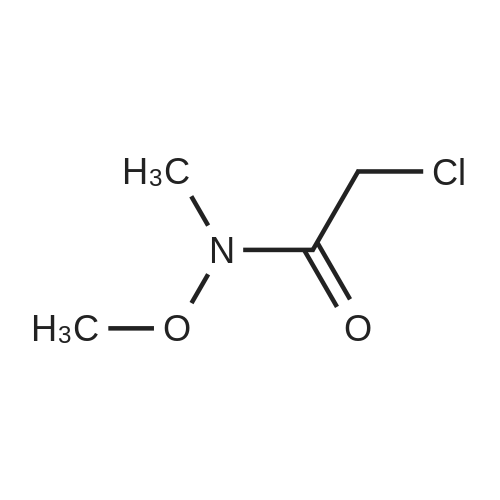
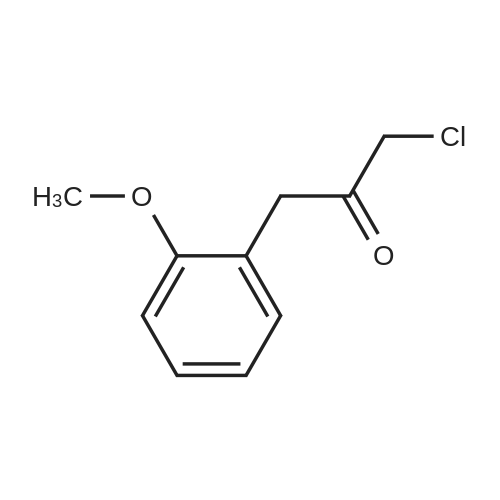
|
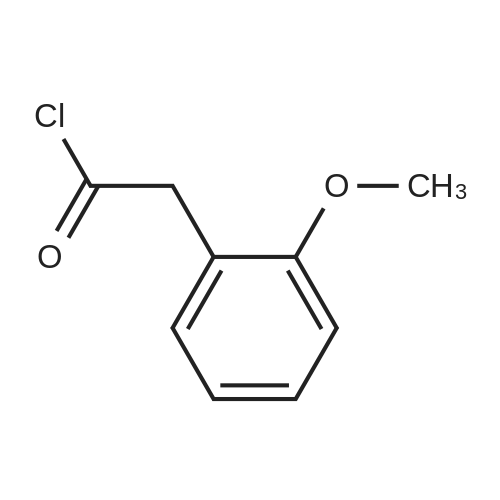
With thionyl chloride; In tetrahydrofuran; for 1.5h;Reflux; |
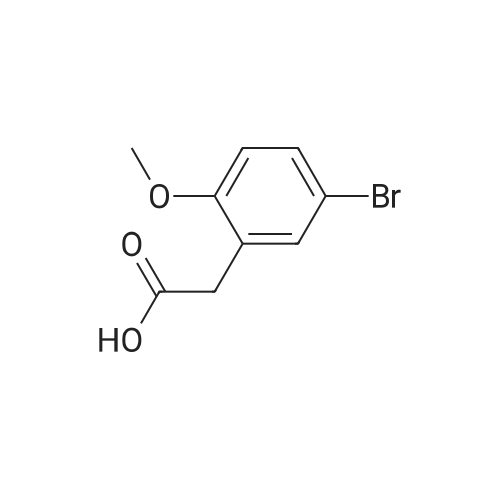
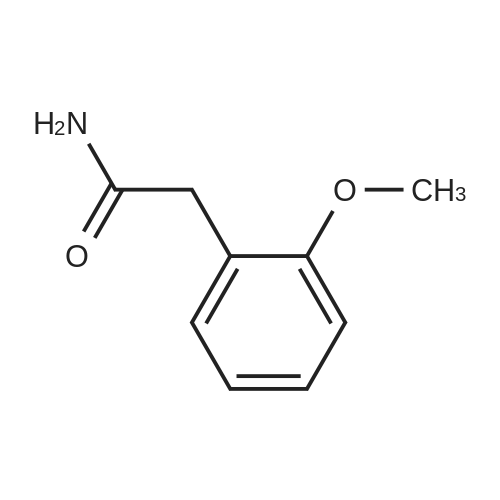
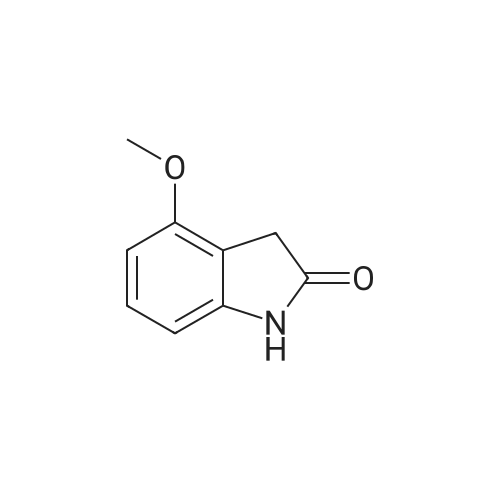
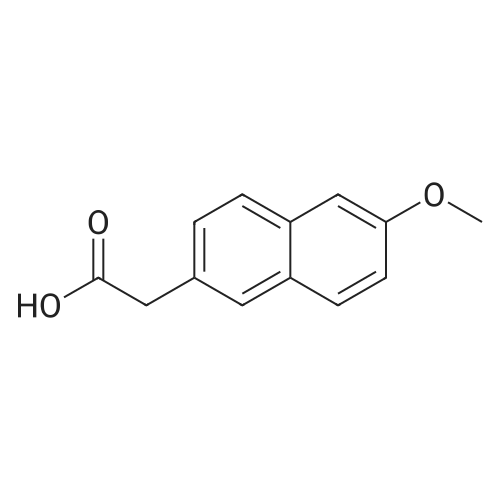
General procedure: 2-Methoxyphenyl acetic acid (465 mg, 2.8 mmol) and thionyl chloride (204 muL, 2.8 mmol) were refluxed in anhydrous THF (1 mL) for 1.5 hours, and the resulting reaction solution was added to a solution containing 9d (200 mg, 0.67 mmol) and DIPEA (210 muL, 1.2 mmol) in THF (0.5 mL). After stirring for 1 day, the reaction mixture was taken up in EtOAc, washed with 1M aqueous HCl, washed with 5% aqueous Na2CO3, dried (Na2SO4) and evaporated. The residue was chromatographed on silica gel (hexane/EtOAc 4:1) to yield an oil (161 mg, 54%). An 81 mg sample was further purified by HPLC (4.8 mg). 1H NMR (300 MHz, CDCl3) delta 11.2 (br s, 1H, NH), 7.31 (m, 2H, Ar-H), 6.95 (m, 2H, Ar-H), 4.53 (s, 2H, 7-CH2), 4.27 (q, J = 7.1 Hz, 2H, CH2CH3), 4.17 (q, J = 7.1 Hz, 2H, CH2CH3), 3.89 (s, 3H, OCH3), 3.82 (s, 2H, CH2Ar), 3.68 (m, 2H, 5-CH2), 2.86 (m, 2H, 4-CH2), 1.30 (t, J = 7.2 Hz, 3H, CH2CH3), 1.30 (t, J = 6.9 Hz, 3H, CH2CH3). ESI-MS m/z 447.3 [M + H]+. |
|
With thionyl chloride; N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 4h; |
General procedure: To a solution of substituted phenyl acetic acid or naphthylacetic acid (200 mg, 1.1 mmol) in dry DCM (10 ml) was added SOCl2 (128 mg, 1.1 mmol) and DMF (1 drop). The mixture was stirred at room temperature for 4 hours, following which the solvent was removed in vacuo. Dry DCM (10 ml) was added to re-dissolve the residue. After that, the solution was dropwise added into the mixture of 4-methoxybenzyl hydroxylamine (3) (165 mg, 1.1 mmol) and Et3N (223 mg, 2.2 mmol) in DCM (10 ml) and stirred at room temperature for 1h. The reaction mixture was quenched with water (20 ml) and then extracted with DCM (3 x 20ml). The combined organic layers was washed with brine and dried with anhydrous Na2SO4. The solvent was removed in vacuo. The crude product was purified with column chromatography (33% EA/Petroleum ether) to obtain a white solid in yields range from 68 to 88% yield. |
|
With trichlorophosphate; In 1,2-dichloro-ethane; for 3h;Reflux; |
General procedure: Aralkanoic acid chlorides 2a-g were synthesized by the reaction of aralkanoic acid 1a-g (1 mmol) in the presence of 1,2-dichloroethane (12 mL) solvent and phosphorous oxychloride(0.4 mL) chlorinating agent under reflux for 3hours. Then, the resulting solution was cooled to room temperature, and the solvent was removed under reduced pressureto afford aralkanoic acid chloride 2a-g, which was directly used in the next step without further purification. Acid chloride 2a-g was dissolved in acetonitrile (80 mL), addeddropwise to a solution containing hydrazine hydrate(1 mmol), TEA (0.5 mL) and acetonitrile (20 mL) and allowed to reflux for 3 hours with monitoring by TLC. After consumption of the starting material, the reaction mixture was cooled to room temperature. Evaporation of the solvent under reduced pressure yielded crude acid hydrazide 3a-g as a white solid on cooling, which was purified by column chromatography and crystallized in methanol [46]. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; for 3h;Inert atmosphere; |
General procedure: To an over-dried 100 mL three-necked flask, the carboxylic acid (10 mmol), DMF (5 drops) and DCM (30 mL) were added under a N2 atmosphere. Oxalyl chloride (1.0 mL, 12 mmol) was added dropwise at 0 C resulting in vigorous bubbling. The mixture was stirred for 3 h at room temperature, and the solvent was then removed in vacuo. The resulting acid chloride was used immediately without further purification. To a solution of the acid chloride in DCM (30 mL) ,a solution of 1,1,1,3,3,3-hexamethyldisilazane (30 mmol) in DCM (10 mL) was added dropwise at 0 C, and the solution was then allowed to warm to room temperature. After stirring overnight, the reaction system was quenched with 1 M HCl aq. and saturated aqueous NH4Cl (excess amount) and the organic layer was separated. The aqueous layer was extracted with DCM (2x15 mL). The combined organic layers were washed with saturated aqueous NH4Cl (30 mL) and brine (30 mL), dried over MgSO4, filtered and evaporated in vacuo. The resulting crude material was purified by recrystallization from EtOAc and hexane. The resulting product (5 mmol), 8-bromomethylquinoline (6 mmol), Al2O3 (50 mmol), KOH (25 mmol) and dioxane (30 mL) were added to an over-dried 100 mL three-necked flask. The mixture was stirred for 8 h at 60 C and then was filtered through a celite pad. The filtrate was washed with H2O (30 mL) and the organic layer was separated. The aqueous layer was extracted with EtOAc (2x15 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous Na2SO4, filtrated and evaporated in vacuo. The resulting crude amide was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 1/1). |
|
With trichlorophosphate; In 1,2-dichloro-ethane; for 3h;Reflux; |
General procedure: The substituted aromatic acid chloride 2 was synthesizedthrough the reaction of substituted aromatic acid 1(1 mmol) in the presence of 1,2-dichloroethane (12 mL)solvent and phosphorous oxychloride (0.4 mL) chlorinatingagent under reflux for 3 h. Then, the resulting solutionwas cooled to room temperature, and the solvent wasremoved under reduced pressure to yield substituted aromaticacid chloride 2, which was directly used in the nextstep without any further purification. The substituted aromaticacid chloride 2 was dissolved in acetonitrile(80 mL), added dropwise to a solution containing hydrazinehydrate (1 mmol), TEA (0.5 mL), and acetonitrile(20 mL), and allowed to reflux for 3 h with monitoring byTLC. After consumption of the starting material, thereaction mixture was cooled to room temperature. Evaporationof the solvent under reduced pressure yielded crudesubstituted aromatic acid hydrazide 3 as a white solid on cooling, which was purified by column chromatography ifneeded and crystallized with methanol. |
|
With thionyl chloride; |
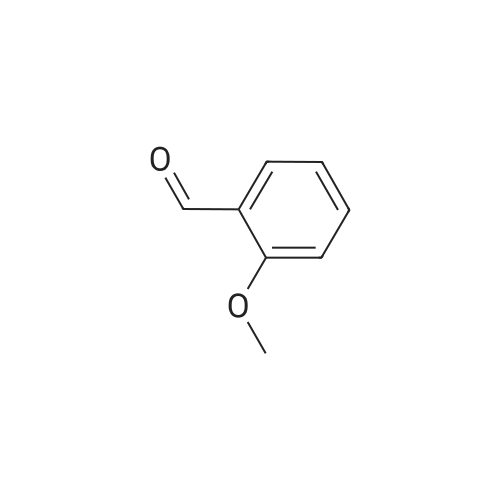
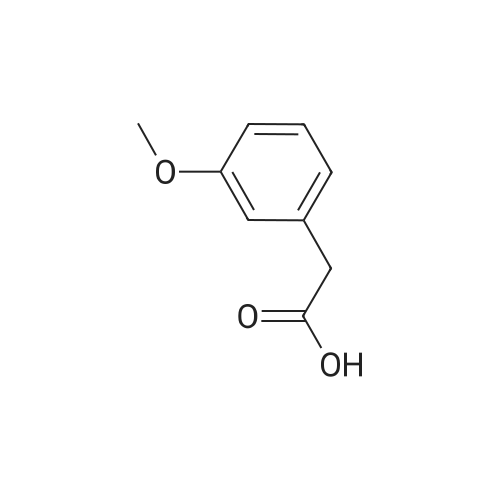
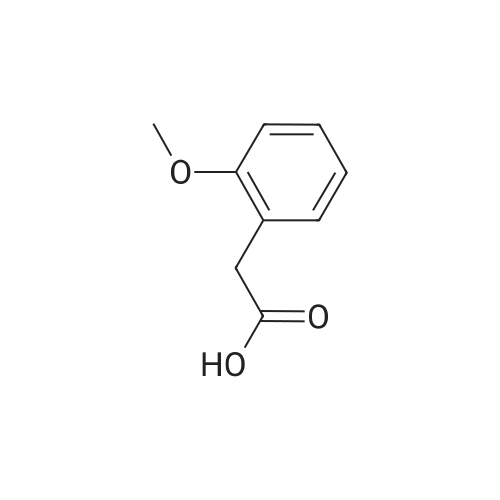
[0081] <strong>[93-25-4]2-Methoxyphenylacetic acid</strong> (75 mg, 0.45 mmol) was treated with thionyl chloride, and thereby made into 2-methoxyphenylacetyl chloride, and the resultant and 5-(4-aminophenyl)-1H-naphtho[1,2-b][1,4]diazepine-2,4(3H,5H)-dione(95 mg, 0.3 mmol) were heated in pyridine to obtain the title compound (83 mg, yield 60%) as pale yellow crystals.1H NMR (DMSO-d6, 400 MHz) delta: 3.14 (1H, d, J=12Hz), 3.64 (2H, s), 3.69 (1H, d, J=12Hz), 3.77 (3H, s), 6.90 (1H, t,J=7Hz), 6.9-7.0 (2H, m), 7.15 (2H, d, J=8Hz), 7.2-7.3 (2H, m), 7.59 (1H, t, J=8Hz), 7.6-7.7 (4H, m), 7.91 (1H, d, J=8Hz),8.25 (1H, d, J=8Hz), 10.17 (1H, s), 10.86 (1H, s) |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping