* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Organic Chemistry, 2014, vol. 79, # 3, p. 1254 - 1264
2
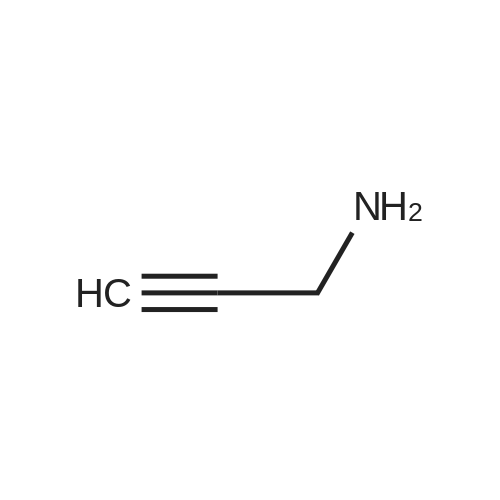
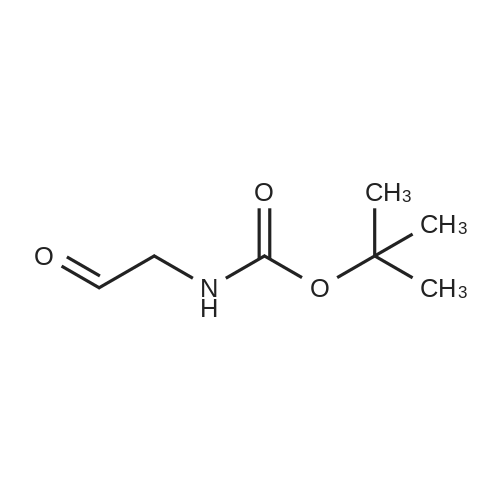
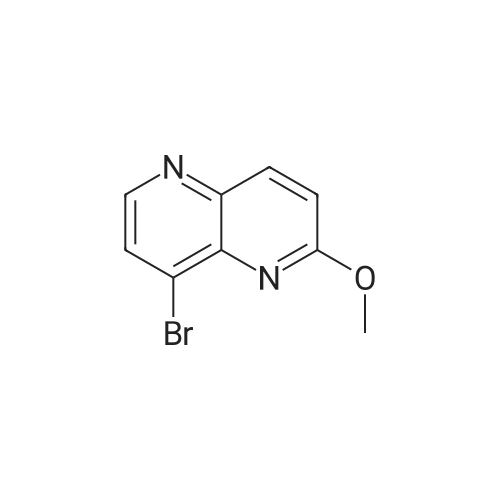
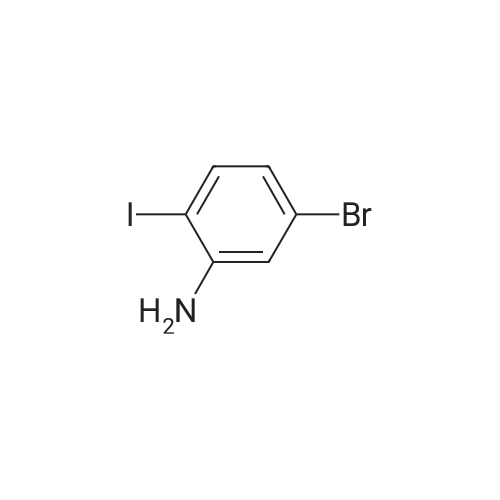
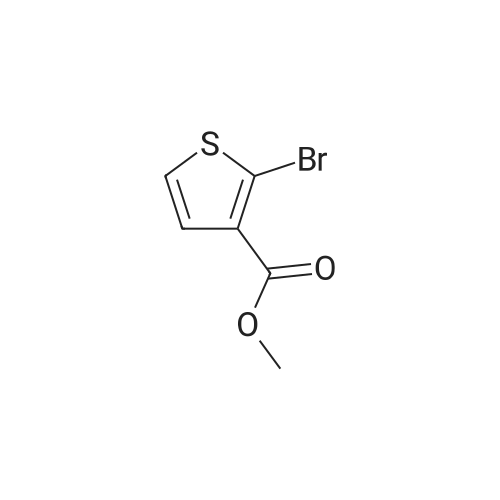
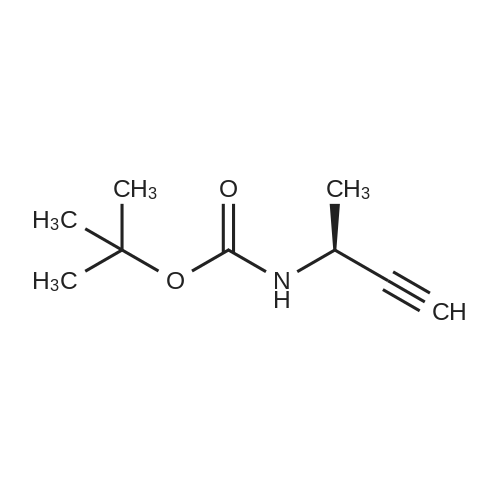
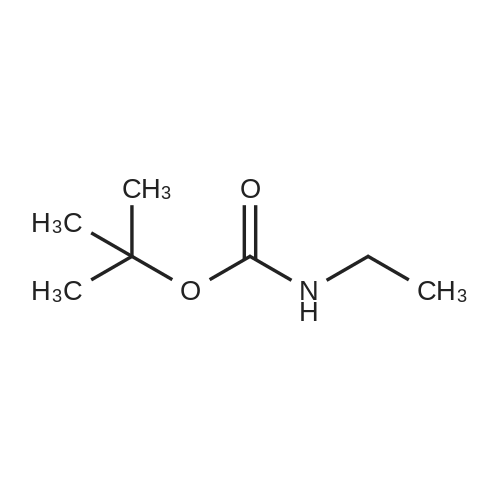
[ 92136-39-5 ]
[ 78888-18-3 ]
Yield
Reaction Conditions
Operation in experiment
63%
Stage #1: With n-butyllithium; copper(l) cyanide In tetrahydrofuran at -78℃; for 0.25 h; Stage #2: With tri-n-butyl-tin hydride In tetrahydrofuran at -78℃; for 0.333333 h; Stage #3: at -78℃; for 1 h;
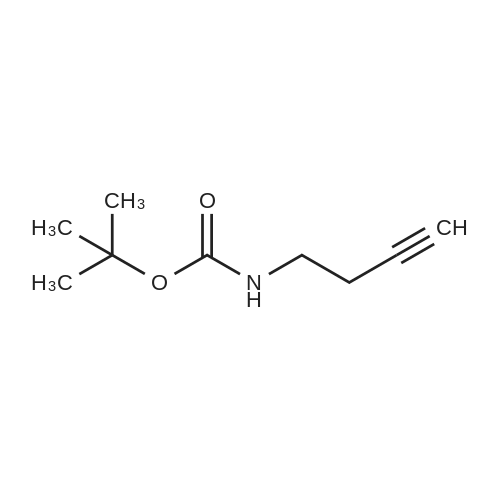
A solution of copper cyanide (1.1Sg, 12.9 mmol) in THF (30 mL) at ?78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at ?78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at ?780C for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2percent ethyl acetate/heptane to provide the desired product (3.66 g, 63percent). 1H NMR (400 MHz, CDCl3) ? 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
With ammonium hydroxide; n-butyllithium; ammonium chloride; tri-n-butyl-tin hydride In tetrahydrofuran; n-heptane; dichloromethane; ethyl acetate
EXAMPLE 314A tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78° C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78° C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78° C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite.(R).). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2percent ethyl acetate/heptane to provide the desired product (3.66 g, 63percent). 1H NMR (400 MHz, CDCl3) δ 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
Stage #1: With n-butyllithium; copper(I) cyanide In tetrahydrofuran at -78℃; for 0.25 h; Stage #2: With tri-n-butyl-tin hydride In tetrahydrofuran for 0.333333 h;
tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78° C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78° C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78° C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite.(R).). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2percent ethyl acetate/heptane to provide the desired product (3.66 g, 63percent). 1H NMR (400 MHz, CDCl3) δ 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
Di-tert-butyl dicarbonate (17.5 g, 80.0 mmol, 1.0 equiv) was added dropwise at 0 °C to a soln of prop-2-yn-1-amine (5.49 mL, 80.0 mmol, 1.0 equiv) in CH2Cl2 (160 mL). After 1 h of stirring, the solvent was removed in vacuo and the resulting colorless oil was dried under high vacuum overnight to yield a white solid (12.4 g, quantitative yield), which was used as such without further purification. The spectral data corresponds to that reported in the literature.1Rf = 0.38 (Hexanes/AcOEt 9:1). mp = 41-42 °C (lit.:[1] 40-44 °C). 1H NMR (500 MHz, CDCl3): d (ppm) 4.94 (br s, 1H), 3.91 (br d, J = 2.5 Hz, 2H), 2.23 (t, J = 2.5 Hz, 1H), 1.45 (s, 9H). 13C NMR (126 MHz, CDCl3): d (ppm) 155.2, 80.1, 79.9, 71.1, 28.2.
99.4%
With triethylamine In dichloromethane
(7a): To a solution of prop-2-ynylamine (5.50 g, 0.10 mmol), Et3N (14 ml, 1.5 eq) in CH2Cl2 (200 mL) was added (Boc)2O (21.8 g, 1.0 eq). The reaction mixture was stirred at rt for 2.5 h, then concentrated, the residue was passed through a silica gel pad, eluted with 20percent ethyl acetate-hexanes (500 mL), the eluant was concentrated to give prop-2-ynyl-carbamic acid tert-butyl ester (15.40 g, 99.4percent) as a crystalline solid.
99%
at 20℃; Cooling with ice
5.5 g of propargylamine (100 mmol) was dissolved in 50 mL of ethyl acetate,Boc anhydride (32 g, 147 mmol) was added under ice-water bath,The reaction was allowed to warm to room temperature overnight,The organic phase was washed with 1percent hydrochloric acid solution,Saturated sodium bicarbonate solution and saturated brine, dried over anhydrous sodium sulfate and spun dry to give product 13 as a yellow solid (15.3 g, 99 mmol, 1 99percent).
98%
at 0℃; for 0.416667 h;
test-Butyl prop-2-ynylcarbamate (46). To a solution of propargylamine (803 mg, 14.6 mmol) in CH2C12 (15 mL) at 0 °C was added a solution of di-tert-butyl dicarbonate (2.67 g, 15.3 mmol) in CH2C12 (20 mL) via dropping funnel over 25 min, the ice bath was removed and the resultant solution was stirred at ambient temperature for 30 min. The solvent was removed in vacuo and the crude material was chromatographed on silica gel (EtOAc/Hex, 10/90, Rf= 0.28) to afford the title compound 46 (2.23 g, 98percent yield) as a white solid: mp = 39-40 °C ; 1H NMR (CDC13) 8 4.79 (s, 1H), 3.90 (br s, 2H), 2.20 (m, 1H), 1.43 (s, 9H); LRMS (ESI) m/z calcd for CsHl3NNa02 [M + Na] + 178, found 178.
90%
for 12 h;
Step A: tert-Butyl prop-2-ynylcarbamate: A solution of propargylamine (5.00 gs 90.8 mmol) and BoC2O (18.8 g, 86.2 mmol) in DCM (200 mL) was stirred for 12 hours. The mixture was washed with dilute aqueous HCl, and the organic layer was dried (Na2SOa), filtered, and concentrated in vacuo. The resulting oil crystallized upon standing to give 12.0 g (90percent) of the title compound. 1H NMR (400 MHz, CDCl3) δ 4.78 (br s, IH), 3.92 (s, 2H), 2.22 (s, IH), 1.46 (s. 9H).
90%
With triethylamine In dichloromethane at 20℃;
Prop-2-ynyl-carbamic acid tert-butyl ester 2:BocHNDi-tert-butyl dicarbonate (9.0g, 40.0mmol) was added to a solution ofpropargylamine (2.0g, 36.0mmol) and triethylamine (7.6ml_, 54.5mmol) in dichloromethane (20ml_) at rt. After overnight stirring, the reaction was washed with saturated solution of NH4CI and brine. The organic layer was dried over Na2S04, filtered and evaporated to provide Boc-propargylamine 2 as a brown viscous oil (5.1 g, 36.0mmol, 90percent yield).
90%
at 20℃; for 4 h; Inert atmosphere
To a stirred solution of propargyl amine (2.0 g, 36.3 mmol) in THF (30 mL) was added di-tert-butyl dicarbonate (8.78 g, 40.2 mmol, 1.1 equiv) at rt. The solution was stirred at the same temperature for 4h, and then concentrated in vacuo. The resulting residue was dissolved in EtOAc (100 mL), washed with water (3×20 mL) and brine (20 mL) and then dried over anhydrous Na2SO4. After removal of the solvent in vacuo, N-Boc-propargylamine (5.11 g, 32.9 mmol, 90percent) was obtained as a yellow solid, which was pure enough and used in the subsequent reaction without further purification. mp 38-39 °C (Lit.13 41-42 °C). 1H NMR (500 MHz, CDCl3) δ 4.81 (brs, 1H), 3.89 (s, 2H), 2.19 (t, J = 2.5 Hz, 1H), 1.42 (s, 9H); 13C NMR (125 MHz, CDCl3, ppm): δ 155.92, 80.76, 71.86, 30.99, 28.97, 28.04.
90%
Stage #1: With triethylamine In dichloromethane at 20℃; for 0.25 h;
Preparation of Compound 29 A solution of amine 28 (10.0 g, 0.181 mmol) in CH2Cl2 (100 mL) was charged with triethylamine (24.0 g, 0.237 mmol) at room temperature, and the reaction mixture was stirred at room temperature for 15 min. (Boc)2O (43.5 g, 0.199 mmol) was added dropwise to the stirring solution. The reaction mixture was partitioned between CH2Cl2 (100 mL) and water (100 mL). The aqueous layer was separated and extracted with CH2Cl2 (100 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated to afford compound 29 (25.0 g, 90percent) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 4.82-4.72 (br s, 1H), 3.92-3.91 (m, 2H), 2.21 (t, J=2.4 Hz, 1H), 1.67 (s, 1H), 1.45 (s, 9H).
90%
for 12 h;
Step B: Preparation of tert-butyl prop-2-ynylcarbamate: A solution of propargylamine (5.00 g, 90.8 mmol) and BoC2O (18.8 g, 86.2 mmol) in DCM (200 mL) was stirred for 12 hours. The mixture was washed with dilute aqueous HCl, and the organic layer <n="20"/>was dried (Na2SO4), filtered, and concentrated in vacuo. The resulting oil crystallized upon standing to give 12.0 g (90percent) of the title compound. 1H NMR (400 MHz, CDCl3) δ 4.78 (br s, IH), 3.92 (s, 2H), 2.22 (s, IH)5 1.46 (s, 9H).
90%
for 12 h;
Step B: Preparation of tert-butyl prop-2-ynylcarbamate: A solution of propargylamine (5.00 g, 90.8 mmol) and BoC2O (18.8 g, 86.2 mmol) in DCM (200 mL) was stirred for 12 hours. The mixture was washed with dilute aqueous HCl, and the organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The resulting oil crystallized upon standing to give 12.0 g (90percent) of the title compound. 1H NMR (400 MHz5 CDCl3) δ 4.78 (br s, IH), 3.92 (s, 2H), 2.22 (s, IH)5 1.46 (s, 9H).
86%
With triethylamine In dichloromethane at 0 - 20℃; for 3.5 h;
To a solution of compound 154 (500 mg, 9.00 mmol) in CH2Cl2 (50 mL) was added TEA (1.63 mL, 11.7 mmol) and Boc2O (2.16 g, 9.90 mmol) at 0° C. The reaction mixture was continued to be stirred at 0° C. for 0.5 h, allowed to be warmed to room temperature and stirred for 3 h. Then the mixture was partitioned between CH2Cl2 (50 mL) and water (50 mL). The aqueous layer was separated and extracted with CH2Cl2 (2×50 mL). The combined organic extracts were washed with brine, dried over Na2SO4, concentrated, the residue was purified by column chromatography (silica gel, 2:3 hexanes/EtOAc) to afford desired compound 155 (1.20 g, 86percent) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 4.70 (br s, 1H), 3.91 (dd, J=5.3, 2.2 Hz, 2H), 2.21 (t, J=2.7 Hz, 1H), 1.45 (s, 9H).
82%
at 0 - 20℃; for 2.5 h;
Di-tert-butyl-dicarbonate (10 g, 47 mmol, 1 equiv.) was dissolved in CH2Cl2 (0.2 M) and cooledd own to 0 C. 3-Amino-1-propyne (2.6 g, 47 mmol, 1 equiv.) was added at 0 C over a period of 30 min. Then, the mixture was allowed to warm to room temperature and stirred for 2 hours. When the reaction was completed by TLC, the solvent was evaporated and n-Pentane was added. The solution was left on standing in the refrigerator for 12 h. The precipitate was collected by filtration to afford 6 g of the N-Boc-propargylamine (82percent) as a white solid.
80%
for 2 h; Inert atmosphere
To a stirred solution of propargylamine (0.83 mL, 12.9mmol, 1 eq.) in dry DCM (13 mL) under nitrogen, was added dropwise a solution of di-tert-butyldicarbonate (2.81 g, 12.9 mmol, 1 eq.) in dry DCM (7 mL) and the reaction was stirred for 2 h.The organic layer was washed with an aqueous solution of HCl (1 N), then with a saturatedaqueous solution of NaHCO3. The aqueous layer was extracted with DCM, then the combinedorganic layers were dried over MgSO4, filtered and concentrated under vacuum. The cruderesidue was purified by flash column chromatography on silica gel (cyclohexane/EtOAc: 80/20)to afford tert-butyl prop-2-yn-1-ylcarbamate (1.6 g 10.4 mmol, 80percent) as a white solid. Thiscompound has been previously reported.S1
79%
at 0 - 20℃; for 1 h;
A solution of 1.28 ml (6.00 mmol, 1.1 eq.) of Boc2O in 15 ml of dry CH2Cl2 was addeddropwise to a solution of 0.348 ml (5.45 mmol, 1.0 eq.) of propargyl amine in 15 ml of dryCH2Cl2 at 0°C. The reaction mixture was stirred at r.t. for 1 h. After TLC showed completeconsumption of starting material, solvent was removed under reduced pressure. The crudeproduct was purified by flash column chromatography (cHex/EtOAc 20:1 – 10:1). Theproduct was obtained as an off-white solid (yield: 0.667 g, 4.303 mmol, 79 percent).
75%
at 0 - 20℃; for 1.5 h;
Di-tert-butyl-dicarbonate (21.8 mg, 100.0 mmol) was dissolved in THF (25 mL) and the solution cooled to 0° C. and treated dropwise with a solution of propargylamine (Aldrich, 5.0 g, 90.0 mmol) keeping the temperature below 15° C. The mixture was stirred at rt for 1.5 h then concentrated under vacuum. The residue was dissolved in hexanes and filtered through a column of silica gel using 0-100percent CH2Cl2/hexanes to elute the product. The eluent containing the product was concentrated in vacuo to give a colorless oil which was dissolved in hexanes (150 mL) and cooled to 0° C. to give white crystals. The crystals were collected by filtration and dried under vacuum to give the title compound (10.5 g, 75percent). 1H NMR (CDCl3) δ 4.75 (s, 1H), 3.95 (s, 2H), 2.25-2.24 (m, 1H), 1.48 (s, 9H).
74%
at 23 - 28℃; for 2 h;
tert -butyl prop-2-yn-l-ylcarbamate (7) [0153] Into a reactor was added propagylamine (10.0kg, 182mol) and MTBE (154L). A B0C2O solution was prepared by dissolving B0C2O (41.3kg, 190 mol) in MTBE (61L) and transferred over a minimum of 60min to the propargyamine solution while maintaining a temperature between 23 and 28 °C. The reaction mixture was stirred for at least lh until >98.0percent conversion was obtained by GC analysis. A solution of sodium bisulfate (5.6kg of NaHSC>4 n 44L water) was added over a minimum of 15min while maintaining the temperature between 20 and 25 °C and stirred for 20min. The phases were separated and washed as before with a solution of sodium bisulfate (5.6kg of NaHSC>4 m 44L water). The resulting organic phase was washed with a sodium bicarbonate solution (4.0kg in 44L water) and water (2 x 47L). The organic phase was concentrated using a maximum jacket temperature of 40 °C until 62kg remained in the reactor. Heptane (186L) is added over a minimum of 20min while maintaining the temperature between 35 and 40 °C. The mixture was concentrated using a maximum jacket temperature of 40 °C until 70kg remained in the reactor. The mixture was cooled to 0 to 5 °C over a minimum of 3h and stirred for lh at 0 to 5 °C. The mixture was filtered and washed with heptane at 0 to 5 °C (2 x 15L). The wet cake was dried under vacuum at 25 to 30 °C to provide 20.9kg (74percent yield) of 7. 3/4 NMR (300 MHz, CDCLj δ 4.70 (s, 1H), 3.92 (d, J= Hz, 2H), 2.22 (m, 1H), 1.45 (s, 9H); Elemental Anal. Calcd. for C8H13N02: C, 61.91; H, 8.44; N, 9.03; 0, 20.62. Found: C, 61.99; H, 8.36; N, 9.11; 0, 20.54.
72%
With triethylamine In dichloromethane at 20℃; for 3 h;
To a solution of 2-PROPYN-1-AMINE (2 g, 36.4 mmol, 1 equiv) in CH2CI2 (20 mi) were added NEt3 (5.3 MI, 38.18 MMOL, 1.05 equiv) and bis (1, 1-dimethylethyl) dicarbonate (8.32 g, 38.18 mmol, 1.05 equiv). The resulting mixture was stirred at room temperature for 3 h then poured in a 2N aqueous HCI solution. The two layers were separated and the organic phase was washed with a saturated aqueous NAHC03 solution then dried over MGS04 and concentrated in VACUO TO give 1, 1-dimethylethyl 2-propyn-1-ylcarbamate (D317) (4.05 g, 72percent) as a colourless crystal.
72%
With triethylamine In dichloromethane at 20℃; for 3 h;
To a solution OF 2-PROPYN-1-AMINE (2 g, 36.36 mmol, 1 equiv) in CH2CI2 (20 ML) at room temperature were added NEt3 (5.3 ml, 38.18 mmol, 1.05 equiv) and bis (1,1- dimethylethyl) dicarbonate (8.32 g, 38.18 mmol, 1.05 equiv) and the resulting mixture was stirred at room temperature for 3 h then washed with a 2N aqueous HCI solution and a saturated NAHC03 aqueous solution, dried over MGS04 and concentrated in vacuo to give 1, 1-DIMETHYLETHYL 2-PROPYN-1-YLCARBAMATE (D38) (4.05 g, 72percent) as colourless needles which were used in the next step without further purification.
71%
With triethylamine In dichloromethane at 20℃; for 16 h; Cooling with ice
(0049) To a solution of prop-2-yn-1-amine (5.0 g, 90.9 mmol) and Et3N (18.4 g, 181.8 mmol) in DCM (100 mL) was added (Boc)2O (23.8 g, 109.1 mmol) dropwise while cooling the reaction mixture with an ice bath. The resulting mixture was removed from the ice bath once the addition was completed, and was then stirred at room temperature for 16 h. When the reaction was complete, the mixture was diluted with DCM (200 mL), washed with brine (100 mL_3), and the organic layer was then dried over Na2SO4 and then concentrated in vacuo. The residue was purified by column chromatography on silica gel (PE:EtOAc=100:1÷10:1) to give 1672-1 (10 g, 71percent) as a colorless oil. MS 178.3 [M+23]+, 100.3 [M_56]+.
67%
With sodium hydrogencarbonate In tetrahydrofuran; water
EXAMPLE 516A tert-butyl prop-2-ynylcarbamate A solution of propargyl amine (2.32 g, 42.1 mmol) in THF (75 mL) and water (200 mL) was treated with a saturated sodium bicarbonate solution (5 mL), followed by the dropwise addition of a solution of di-tert-butyl-dicarbonate (9.19 g, 42.1 mmol) in THF (20 mL). The solution was stirred overnight at room temperature, concentrated in vacuo to remove THF, extracted with ethyl acetate. The combined organics were washed with brine, dried (MgSO4) and concentrated to provide 4.37 g (67percent yield) of the desired product. 1H NMR (300 MHz, CHLOROFORM-D) δ ppm 1.46 (s, 9H), 2.22 (t, J=2.54 Hz, 1H), 3.92 (dd, J=5.26, 2.20 Hz, 2H), 4.68 (s, 1H).
67.6%
With triethylamine In dichloromethane at 20℃; for 2.5 h;
Propargyl amine (25.18 g, 0.448 mol), triethylamine (55.52 g, 0.549 mol) and dichloromethane 400 ml were added to a four-necked flask, and while cooling the reaction solution in a water bath (20 ° C.) Di-tert-butyl carbonate (118.15 g, 0.541 mol) was added dropwise over 30 minutes. After completion of the dropwise addition, after stirring for 2 hours, 300 ml of saturated brine and 200 ml of dichloromethane were added to the reaction solution and extracted. The obtained organic layer was dried with magnesium sulfate. After removing the desiccant, the solvent of the obtained solution was distilled off to obtain a pale yellow oil. Purification by recrystallization (hexane) gave N-Boc-propargylamine as a white solid (yield: 47.01 g, yield: 67.6percent).
65%
With sodium hydrogencarbonate In water
Propargylamine (5.50 g, 0.1 mol) and di-tert-butyl dicarbonate (4.36 g, 2 eq.) were suspended together in 100 mL of a 10percent aqueous solution of NaHCO3. Reaction mixture was stirred overnight and extracted by EtOAc (3x20 mL). The organic phases were combined together, washed with citric acid 10percent aq., dried over MgSO4, filtered and evaporated, providing compound 18d as white solid (10.1 g, 65percent yield). 1H N MR (CDCI3) δ 4.72 (bs, 1 H), 3.91 (d, J= 3.0 Hz1 2H), 2.22 (t, J= 2.9 Hz, 1 H), 1.51 (s, 9H).
17%
at 0 - 20℃; for 3.25 h;
14C. Prop-2-ynyl-carbamic acid tert-butyl ester; Di-fert-butyl dicarbonate (19.8g, 90.8 mmol) was dissolved in anhydrous dichloromethane (36 ml) and then added dropwise over 15 minutes to a solution of prop-2-ynylamine (6.22 ml, 90.8 mmol) in anhydrous dichloromethane (36 ml) at 0 0C. The resulting solution was stirred at room temperature for 3 hours. The solvent was removed under reduced pressure to leave a liquid that crystallized on standing. The solid was triturated with petroleum ether, filtered then dried to yield the title compounds as a yellow crystalline solid (2.45g, 17percent yield). 1H NMR (CDCl3) δ , 1.47 (9H, s), 2.23 (IH, t), 3.94 (2H, br s).
4.1 g
at 20℃; for 1 h;
To a solution of prop-2-yn-1-amine (245, 2.1 g, 38.2 mmol) in THF (30 mL) was added(Boc)20 (15 g, 68.8 mmol ). After stirring at room temperature for 1 h, the reaction mixturewas concentrated in vacuo to afford the residue, which was purified by column chromatography with a gradient elution of hexane (100percent) to hexane (80percent) and EtOAc (20percent) to provide tertbutyl prop-2-yn-1-ylcarbamate (246, 4.1 g, 26.4 mmol); ‘H NMR (300 MHz, CDC13): ö 4.70 (s, 1H), 3.85 (d, J= 3.0 Hz, 2H), 2.15 (t, J 2.7 Hz, 1H), 1.38 (s, 9H).
Reference:
[1] Tetrahedron Letters, 2011, vol. 52, # 17, p. 2199 - 2202
[2] Organic Letters, 2014, vol. 16, # 9, p. 2430 - 2433
[3] European Journal of Organic Chemistry, 2015, vol. 2015, # 32, p. 7091 - 7113
[4] Patent: US2004/254231, 2004, A1,
[5] Angewandte Chemie - International Edition, 2011, vol. 50, # 6, p. 1338 - 1341
[6] Arkivoc, 2013, vol. 2013, # 4, p. 334 - 345
[7] Patent: CN104250255, 2016, B, . Location in patent: Paragraph 0055; 0056; 0057; 0058; 0059
[8] Journal of Medicinal Chemistry, 2005, vol. 48, # 1, p. 224 - 239
[9] Patent: WO2005/66162, 2005, A1, . Location in patent: Page/Page column 105
[10] Journal of Organic Chemistry, 2015, vol. 80, # 10, p. 5287 - 5295
[11] Bioorganic and Medicinal Chemistry, 1996, vol. 4, # 5, p. 727 - 737
[12] Journal of Medicinal Chemistry, 2013, vol. 56, # 12, p. 5151 - 5172
[13] Synthesis, 2006, # 18, p. 3080 - 3084
[14] Archives of Pharmacal Research, 2013, vol. 36, # 7, p. 832 - 839
[15] Advanced Synthesis and Catalysis, 2013, vol. 355, # 11-12, p. 2353 - 2360
[16] Organic and Biomolecular Chemistry, 2016, vol. 14, # 24, p. 5606 - 5611
[17] ChemMedChem, 2017, vol. 12, # 24, p. 2044 - 2053
[18] Organic Letters, 2006, vol. 8, # 20, p. 4389 - 4392
[19] Chemical Communications, 2002, # 1, p. 22 - 23
[20] Synlett, 2006, # 7, p. 1110 - 1112
[21] Tetrahedron Letters, 1999, vol. 40, # 19, p. 3811 - 3814
[22] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 19, p. 2713 - 2723
[23] Patent: WO2008/24784, 2008, A1, . Location in patent: Page/Page column 18-19
[24] Patent: WO2012/175962, 2012, A1, . Location in patent: Page/Page column 94
[25] Synthetic Communications, 2014, vol. 44, # 16, p. 2416 - 2425
[26] Patent: US2015/166487, 2015, A1, . Location in patent: Paragraph 0390
[27] Antimicrobial Agents and Chemotherapy, 2016, vol. 60, # 12, p. 7146 - 7152
[28] Patent: WO2008/147764, 2008, A1, . Location in patent: Page/Page column 18-19
[29] Patent: WO2008/147763, 2008, A1, . Location in patent: Page/Page column 19
[30] Advanced Synthesis and Catalysis, 2015, vol. 357, # 18, p. 3943 - 3948
[31] Journal of the American Chemical Society, 1999, vol. 121, # 19, p. 4704 - 4705
[32] Patent: US2014/171447, 2014, A1, . Location in patent: Paragraph 0445; 0446
[33] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1985, p. 2201 - 2208
[34] Tetrahedron, 2009, vol. 65, # 31, p. 6156 - 6168
[35] Synlett, 2003, # 15, p. 2353 - 2353
[36] Synthesis (Germany), 2017, vol. 49, # 17, p. 3945 - 3951
[37] Synlett, 2016, vol. 27, # 18, p. 2575 - 2580
[38] Synlett, 2018, vol. 29, # 6, p. 785 - 792
[39] Advanced Synthesis and Catalysis, 2016, vol. 358, # 18, p. 2912 - 2922
[40] Journal of Organic Chemistry, 2007, vol. 72, # 8, p. 2897 - 2905
[41] Patent: US2005/245530, 2005, A1, . Location in patent: Page/Page column 53-54
[42] Patent: WO2011/103089, 2011, A1, . Location in patent: Page/Page column 42-43
[43] Patent: WO2004/50619, 2004, A1, . Location in patent: Page 74-75
[44] Patent: WO2004/94430, 2004, A1, . Location in patent: Page 23
[45] Patent: US9951069, 2018, B1, . Location in patent: Page/Page column 11-12
[46] Chemistry - A European Journal, 2016, vol. 22, # 29, p. 10150 - 10154
[47] Patent: US2005/26944, 2005, A1,
[48] Patent: JP5846230, 2016, B2, . Location in patent: Paragraph 0170; 0171
[49] Patent: WO2006/104713, 2006, A1, . Location in patent: Page/Page column 98
[50] Journal of Medicinal Chemistry, 2015, vol. 58, # 8, p. 3593 - 3610
[51] Journal of the American Chemical Society, 2013, vol. 135, # 35, p. 13193 - 13203
[52] Patent: WO2006/51290, 2006, A2, . Location in patent: Page/Page column 112
[53] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 7, p. 1241 - 1244
[54] Journal of the American Chemical Society, 2004, vol. 126, # 25, p. 8038 - 8045
[55] Patent: US2003/162788, 2003, A1,
[56] Patent: US2003/73836, 2003, A1,
[57] Patent: US6541505, 2003, B1,
[58] Patent: US5719193, 1998, A,
[59] Patent: US2007/254879, 2007, A1, . Location in patent: Page/Page column 29; 30
[60] Patent: US2009/48277, 2009, A1, . Location in patent: Page/Page column 12
[61] Patent: US2009/36450, 2009, A1, . Location in patent: Page/Page column 34
[62] Patent: WO2005/19174, 2005, A1, . Location in patent: Page/Page column 49
[63] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 15, p. 4635 - 4638
[64] Tetrahedron Letters, 2010, vol. 51, # 45, p. 5915 - 5918
[65] Patent: US2006/223797, 2006, A1, . Location in patent: Page/Page column 90
[66] Patent: WO2007/75896, 2007, A2, . Location in patent: Page/Page column 146
[67] Journal of Fluorine Chemistry, 2011, vol. 132, # 10, p. 850 - 857
[68] Patent: US2011/319418, 2011, A1, . Location in patent: Page/Page column 37
[69] Patent: WO2012/70024, 2012, A1, . Location in patent: Page/Page column 34-65
[70] European Journal of Medicinal Chemistry, 2013, vol. 69, p. 244 - 261
[71] Organic Process Research and Development, 2005, vol. 9, # 4, p. 440 - 450
[72] Patent: US2014/171403, 2014, A1, . Location in patent: Page/Page column
[73] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 2, p. 104 - 112
[74] PLoS ONE, 2017, vol. 12, # 2,
[75] Patent: WO2018/5860, 2018, A1, . Location in patent: Page/Page column 172; 190; 191
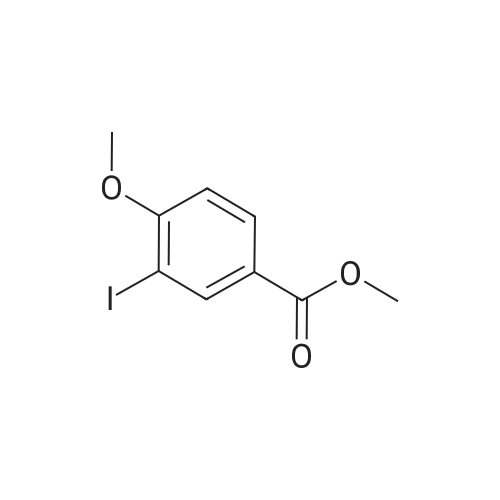
4
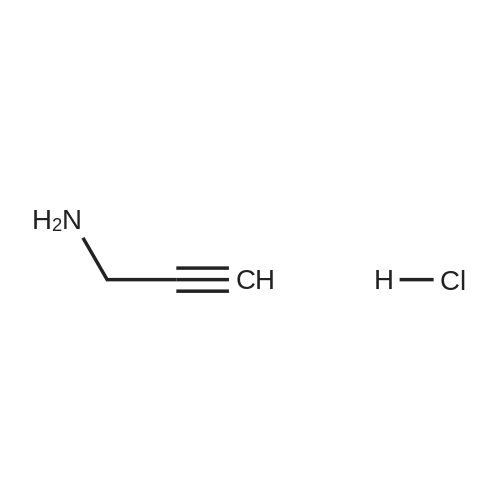
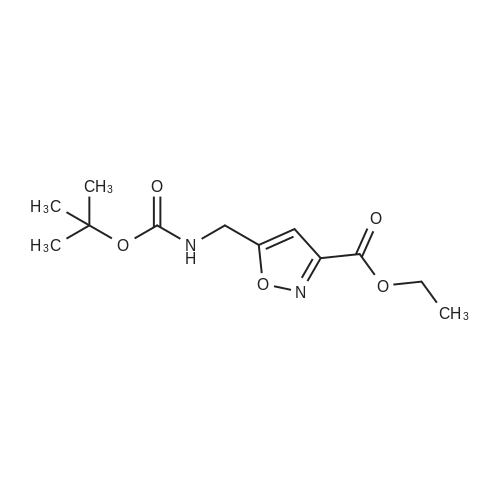
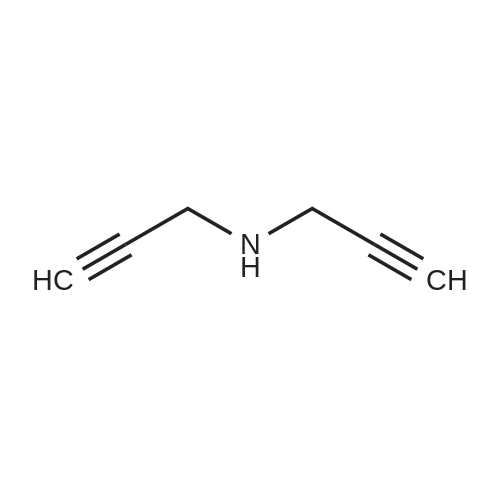
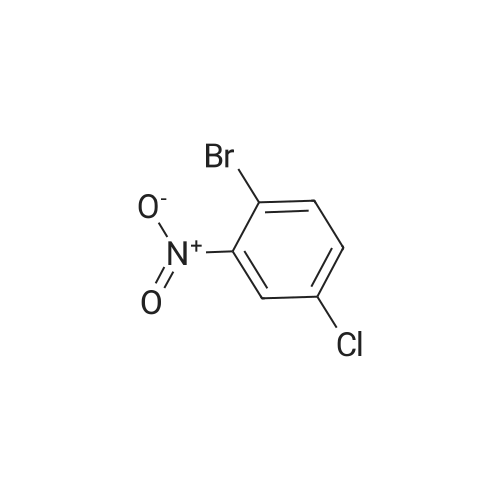
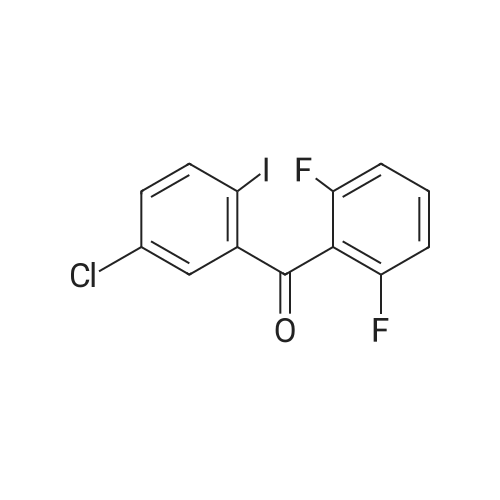
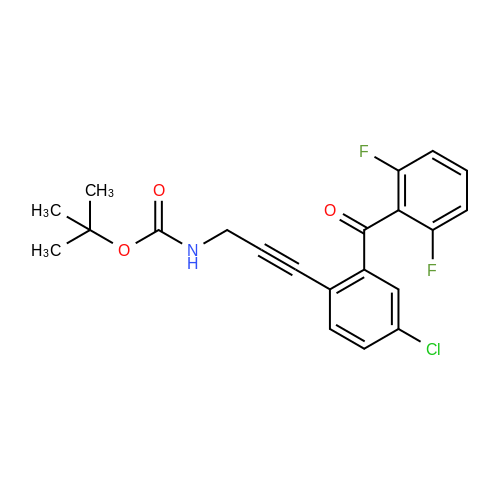
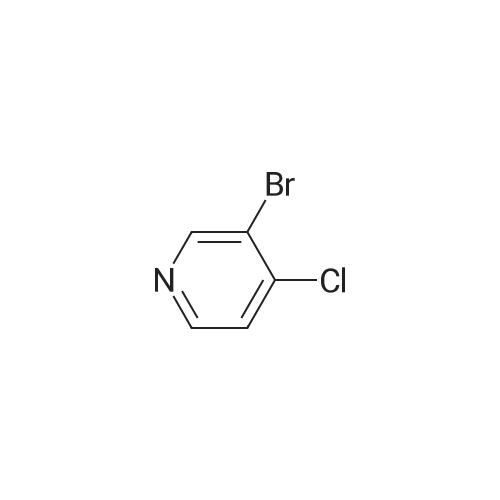
[ 24424-99-5 ]
[ 15430-52-1 ]
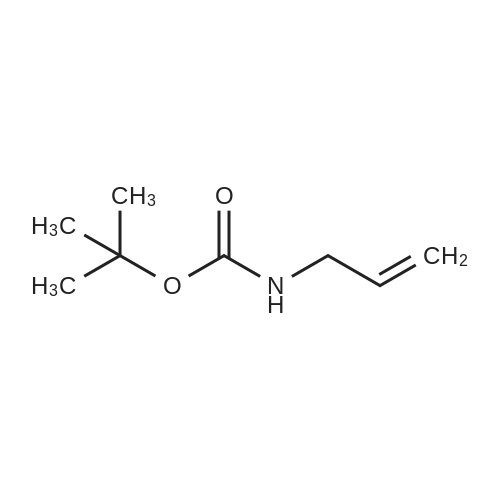
[ 92136-39-5 ]
Yield
Reaction Conditions
Operation in experiment
96%
With triethylamine In dichloromethane for 2 h; Cooling with ice
Propargylamine hydrochloride (3.6 g, 39 mmol) and triethylamine (11.5 mL, 83 mmol, 2.13 equiv) were dissolved in CH2Cl2 (100 mL) and this solution was cooled on ice. Then, a solution of Boc2O (8.5 g, 40 mmol), (1.03 equiv) in CH2Cl2 (50 mL) was added dropwise and the obtained reaction mixture was stirred for 2 h. After removing the solvent under reduced pressure, the residue was redissolved in EtOAc (150 mL) and this solution was washed with 1 N KHSO4 (3 x 75 mL) and brine (150 mL). The EtOAc solution was dried (Na2SO4) and concentrated in vacuo to give 8 as pink crystals in 96percent yield (5.8 g). Rf 0.70 (CH2Cl2/MeOH 95:5); 1H NMR (300 MHz, CDCl3, 25 °C): d 4.71 (broad s, 1H; urethane NH), 3.93(m, 2H; CH2), 2.22 (s, 1H; ^CH), 1.46 (s, 9H; C(CH3)3); 13C NMR(75.5 MHz, CDCl3, 25 °C): d 155.3, 80.0, 69.3, 30.4, 28.3, 27.4.
Reference:
[1] European Journal of Medicinal Chemistry, 2014, vol. 88, p. 55 - 65
[2] Organic Letters, 2014, vol. 16, # 12, p. 3196 - 3199
5
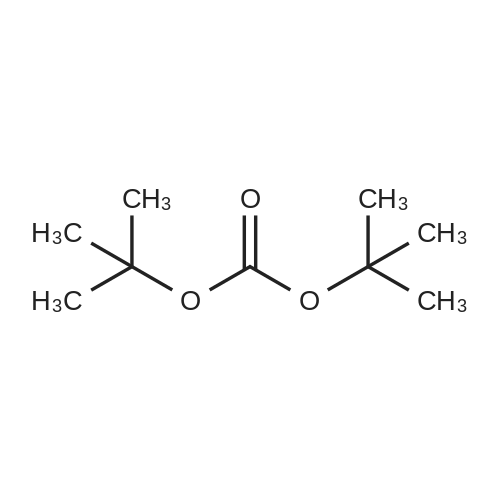
[ 34619-03-9 ]
[ 2450-71-7 ]
[ 92136-39-5 ]
Yield
Reaction Conditions
Operation in experiment
97%
for 12 h;
Propargyl amine (9.6 g, 174.4 MMOL) was added dropwise to a solution of di-tert-butyl dicarbonate (46.1 g, 211.0 MMOL) in THF (70 mL). After 12 h the reaction was concentrated, the residue was dissolved in diethyl ether and washed with water (1 x) and brine (1 x). The organic layer was dried over NA2SO4 then concentrated to afford the title compound as a yellow oil (26 g, 97percent).
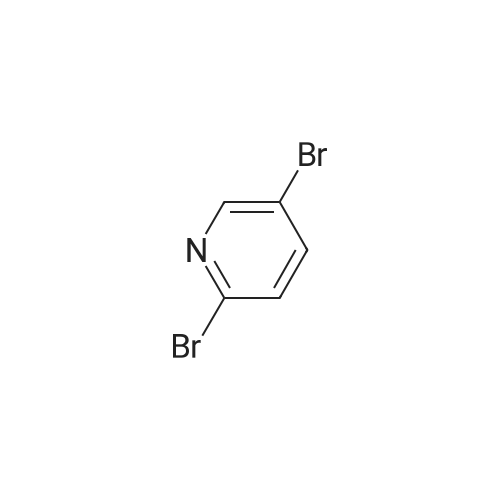
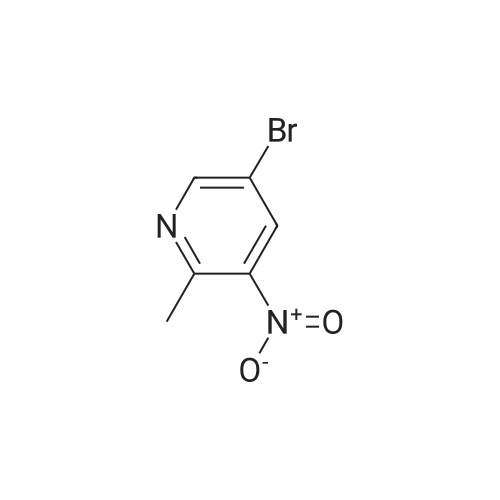
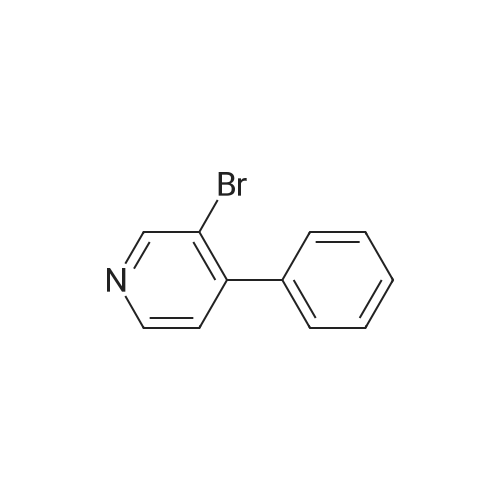
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); sodium carbonate In 1,2-dimethoxyethane; ethanol at 90℃; for 1h;
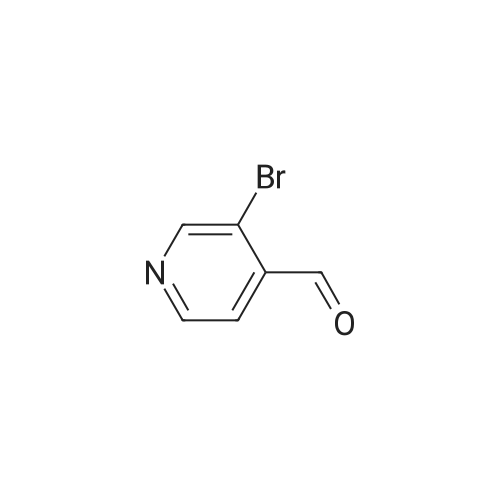
97%
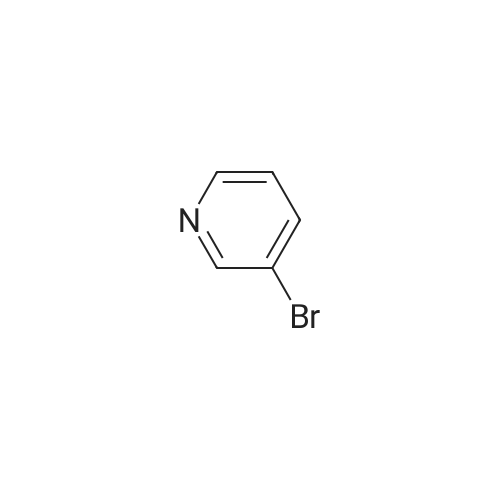
Stage #1: N-tert-Butoxycarbonyl-1-amino-3-propyne With copper(l) iodide; sodium carbonate In 1,2-dimethoxyethane; ethanol; water at 20℃; for 0.0833333h;
Stage #2: 3-Bromopyridine In 1,2-dimethoxyethane; ethanol; water at 90℃; for 1h;
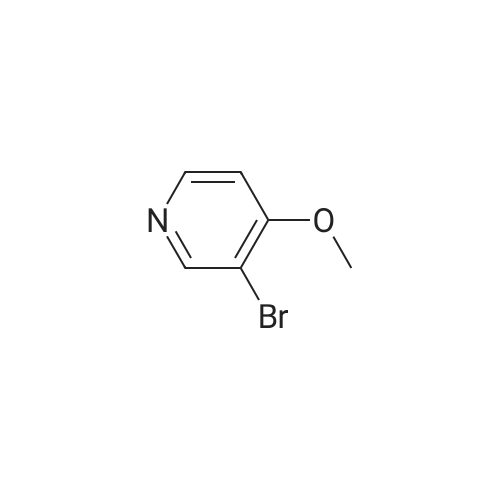
[0325] tet-Butyl 3- (pyridin-3-yl) prop-2-ynylcarbamate (47). To a pressure tube containing a magnetic stir bar was added 46 (202 mg, 1.3 mmol) and the vial was purged with argon. To the tube is added a solution of tetrakis (triphenylphosphine) palladium (0) (45 mg, 0.04 mmol) in ethanol/dimethoxyethane (1: 1,2 mL), sodium carbonate (aq) (2 M, 4 mL, 4 mmol), copper (I) iodide (46 mg, 0.24 mmol) and and the vial was once again purged with argon. The resultant solution was stirred at room temperature for 5 min when 3- bromopyridine (483 1L, 5 mmol) was added as a neat oil. The tube was purged with argon, capped, heated to 90 °C and stirred for 1 h. The solution was cooled to room temperature and poured into a flask containing anhydrous sodium sulfate (5 g). The solution was dried for 10 min, filtered and the solvent was removed in vacuo. The crude material was chromatographed on silica gel (CH30H/CHC13, 5/95, Rf= 0.43) to afford the title compound 47 (295 mg, 97% yield) as a brown solid: mp = 74-78 °C ; lH NMR (CDCl3) 6 8.63 (m, 1H), 8.51 (m, 1H), 7.67 (m, 1H), 7.22 (m, 1H), 5.05 (br s, 1H), 4.15 (m, 2H), 1.45 (s, 9H); LRMS (ESI) m/z calcd for C13Hl6N202 [M + H] + 233, found 233; m/z calcd for C9H9N202 [M + H- CH2C (CH3) 2] + 177, found 177; mlz calcd for C8H9N2 [M + H-t-Boc] + 133, found 133.
84%
With copper(l) iodide; sodium carbonate In 1,2-dimethoxyethane at 90℃; for 1h;
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; sodium carbonate In 1,2-dimethoxyethane; ethanol; water
With triethylamine In tetrahydrofuran; diethyl ether at 20℃; for 16h;
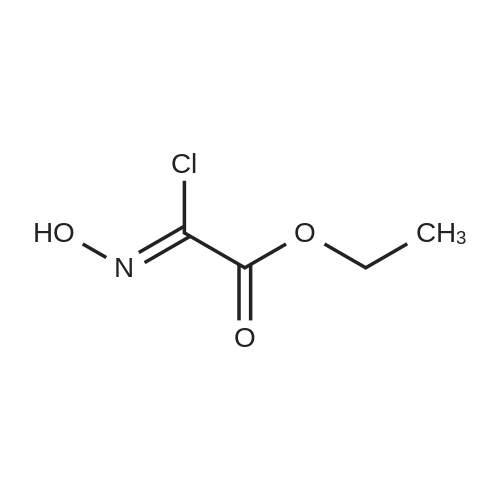
5.3.35 Ethyl 5-[(tert-butoxycarbonyl)amino]methyl}isoxazole-3-carboxylate (33) [29]
Ethyl 2-chloro-2-(hydroxyimino)acetate 32 (0.1g, 0.66mmol, 1.0 equiv.) was dissolved in THF (15mL). A solution of N-boc-propargylamine (0.11g, 0.73mmol, 1.1 equiv.) and Et3N (0.14mL, 1.0mmol, 1.5 equiv.) in Et2O was added and stirred at rt overnight. An aqueous solution of NH4Cl was added. The organic phase was separated, and the aqueous layer was extracted with ethyl acetate (3×40mL). The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by FCC (=2cm, l=10cm, V=20mL, cyclohexane/ethyl acetate 7:3). Rf=0.25 (cyclohexane/ethyl acetate 8:2). Pale yellow oil, yield 0.12g (69%). C12H18N2O5 (270.3g/mol). 1H NMR (600MHz, DMSO-D6): δ (ppm)=1.29 (t, J=7.1Hz, 3H, OCH2CH3), 1.37 (s, 9H, (H3C)3C-O-C (=O)), 4.30 (d, J=6.0Hz, 2H, C (=O)NHCH2), 4.33 (q, J=7.1Hz, 2H, OCH2CH3), 6.61 (s, 1H, 4-Harom), 7.57 (t, J=6.0Hz, 1H, C(=O)NHCH2). 13C NMR (151MHz, DMSO-D6): δ (ppm)=17.0 (OCH2CH3), 31.2 (3C, (CH3)3CO), 39.1 (CONHCH2), 64.9 (OCH2CH3), 81.7 ((CH3)3C-O-C (=O)), 105.2 (C-4B), 158.6 (C-5B), 159.1 (C-3B), 162.4 (C(=O)OCH2CH3), 176.3 (CONHCH2).
56%
With triethylamine In tetrahydrofuran at 18 - 25℃;
61; 64
Tert-butyl N-prop-2-ynylcarbamate (40.97 g, 0.26mol, 1 eq) was dissolved in anhydrous THF (150 mL) and N,N-diethylethanamine (22 mL, 0.16 mol, 1.2 eq) added. A solution of ethyl-2-chloro-2-hydroxyimino-acetate (20 g, 0. 13mol, 1 eq) in anhydrous THF (350mL) was added dropwise over 7 h. The reaction was stirred at room temperature overnight then evaporated to dryness. The residue was dissolved in DCM and washed with water, brine and dried (MgSO4). After filtration, the solution was evaporated to give the crude product as a yellow oil. This was purified by silica column chromatography, eluting with 20% -60% ether in iso-hexane to give ethyl 5-[[(2-methylpropan-2-yl)oxycarbonylamino]methyl]-1,2-oxazole-3-carboxylate as a white solid (20.12 g, 56%).1H NMR (CDCl3 400.13 MHz) δ 1.39-1.47 (12H, m), 4.40-4.49 (5H, m), 5.0 (1H, s), 6.58 (1H, s). MS m/z 269 (M-H).; Ethyl 5-[[(2-methylpropan-2-yl)oxycarbonylamino]methyl]-1,2-oxazole-3-carboxylate used as starting material was prepared as shown in Example 61.
With triethylamine In tetrahydrofuran; diethyl ether at 20℃;
22.2
(Step 2) Production of ethyl 5-(tert-butoxycarbonylamino-methyl)-isoxazole-3-carboxylate To a solution of ethyl 2-chloro-2-hydroxyiminoacetate (4.77 g) in tetrahydrofuran (35 mL), a solution of N-(tert-butoxycarbonyl)propargylamine (5.62 g) and triethylamine (5.0 mL) in diethyl ether (120 mL) was added dropwise. After stirring overnight at room temperature, 5% aqueous ammonium chloride was added to the reaction mixture. The organic layer was separated, successively washed with water and saturated brine, dried over anhydrous sodium sulfate, and then concentrated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate, 3:1) to obtain the title compound (1.89 g) as a light yellow oily matter.
Stage #1: dipropargylamine With pyridine In dichloromethane at 20℃; for 12h;
Stage #2: N-tert-Butoxycarbonyl-1-amino-3-propyne In ethanol; dichloromethane at 60℃; for 48h;
Stage #3: With hydrogenchloride In methanol; dichloromethane at 20℃; for 1h; Further stages.;
A solution of copper cyanide (1.1Sg, 12.9 mmol) in THF (30 mL) at ?78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at ?78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at ?780C for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2% ethyl acetate/heptane to provide the desired product (3.66 g, 63%). 1H NMR (400 MHz, CDCl3) ? 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
With ammonium hydroxide; n-butyllithium; ammonium chloride; tri-n-butyl-tin hydride; In tetrahydrofuran; n-heptane; dichloromethane; ethyl acetate;
EXAMPLE 314A tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78 C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2% ethyl acetate/heptane to provide the desired product (3.66 g, 63%). 1H NMR (400 MHz, CDCl3) delta 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78 C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2% ethyl acetate/heptane to provide the desired product (3.66 g, 63%). 1H NMR (400 MHz, CDCl3) delta 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
With copper(l) iodide; triethylamine;bis-triphenylphosphine-palladium(II) chloride; at 50℃; for 2h;
A mixture of L- OROMO-4-CHLORO-2-NITRO-BENZENE (0.15 mol), DICHLOROBIS (TRIPHENYLPHOSPHINE)-PALLADIUM (0. 0075 mol) and copper (I) iodide (0. 0075 mol) in triethylamine (300 ml) was stirred at 50C and 2-propynyl-carbamic acid, 1, 1-dimethylethyl ester (0.375 mol) was added portionwise, then the reaction mixture was stirred for 2 hours at 50C and the solvent was evaporated. The residue was taken up in water and the mixture was extracted with EtOAc. The organic layer was separated, dried, filtered off and the solvent was evaporated. The residue was purified twice by column chromatography (eluent : Hexane/EtOAc 80/20). The product fractions were collected and the solvent was evaporated. The obtained residue (31. 8 g) was stirred in hexane and then the resulting precipitate was filtered off and dried, yielding 31.5 g (67. 6 %) of intermediate 173.
{3-[4-Chloro-2-(2,6-difluoro-benzoyl)-phenyl]-prop-2-ynyl}-carbamic acid tert-butyl ester (3aa) <strong>[869365-97-9](5-Chloro-2-iodo-phenyl)-(2,6-difluoro-phenyl)-methanone</strong> (2aa) (5.5 g, 14.5 mmol), prop-2-ynyl-carbamic acid tert-butyl ester (2.5 g, 16 mmol), PdCl2(PPh3)2 (0.6 g, 0.9 mmol) and Cu(I)I (0.2 g, 0.9 mmol) were suspended in anhydrous CH2Cl2 (50 mL) and the mixture was sparged with nitrogen for 30 min. Diethylamine (8 mL) was added and the solution was stirred at room temperature for 16 h. The solution was concentrated in vacuo and the resulting residue purified by column chromatography (silica gel, 0 to 15% EtOAc/hexanes) to afford 3aa (3.6 g, 61%) as a white solid, MS m/z=406 (M+H).
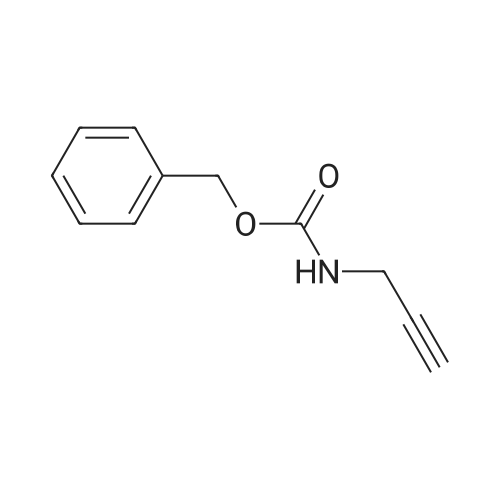
9 Benzyl 3-(6-{3-[(tert-butoxycarbonyl)amino]prop-1-ynyl}pyridin-3-yl)prop-2-ynylcarbamate (6-1)
Example 9 Benzyl 3-(6-{3-[(tert-butoxycarbonyl)amino]prop-1-ynyl}pyridin-3-yl)prop-2-ynylcarbamate (6-1) A mixture of 2,5-dibromopyridine (1.19 g), N-Boc-propargylamine (781 mg), Pd(PPh3)2Cl2 (73 mg), and CuI (10 mg) in diisopropylamine (20 mL) was stirred at room temperature overnight under nitrogen atmosphere. To the mixture was added N-Cbz-propargylamine (1.14 g) and the mixture was heated under reflux temperature for 5 hours under nitrogen atmosphere. The mixture was cooled to room temperature. To the mixture was added ethyl acetate (50 mL). The precipitates were filtered and washed with ethyl acetate. The filtrate and washings were combined and purified by silica gel column to give 6-1 (250 mg) as a crystalline compound.
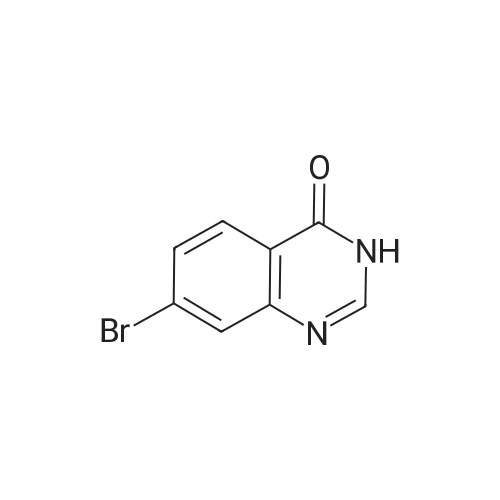
With triethylamine;bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; In N,N-dimethyl-formamide; at 55℃; for 18h;
14D. [3-(4-Oxo-3,4-dihvdro-quinazolin-7-yl)-prop-2-vnyl]-carbamic acid tert butyl ester; 7-Bromo-3H-quinazolin-4-one (0.38 g, 1.69 mmol) was mixed with copper iodide (0.0456g, 0.239 mmol). The mixture was suspended in anhydrous N5N- dimethylformamide (9.12 ml) and triethylamine was added (6.08 ml, 43.3 mmol). The solution was degassed and bis (triphenylphosphine)palladium(II)chloride (0.0228g, 0.032 mmol) was added followed by prop-2-ynyl-carbamic acid fert-butyl ester (0.258g, 1.66 mmol). The solution was heated at 55 0C with stirring for 18 hours under nitrogen. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane and washed with water. A solid precipitated out and was filtered then washed with dichloromethane and water. The EPO <DP n="115"/>solid was dried under vacuum to yield the title compound that was used in the next step without purification (0.178g, 35% yield). LC/MS: (PS-A2) Rt 2.50 [MH-H]+ 300.05.
With copper(l) iodide; diisopropylamine;bis-triphenylphosphine-palladium(II) chloride; In tetrahydrofuran; at 0℃; for 2h;
XlV.i .a [3-(6-Chloro-pyridazin-3-yl)-prop-2-vnyl1-carbamic acid tert-butyl ester; 19.2 g (80.0 mmol) 3-Chloro-6-iodo-pyridazine (Tetrahedron 55, 1999, 15067) and 13.7 g (88.0 mmol) prop-2-ynyl-carbamic acid tert-butyl ester are dissolved in 200 ml THF and 2.50 g (4.0 mmol) bis-(triphenylphosphine)palladiumdichloride, 2.80 g (14.8 mmol) copper-(l)- iodide and finally 60 ml diisopropylamine are added at 0C. The mixture is stirred for 2 hours at 0C. After that time ice-water is added and the mixture is extracted with ethylacetate. The organic phase is separated and dried over sodium sulphate. The solvent is evaporated and the residue is purified by silica gel column chromatography with methylene chloride/ethyl acetate (5:1 ) as eluent. The product is dried in vacuo at 50C. Yield: 12.8 g (60% of theory), Rf value: 0.50 (silica gel, methylene chloride/ethyl acetate = 5:1 ) Ci2H12CIN3O2EII Mass spectrum: m/z = 268/270 [M+H]+ M. p. 102-105 C
With copper(l) iodide; triethylamine;bis-triphenylphosphine-palladium(II) chloride; In N,N-dimethyl-formamide; at 20℃;Inert atmosphere;
51.i. [3-(6-methoxy-[1,5]naphthyridin-4-yl)-prop-2-ynyl]-carbamic acid tert-butyl ester To a solution of N-Boc propargyl amine (3.25 g, 20.9 mmol) and <strong>[881658-92-0]8-bromo-2-methoxy-[1,5]naphthyridine</strong> (5.00 g, 20.9 mmol; prepared as in WO 2006/032466) and TEA (17.5 mL, 6 eq.) in DMF (120 mL) were added Pd(PPh3)2Cl2 (755 mg, 1.08 mmol) and CuI (432 mg, 2.27 mmol). The mixture was degassed with a stream of N2 for 15 min and then stirred at rt for 5 h. The mixture was partitioned between water and EA, the org. layer was washed several times with water and a sat. NH4Cl solution, dried over MgSO4 and concentrated. Chromatography on SiO2 (Hex/EA 1:1, EA) gave the coupling product as a beige solid (2.90 g, 44% yield).
44%
With triethylamine;bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; In N,N-dimethyl-formamide; at 20℃; for 5.25h;
Example 51: (S>3-(2,3-dihydro-benzo[l,4]dioxin-6-yl)-2-oxo-oxazolidine- 5-carboxylic acid [3-(6-methoxy-[l,5]naphthyridin-4-yl)-propyl]-amide:; 57. i. [3-(6-methoxy- [ 1 ,5] naphthyridin-4-yl)-prop-2-ynyl] -carbamic acid tert-butyl ester:; To a solution of JV-BOC propargyl amine (3.25 g, 20.9 mmol) and 8-bromo-2-methoxy- [l,5]naphthyridine (5.0O g, 20.9 mmol; prepared as in WO 2006/032466) and TEA(17.5 mL, 6 eq.) in DMF (120 mL) were added Pd(PPh3)2Cl2 (755 mg, 1.08 mmol) and CuI(432 mg, 2.27 mmol). The mixture was degassed with a stream of N2 for 15 min and then stirred at rt for 5 h. The mixture was partitioned between water and EA, the org. layer was washed several times with water and a sat. NH4Cl solution, dried over MgSO4 and concentrated. Chromatography on SiO2 (Hex/EA 1 :1, EA) gave the coupling product as a beige solid (2.90 g, 44% yield).
With potassium carbonate;copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 60℃; for 1h;
Method for svnthesising A.2c and A.2 d; Bromoindazole A.4q (1.50 g, 7.61 mmol), K2CO3 (2.60 g, 19.0 mmol), CuI (304 mg, 1.60 mmol) and Pd(PPh3)4 (1.76 g, 1.60 mmol) are taken up in DME/H2O (30 mL, 1 :1), combined with alkyne A.3b (1.18 g, 7.61 mmol) and stirred for 1 h at 600C. The solvent is removed, the reaction mixture is purified by column chromatography (cyclohexane/EtOAc, 10% to 70%) and A.2c-PG (HPLC-MS: tRet. = 1.78 min; MS(M+H)+ = 272; method LCMSBASl) is obtained.
With potassium carbonate;copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 60℃; for 1h;
Method for svnthesising A.2c and A.2; *d(Boc)HNk 1 h, 60 0C A.2C-PG A.2*d-PGA.2c A.2*d; Bromoindazole A.4q (1.50 g, 7.61 mmol), K2CO3 (2.60 g, 19.0 mmol), CuI (304 mg, 1.60 mmol) and Pd(PPh3)4 (1.76 g, 1.60 mmol) are taken up in DME/H2O (30 mL, 1 :1), combined with alkyne A.3b (1.18 g, 7.61 mmol) and stirred for 1 h at 60C. The solvent is removed, the reaction mixture is purified by column chromatography (cyclohexane/EtOAc, 10% to 70%) and A.2c-PG (HPLC-MS: tRet. = 1.78 min; MS(M+H)+ = 272; method LCMSBASl) is obtained.
With CuCl; NaOC(CH3)3; (O3SC6H4)(((CH3)2CH)3C6H2)(C6H5)2C3H3N2 In tetrahydrofuran treatment of acetylene deriv. with 0.9 equiv. of boron compd. in presence of imidazolium deriv., copper chloride, sodium tert-butoxide and 1 equiv. of methanol in THF at 0°C for 12 h under N2; NMR;
With (((CH3)3C6H2)2C3H4N2)CuCl; NaOC(CH3)3; CH3OH In tetrahydrofuran treatment of acetylene deriv. with 0.9 equiv. of boron compd. in presence of copper compd., sodium tert-butoxide and 1 equiv. of methanol in THFat 0°C for 12 h under N2; NMR;
With C27H19N2(1+)*Cl(1-); 1,8-diazabicyclo[5.4.0]undec-7-ene In methanol at 20℃; for 24h; Overall yield = 72 %; Overall yield = 407 mg; regioselective reaction;
With (1,3-bis(2,6-diisopropylphenyl)-2,3-dihydro-1H-imidazol-2-yl)copper(I) chloride; SPGS-550-M; NOK; sodium hydroxide In lithium hydroxide monohydrate at 20℃; for 20h; Green chemistry; Overall yield = 85 %; regioselective reaction;
With chloro[1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene]copper(I) In lithium hydroxide monohydrate at 30℃; for 48h; Overall yield = 75 percent;
With NaOC(CH3)3; methanol In tetrahydrofuran 5.0 mol% Cu-complex, alkyne, diborane, 5.0 mol% NaOC(CH3)3, 1.1 equiv CH3OH, THF, -50°C, 12 h, N2 atm.; >98:2 molar ratio of isomers;
1: 6%
2: 15%
With bromobenzene; palladium diacetate; tricyclohexylphosphine In 2,2,2-trifluoroethanol; toluene at 60℃; for 6h; Inert atmosphere; Overall yield = 1.9 g;
With methanol; (1,3-dimesitylimidazolidin-2-yl)copper(I) chloride; sodium t-butanolate In tetrahydrofuran at 0℃; for 12h; Inert atmosphere;
With methanol; C36H40N2O3S; copper(l) chloride; sodium t-butanolate In tetrahydrofuran at 0℃; for 12h; Inert atmosphere;
With chloro[1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene]copper(I); SPGS-550M; sodium hydroxide In water at 20℃; for 20h; Green chemistry; Overall yield = 85 %; Overall yield = 120 mg; regioselective reaction;
With triethylamine;bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; at 70℃; for 2.5h;Inert atmosphere;
fert-Butyl r3-(5-{ramino(imino)methyllamino)-6-methylpyridin-3-yl)propyncarbamate; Step 1 : te^Butyl[3-(6-methyl-5-nitropyridin-3-yI)prop-2-yn-l-yl]carbamate; To a solution of <strong>[911434-05-4]5-bromo-2-methyl-3-nitropyridine</strong> (2.81 g, 12.9 mmol) in TEA (25 mL) was added tert-buty prop-2-yn-l -ylcarbamate (2.58 g, 16.6 mmol). The solution was degassed with argon.To the reaction mixture were added bis(triphenylphosphine)palladium(II) chloride (0.18 g, 0.26 mmol) and copper(I) iodide (0.098 g, 0.52 mmol). The reaction mixture was allowed to stir at 70 0C for 2.5 h and was then concentrated and diluted with EtOAc (100 mL). The mixture was filtered over a pad ofCelite and the resulting filtrate was washed with sat. aq. NaHCC>3 (100 mL). The organic solution was dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography to give tert-buty [3-(6-methyl-5-nitropyridin-3-yl)prop-2-yn-l-yl]carbamate (2.98 g, 79%) as a yellow solid. LCMS (FA): R1= 1.81 min, m/z = 292.0 (M+H).
Stage #1: N-tert-Butoxycarbonyl-1-amino-3-propyne With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.5h;
Stage #2: 3-chloro-1,1-diethoxy-propane With tetra-(n-butyl)ammonium iodide In N,N-dimethyl-formamide; mineral oil at 0 - 20℃;
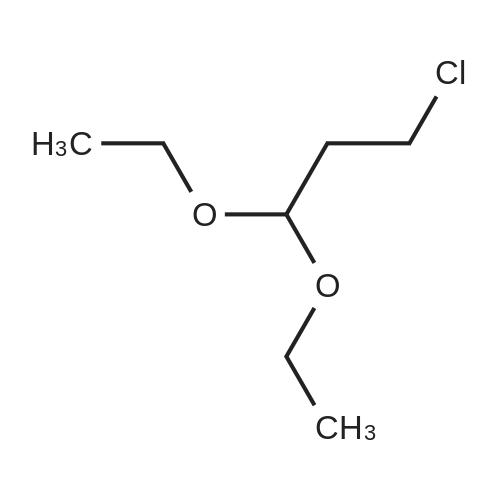
tert-Butyl 3,3-diethoxypropyl(prop-2-ynyl)carbamate 2.
Amine 1 (12.4 g, 80.0 mmol, 1.0 equiv) was dissolved in DMF (160 mL) and NaH (60% in mineral oil, 3.84 g, 96.0 mmol, 1.2 equiv) was carefully added by portions at 0 °C to the soln that was stirred for 30 min at 0 °C. In a separate flask, n-Bu4NI (5.91 g, 16.0 mmol, 0.2 equiv) was added to a soln of 3-chloro-1,1-diethoxypropane (26.8 mL, 160 mmol, 2.0 equiv) in DMF (40 mL) and the mixture was stirred for 20 min at rt. The latter mixture was then transferred via canula to the soln of the amide at 0 °C. The ice bath was removed and, after 16 h of stirring at rt, distilled H2O (250 mL) and Et2O (250 mL) were added. The organic layer was separated and the aq phase was extracted with Et2O (2'250 mL). The combined organic layers were then washed with distilled H2O (3'400 mL), dried (Na2SO4) and concentrated in vacuo to give a dark orange oil, that was distilled under reduced pressure to yield the pure alkyne 2 (16.2 g, 71%, bp = 155 °C under 5 mm Hg) as a colorless oil. Some starting chloride was also recovered (13.7 g, 82.3 mmol, bp = 65 °C under 5 mm Hg).Rf = 0.30 (Hexanes/AcOEt 9:1). 1H NMR (500 MHz, CDCl3): d (ppm) 4.53 (t, J = 5.5 Hz, 1H), 4.04 (m, 2H), 3.65 (m, 2H), 3.50 (m, 2H), 3.38 (t, J = 7.2 Hz, 2H), 2.20 (t, J = 2.1 Hz, 1H), 1.91 (m, 2H), 1.47 (s, 9H), 1.21 (t, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3): d (ppm) 154.8, 100.9, 80.0, 79.7, 71.2, 61.0, 42.8, 36.3, 32.1, 28.2, 15.2.[i] IR (neat): n (cm-1) 3309, 3257, 2975, 2931, 2883, 1698, 1411, 1367, 1248, 1170, 1062. HRMS (ES+): m/z calcd for C15H27NO4Na ([MNa]+) 308.1838, found 308.1848.
Stage #1: bis(pinacol)diborane With C23H37ClCuN; sodium t-butanolate In tetrahydrofuran at 22℃; for 0.0833333h; Inert atmosphere;
Stage #2: N-tert-Butoxycarbonyl-1-amino-3-propyne In tetrahydrofuran; methanol at 22℃; for 12h; Inert atmosphere;
93%
Stage #1: bis(pinacol)diborane With (1,3-dimesitylimidazolidin-2-yl)copper(I) chloride; sodium t-butanolate In tetrahydrofuran at 22℃; for 0.5h; Inert atmosphere;
Stage #2: N-tert-Butoxycarbonyl-1-amino-3-propyne With methanol In tetrahydrofuran at -50℃; for 9h; Inert atmosphere; regioselective reaction;
91%
With chloro[1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene]copper(I); sodium t-butanolate In methanol; toluene at -50℃;
76%
With methanol; tri-tert-butyl phosphine; copper dichloride; sodium t-butanolate In toluene at 0 - 20℃; for 5h; Inert atmosphere; regioselective reaction;
45%
With bromobenzene; 2,2,2-trifluoroethanol; palladium diacetate; tricyclohexylphosphine In toluene at 60℃; for 3h; Inert atmosphere; Darkness;
Synthesis of compound 4d
To a flame-dried two-neck flask under argon equipped with a magnetic stirring bar were added sequentially bis(pinacolato)diboron (1.1 eq., 2.717 g, 10.7 mmol), tricyclohexylphosphine (10 mol%, 272 mg, 970 μmol), Palladium(II) acetate (5 mol%, 109 mg, 485 μmol), anhydrous degassed toluene (16 mL), N-Boc propargylglycine (1 eq., 1.5 g, 9.7 mmol), bromobenzene (1.1eq., 1.1 mL, 10.7 mmol) and then anhydrous degassed trifluoroethanol (2 eq., 1.5 mL, 19.4 mmol). The dark mixture was stirred at 60°C for 3h. The mixture was cooled to room temperature and filtered through a pad of silica gel (2 cm wide, 2 cm high), eluting with Et2O (200 mL). The greenish filtrate was concentrated. The crude residue (a greenish oil) was purified by silica gel column chromatography (Et2O/hexanes 0% to 20%) to afford pure 5-phenylpent-1-en-2-yl pinacol boronate ester (1.25 g, 4.4 mmol, 45% yield) as a yellowish oil. The characterization data matches the reported data.11H-NMR (300 MHz, CDCl3) δ 5.80 (dt, J = 2.9, 1.5 Hz, 1H), 5.69 (q, J = 2.0 Hz, 1H), 4.82 (s,1H), 3.77 (d, J = 6.1 Hz, 2H), 1.38 (s, 9H), 1.20 (s, 12H).
With tri-tert-butyl phosphine; copper(l) chloride; sodium t-butanolate In methanol; toluene at 0 - 20℃; for 16h;
2.1 1. Preparation of tert-butyl (2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)allyl)carbamate
At 0°C, to N-(tert-butoxycarbonyl)aminopropyne (150.0g, 967.7mmol), B2(Pin)2 (300.0g, 1181.1mmol), CuCl (10.0g, 100.8mmol), t-BuONa (15.0g, 156.1mmol), And P(t-Bu)3 (25.0g, 123.6mmol) in a 3.0L toluene suspension, slowly adding MeOH (75.0mL, 1875.0mmol) dropwise, After the addition was completed, the system was heated to 20°C and stirred for 16 hours. After silica gel column chromatography (petroleum ether: ethyl acetate = 5:1), The crude compound (330.0 g) was obtained and used directly in the next step.
With tri-tert-butyl phosphine; copper(l) chloride; sodium t-butanolate In methanol; toluene at 0 - 20℃; for 16h;
3.1 1. Preparation of tert-butyl (2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)allyl)carbamate
At 0°C, to N-tert-butoxycarbonylaminopropyne (150.0g, 967.7mmol),B2(Pin)2 (300.0g, 1181.1mmol), CuCl (10.0g, 100.8mmol), t-BuONa (15.0g, 156.1mmol),MeOH (75.0 mL, 1875.0 mmol) was slowly added dropwise to a 3.0L toluene suspension of P(t-Bu)3 (25.0g, 123.6mmol), and after the addition was completed, the system was heated to 20°C and stirred for 16h. After silica gel column chromatography (petroleum ether: ethyl acetate = 5:1), the crude compound (330.0 g) was obtained, which was directly used in the next step.
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In N,N-dimethyl-formamide; at 50℃;Inert atmosphere;
To a mixture of <strong>[149965-40-2](5-bromo-2-chlorophenyl)methanol</strong> (1.00 g, 4.51 mmol), t-butyl prop-2-yn-l-ylcabamate (0.84 g, 5.4 mmol) and TEA (5.2 niL) in DMF (3.2 mL) were added Pd(PPh3)2Cl2 (158 mg, 0.226 mmol) and Cul (86 mg, 0.45 mmol). The mixture was stirred under N2 at 50 C overnight. The reaction mixture was diluted with EtOAc, washed with water (2x) and brine (lx), dried over anhydrous sodium sulfate, concentrated under reduced pressure and purified by flash-column chromatography to give 477 mg (36 %) of 68a as a pale yellow syrup.
Stage #1: 2-methylpropanehydroxamoyl chloride; N-tert-Butoxycarbonyl-1-amino-3-propyne With triethylamine In dichloromethane at 20℃; for 2h; Large scale;
Stage #2: With hydrogenchloride In isopropyl alcohol at 0℃; for 12h; Large scale; regioselective reaction;
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 20℃; for 48h;
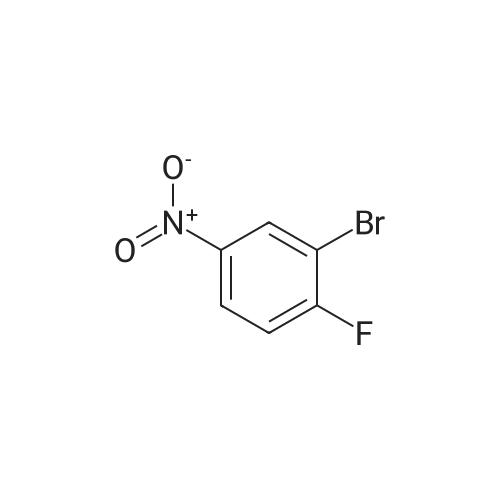
To a solution of N-Boc-propargylamine (1.74 g, 11.2 mmol) in acetonitrile (14 mL) was sequentially added triethyamine (1.53 mL, 11.2 mmol), CuI (61 mg, 0.32 mmol), and <strong>[701-45-1]3-bromo-4-fluoronitrobenzene</strong> (880 mg, 4.00 mmol). The mixture was de-aired, followed by addition of Cl2Pd(PPh3)4 (112 mg, 0.16 mmol), and de-aired. The resulted mixture was stirred at ambient temperature for 48 hours, diluted with EtOAc (30 mL), filtered through a short path silica gel plug, washed with EtOAc (15 mL*3). The filtrates and washings were combined, concentrated, and flash chromatographed on a silica gel column, eluted with DCM/hexanes (50-100%) to give the title compound (739 mg, 63% yield) as yellow oil. 1H NMR (CDCl3) was consistent with the structure
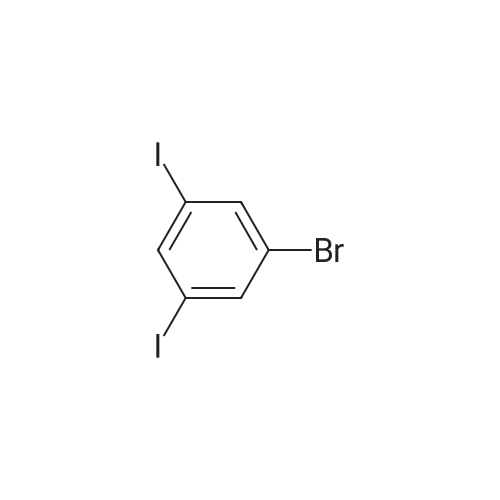
General procedure: Intermediate C was prepared from <strong>[149428-64-8]1-bromo-3,5-diiodobenzene</strong> and Boc amino propyne using the general procedure for Sonogashira coupling. General procedure for Sonogashira coupling reaction The iodobenzene (1 mmol), Pd(PPh3)2C12 (0.36 mmol), CuT (0.369 mmol) and DIEA (4.5 mL) were added in 6 mL toluene. The mixture was flushed with argon for 5 minutes and then heated to 70C. The ethynylbenzene (1 mmol) in 2 mL of toluene was added over ten5 minutes. The mixture was stirred at 70C for another 3 hours. The mixture was filtered and evaporated. The product was purified by flash column.
With triethylamine; In dichloromethane; for 2h;Cooling with ice;
Propargylamine hydrochloride (3.6 g, 39 mmol) and triethylamine (11.5 mL, 83 mmol, 2.13 equiv) were dissolved in CH2Cl2 (100 mL) and this solution was cooled on ice. Then, a solution of Boc2O (8.5 g, 40 mmol), (1.03 equiv) in CH2Cl2 (50 mL) was added dropwise and the obtained reaction mixture was stirred for 2 h. After removing the solvent under reduced pressure, the residue was redissolved in EtOAc (150 mL) and this solution was washed with 1 N KHSO4 (3 x 75 mL) and brine (150 mL). The EtOAc solution was dried (Na2SO4) and concentrated in vacuo to give 8 as pink crystals in 96% yield (5.8 g). Rf 0.70 (CH2Cl2/MeOH 95:5); 1H NMR (300 MHz, CDCl3, 25 C): d 4.71 (broad s, 1H; urethane NH), 3.93(m, 2H; CH2), 2.22 (s, 1H; ^CH), 1.46 (s, 9H; C(CH3)3); 13C NMR(75.5 MHz, CDCl3, 25 C): d 155.3, 80.0, 69.3, 30.4, 28.3, 27.4.
tert-butyl (5-bromofuro[2,3-b]pyridin-2-yl)methylcarbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In N,N-dimethyl-formamide; at 90℃; for 3h;Inert atmosphere;
Synthesis of tert-butyl (5-bromofuro[2,3-Z]pyridin-2-yl) methylcarbamate (219): 5-Bromo-3-iodopyridin-2-ol (218); (200 mg, 0.67 mmol), tert-butyl prop-2- ynylcarbamate (104 mg, 0.67 mmol), Pd(PPh3)2Cl2 (47 mg, 0.07 mmol), Cul (13 mg, 0.07 mmol), and triethylamine (135 mg, 1.34 mmol) were added in 10 mL of DMF and degassed. The reaction mixture was heated at 90 C under nitrogen atmosphere for 3 h. After cooling down to room temperature, the reaction mixture was filtered. The filtrate was concentrated under reduced pressure to give the crude product, which was purified by silica gel chromatography (30% EtO Ac/petroleum ether) to yield 150 mg of tert-butyl (5- bromofuro[2,3-&]pyridin-2-yl) methylcarbamate (219) as a yellow solid (69% yield). LCMS: m/z 329.0 [M+H]+; tR = 1.76 min.
methyl 5-bromo-2-(((tert-butoxycarbonyl)amino)methyl)-benzofuran-7-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; triethylamine at 90℃; for 2h; Inert atmosphere;
1 Synthesis of methyl 5-bromo-2-((fert- butoxycarbonylamino)methyl)benzofuran-7-carboxylate (231):
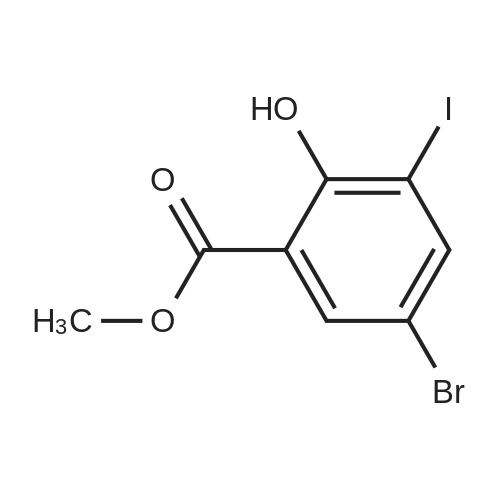
Synthesis of methyl 5-bromo-2-((fert- butoxycarbonylamino)methyl)benzofuran-7-carboxylate (231): A mixture of methyl 5- bromo-2-hydroxy-3-iodobenzoate (230) (7.5 g, 21 mmol), prop-2-ynylcarbamate (3.6 g, 23 mmol), Pd(PPh3)2Cl2 (1.5 g, 2.1 mmol), Cul (800 mg, 4.2 mmol) in 80 mL of Et3N was heated at 90 °C under nitrogen atmosphere for 2 h. After cooling down to room temperature, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure to give the crude product, which was purified by silica gel chromatography (33%- 59% EtOAc/petroleum ether) to give 6.1 g of methyl 5-bromo-2-((tert- butoxycarbonylamino)methyl)benzofuran-7-carboxylate (231) as a yellowish solid (76% yield). LCMS: m/z 408.0 [M+Na ; tR = 1.82 min.
75%
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; triethylamine at 85℃; for 2h; Sealed tube; Inert atmosphere;
75%
With [1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II); copper (I) iodide; triethylamine at 85℃; for 2h; Sealed tube;
1.B; 80.B B) Methyl 5-bromo-2-(((tert-butoxycarbonyl)amino)methyl) benzofuran- 7-carboxylate
Methyl 5-bromo-2-hydroxy-3-iodobenzoate (9.26 g, 25.9 mmol) was dissolved in triethylamine (130 ml) in a 350 mL sealed tube. tert-Butyl prop-2-yn-1-ylcarbamate (3.62 g, 23.4 mmol) was added followed by cuprous iodide (0.247 g, 1.30 mmol) and bis(triphenylphosphine)palladium(II)dichloride (1.82 g, 2.59 mmol). The reaction was degassed with dry nitrogen for 10 minutes before sealing and heating to 85 °C for 2 hours. The reaction was then cooled, filtered, and concentrated in vacuo. The resulting crude residue was purified via ISCO (120g column, 0-60% EtOAc in hexanes) to give methyl 5-bromo-2-(((tert-butoxycarbonyl)amino) methyl)benzofuran-7-carboxylate (7.52 g, 19.6 mmol, 75 % yield).1H NMR (400 MHz, Chloroform-d) δ 8.02 (d, J = 2.0 Hz, 1H), 7.84 (d, J = 2.0 Hz, 1H), 6.63 (s, 1H), 5.11 (br s, 1H), 4.51 (br d, J = 5.9 Hz, 2H), 4.00 (s, 3H), 1.47 (s, 9H); MS (ESI+) m/z = 328.1 (M- tBu+H)+.
tert-butyl (E)-{4-[2-(dimethylamino)phenyl]-4-oxobut-2-en-1-yl}carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
With bis(norbornadiene)rhodium(l)tetrafluoroborate; 1,1-dicyclohexyl-N-(dicyclohexylphosphino)-N-methylphosphinamine In acetone at 20℃; for 6h; Inert atmosphere;
tert-butyl N-[3-(5-nitro-3-pyridyl)prop-2-ynyl]carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1 g
With palladium diacetate; caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tetrahydrofuran; for 4.5h;Inert atmosphere; Reflux;
a) Preparation of Compound 48B (0390) To a solution of <strong>[15862-30-3]3-bromo-5-nitropyridine</strong> (1 g, 4.93 mmol) in THF (30 mL) was added successively tert-butyl prop-2-yn-1-ylcarbamate (1.15 g, 7.39 mmol), Pd(OAc)2 (55 mg, 246 mumol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (285 mg, 493 mumol), and Cs2CO3 (4.82 g, 14.80 mmol). After the mixture was degassed and recharged with argon for five times, it was heated to reflux for 4.5 hrs. The reaction mixture was filtered through celite and the filtrate was concentrated to give a dark oil, which was purified by silica gel chromatography (eluting with EA/PE=O-20%30%) to give a brown oil. The oil was triturated with PE to give tert-butyl N-[3-(5-nitro-3-pyridyl)prop-2-ynyl]carbamate (compound 48B, 1 g) as brown solid. MS: calc'd 278 (M+H)+, measured 278 (M+H)+.
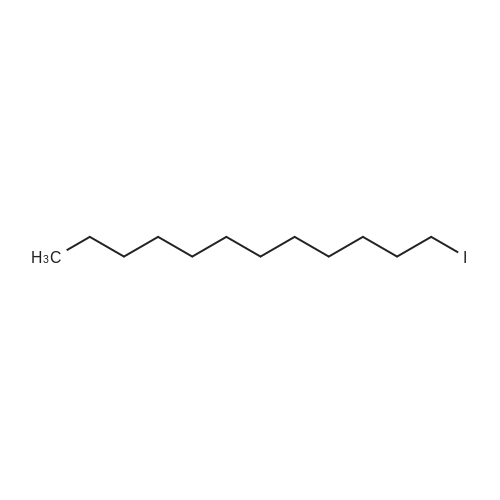
tert-butyl dodecyl(prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
Stage #1: N-tert-Butoxycarbonyl-1-amino-3-propyne; propargyl bromide With sodium hydride In N,N-dimethyl-formamide; mineral oil for 0.25h; Inert atmosphere;
Stage #2: 1-Iodododecane In N,N-dimethyl-formamide; mineral oil at 20℃; for 15h; Inert atmosphere;
tert-Butyl dodecyl(prop-2-yn-1-yl)carbamate 1a
General procedure: To a stirred solution of tert-Butyl prop-2-yn-1-ylcarbamate (300 mg, 1.93 mmol, 1 eq.) in dry DMF (1.93 mL) under nitrogen at 0 °C, was addedNaH (93 mg, 60 % in oil, 2.32 mmol, 1.2 eq.) in small portions. After stirring for 15 min, 1-iodododecane (570 μL, 2.32 mmol, 1.2 eq.) was added dropwise and the reaction was warmedup to room temperature for 15 hours. After completion of the reaction, water and brine wereadded and the aqueous layer was extracted with Et2O (three times). Then, the combinedorganic layers were dried over MgSO4, filtered and concentrated under vacuum. The crude was purified by flash column chromatography on silica gel (cyclohexane/DCM: 50/50) to afford 1a(531 mg, 1.63 mmol, 84%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 4.03 (br. s, 2H), 3.29 (t,J = 7.3 Hz, 2H), 2.17 (t, J = 2.4 Hz, 1H), 1.54 (m, 2H), 1.46 (s, 9H), 1.33-1.21 (m, 18H), 0.88 (t, J =6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 155.2 (br. s), 80.1, 80.0 (br. s), 71.3 (br. s), 46.6, 36.4and 35.9 (br. s, rot.), 32.0, 29.8, 29.74, 29.71 (2C), 29.5 (2C), 28.5 (3C), 28.0 (br. s), 26.9, 22.8,14.2.
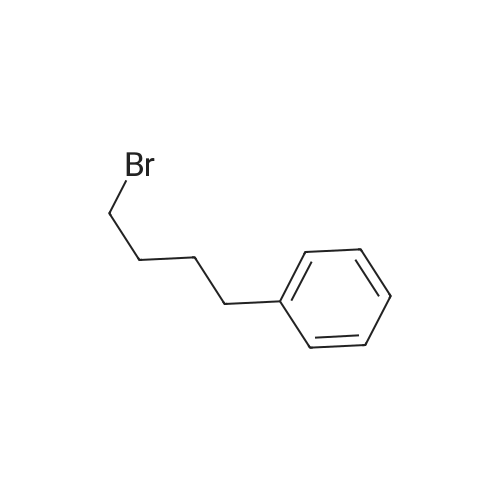
tert-butyl (4-phenylbutyl)(prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
To a stirred solution of tert-Butylprop-2-yn-1-ylcarbamate (300 mg, 1.93 mmol, 1 eq.) in dry DMF (1.93 mL) under nitrogen at 0C, was added NaH (93 mg, 60% in oil, 2.32 mmol, 1.2 eq.) in small portions. After stirring for 15min, (4-bromobutyl)benzene (410 muL, 2.34 mmol, 1.2 eq.) was added dropwise followed by KI(32 mg, 0.19 mmol, 0.1 eq.) and the reaction was warmed up to room temperature for 15 h.After completion of the reaction, water and brine were added and the aqueous layer wasextracted with Et2O (three times). Then, the combined organic layers were dried over MgSO4,filtered and concentrated under vacuum. The crude residue was purified by flash columnchromatography on silica gel (cyclohexane/DCM: 50/50) to afford 1h (446 mg, 1.54 mmol, 80%)as a colorless oil. 1H NMR (300 MHz, CDCl3): delta 7.31-7.24 (m, 2H), 7.21-7.14 (m, 3H), 4.01 (br. s,2H), 3.33 (t, J = 6.6 Hz, 2H), 2.64 (t, J = 7.1 Hz, 2H), 2.16 (t, J = 2.4 Hz, 1H), 1.67-1.56 (m, 4H),1.45 (s, 9H); 13C NMR (100 MHz, CDCl3): delta 155.2 (br. s), 142.4 (br. s), 128.5 (2C), 128.4 (2C),125.8, 80.2, 80.0 (br. s), 71.4 (br. s), 46.4, 36.4 and 36.0 (br. S, rot.), 35.6, 28.6 (br. s), 28.5 (3C),27.6 (br. s).
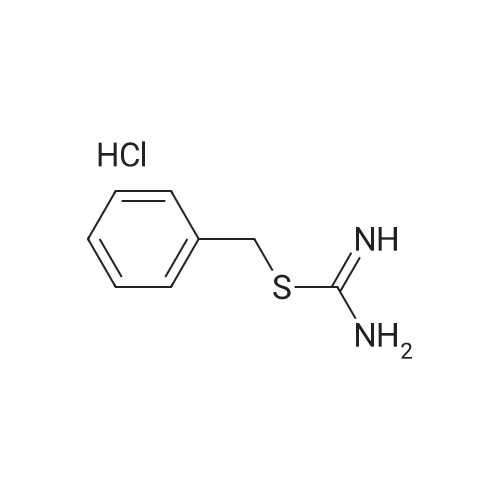
To a solution of 790 tert-butyl N-prop-2-ynyl carbamate (5.0 g, 32 mmol) in 20 tetrahydrofuran (100mL) were added 768 4-trifluoromethylbenzoylchloride (4.3 mL, 29 mmol), 791 dichlorobis(triphenylphosphine)palladium(II) (0.25 g, 0.36 mmol) and 18 copper(I) iodide (0.25 g, 1.3 mmol) and themixture was stirred at room temperature for 5 min. 14 Triethylamine (5.5 mL, 39 mmol) was added andthe mixture was stirred for 30 min. Using a small amount of silica gel, the insoluble material was filtered off,and the filtrate was concentrated under reduced pressure. The obtained residue was dissolved in 79acetonitrile (300 mL), and 792 <strong>[538-28-3]S-benzylisothiourea hydrochloride</strong> (7.5 g, 37 mmol) and 106 potassiumcarbonate (6.0 g, 43 mmol) were added. The reaction mixture was stirred at 70° C. overnight, 12dichloromethane was added and the mixture was washed with water. The organic layer was dried oversodium sulfate, and the desiccant was filtered off. The solvent was evaporated and the obtained residue waspurified by silica gel column chromatography (hexane/ethyl acetate) to give the 793 title compound ( 4.0g , 8.4 mmol, 26percent). MS (ESI) m/z 476 (M+H)+
26%
To a solution of tert-butyl N-prop-2-ynylcarbamate (5.0 g, 32 mmol) in tetrahydrofuran (100 mL) were added 4-trifluoromethylbenzoylchloride (4.3 mL, 29 mmol), dichlorobis(triphenylphosphine)palladium(II) (0.25 g, 0.36 mmol) and copper(I) iodide (0.25 g, 1.3 mmol) and the mixture was stirred at room temperature for 5 min. Triethylamine (5.5 mL, 39 mmol) was added and the mixture was stirred for 30 min. Using a small amount of silica gel, insoluble material was filtered off, and the filtrate was concentrated under reduced pressure. To the obtained residue was added acetonitrile (300 mL) for dissolution and <strong>[538-28-3]S-benzylisothiourea hydrochloride</strong> (7.5 g, 37 mmol) and potassium carbonate (6.0 g, 43 mmol) were added. The reaction mixture was stirred at 70°C overnight, dichloromethane was added and the mixture was washed with water. The organic layer was dried over sodium sulfate and the desiccant was filtered off. The solvent was evaporated and the obtained residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give the title compound (4.0 g, 8.4 mmol, 26percent). MS (ESI) m/z 476 (M+H)+
With copper(II) bis(trifluoromethanesulfonate); copper(l) chloride In toluene at 100℃; for 20h; Inert atmosphere;
Experimental procedure for the synthesis of C-4a (method A)
2-Amino-isonicotinic acid methyl ester C-la (1.00 g, 6.572 mmol), N-Boc prop-2-ynylamine C-2a (1.12 g, 7.230 mmol), E-3-(3-chloro-2-fluorophenyl) propenal C-3b (1 .34 g, 7.23 mmol), Cu(OTf)2 (0.24 g, 0.66 mmol) and CuCI (0.06 g, 0.07 mmol) are dissolved in toluene under argon and stirred at 100 00 for 20 h. The solvent is removed under vacuum and the crude product is purified by chromatography to deliver intermediateC-4a.
tert-butyl (3-(4-methoxypyridin-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
To a Biotage 2.0-5.0 mL microwave tube containing N-Boc-propargylamine (212 mg, 1.37 mmol) under a blanket of argon was added degassed DME/EtOH 50:50 (0.3 mL) followed by a slurry of tetrakis(triphenylphosphine)palladium(0) (109 mg, 0.094 mmol) in degassed DME/EtOH 50:50 (2.0 ml_), followed by cuprous iodide (29.0 mg, 0.152 mmol). To the vigorously stirred suspension was added sodium carbonate (290 mg, 2.73 mmol) dissolved in a minimum amount of degassed water (ca. 1.5 ml_), the tube was purged with argon, stirred at ambient temperature for ten minutes, followed by the addition of a solution of 3-bromo-4- methoxypyridine (257. mg, 1.37 mmol). The tube was purged with argon, capped and placed in a Biotage Initiator-·- microwave and heated to 150 C for 15 minutes on normal absorption level. The contents of the flask were transferred to a sintered glass funnel containing anhydrous Na2SC>4 and the crude material was eluted with dichloromethane. The solvent was removed in vacuo and the residue was chromatographed on silica gel (EtOAc/Hex, 25:75, v/v Rf = 0.23) to afford K-Boc (272. mg, 76% yield) as a yellow oil: 1 H NMR (500 MHz ,CDCI3) d = 8.48 (s, 1 H), 8.40 (d, J = 5.7 Hz, 1 H), 6.77 (d, J = 6.0 Hz, 1 H), 4.93 (br s, 1 H), 4.20 (br s, 2 H), 3.93 - 3.88 (m, 3 H), 1.46 (s, 9 H).
tert-butyl (3-(5-methylpyridin-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
59%
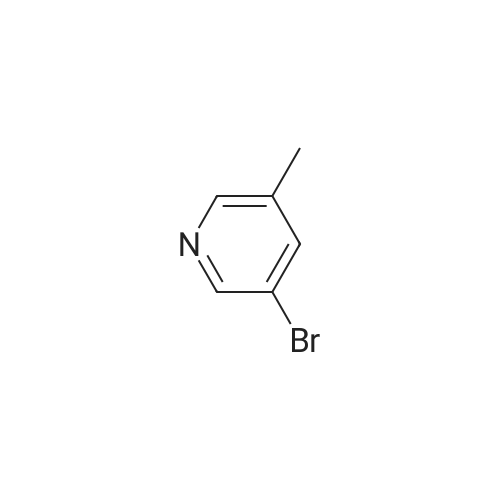
To a Biotage 2.0-5.0 ml_ microwave tube containing N-Boc-propargylamine (372. mg, 2.34 mmol) under a blanket of argon was added cuprous iodide (44.6 mg, 0.234 mmol) followed by tetrakis(triphenylphosphine)palladium(0) (135. mg, 0.117 mmol) followed by degassed 1-propanol (2.0 ml_). To the vigorously stirred suspension was added sodium carbonate (410. mg, 3.87 mmol) dissolved in a minimum amount of water (ca. 1.3 ml_). The tube was purged with argon, stirred at ambient temperature for ten minutes followed by the addition of a solution of <strong>[3430-16-8]3-bromo-5-methylpyridine</strong> (402. mg, 2.34 mmol) in degassed 1- propanol. The tube was purged with argon, capped and placed in a Biotage Initiator? microwave and heated to 100 C for 15 minutes on normal absorption level. The contents of the flask were transferred to a sintered glass funnel containing anhydrous Na2S04. The crude material was eluted with dichloromethane followed by methanol. The solvent was removed in vacuo and the residue was chromatographed on silica gel (EtOAc/Hex, 25:75, v/v, TLC: (0233) EtOAc/Hex, 25:75, Rf = 0.116) to afford D-Boc (342. mg, 59.0% yield) as a yellow solid: 1 H NMR (500 MHz ,CDCI3) d 8.45 (s, 1 H), 8.35 (s, 1 H), 7.49 (s, 1 H), 5.05 (br. s., 1 H), 4.14 (br. s., 2 H), 2.28 (s, 3 H), 1.45 (s, 9 H).
tert-butyl (3-(2-methylpyridin-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
32%
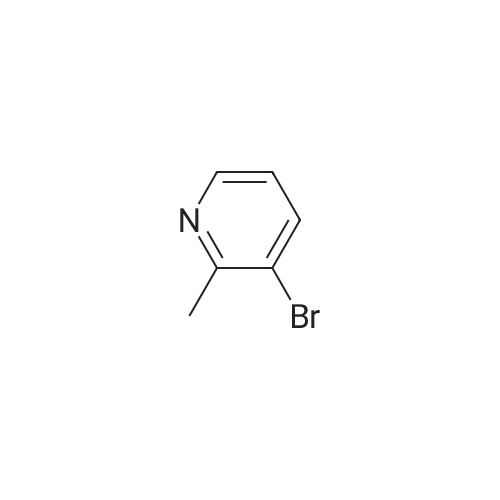
To a Biotage 2.0-5.0 ml_ microwave tube containing N-Boc-propargylamine (413 mg, 2.66 mmol) under a blanket of argon was added degassed DME/EtOH 50:50 (2.5 ml_) followed by cuprous iodide (46.0 mg, 0.242 mmol) followed by (0248) tetrakis(triphenylphosphine)palladium(0) (290 mg, 0.25 mmol). To the vigorously stirred suspension was added triethylamine (734 mg, 7.26 mmol). The tube was purged with argon, stirred at ambient temperature for ten minutes followed by the addition of a solution of 3-bromo- 2-methylpyridine (416 mg, 2.42 mmol). The tube was purged with argon, capped and placed in a Biotage Initiator-·- microwave and heated to 150 C for 15 minutes on normal absorption level. The contents of the flask were filtered through a compressed bed of celite and the crude material was eluted with dichloromethane. The solvent was removed in vacuo and the residue was chromatographed on silica gel (EtOAc/Hex, 25:75, v/v Rf = 0.167) to afford F-Boc (192 mg, 32.0% yield) as a red oil: 1 H NMR (500 MHz, CDCI3) d = 8.34 (d, J = 3.6 Hz, 1 H), 7.55 (dd, J = 1.5, 7.7 Hz, 1 H), 6.98 (dd, J = 4.9, 7.5 Hz, 1 H), 5.31 (br. s., 1 H), 4.13 (d, J = 4.9 Hz, 2 H), 2.56 (s, 3 H), 1.40 (s, 9 H).
tert-butyl (3-(4-ethylpyridin-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50.6%
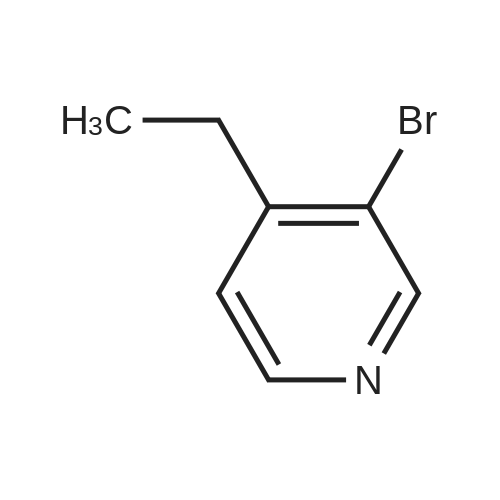
To a Biotage 2.0-5.0 mL microwave tube containing N-Boc-propargylamine (224 g, 1.44 mmol) under a blanket of argon was added degassed DME/EtOH 50:50 (0.5 mL) followed by a slurry of tetrakis(triphenylphosphine)palladium(0) (280. mg, 0.242 mmol) in degassed DME/EtOH 50:50 (1.5 mL), followed by cuprous iodide (27.0 mg, 0.144 mmol). To the vigorously stirred suspension was added sodium carbonate (300. mg, 1.57 mmol) dissolved in a minimum amount of degassed water (ca. 1.5 mL), the tube was purged with argon, stirred at ambient temperature for ten minutes, followed by the addition of a solution of 3-bromo-4- ethylpyridine (269. mg, 1.44 mmol). The tube was purged with argon, capped and placed in a Biotage Initiator-·- microwave and heated to 150 C for 15 minutes on normal absorption level. The contents of the flask were transferred to a sintered glass funnel containing anhydrous Na2SC>4 and the crude material was eluted with dichloromethane. The solvent was removed in vacuo and the residue was chromatographed on silica gel (EtOAc/Hex, 25:75, v/v Rf = 0.2) to afford H-Boc (141. mg, 50.6% yield) as a yellow oil: 1 H NMR (500 MHz ,CDCI3) d = 8.54 (s, 1 H), 8.39 (d, J = 5.4 Hz, 1 H), 7.10 (d, J = 5.0 Hz, 1 H), 5.10 (br, s, 1 H), 4.17 (s, 2H), 2.74 (q, J = 7.6 Hz, 2 H), 1.45 (s, 9 H), 1.21 (t, J = 7.6 Hz, 3 H).
tert-butyl (3-(4-phenylpyridin-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66.3%
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); sodium carbonate In propan-1-ol; water at 120℃; for 0.25h; Inert atmosphere; Microwave irradiation;
66%
Stage #1: N-tert-Butoxycarbonyl-1-amino-3-propyne With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); sodium carbonate In 1,2-dimethoxyethane; ethanol; water at 20℃; for 0.166667h; Inert atmosphere;
Stage #2: 3-bromo-4-phenylpyridine In 1,2-dimethoxyethane; ethanol; water at 120℃; for 0.25h; Microwave irradiation; Inert atmosphere;
1 Compound J-Boc: tert-butyl (3-(4-phenylpyridin-3-yl)prop-2-yn-1-yl)carbamate.
To a Biotage 2.0-5.0 mL microwave tube containing N-Boc-propargylamine (167 mg, 1.07 mmol) under a blanket of argon(g) was added degassed DME/EtOH 50:50 (1.0 mL) followed by a slurry of tetrakis(triphenylphosphine)palladium(0) (102 mg, 0.080 mmol) in degassed DME/EtOH 50:50 (1.0 mL) followed by cuprous iodide (25.0 mg, 0.131 mmol). To the vigorously stirred suspension was added sodium carbonate (253 mg, 2.38 mmol) dissolved in degassed water (ca. 1.5 ml_). The tube was purged with argon, stirred at ambient temperature for ten minutes followed by the addition of a solution of 3-bromo-4-phenylpyridine (252 mg, 1.07 mmol). The tube was purged with argon, capped and placed in a Biotage Initiator-·- microwave and heated to 120 °C for 15 minutes on normal absorption level. The contents of the flask were transferred to a sintered glass funnel containing anhydrous Na2S04. The crude material was eluted with dichloromethane. The solvent was removed in vacuo and the residue was chromatographed on silica gel (EtOAc/Hex, 25:75, v/v Rf = 0.16) to afford J-Boc (219.9 mg, (0277) 66% yield) as a red oil: 1 H NMR (500 MHz ,CDCI3) d = 8.73 (br. s., 1 H), 8.55 (br. s., 1 H), 7.68 - 7.57 (m, 2 H), 7.53 - 7.38 (m, 3 H), 7.36 - 7.27 (m, 1 H), 4.73 (br. s., 1 H), 4.11 - 3.99 (m, 2 H), 1.48 - 1.42 (m, 9 H).
In a nitrogen-purged four-necked flask, <strong>[6293-83-0]2-iodo-4-nitroaniline</strong>(5.11 g, 19.4 mmol), Bis(triphenylphosphine)palladium(II) dichloride (281.7 mg, 0.401 mmol), Copper iodide (160.7 mg, 0.844 mmol),And 30 ml of diethylamine, and the mixture was stirred at room temperature (20 C.) for 10 minutes.Thereafter, N-Boc-propargylamine (3.72 g, 24.0 mmol) was added and the mixture was stirred at room temperature (20 C.) for 4 hours. After disappearance of the raw material was confirmed by HPLC (high performance liquid chromatography), 200 ml of ethyl acetate and 200 ml of 1 M ammonium chloride aqueous solution were added and extracted. The obtained organic layer was washed twice with 1 M ammonium chloride aqueous solution and dried over anhydrous magnesium sulfate. After removing the drying agent, the filtrate was concentrated and purified by silica gel column chromatography (distillate: ethyl acetate: hexane = 3: 7 (volume ratio)). The yield was 4.97 g, and the yield was 88.0%.
Stage #1: 2,6-diiodo-4-nitroaniline With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; diethylamine In tetrahydrofuran at 20℃; for 0.166667h;
Stage #2: N-tert-Butoxycarbonyl-1-amino-3-propyne In tetrahydrofuran at 20℃; for 12h;
18.1 Precursor Synthesis 1
In a 1 L four-necked flask, 2,6-diiodo-4-nitroaniline (50.00 g, 128 mmol), bis(triphenylphosphine)palladium(II) dichloride (900 mg, 1.28 mmol), copper iodide (488 mg, 2 .56 mmol),32 ml of diethylamine and 165 ml of THF were added, and the mixture was cooled to room temperature (20 °C.) For 10 minutes. Thereafter, while stirring the solution in the four-necked flask, A THF solution (90 ml) of N-Boc-propargylamine (47.60 g, 307 mmol) was added dropwise over 15 minutes. After completion of the dropwise addition, the mixture was stirred at room temperature (20 °C.) for 12 hours. After 12 hours,The reaction solution was added to 1.28 L of pure water with stirring, and the precipitated black precipitate was collected by suction filtration and washed twice with 50 ml of water. The obtained solid was dried under reduced pressure to obtain a brown solid. The brown solid was purified by recrystallization (toluene) to obtain a yellow solid. It was confirmed by 1H-NMR measurement that the obtained yellow solid was the nitro compound. The yield was 47.09 g, and the yield was 83.0%.
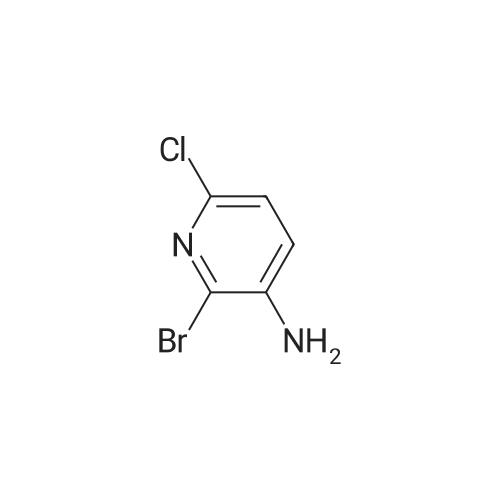
tert-butyl (3-(3-amino-6-chloropyridin-2-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; copper(l) iodide; triethylamine; In tetrahydrofuran; at 70℃; for 16h;Inert atmosphere;
A mixture of <strong>[1050501-88-6]2-bromo-6-chloropyridin-3-amine</strong> (1 g, 4.82 mmol, Fluorochem), 2-bromo-6- chloropyridin-3-amine (1 g, 4.82 mmol, Alfa Aesar), copper I iodide (0.092 g, 0.482 mmol), PdCI2(dppf) (0.353 g, 0.482 mmol) and TEA (1.008 mL, 7.23 mmol) in Tetrahydrofuran (THF) (20mL) was degassed under a flow of nitrogen for 10 min. The reaction mixture was heated to 70 C for 16h. The mixture was cooled down to RT. EtOAc (20 mL) was added and the resulting mixture was filtered through Celite (2.5 g cartridge). The residual solild was washed with additional EtOAc (2 x 20 mL) The filtrate was collected and washed with saturated ammonium chloride solution (50 mL), water (50 mL), brine (50 ml.) and dried over MgS04. The volatiles were removed under reduced pressure to give a residue that was purified by column chromatography on Silica (120 g Cartridge, dry load on Florisil) using the elution gradient EtOAc in Cyclohexane 0-100% to give tert-butyl (3-(3-amino-6- chloropyridin-2-yl)prop-2-yn-1-yl)carbamate (1.1635 g, 4.13 mmol, 86% yield) as a orange solid. LCMS (System B, UV, ESI): Rt = 0.96 min, [M+H]+ 282
tert-butyl N-[3-[2-(2,6-dioxo-3-piperidyl)-1,3-dioxoisoindolin-5-yl]prop-2-ynyl]carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With copper(l) iodide; trans-bis(triphenylphosphine)palladium dichloride; triethylamine In N,N-dimethyl-formamide at 80℃; for 0.5h; Microwave irradiation;
1 Step 1-Tert-butyl N-[3-[2-(2,6-dioxo-3-piperidyl)-1,3-dioxo-isoindolin-5-yl]prop-2-ynyl]carbamate
To a solution of 5-bromo-2-(2,6-dioxo-3-piperidyl)isoindoline-1,3-dione (1 g, 2.97 mmol Intermediate GA) and tert-butyl N-prop-2-ynylcarbamate (920 mg, 5.93 mmol) in DMF (4 mL) was added Pd(PPh3)2Cl2 (208 mg, 296 umol) TEA (5.40 g, 53 mmol, 7.43 mL) and CuI (57 mg, 296 umol). The mixture was stirred at 80° C. for 0.5 hr under microwave. On completion, the mixture was concentrated in vacuo, the residue was purified by silica gel chromatography to give the title compound (1.3 g, 95% yield) as yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 8.01-7.81 (m, 3H), 5.17 (dd, J=5.2, 12.8 Hz, 1H), 4.10-4.03 (m, 2H), 3.33 (s, 1H), 2.94-2.84 (m, 1H), 2.73-2.53 (m, 2H), 2.13-2.02 (m, 1H), 1.47-1.36 (m, 9H); LC-MS (ESI+) m/z 434.0 (M+Na)+.
3-(3-((tert-butoxycarbonyl)amino)-1-propynyl)-2-pyridinecarboxylic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
41.7%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; copper(l) iodide; N-ethyl-N,N-diisopropylamine; In acetonitrile; for 6h;Reflux; Inert atmosphere;
to<strong>[53636-56-9]3-bromo-2-pyridinecarboxylic acid methyl ester</strong>(2.5g, 11.6mmol),To a solution of N-tert-butoxycarbonylaminopropyne (2.0 g, 12.9 mmol) and diisopropylethylamine (5 mL) in acetonitrile (30 mL) was added (1,1'-bis (diphenylphosphino) di) Ferrocene) palladium dichloride (0.90 g, 1.0 mmol) and cuprous iodide (0.2 g, 1.5 mmol), and the resulting mixture was heated under reflux under nitrogen for 6 hours.The reaction solution was cooled to room temperature, filtered through celite, the filtrate was concentrated to remove the solvent, and the residue was purified by a silica gel column to obtain a yellow solid compound 2 (1.4 g, yield: 41.7%).
2-(3-((tert-butoxycarbonyl)amino)-1-propynyl)-3-thiophenecarboxylic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84.9%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; copper(l) iodide; N-ethyl-N,N-diisopropylamine; In acetonitrile;Reflux; Inert atmosphere;
Acetonitrile (30 mL) to <strong>[76360-43-5]methyl 2-bromo-3-thiophenecarboxylate</strong> (3.0 g, 13.5 mmol), N-tert-butoxycarbonylaminopropyne (2.5 g, 16.3 mmol) and diisopropylethylamine (5 mL) To the solution, (1,1'-bis (diphenylphosphino) ferrocene) palladium dichloride (0.50 g, 0.67 mmol) and cuprous iodide (0.13 g, 0.67 mmol) were added. The resulting mixture was Heat to reflux under nitrogen and stir overnight.The reaction solution was cooled to room temperature, filtered through celite, and the filtrate was concentrated to remove the solvent. The residue was purified by a silica gel column to obtain a yellow oily compound 3 (3.4 g, yield: 84.9%).
tert-butyl (3-(4-chloropyridin-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In 1,2-dimethoxyethane; ethanol; at 140℃; for 0.25h;Microwave irradiation; Inert atmosphere;
To a 5 mL microwave vial (Biotage) was added <strong>[36953-42-1]3-bromo-4-chloropyridine</strong> (267 g, 1.39 mmol), tert-butyl prop-2-ynyl-carbamate (232 mg, 1.49 mmol), Cul (23.0 mg, 0.121 mmol) and bis(triphenylphosphine)palladium(ll) dichloride (43.0 mg, 0.0613 mmol). The vial was purged with argon for 5 minutes followed by addition of degassed DME/EtOH 50:50 (2.0 mL) and degassed Et3N (600. mL, 4.30 mmol). The vial was capped and placed in a Biotage Initiator-·- microwave and heated to 140 C for 15 minutes on normal absorption. The contents of the flask were cooled to rt, transferred to a separatory funnel, diluted with EtOAc (30 mL), washed with water (15 mL), followed by saturated NaCI (15 mL), dried over Na2S04, gravity filtered, the solvent was removed in vacuo and the residue was chromatographed on silica gel (EtOAc/Hex, 0: 100, v/v to EtOAc/Hex, 100:0, v/v, TLC: 50% EtOAc/hexane, Rf = 0.52) to afford L-Boc (113. mg, 30% yield) as a brown syrup: 1 H NMR (500 MHz, CDCIs) d 8.67 (bs, 1 H), 8.45 (bs, 1 H), 7.35 (s, 1 H), 5.30 (bs, 1 H), 4.24 (m, 2 H), 1.48 (s, 9 H).
tert-butyl (3-(1-benzyl-4-(12-hydroxydodec-1-yn-1-yl)-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
17.7%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; potassium carbonate; In tetrahydrofuran; toluene; at 20℃; for 18h;Inert atmosphere;
Compound 2 (219.5 mg, 0.5 mmol), t-Boc protected propargyl amine (77.6 mg, 0.5 mmol), CuI (38 mg, 40 %), Pd(PPh3)2Cl2 (35 mg, 10 %), K2CO3 (207.3 mg, 1.5 mmol) were successively added to a solvent mixture of dry THF (1 mL) and toluene (4 mL) under nitrogen. Then, <strong>[18202-10-3]dodec-11-yn-1-ol</strong> (91 mg, 0.5 mmol) in THF (1 mL) was added dropwisely. The mixture was stirred at 20 C for 18 h. After the completion of the reaction as detected by TLC, the mixture was directly purified by column chromatography over silica gel (hexane/ethyl acetate = 3 : 1) to yield a yellow liquid (46 mg, 17.7 %). 1H NMR (600 MHz, Chloroform-d) delta 7.34-7.28 (m, 5H), 4.85 (s, br, 1H), 4.67 (s, 2H), 4.26 (s, br, 2H), 3.63 (t, 2H), 2.55 (t, 2H), 1.56 (m, 2H), 1.45 (s, 13H), 1.29 (m, 10H). 13C NMR (151 MHz, CDCl3) delta 167.2, 166.9, 155.2, 135.8, 130.0, 128.8, 128.7, 128.1, 126.8, 114.0, 105.1, 80.5, 73.6, 71.8, 63.2, 42.5, 32.9, 31.8, 29.6, 29.5, 29.1, 28.9, 28.5, 28.1, 25.8, 20.7. HR-MS (EI): m/z calcd. for C31H40N2O5Na (M + Na)+: 543.2835; found: 543.2834.
tert-butyl (3-(1-benzyl-4-(8-hydroxyoct-1-yn-1-yl)-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)prop-2-yn-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
18.1%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; potassium carbonate In tetrahydrofuran; toluene at 20℃; for 18h; Inert atmosphere;
Tert-butyl-(3-(1-benzyl-4-(8-hydroxyoct-1-yn-1-yl)-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)-prop-2-yn-1-yl)-carbamate (EDY 3)
Compound 2 (219.5 mg, 0.5 mmol), t-Boc protected propargyl amine (77.6 mg, 0.5 mmol), CuI (38 mg, 40 %), Pd(PPh3)2Cl2 (35 mg, 10 %), K2CO3 (207.3 mg, 1.5 mmol) were successively added to a solvent mixture of dry THF (1 mL) and toluene (4 mL) under nitrogen. Then, oct-7-yn-1-ol (63.1 mg, 0.5 mmol) in THF (1 mL) was added dropwisely. The mixture was stirred at 20 °C for 18 h. After the completion of the reaction as detected by TLC, the mixture was directly purified by column chromatography over silica gel (hexane/ethyl acetate = 2 : 1) to yield a yellow liquid (42 mg, 18.1 %). 1H NMR (600 MHz, Chloroform-d) δ 7.34-7.27 (m, 5H), 4.96 (s, br, 1H), 4.67 (s, 2H), 4.26 (s, br, 2H), 3.65 (t, 2H), 2.57 (t, 2H), 1.65 (m, 2H), 1.59 (m, 2H), 1.45 (s, 13H). 13C NMR (151 MHz, CDCl3) δ 167.2, 166.9, 155.3, 135.8, 130.0, 128.8, 128.7, 128.1, 127.0, 113.7, 105.2, 80.5, 73.4, 72.0, 62.8, 42.5, 32.6, 28.5, 27.9, 25.2, 20.6. HR-MS (EI): m/z calcd. for C27H32N2O5Na (M + Na)+: 487.2209; found: 487.2208.
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine In acetonitrile at 80℃; for 2h; Inert atmosphere;
32.1 Step 1: fert-Butyl (3-(4-formylpyridin-3-yl)prop-2-yn-1-yl)carbamate
A mixture of 3-bromopyridine-4-carbaldehyde (15 g, 80.64 mmol), fe/f-butyl prop-2-yn-1- ylcarbamate (13.14 g, 84.67 mmol), Et3N (81.60 g, 806.43 mmol, 112.25 ml_), Cul (1.54 g, 8.06 mmol) and Pd(PPh3)2Cl2(5.66 g, 8.06 mmol) in CH3CN (120 ml_) was stirred at 80 °C for 2 hrs under N2. The reaction mixture was quenched by addition of water (300 ml_) and extracted with ethyl acetate (300 ml_ * 3). The combined organic phase was washed with brine (300 ml_), dried over Na2SC>4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (S1O2, Petroleum ether/Ethyl acetate=1/1) to give the title compound (18 g, 69.15 mmol, 85.75% yield) as a brown oil.LCMS (ESI) m/z: [M+H]+= 261 .1 .1HNMR (400 MHz, CDCb) d = 10.48 (s, 1 H), 8.66 (s, 1 H), 8.78 - 8.68 (m, 1 H), 7.78 - 7.62 (m, 1 H), 4.88 (br s, 1 H), 4.32 - 4.18 (m, 2H), 1.46 (s, 9H) ppm.
ethyl 2-[4-(3-[(tert-butoxy)carbonyl]amino}prop-1-yn-1-yl)phenyl]acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; diisopropylamine; In tetrahydrofuran; at 20℃;Inert atmosphere; Sealed tube;
Ethyl 2-(4-iodophenyl)acetate (1.25 mL, 6.90 mmol), tert-butyl N-(prop-2-yn-1-yl)carbamate (1.17 g, 7.59 mmol), bis(triphenylphosphine) palladium dichloride (245 mg, 0.35 mmol), and copper iodide (66 mg, 0.35 mmol) was dissolved in 10 mL of anhydrous THF, sealed with a septum, and degassed with nitrogen for 20 minutes. To the reaction mixture was added 5 mL of di-isopropyl amine. The mixture was allowed to stir at room temp overnight. The solvent was removed in vacuo, and the reaction mixture was filtered through celite, rinsing with methanol. The methanol was removed in vacuo and the remaining oil was extracted with ethyl acetate, rinsing twice with water and once with brine. The organic layer was dried over magnesium sulfate, filtered and concentrated in vacuo. The remaining oil was purified via Normal Phase flash chromatography (0-35% Ethyl Acetate/Hexanes) to give the title compound (1.00 g, 46%). LC-MS: m/z 317.2 (MH)+.
46%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; diisopropylamine; In tetrahydrofuran; at 20℃;Inert atmosphere; Sealed tube;
Ethyl 2-(4-iodophenyl)acetate (1.25 mL, 6.90 mmol), tert-butyl N-(prop-2-yn-1-yl)carbamate (1.17 g, 7.59 mmol), bis(triphenylphosphine) palladium dichloride (245 mg, 0.35 mmol), and copper iodide (66 mg, 0.35 mmol) was dissolved in 10 mL of anhydrous THF, sealed with a septum, and degassed with nitrogen for 20 minutes. To the reaction mixture was added 5 mL of di-isopropyl amine. The mixture was allowed to stir at room temp overnight. The solvent was removed in vacuo, and the reaction mixture was filtered through celite, rinsing with methanol. The methanol was removed in vacuo and the remaining oil was extracted with ethyl acetate, rinsing twice with water and once with brine. The organic layer was dried over magnesium sulfate, filtered and concentrated in vacuo. The remaining oil was purified via Normal Phase flash chromatography (0-35% Ethyl Acetate/Hexanes) to give the title compound (1.00 g, 46%). LC-MS: m/z 317.2 (MH)+.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






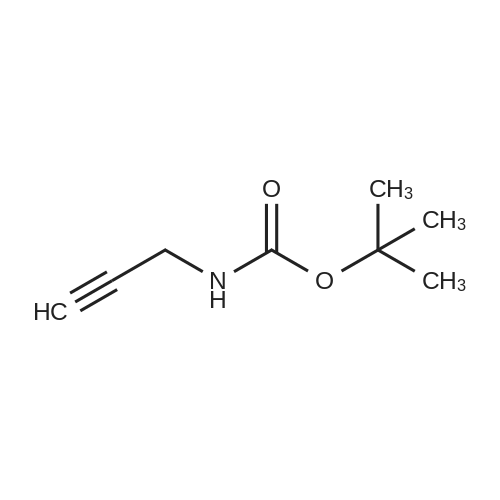

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping