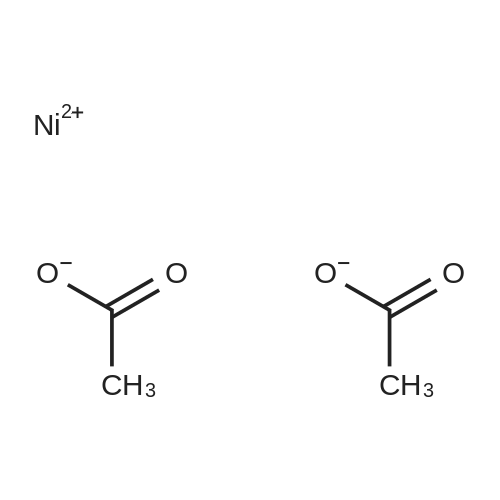
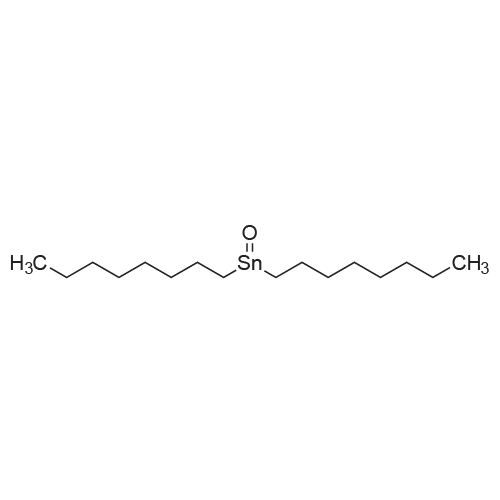
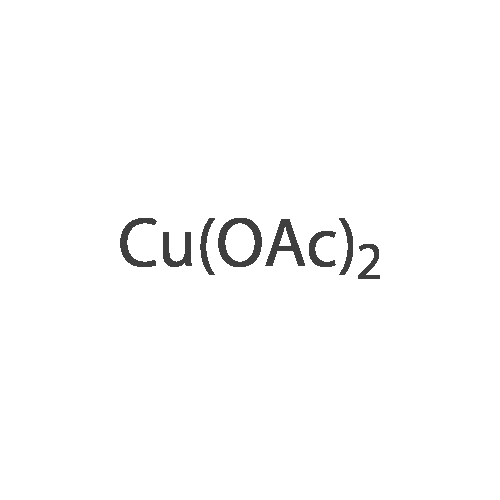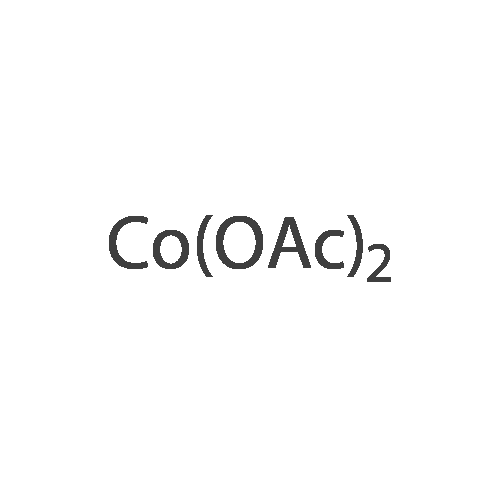* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
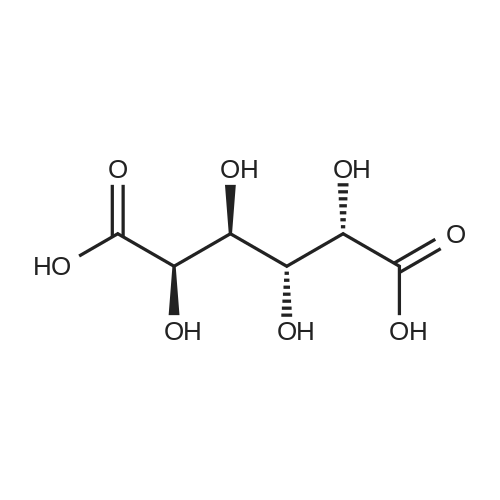
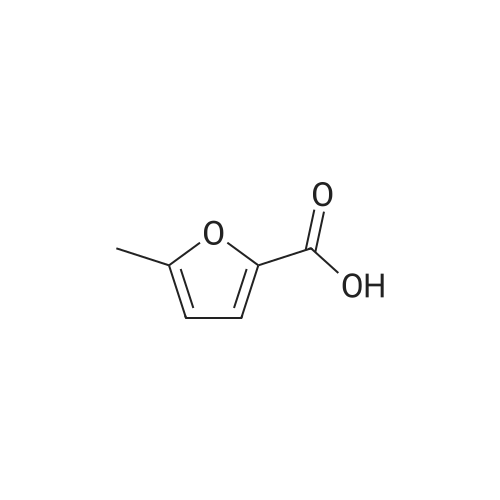
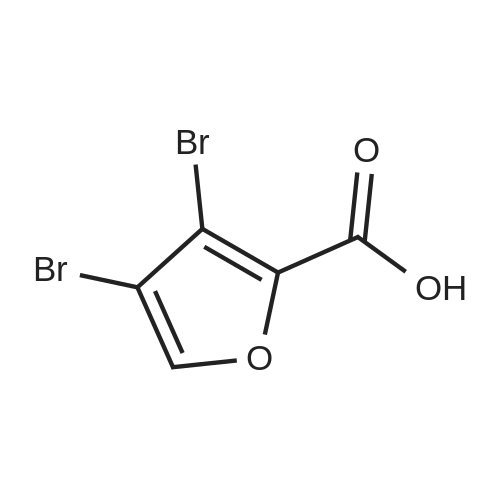
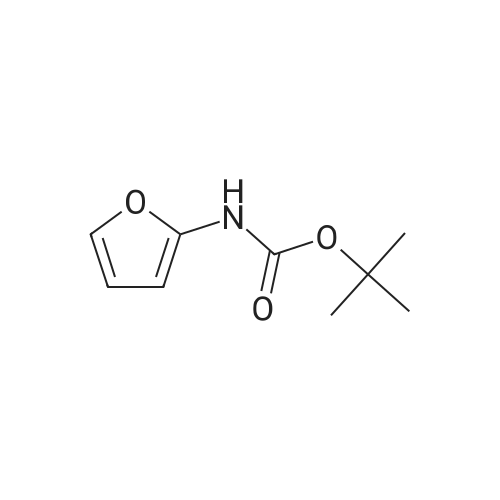
[1] Acta Chemica Scandinavica, Series B: Organic Chemistry and Biochemistry, 1984, vol. 38, # 8, p. 689 - 694
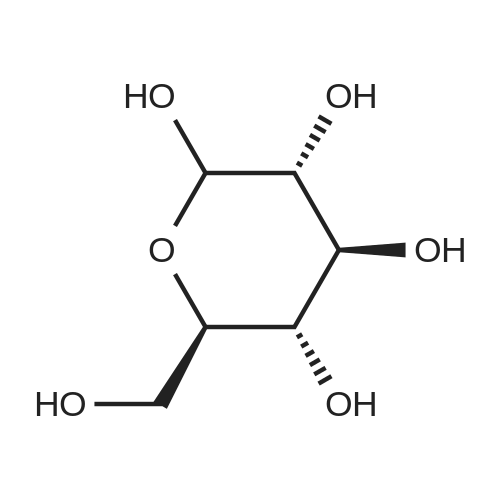
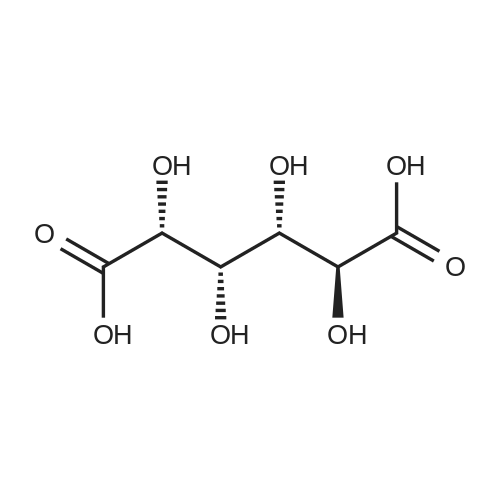
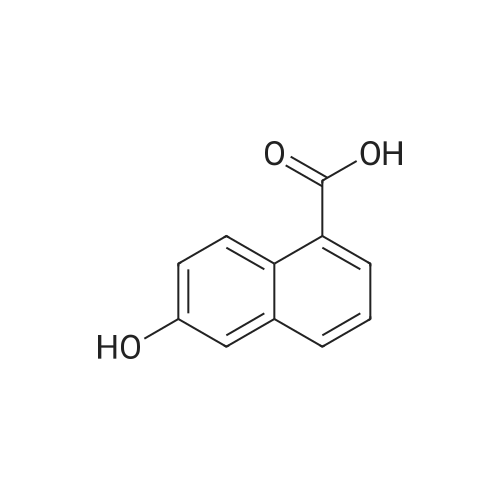
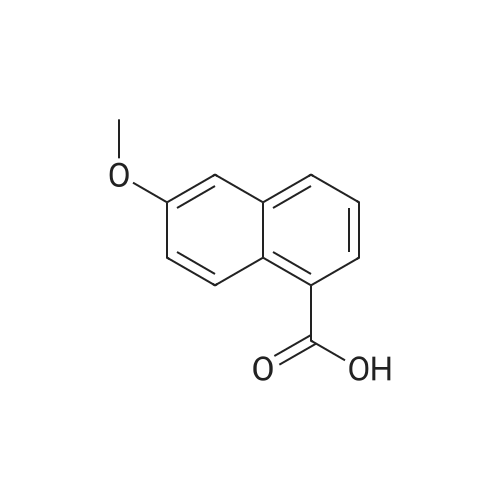
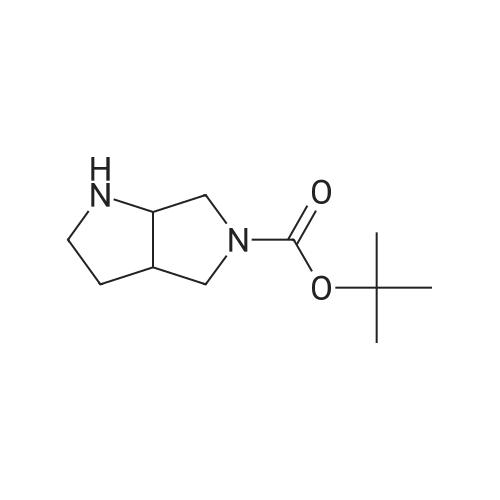
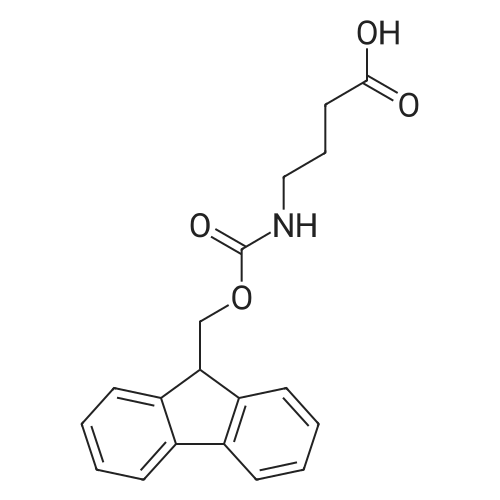
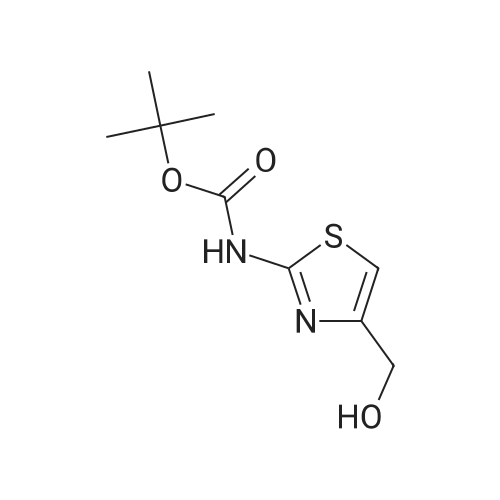
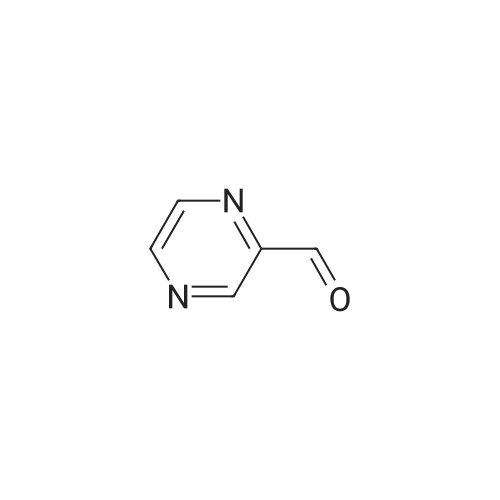
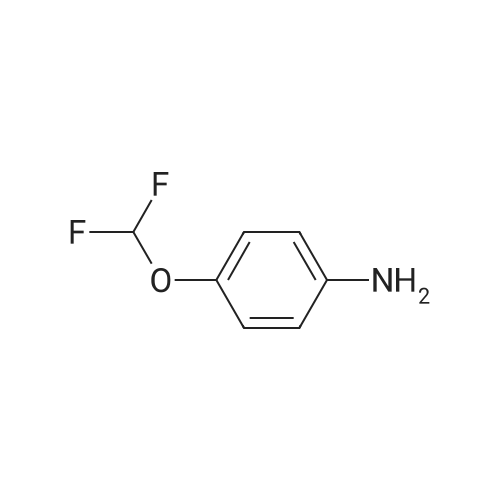
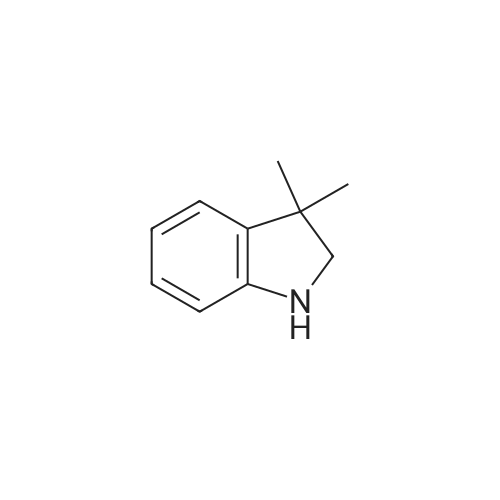
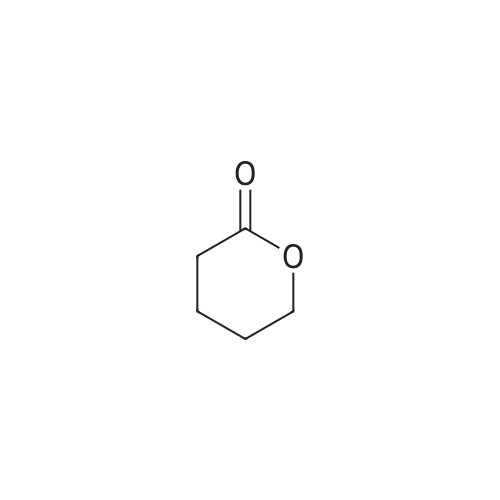
2
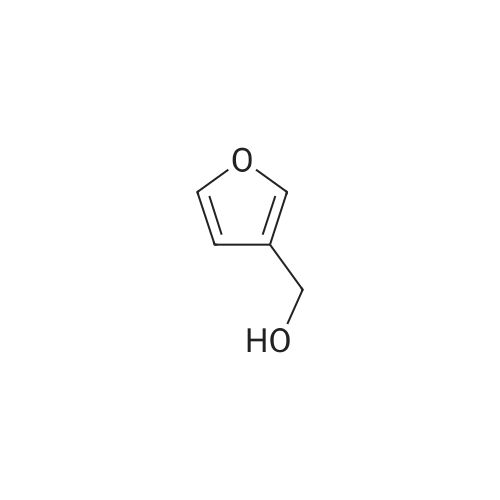
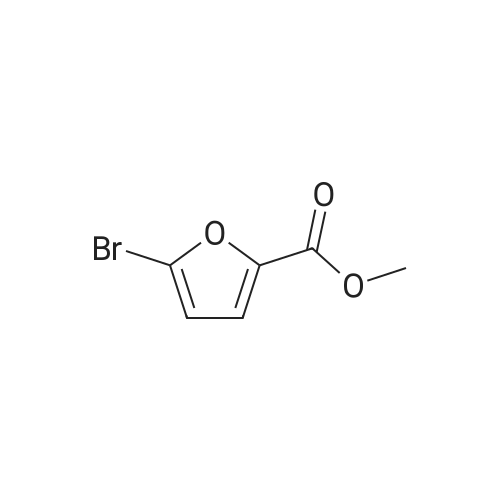
[ 88-14-2 ]
[ 4412-91-3 ]
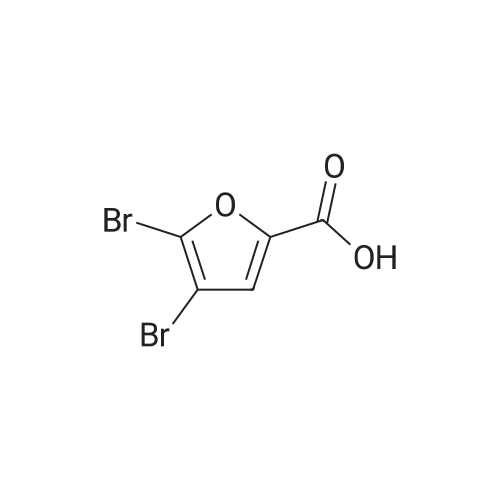
Reference:
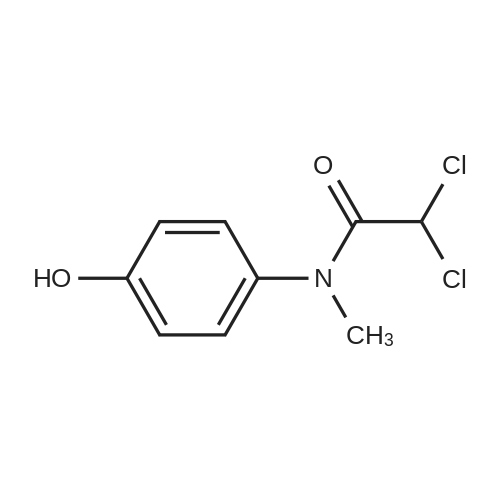
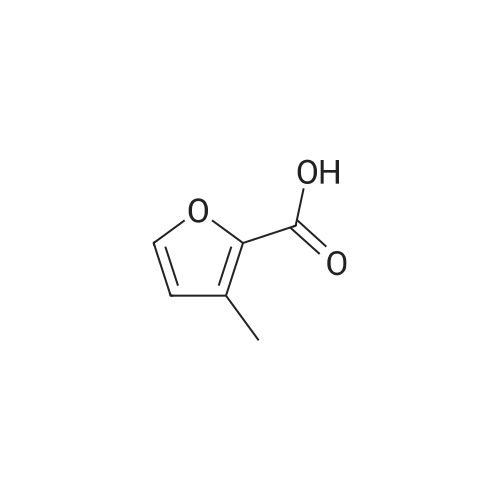
[1] Beilstein Journal of Organic Chemistry, 2013, vol. 9, p. 2925 - 2933
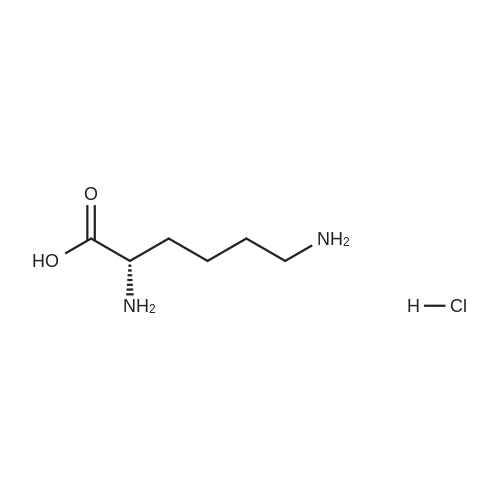
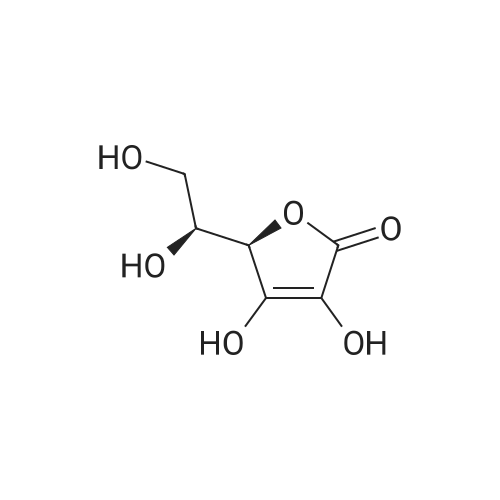
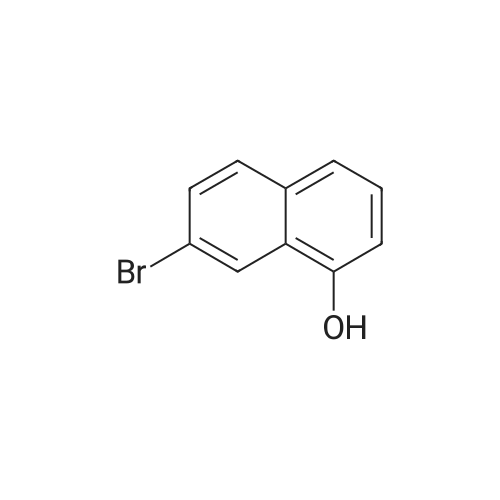
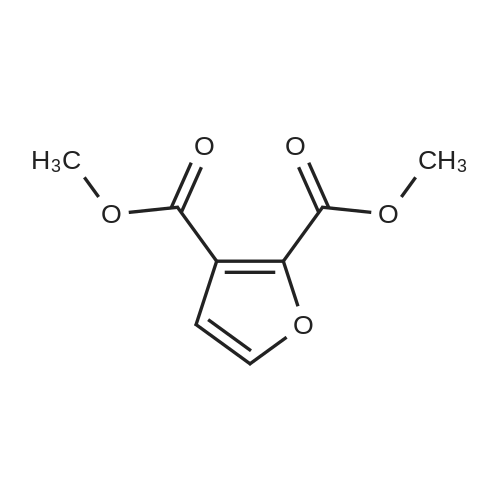
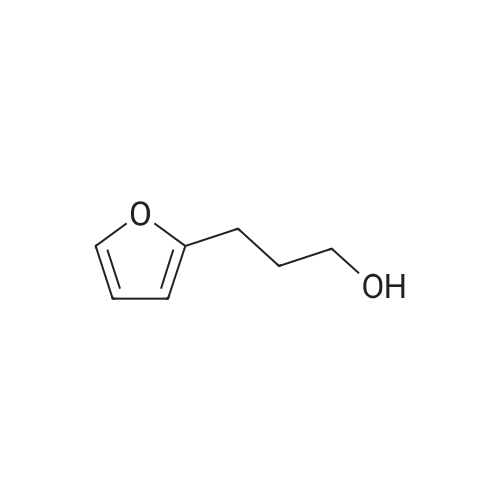
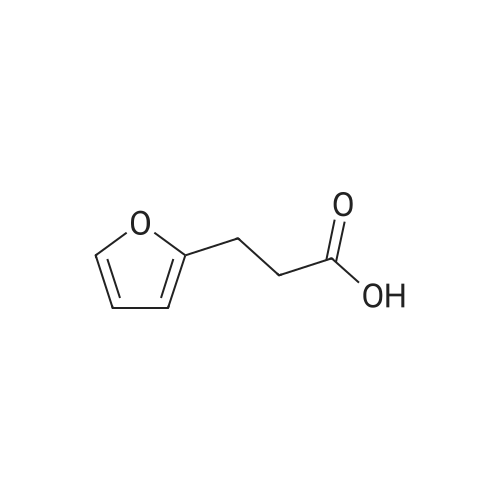
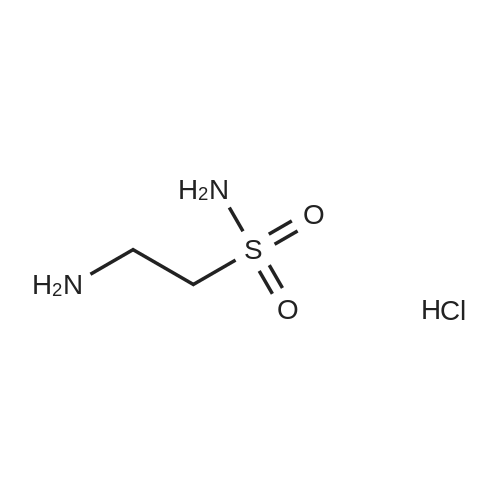
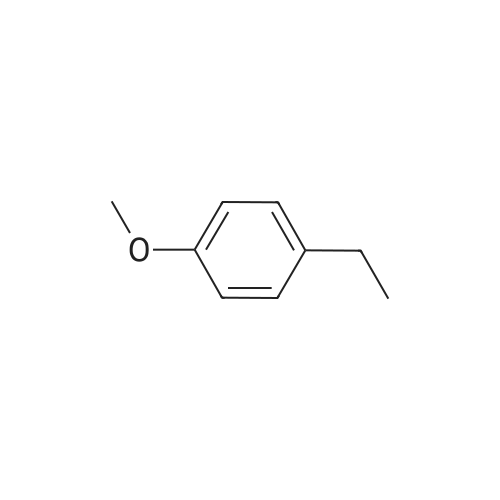
3
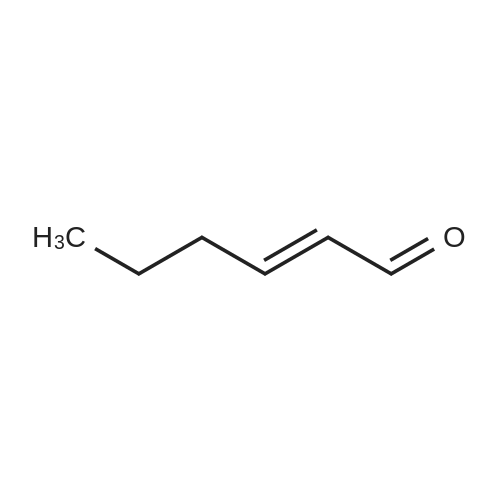
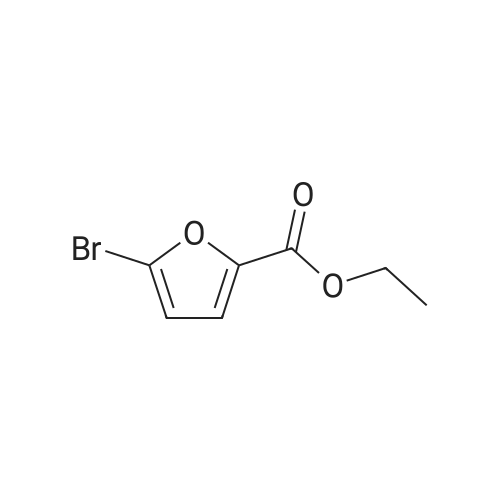
[ 6728-26-3 ]
[ 1129294-89-8 ]
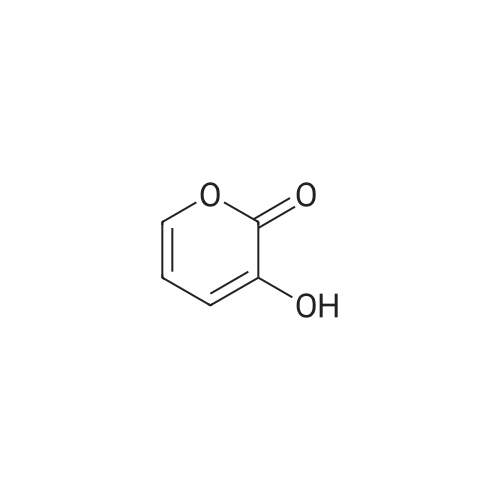
[ 98-01-1 ]
[ 496-64-0 ]
[ 88-14-2 ]
[ 13419-69-7 ]
[ 1401094-48-1 ]
[ 1401094-49-2 ]
[ 1309945-34-3 ]
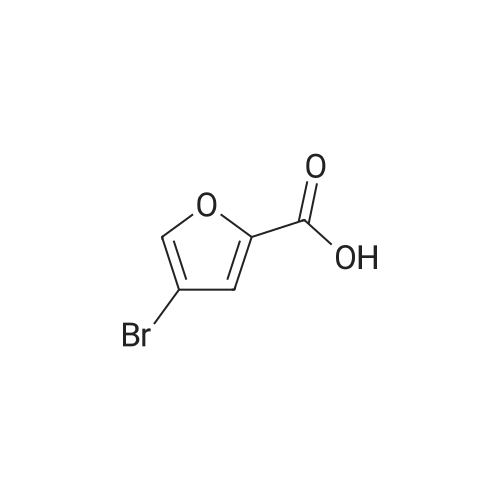
Reference:
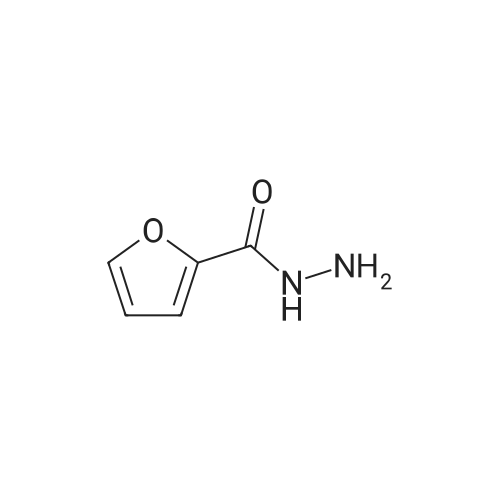
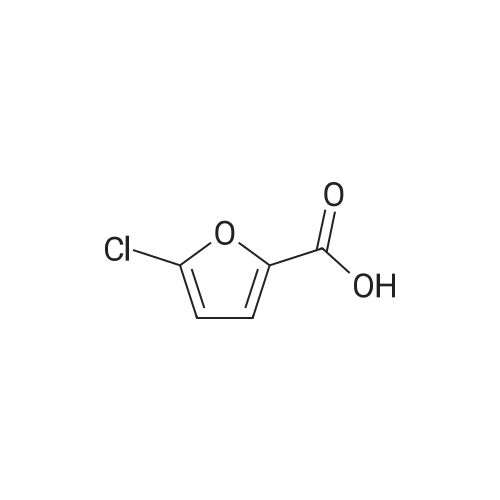
[1] Journal of Agricultural and Food Chemistry, 2012, vol. 60, # 40, p. 9967 - 9973,7
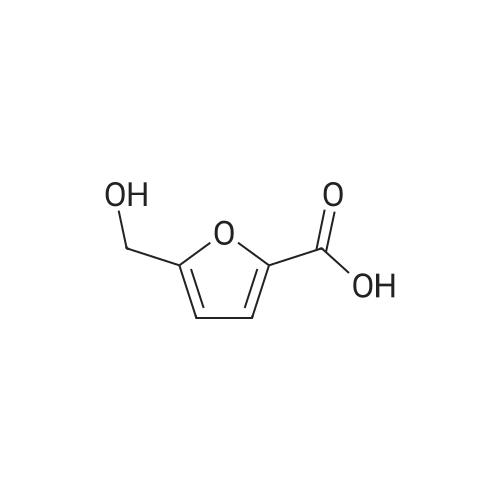
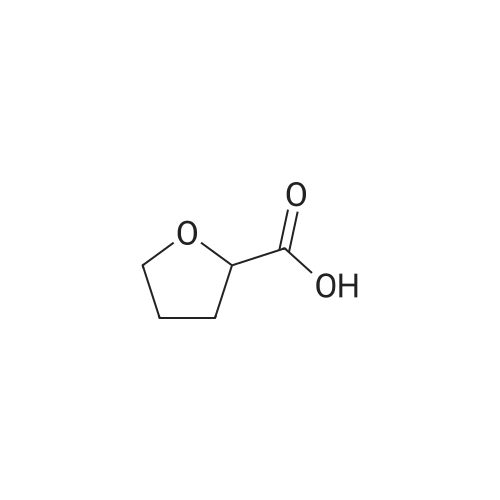
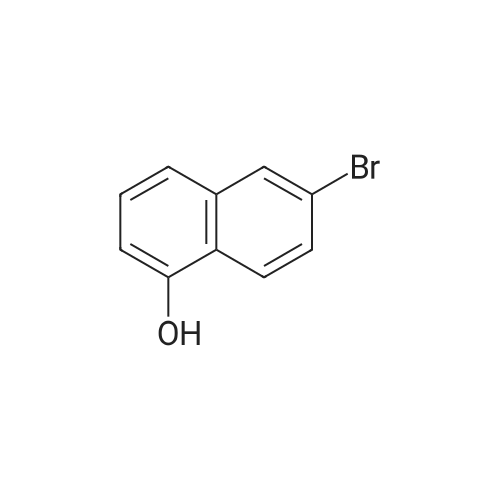
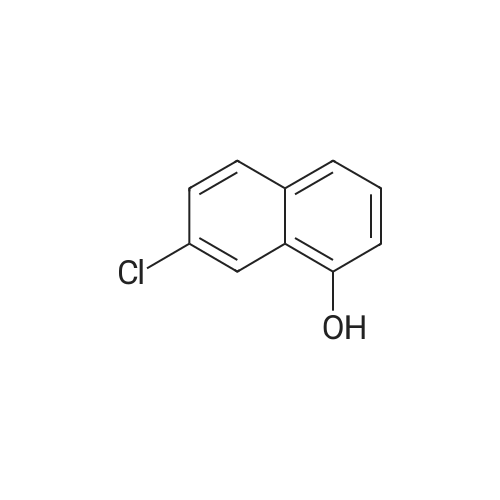
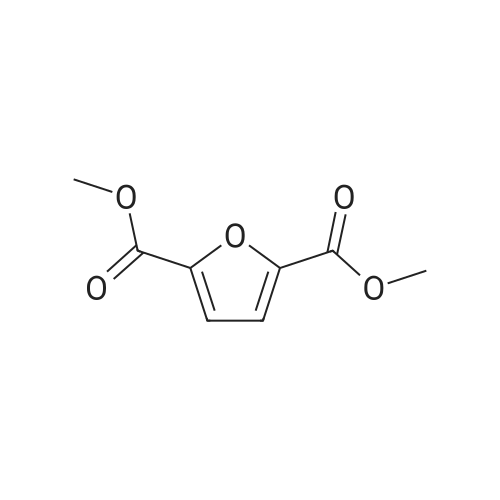
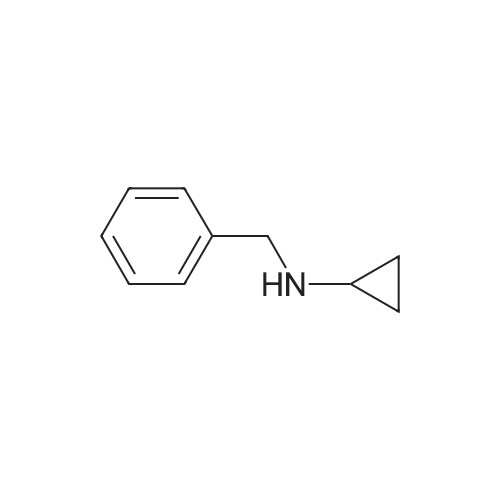
4
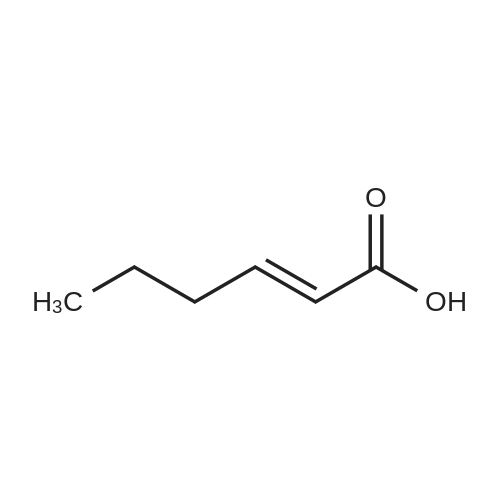
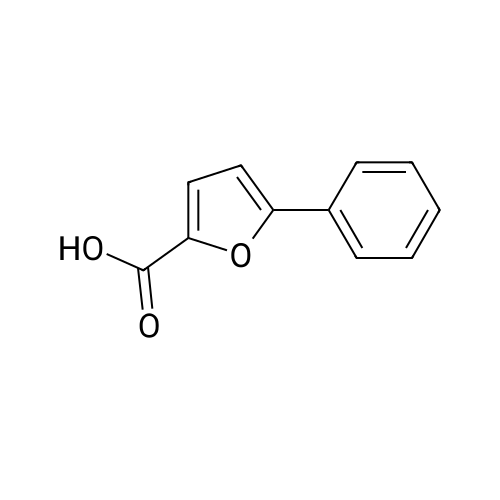
[ 87-73-0 ]
[ 496-64-0 ]
[ 88-14-2 ]
Reference:
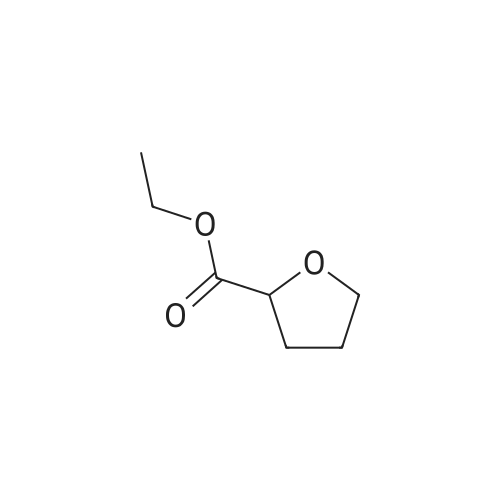
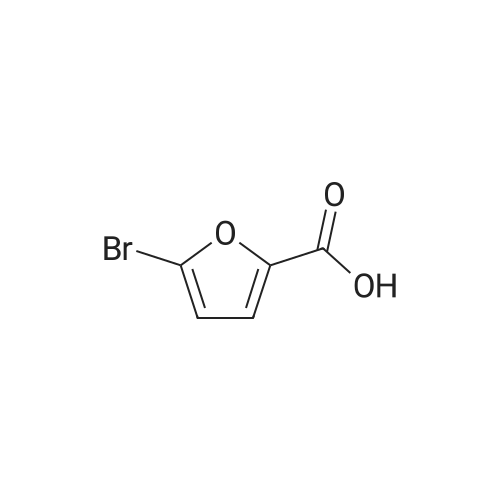
[1] Annales de Chimie (Cachan, France), 1904, vol. <8> 3, p. 548[2] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1903, vol. 137, p. 993
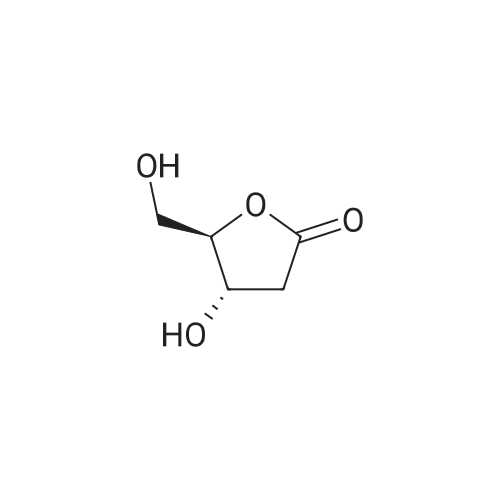
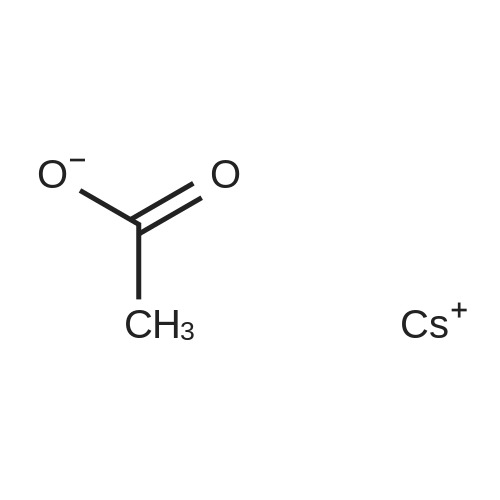
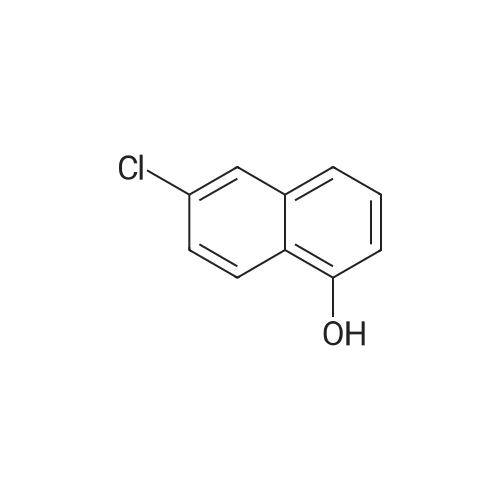
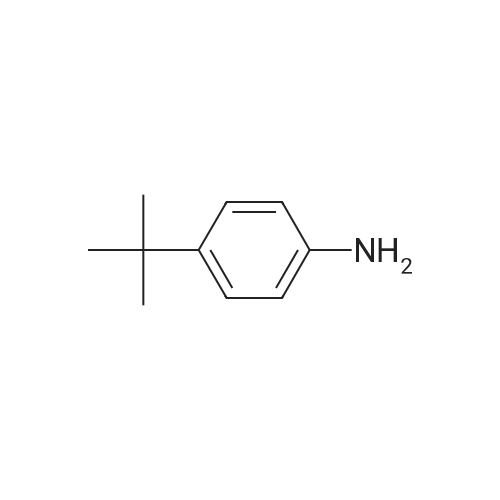
5
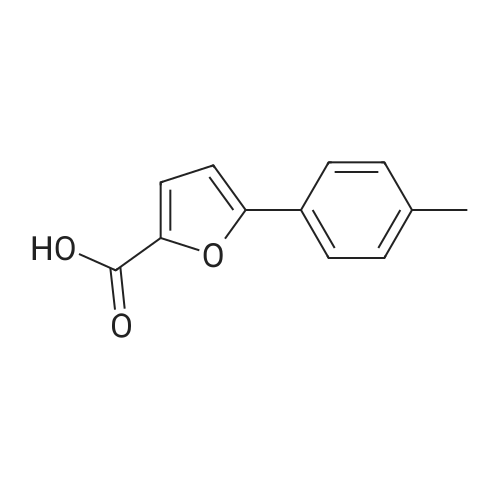
[ 15909-67-8 ]
[ 496-64-0 ]
[ 88-14-2 ]
Reference:
[1] Annales de Chimie (Cachan, France), 1904, vol. <8> 3, p. 548[2] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1903, vol. 137, p. 993
6
[ 50-81-7 ]
[ 496-64-0 ]
[ 88-14-2 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2012, vol. 60, # 42, p. 10696 - 10701
7
[ 88-14-2 ]
[ 16874-33-2 ]
Reference:
[1] Monatshefte fur Chemie, 2000, vol. 131, # 12, p. 1335 - 1343
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1994, # 20, p. 2931 - 2942
[3] Chemische Berichte, 1913, vol. 46, p. 1929
[4] Yakugaku Zasshi, 1925, # 515, p. 1[5] Chem. Zentralbl., 1925, vol. 96, # I, p. 2377
[6] Journal of the American Chemical Society, 1923, vol. 45, p. 3041
[7] Kogyo Kagaku Zasshi, 1954, vol. 57, p. 149,150[8] Chem.Abstr., 1955, p. 1484
[9] Journal of the Chemical Society, 1945, p. 52[10] Journal of the American Chemical Society, 1947, vol. 69, p. 3002,3003
[11] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1939, vol. 208, p. 359
[12] Catalysis Science and Technology, 2017, vol. 7, # 24, p. 5982 - 5992
8
[ 88-14-2 ]
[ 108-59-8 ]
[ 611-13-2 ]
Yield
Reaction Conditions
Operation in experiment
90%
With potassium bromide In N,N-dimethyl-formamide at 130℃; for 12 h; Schlenk technique
General procedure: To a Schlenk tube equipped with a magnetic stir bar were added under air, carboxylic acid (0.3 mmol), dimethyl malonate (1.8 mmol) and KBr (0.09 mmol) in DMF (2 mL). The resultant reaction mixture was kept stirring at the required temperature for 12 h. After indicated reaction time, the mixture was cooled down to room temperature. It was poured into ethyl acetate, then washed with water, extracted with ethyl acetate, dried by anhydrous Na2SO4, then filtered and evaporated under vacuum, the residue was purified by flash column chromatography (petroleum ether or petroleum ether/ethyl acetate) to afford the corresponding coupling products with high purity.
Reference:
[1] Journal of Organic Chemistry, 1995, vol. 60, # 26, p. 8336 - 8340
10
[ 67-56-1 ]
[ 526-99-8 ]
[ 88-14-2 ]
[ 611-13-2 ]
Yield
Reaction Conditions
Operation in experiment
9.6 %Chromat.
With hydrogen; methyltrioxorhenium(VII) In methanol at 200℃; for 1 h;
Example 1 Catalytic dehydroxylation of galactaric acid (I) for the production of compounds II- VII. Galactaric acid (1.0 g, 4.76 mmol), methyl trioxo rhenium (0.12 g, 0.47 mmol, 10 molpercent) and methanol (10 ml) were charged in the reaction vessel. The reaction vessel was pressurised with hydrogen and heated up to the reaction temperature (Table 1). After the indicated reaction time the mixture was cooled down to room temperature, any solid precipitate was filtered, washed with methanol (5 ml) and dried. The solvent fraction was concentrated in a rotary evaporator. Purification was by flash silica column chromatography. The different fractions were analysed with GC-FID, GC-MS and NMR.
General procedure: Carboxylic acid (0.5 mmol, 1.0 equiv), K2CO3 (0.5 mmol, 1.0 equiv),and CaCl2 powder (0.5 mmol, 1.0 equiv) were added to a 25-mL tube.10percent CuCl2·2 H2O in DMSO (2.0 mL) was added, followed by the dropwise addition of 30percent aq H2O2 (1.5 mmol, 3.0 equiv). The tube was sealed with a Teflon-lined cap and the mixture was stirred at 80 °C under O2 for 15 h. The mixture cooled and water (10 mL) was added; the mixture was extracted with EtOAc (3 × 10 mL). After evaporationof the solvent, the residue was purified by column chromatography(silica gel) to obtain the product.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 19, p. 5213 - 5224
30
[ 88-14-2 ]
[ 585-70-6 ]
Yield
Reaction Conditions
Operation in experiment
86%
With bromine; acetic acid In tetrachloromethane at 60℃; for 24 h;
The starting raw materials furan -2 - carboxylic acid (2 . 50 g, 22.3 mmol) is dissolved in glacial acetic acid (2.0 ml) and carbon tetrachloride (20.0 ml) in the mixed solvent, divided into 3 times for every six hours 20 degree Celsius, 5 minutes is slowly added bromine (0.8 ml, 13.3 mmol), keeping the reaction temperature is 60 degrees centigrade, the total stirring 24 hours, concentrated under reduced pressure to remove the solvent, the hot deionized water washing the solid, filtered and dried to obtain compound 3 (3 . 46 g, 18.1 mmol), yield 86percent.
Reference:
[1] Patent: CN106977476, 2017, A, . Location in patent: Paragraph 0073-0093
[2] Journal of Materials Chemistry A, 2014, vol. 2, # 18, p. 6589 - 6597
[3] Journal of Pharmaceutical Sciences, 1981, vol. 70, # 10, p. 1095 - 1100
[4] Heterocycles, 1994, vol. 38, # 4, p. 759 - 768
[5] Justus Liebigs Annalen der Chemie, 1953, vol. 580, p. 169,187
[6] Bulletin de la Societe Chimique de France, 1946, p. 669,672
[7] Vestnik Moskovskogo Universiteta, 1958, vol. 13, # 5, p. 119,121[8] Chem.Abstr., 1959, p. 12267
[9] Recueil des Travaux Chimiques des Pays-Bas, 1933, vol. 52, p. 352,354
[10] Zhurnal Obshchei Khimii, 1946, vol. 16, p. 751,759[11] Chem.Abstr., 1947, p. 2033
[12] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1985, p. 45 - 52
[13] European Journal of Medicinal Chemistry, 2009, vol. 44, # 11, p. 4726 - 4733
[14] Molecules, 2011, vol. 16, # 6, p. 4897 - 4911
31
[ 88-14-2 ]
[ 585-70-6 ]
[ 32460-04-1 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1886, vol. 232, p. 86
32
[ 88-14-2 ]
[ 7726-95-6 ]
[ 64-19-7 ]
[ 585-70-6 ]
Reference:
[1] Chemische Berichte, 1883, vol. 16, p. 1132
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1974, vol. 39, p. 767,768, 770
41
[ 88-14-2 ]
[ 106-49-0 ]
[ 52938-98-4 ]
Reference:
[1] Kogyo Kagaku Zasshi, 1952, vol. 55, p. 271[2] Chem.Abstr., 1954, p. 3953
[3] Collection of Czechoslovak Chemical Communications, 1974, vol. 39, p. 767,768, 770
[4] Phosphorus, Sulfur and Silicon and the Related Elements, 2007, vol. 182, # 5, p. 1083 - 1091
[5] European Journal of Medicinal Chemistry, 2009, vol. 44, # 2, p. 551 - 557
[6] Molecules, 2010, vol. 15, # 6, p. 4267 - 4282
[7] Chemical Biology and Drug Design, 2012, vol. 79, # 1, p. 121 - 127
[8] Letters in Drug Design and Discovery, 2014, vol. 11, # 7, p. 877 - 885
[9] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 15, p. 3632 - 3635
[10] European Journal of Medicinal Chemistry, 2018, vol. 151, p. 546 - 556
[11] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 19, p. 3276 - 3280
[12] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 19, p. 3271 - 3275
42
[ 88-14-2 ]
[ 53662-83-2 ]
Reference:
[1] Patent: CN106977476, 2017, A,
43
[ 88-14-2 ]
[ 488-11-9 ]
Reference:
[1] Journal of the Chemical Society [Section] C: Organic, 1969, p. 728 - 730
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1985, p. 45 - 52
With aluminum (III) chloride; In chlorobenzene; at 35 - 80℃; for 24h;Catalytic behavior;
In a 250ml four-necked bottle, add 20g of furoic acid, 23.2g of anisole, 100ml chlorobenzene, stirred and mixed; oil bath heated to internal temperature 35 ~ 40 C, slowly added 55.9g anhydrous aluminum trichloride; after the completion of the feed, stirring at 75-80 C for about 24 hours; cooling, concentrated distillation under reduced pressure Chlorobenzene After the distillation was completed, the temperature was lowered to room temperature, 60 ml of N,N-dimethylformamide was added to the reaction flask, and the mixture was stirred and mixed. At room temperature, 32.1 g of anhydrous aluminum trichloride was slowly and separately added; after the completion of the feeding, the oil bath was heated to The internal temperature is 135 C, and the reaction is kept for 6 hours; The water is cooled to room temperature; the reaction solution is slowly added to dilute hydrochloric acid solution for hydrolysis; extracted with ethyl acetate several times, and the ethyl acetate extract is combined, washed with water, and Ph is weakly acidic; The organic layer was concentrated under reduced pressure to recover ethyl acetate; The residue is decolorized and recrystallized with dilute alcohol water. Yielding 19.7 g of 6-hydroxy-1-naphthoic acid as a gray to pale yellow powder. Yield: 58.7%, melting point: 205.5 to 208.6 C, purity: 98.5%.
With diphenyl phosphoryl azide; triethylamine;Reflux;
[00193] Preparation of tert-butylfuran-2-ylcarbamate (?3-2. A solution of furan-2- carboxylic acid (35.0 g, 312.2 mmol), triethylamine (86 mL, 624.5 mmol), and diphenyl phosphorazidate (DPPA) (135 mL, 624.5 mmol) in tert-butyl alcohol (400 rnL) was retluxed overnight. The volatile was removed in vacuo and the residue was purified by chromatography (silica gel, PE to EA:PE (1:30, v:v)) to give compound C3-2 as a white solid (55.0g. yield: 96%). ?H NMR(DMSO-d6, 400 MHz) : 1.43 (s, 9H), 5.91 (s, 1H), 6.37 (t, J 2.6 Hz, IH), 7.26 (s, 1H), 9.81 (s, 1H).
2-[[<i>tert</i>-butylcarbamoyl-(2-iodo-phenyl)-methyl]-(furan-2-carbonyl)-amino]-methyl}-pyrrolidine-1-carboxylic acid <i>tert</i>-butyl ester[ No CAS ]
Commercially available <strong>[2434-03-9]4,5-dibromofuran-2-carboxylic acid</strong> (6.1 g, 22.6 mol) was suspended in 100 ml of ammonium hydroxide and treated portion-wise with zinc dust (1.48 g, 22.6 mol) and stirred at room temperature for a few minutes. The reaction was filtered and the filtrate acidified with 5N HCl and extracted several times with methylene chloride. The extract was washed with brine and concentrated to give 2.93 g of a white solid consisting mainly of 4-bromofuran-2-carboxylic acid. MS (ES-)found: (M-H)-=190.95 and 188.95. NMR (500 MHz, DMSO-D6) delta 13.3 (bs, 1 H), 8.14 (s, 1 H), 7.36 (s, 1 H). Product was contaminated with 25% furan-2-carboxylic acid by-product. NMR (500 MHz, DMSO-D6) delta 13.3 (bs, 1 H), 7.90 (m, 1 H), 7.19 (m, 1 H), 6.64 (m, 1 H).
Preparation F1: 4-phenylfuran-2-carboxylic Acid Preparation F1, Step 1: Synthesis of 4-bromofuran-2-carboxylic acid: Commercially available <strong>[2434-03-9]4,5-dibromofuran-2-carboxylic acid</strong> (6.1 g, 22.6 mol) was suspended in 100 ml of ammonium hydroxide and treated portion-wise with zinc dust (1.48 g, 22.6 mol) and stirred at room temperature for a few minutes. The reaction was filtered and the filtrate acidified with 5N HCl and extracted several times with methylene chloride. The extract was washed with brine and concentrated to give 2.93 g of a white solid consisting mainly of 4-bromofuran-2-carboxylic acid. MS (ES-)found: (M-H)-=190.95 and 188.95. NMR (500 MHz, DMSO-D6) delta 13.3 (bs, 1 H), 8.14 (s, 1 H), 7.36 (s, 1 H). Product was contaminated with 25% furan-2-carboxylic acid by-product. NMR (500 MHz, DMSO-D6) delta 13.3 (bs, 1 H), 7.90 (m, 1 H), 7.19 (m, 1 H), 6.64 (m, 1 H).
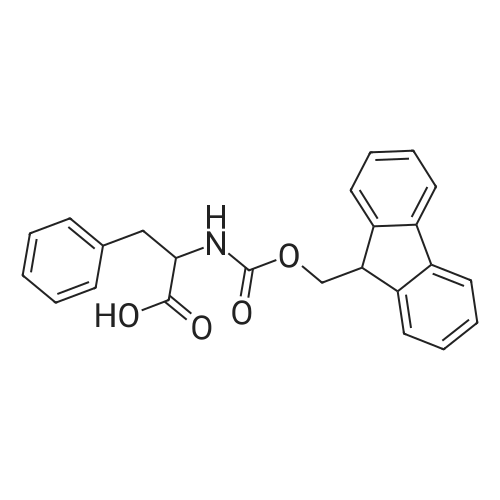
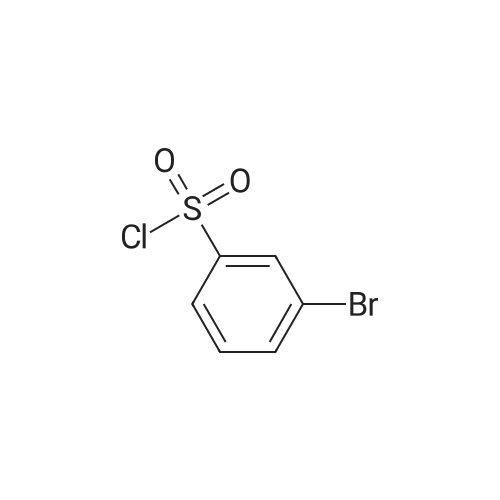
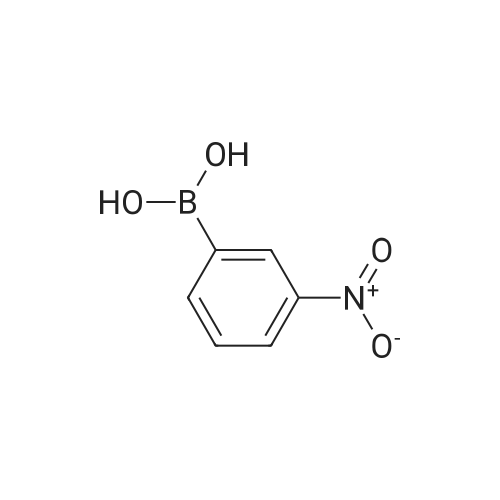
1.2 g of Wang polystyrene resin (Rapp-Polymere, Tuebingen; loading 1.08 mmol/g) are swollen in dimethylformamide (DMF). The solvent is filtered off with suction and a solution of 1005 mg of Fmoc-phenylalanine (amino acid reagent) in 10 ml of dimethylformamide (DMF) is added. After shaking at room temperature for 15 min, the suspension is treated with 345 mul of pyridine and 543 mg of 2,6-dichlorobenzoyl chloride. It is shaken overnight at room temperature. The resin is then washed with dimethylformamide (DMF), methanol and dichloromethane. The resin is treated with 15 ml of a 20% strength piperidine solution dimethylformamide (DMF) and shaken at room temperature for 10 min. It is then washed 3 times with dimethylformamide (DMF) and a further 15 ml of a 20% strength piperidine solution in dimethylformamide (DMF) are added. After shaking for 20 min, it is washed with dimethylformamide (DMF) and tetrahydrofuran (THF). The resin is treated with a solution of 452 ml of diisopropylethylamine in 5 ml of tetrahydrofuran (THF) and a solution of 431 mg of 3-bromobenzenesulfonyl chloride in 5 ml of tetrahydrofuran (THF). It is shaken overnight at room temperature. The resin is then washed with dimethylformamide (DMF), methanol and tetrahydrofuran (THF). The resin is suspended in 7 ml of xylene, treated with 1.08 g of 3-nitrobenzeneboronic acid and a solution of 1.37 g of sodium carbonate in 6 ml of water and shaken for 5 min at room temperature. 227 mg of bis-(triphenylphosphane)-palladium(II) chloride and 170 mg of triphenylphosphane are then added and the mixture is stirred overnight at 85C. The resin is then washed with tetrahydrofuran (THF)/water 1:1, 0.25 M aqueous hydrochloric acid, water, dimethylformamide (DMF), methanol, tetrahydrofuran (THF) and dichloromethane. The resin is treated with a solution of 5.4 g of tin(II) chloride dihydrate in 12 ml of N-methylpyrrolidone (NMP) and shaken overnight at room temperature. The resin is then washed with N-methylpyrrolidone (NMP), methanol, tetrahydrofuran (THF) and dichloromethane. The resin is treated with a solution of 1.45 g 2-furanyl-carboxylic acid (acid reagent) in 20 ml dimethylformamide (DMF). After shaking for 1 minute a solution of 2.64 ml diisopropylcarbodiimide in 5 ml dimethylformamide (DMF) is added and the mixture is shaken for 3 hours at room temperature. The resin is then washed with dimethylformamide (DMF) and is treated with 1.45 g 2-furanyl-carboxylic acid in 20 ml dimethylformamide (DMF) and 2.64 ml diisopropylcarbodiimide in 5 ml dimethylformamide (DMF) again. After shaking for 3 hours the resin is washed with dimethylformamide (DMF), methanol, tetrahydrofurane (THF) and dichloromethane. For removal of the product, the resin is shaken with 10 ml of trifluoroacetic acid (TFA)/dichloromethane 1:1 for 1 hour, filtered off. The filtrate is concentrated in vacuo and purified on silica gel. 201 mg of the title compound are obtained. Mass spectrometry (ESI): 491. Retention time (HPLC): Rt = 9,6.1H-NMR (400 MHz, methanol) delta = 7,99 (s, 1H), 7,91 (s, 1H), 7,81 (d, 1H), 7,75 (m, 2H), 7,66 (m, 1H), 7,52 - 7,43 (m, 2H), 7,41 (d, 1H), 7,29 (d, 1H), 7,11 (s, 5H), 7,68 (m, 1H), 4,10 (dd, 1H, J =5,6 Hz, J = 10,8 Hz, H-2), 3,06 (dd, 1H, J = 5,6 Hz, J = 13,8 Hz, H-3a), 2,85 (dd, 1H, J = 10,8 Hz, J = 13,8 Hz, H-3b).
furan-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-yl-methyl]-amide[ No CAS ]
furan-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
furan-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
furan-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
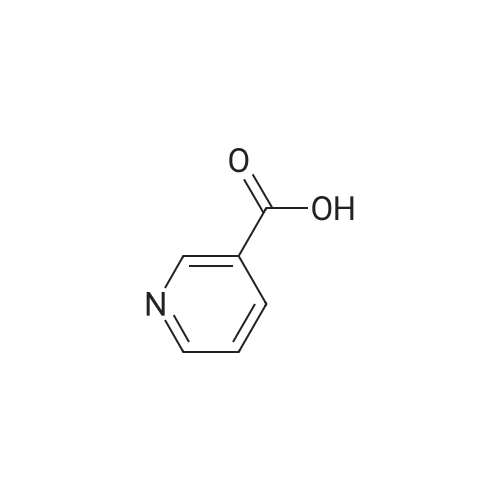
N-[4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-nicotin amide[ No CAS ]
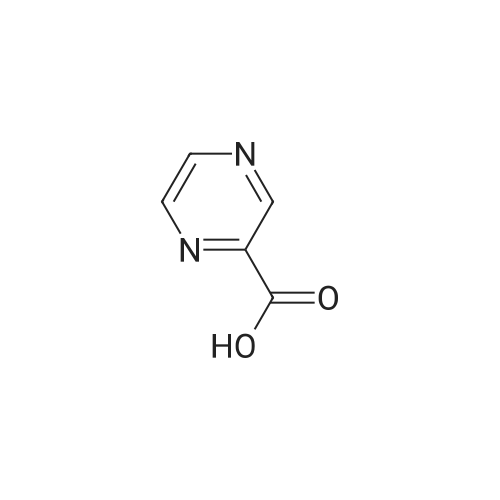
pyrazine-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
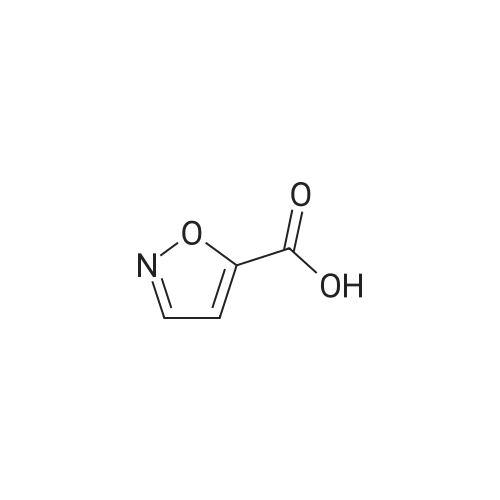
isoxazole-5-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
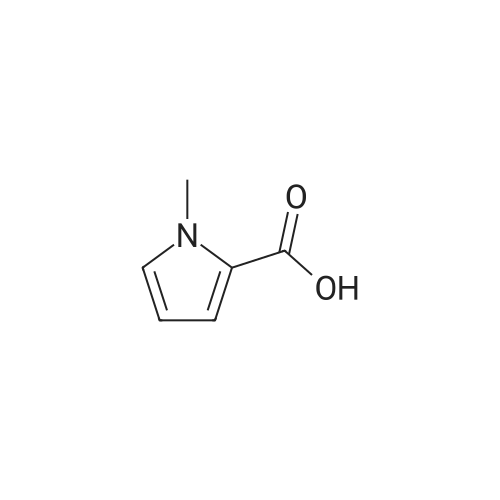
1-methyl-1H-pyrrole-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
isoxazole-5-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
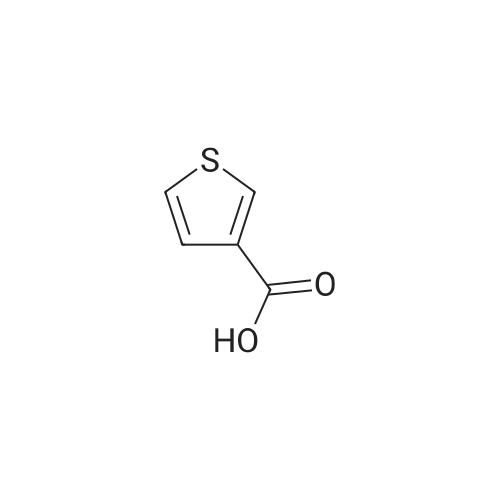
thiophene-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
N-[4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-nicotinamide[ No CAS ]
pyrazine-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
thiophene-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
1-methyl-1H-pyrrole-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
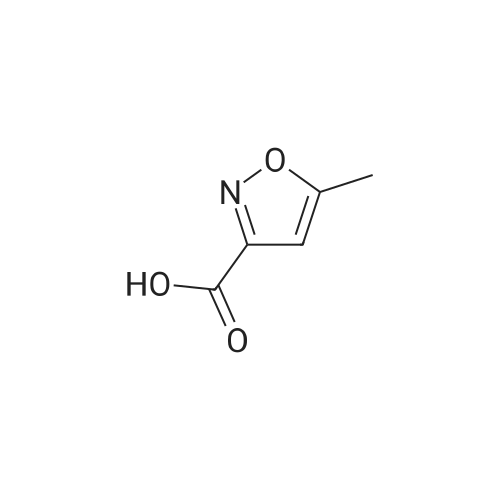
5-methyl-isoxazole-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
5-methyl-isoxazole-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
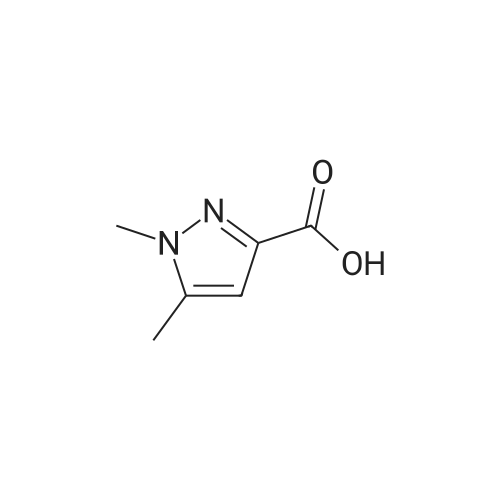
1,5-dimethyl-1H-pyrazole-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
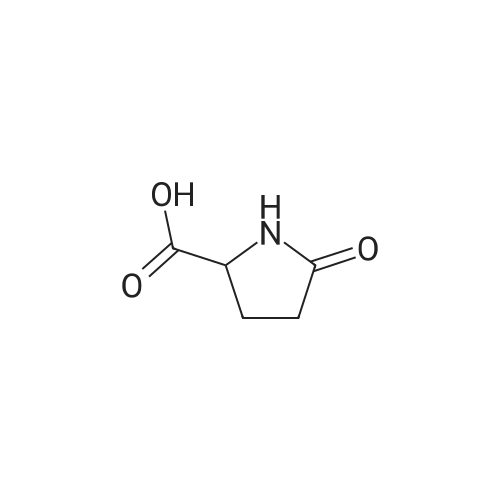
5-oxo-pyrrolidine-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
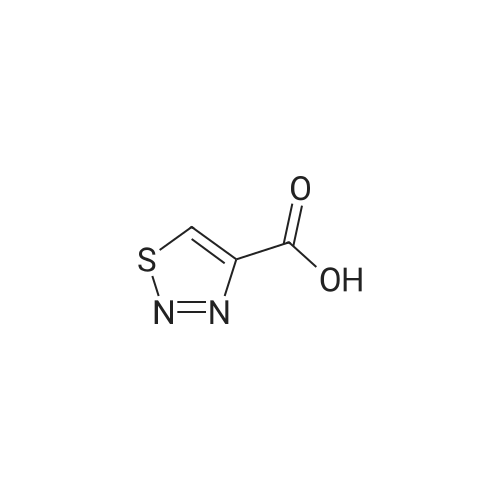
[1,2,3]-thiadazole-4-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
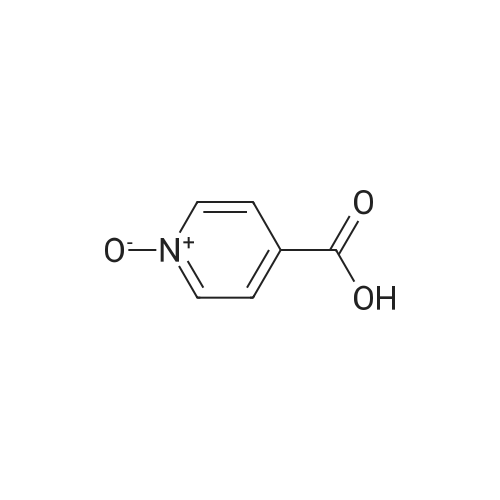
N-[4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-1-hydroxyisonicotin amide N-oxide[ No CAS ]
tetrahydro-furan-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
tetrahydro-furan-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
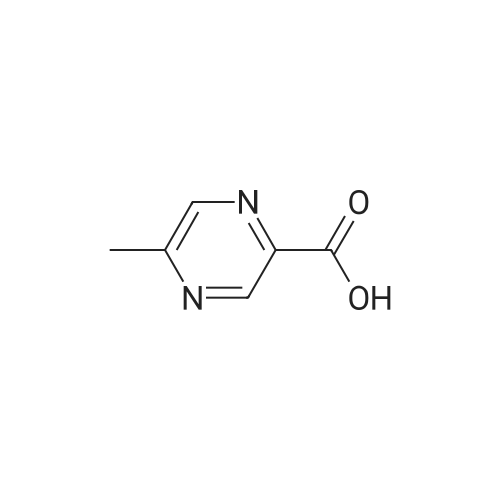
5-methyl-pyrazine-2-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
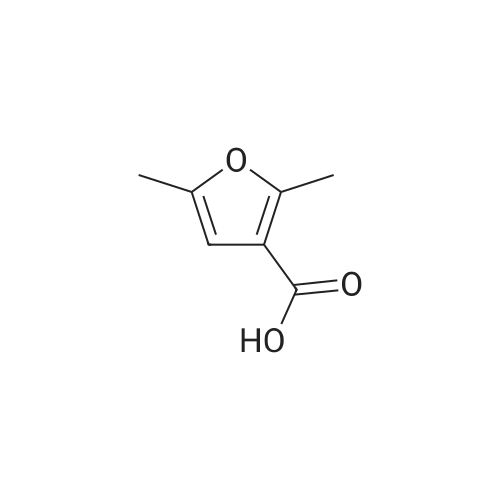
2,5-dimethyl-furan-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-3-ylmethyl]-amide[ No CAS ]
2,5-dimethyl-furan-3-carboxylic acid [4'-(1,1-dimethyl-6-morpholin-4-yl-6-oxo-hexyl)-2'-hydroxy-biphenyl-2-ylmethyl]-amide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Compounds 41-70 were part of a parallel set prepared in library plate format according to General Procedure L, outlined below. ; L. General Procedure for Plate Preparation-Amide Formation XXI: Resin bound deprotected biarylphenol XVII (prepared from intermediate XII, boronates XIVd and XIVe, following general procedures D-F) was distributed into a 96 well plate, 10 mg of resin (0.013 mmol) per well. To the resin 400 mul of dichloromethane was added, followed by 100 mul of DIEA, followed by 0.13 mmol (10 equiv) of heterocyclic carboxylic acid XXa-XXn was added followed by 61 mg (0.13 mmol, 10 equiv) of PyBrop. The plate was shaken at room temperature for 24 hours, then drained and washed with dichloromethane, methanol/dichloromethane, dimethylformamide, methanol/dichloromethane and dichloromethane. The compounds were cleaved with TFA/dichloromethane (600 mul, 1:1) into a 96 deep well plate and submitted for testing without further purification. (Mass spec results obtained are shown in Table 4). Carboxylic Acids Het-COOH XX:
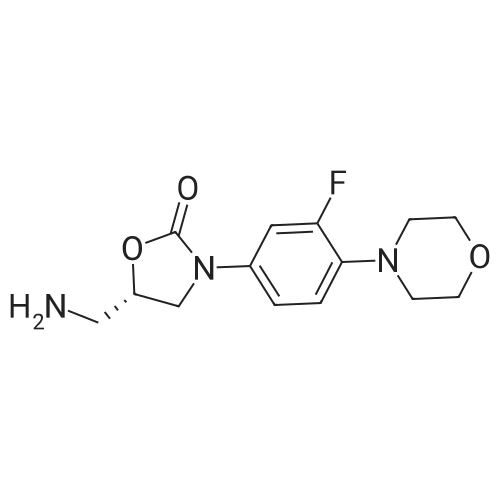
(S)-N-((3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)furan-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 12.5h;
Example 1: N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)-phenyl]-2-oxo-5-oxazolidinyl]methyl]furan-2-yl-amide,QftN"qNOHN .HO F 0CH, _. NHNHA solution of 57 m§ (1.5 eq) of 2-furanoic acid, 21 m§ (0.5 eq) of 4-(dimethylamino)pyridine (DAAAP), 97 m§ of 1-ethyl-3-(3'-dimethylaminopropyl)carbodiimide hydrochloride (EDCI.HCl, 1.5 eq) in 5 ml of dichloromethane (DCM) was stirred at room temperature under argon for 30 minutes. Then, 100 m§ (1 eq) of N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)-phenyl]-2-oxo-5-oxazolidinyl] methyljamine were added in 5 ml of DCM and stirring was continued for 12 hours when complete conversion of the starting amine was observed by TLC. The crude mixture was washed with 5% HOAc solution, saturated NaHCOs and brine. The combined organic layers were dried (MgSCXt) and concentrated in vacuum to afford 125 mg of N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)-phenyl]-2-oxo-5-oxazolidinyl]methyl]furan-2-yl-amide (Yield = 95%).1H NMR (400 MHz, 6, ppm, CDC13): 3.05 (4H, m), 3.79 (2H, m), 3.86 (m,5H), 4.05 (1H, t, J = 8.8 Hz), 4.84 (1H, m), 6.49 (1H, dd, J = 4.5 Hz), 6.81(1H, t, J = 5 Hz), 6.93 (1H, t, J = 6.6 Hz), 7.06 (1H, m), 7.12 (1H, dd, J = 3.2,0.8 Hz), 7.4 (1H, m), 7.44 (1H, m).HPLC (t, %): 6.99 min, 99%.MS(ESI)m/z = 390(M+1)
1-(3-dimethyl amino propyl)-3-ethyl carbodiimide hydrochloride[ No CAS ]
[ 92-36-4 ]
[ 80029-43-2 ]
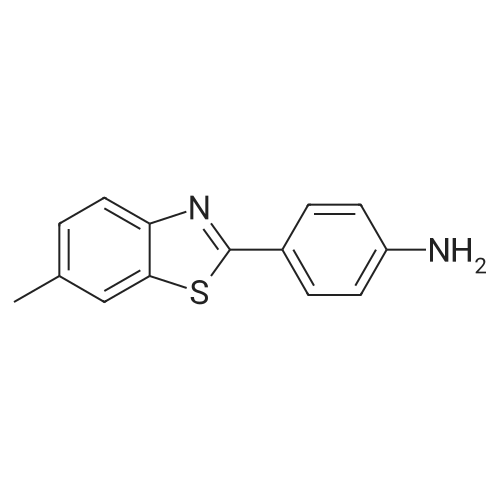
furan-2-yl-N-[4-(6-methylbenzothiazol-2-yl)phenyl]carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In hydrogenchloride; dichloromethane;
EXAMPLE 3 2-Furyl-N-[4-(6-methyl-benzothiazol-2-yl)phenyl]carboxamide First Route: 2-Furoic acid (1.85 gms, 16.5 mmole) was dissolved in anhydrous methylene chloride (30 mls), to which solution was added a suspension of 2-(4-amino-phenyl)-6-methyl benzothiazole (4.76 g., 16.5 mmole) and N-methyl morpholine (2.0 g., 16.5 mmole) in methylene chloride (30 mls, at room temperature). Then, 1-hydroxy-benzotriazole hydrate (2.67 g., 16.5 mmole) and 1-(3-dimethyl amino propyl)-3-ethyl carbodiimide hydrochloride (4.75 g., 16.5 mmole) were added at room temperature. More methylene chloride (20 ml.) was added with stirring at room temperature, and the reaction maintained overnight. The initial clear reaction solution changed to a turbid solution. More methylene chloride (10 ml.) was added to the product mixture, which was then extracted with IN HCl to separate a solid. The solid was filtered and washed with water. The product solid was crystallized from large amount of MeOH to yield 2.19 gm. (33.1%). mp. 238-240 C. 1H and 13C NMR were consistent with the expected product. TLC showed one spot (5% MeOH-CH2Cl2 as developing solvent on silica gel plate). Route 2: 2-(4-aminophenyl)-6-methyl benzothiazole (2.0 gm, 8.3 mmole) and 2-Furoyl chloride (1.086 gm., 8.32 mmole) were suspended in methylene chloride (30 ml, anhydrous). triethylamine (1.24 gm., 12.25 mmole) was added to the reaction mixture with stirring at room temperature for 2 days. (pH 7.0-7.2). Methylene chloride (50 ml) was added to the reaction mixture, and the reaction mixture extracted with 1 N HCl (50 ml) to separate a solid. The solid was filtered and washed with water to yield 1.3 gm. (46%) of the desired compound. The product was crystallized from MeOH to obtain 0.99 gm. mp. 238-240 C. 1H and 13C NMR were consistent with the expected product. TLC showed one spot (5% MeOH-CH2Cl2 as developing solvent on silica gel plate).
With hydrogenchloride; 1,1'-carbonyldiimidazole; In tetrahydrofuran; methanol;
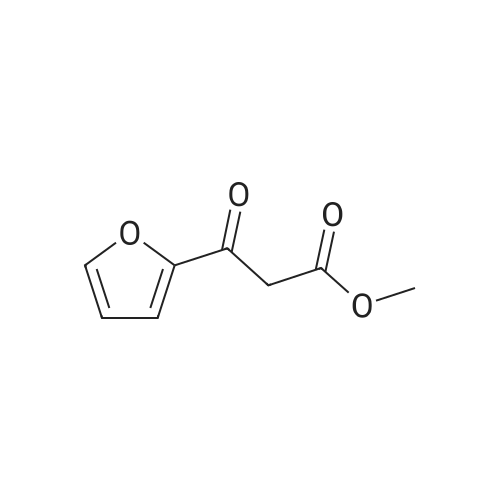
Example 141 Synthesis of Compound 141 In 100 mL of methanol, was dissolved 5.90 g (50.0 mmol) of monomethyl malonate, and the resulting solution was mixed with 2.86 g (25 mmol) of magnesium ethoxide followed by stirring at room temperature for 4 hours. The reaction solution was concentrated and the residue was dried under reduced pressure. In 100 mL of tetrahydrofuran, was dissolved 2.80 g (25 mmol) of 2-furancarboxylic acid and the resulting solution was mixed with 4.45 g (27.4 mmol) of carbonyldiimidazole, followed by stirring for 1 hour. The reaction solution was added to a dry magnesium salt and stirred at room temperature for 19 hours. The reaction solution was concentrated; the residue was mixed with 100 mL of 1.5 N hydrochloric acid, extracted with ethyl acetate, and washed with an aqueous sodium hydrogencarbonate solution and saturated sodium chloride water. The resultant was dried over sodium sulfate anhydride, filtered, and concentrated. The residue was distilled (0.06 mmHg, 69 to 75) to give 2.93 g (17.4 mmol, 69.6%) of methyl 2-furancarbonylacetate as an anhydrous liquid.
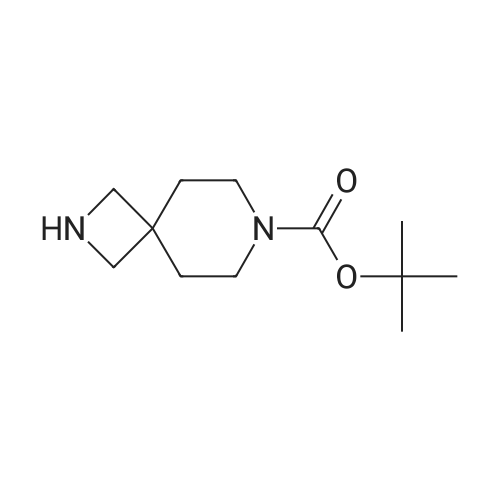
Example 12: Synthesis of 2-(furan-2-ylcarbonyl)-2,7-diazabicyclo[3.3.0]octane; The following procedures are exemplary of those used to produce various 2- (heteroarylcarbonyl)-2,7-diazabicyclo[3.3.0]octanes.To a solution of furan-2-carboxylic acid (0.028 g, 0.25 mmol) in anhydrous THF (2.5 mL) was added DCC (0.064 g, 0.31 mmol) and HOBt (0.041 g, 0.31 mmol). This mixture was stirred at ambient temperature for 10 minutes, and then a solution of racemic tert-butyl 2,7-diazabicyclo[3.3.0]octane-7-carboxylate (commercially available) in THF (1 mL) was added. The reaction mixture was stirred at ambient temperature for 16 h. The solids were removed by filtration, and the residue was purified by reverse phase HPLC to give tert-butyl 2-(furan-2-ylcarbonyl)-2,7-diazabicyclo[3.3.0]octane-7-carboxylate (0.024 g, 32% yield) as colorless syrup. This material was dissolved in 1 :1 mixture of trifluoroacetic acid and dichloromethane (1 mL) and shaken at ambient temperature for 1 h. The volatiles were removed under reduced pressure, and the residue was dried overnight at high vacuum, to give 0.012 g of 2-(furan-2-ylcarbonyl)-2,7-diazabicyclo[3.3.0]octane as a solid (50% yield).
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; triethylamine; In N,N-dimethyl-formamide; at 20℃; for 24h;
Triethylamine (3.91 rnL, 28.0 mmol) and Z4 (1.0 g, 5.61 mmol) are added successively to furan-2-carboxylic acid (1.32 g, 11.79 mmol) and TBTU (3.79 g, 11.79 mmol) in anhydrous DMF (5 mL) and the mixture is stirred for 24 h at RT. The reaction mixture is in poured into 1 N HCl: MeOH = 1 : 1, the precipitate is suction filtered and digested with iPrOH. Yield: 1.60 g (78 percent).Alternatively CH2Cl2 may be used as solvent. If no crystalline product is obtained, the reaction mixture is worked up by extraction and the residue is optionally chromatographed.
To a 0 C. solution of furan-2-carboxylic acid (1.1 g, 9.81 mmol, 1.3 eq.) in dry DMF (20 mL) were added EDC (1.88 g, 9.81 mmol, 1.3 eq.) and HOBt (1.32 g, 9.81 mmol, 1.3 eq.). The mixture was stirred at room temperature for 20 minutes and 1 (1.1 g, 9.81 mmol, 1.0 eq.) was added. The reaction was stirred at room temperature for 16 hours. The mixture was diluted with ethyl acetate and washed first with water and then with a saturated aqueous NaHCO3 solution, the organic extracts were dried over anhydrous magnesium sulphate and were evaporated under reduced pressure. The resulting crude product was purified by flash chromatography on silica gel, eluting with cyclohexane-ethyl acetate (8:2 to 1:1) to afford 11 (1.8 g, 81%) as a colorless gum.
To a well ground intimate mixture of triphenyl phosphine(1.1 mmol) and carboxylic acid, 1 (1.0 mmol) in a microwave vial (10 mL) equipped with a magnetic stirring bar, the organic azide, 2 (1.0 mmol) was added in drops while stirring. Stirring was continued until liberation of nitrogen ceased and the reaction vessel was sealed with a septum. It was then placed into the cavity of a focused monomode microwave reactor (CEM Discover, benchmate) and operated at 1800C (temperature monitored by a built-in IR sensor), power 180W for 15 min. The reaction temperature was maintained by modulating the power level of the reactor. Alternatively, the microwave vial with the mixture was conventionally heated around 180oC by immersing in a preheated glycerol bath for 15 minutes. The reaction vessel was then cooled to room temperature and the residue subjected to column chromatography on silica(pet ether-ethyl acetate). This resulted in the isolation of pure amide, 3 from the by-product viz. triphenylphosphine oxide. Yield of the amide was good and nearly same in the Micro-wave accelerated and Glycerol Bath accelerated protocols.
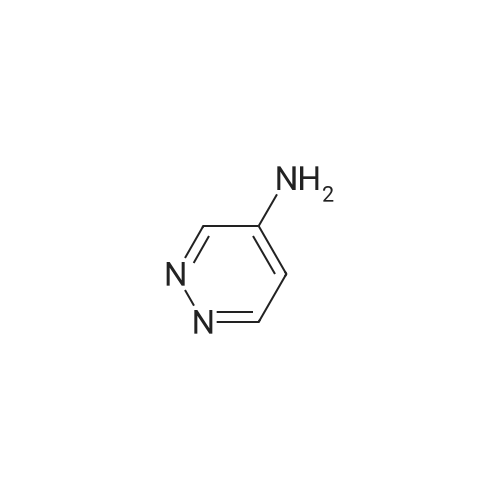
Furan-2-carboxylic acid pyridazin-4-ylamide (example 11.31 )2.00 g (17.8 mmol) of furan-2-carboxylic acid were suspended in 25 ml. of toluene and one drop of dimethylformamide was added to the mixture. 1 .95 ml of thionylchlo- ride (26.8 mmol) were added at room temperature and the reaction mixture was stirred at 80 C for two hours. After removal of the solvent, toluene was added and the evaporation was repeated. The obtained residue was then dissolved in 5 ml of dichloro- methane and the solution was added dropwise to a solution containing 1 .70 g of pyri- dazin-4-yl-amine (17.8 mmol) and 2.73 ml of triethyl amine (19.6 mmol) in 20 ml of di- chloromethane. The mixture was stirred at room temperature for 2 days and washed twice with water. The organic layer was dried over Na2S04, filtered and the solvent was removed under vaccum to give the title compound as a brown solid (1 .9 g, 54%, 95% purity). HPLC-MS: r.t. 1 .146 minutes, m/z [M+H+]189.9
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 0 - 20℃; for 8h;
General procedure: The intermediate (3) (0.5 g, 0.00217 mol), EDCl (0.622 g, 0.00325 mol), DMAP (0.345 g, 0.0028 mol) were stirred in dichloromethane (6 mL) at 0 C, and the substituted acid (0.00217 mol) were dissolved in (4 mL) of dichloromethane and charged to the reaction mixture and stirred at room temperature for 8 h. The reaction completion was monitored by TLC. Reaction was completed. The reaction mixture was diluted with (10 mL) of dichloromethane, and was washed with 10% NaHCO3 (10 mL). Separated the organic layer and was washed with saturated brine solution (10 mL). The organic layer was dried over sodium sulfate and concentrated the organic layer under reduced pressure to afford compounds 4a-t. The spectral data of compounds 4(a-t) are given below.
General procedure: The compounds 1-12 were synthesized usingthe mixed anhydrides method of peptide synthesis(8). Suitable acid (10 mmol) was dissolved in DMF(15 mL) and THF (15 mL) was added. Next, Nmethylmorpholine(10 mmol, 1.1 mL) was addedand the mixture was stirred under nitrogen andchilled to -15OC. Isobutyl chloroformate (10 mmol,1.3 mL) was added dropwise to keep the temperaturebelow -15O. Then, benzylamine (10 mmol) inTHF was added in small portions and the reactionmixture was stirred at -15OC for 30 min and at roomtemperature for 1 h. The solution was concentratedin vacuo and the residue was dissolved in EtOAc (20mL). This solution was washed with 20 mL portionsof 1 M HCl, saturated NaHCO3 solution and saturatedNaCl solution, then dried with anhydrousMgSO4, filtered and concentrated in vacuo. Theobtained compounds were purified by crystallization.All stages of synthesis were controlled by TLC.The purity of the final compound was determined byHPLC and identity by 1H NMR. The compoundsobtained and tested are showed in Figure 1.
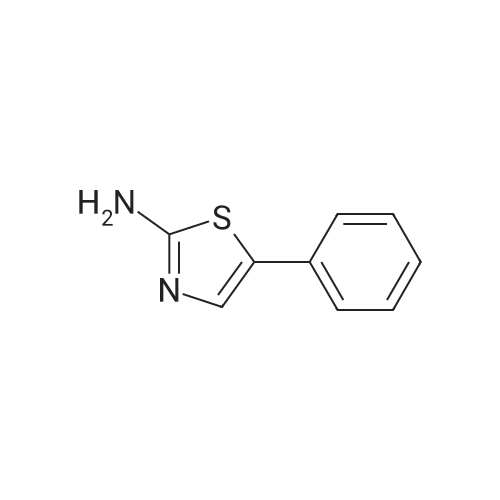
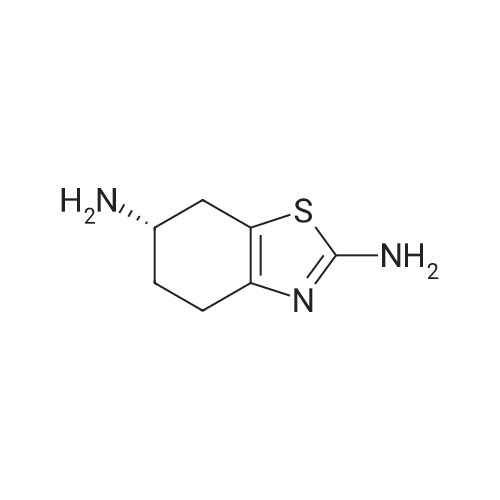
(S)-N-(2-amino-4,5,6,7-tetrahydrobenzo[d]thiazol-6-yl)furan-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
General procedure: To a solution of carboxylic acid (1 mmol) in CH2Cl2 (10 ml) were added Et3N (2 mmol) and TBTU (1.1 mmol) and the mixture was stirred at room temperature for 15 min. Then amine 1 or 3 (1 mmol)and Et3N (2 mmol) were added and the reaction mixture was stirred at room temperature for 2.5 h. The reaction mixture was diluted with CH2Cl2 (15 ml) and washed with saturated aqueous NaHCO3 solution (2x15 ml). Combined water phases were extracted with CH2Cl2 (1 20 ml). Combined organic phases weredried over Na2SO4, filtered and the solvent removed under reduced pressure.
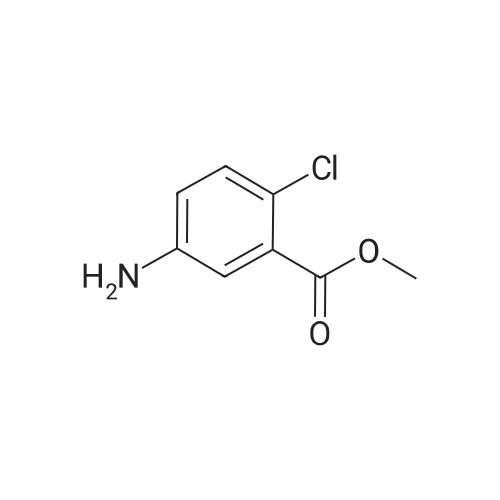
methyl 2-chloro-5-(furan-2-carboxamido)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In dichloromethane;
Step A In a 500 mL round bottom flask, was transferred <strong>[42122-75-8]methyl 5-amino-2-chlorobenzoate</strong> (5 g, 27 mmol) in DCM (Volume: 150 mL) to give a yellow solution. Furan-2-carboxylic acid, (3.78 g, 33.78 mmols) was then added followed by HATU (12.83 g, 33.78 mmols) and DIEA (8.71 g, 67.55 mmols). The reaction mixture was stirred overnight. The reaction mixture was then quenched with water, followed by extraction with ethyl acetate (3*100 mL). The organics were combined, dried over MgSO4 and purified by column chromatography (0-100 EtOAC/Hex) yielding 5.68 g (20.35 mmols, 75%) of methyl 2-chloro-5-(furan-2-carboxamido)benzoate (I-1a).
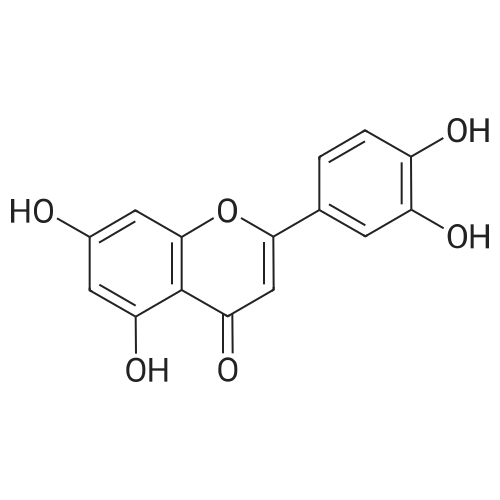
The luteolin1(500mg, 1.74mmol) placed in 50ml round bottom flask, and dissolved in 20 ml of THF, added DMAP (488mg, 4mmol), stirred for 30min at 40 dropwise DCC (537mg, 2.61mmol), stirred for 30min and then added furan - 2- carboxylic acid (430mg, 4.5mmol) the reaction was continued at 40 24h.Join NH4After termination of the reaction Cl saturated solution 10ml, share THF layer, continue with CH2Cl2(10ml × 2) the aqueous layer was extracted, and the combined THF layers CH2Cl2The extract was dried over anhydrous Na2SO4Dried under reduced pressure to recover the solvent residue.The residue (10g) column chromatography through silica gel, chloroform - methanol (220: 1) to give compound5a(Yellow amorphous powder, 80mg, 16% yield) and5b(Yellow amorphous powder, 35mg, 7% yield)
With C21H30ClN2Ru(1+)*Cl(1-); sodium formate; In water; at 60℃; for 12h;Inert atmosphere; Schlenk technique;
General procedure: To a solution of a ruthenium catalyst (0.0168 mmol)in degassed water (8 mL) was added a ketone (0.168mmol) and sodium formate (0.84 mmol). The reactionmixture was kept in a thermostat at 60C. 1 mL of thesolution from the reaction mixture was withdrawn at 1 h,2 h and 12 h; extracted with 5 mL of ethyl acetate;dried over anhydrous sodium sulphate and analyzedby 1H NMR spectroscopy. Conversion was calculatedfrom the 1H NMR spectrum. Chiral alcohols were purifiedby preparative thin layer chromatography. Enantiomericexcess (ee) was determined using the chiralHPLC technique employing the Chiralcel OD column,except in the case of 1-(4-Chlorophenyl)ethanol, 1-(2-Chlorophenyl)ethanol, 1-(4-Nitrophenyl)ethanol), forwhich it was determined using a Chiralcel OJ-Hcolumn.
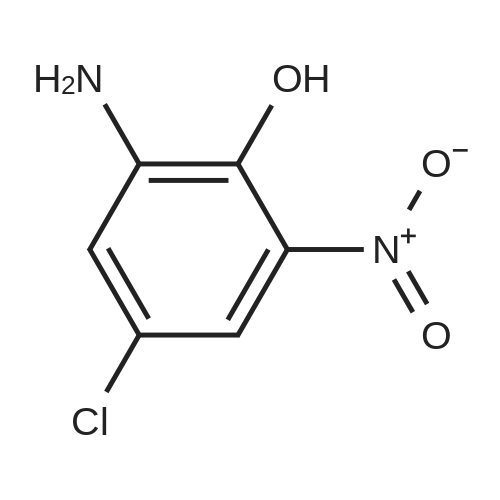
N-(3-chloro-2-hydroxy-5-nitrophenyl)furan-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
97%
To a solution of 2-furoic acid (3.4g, 0.03mol) in DCM (90mL) was added 57 SOCl2 (4.7mL, 0.06mol) dropwise and 4 drops of DMF. The mixture was refluxed for 12h, cooled to room temperature and concentrated in vacuo. The obtained residue was diluted in EtOAc (30mL) and added dropwise to a solution of <strong>[6358-08-3]2-amino-4-chloro-6-nitrophenol</strong> (3.76g, 0.02mol) and Et3N (5.5mL, 0.04mol) in EtOAc (100mL) at 0C. After 2h stirring at room temperature, the mixture was hydrolyzed with water and extracted twice with EtOAc. Combined organic layers were washed with 1M HCl solution, NaHCO3, dried over MgSO4 and concentrated in vacuo. Solid was suspended in H2O/63 EtOH (150/20mL) and NaOH (2.4g, 0.06mol) was added. The mixture was heated at 70C for 3h, cooled to room temperature, acidified with 6M HCl solution up to acid pH, filtered, washed with water and diethyl ether to afford a yellow solid (5.5g, 97%): mp>300C. 1H NMR (300MHz, DMSO-d6): delta 9.73 (br s, 1H), 8.78 (br s, 1H), 8.60 (d, 1H, J= 2.7Hz), 8.14 (d, 1H, J= 2.7Hz), 7.98-7.97 (m, 1H), 7.37 (dd, 1H, J= 0.7Hz and J= 3.5Hz), 6.74 (dd, 1H, J= 1.7Hz and J= 3.5Hz). 13C NMR (DMSO-d6, 75MHz): delta 156.9 (C), 152.8 (C), 147.2 (C), 146.7 (CH), 139.2 (C), 127.6 (C), 122.2 (CH), 121.5 (C), 118.3 (CH), 116.3 (CH), 113.0 (CH). LC-MS (ESI) m/z found: 283 [M+H]+.
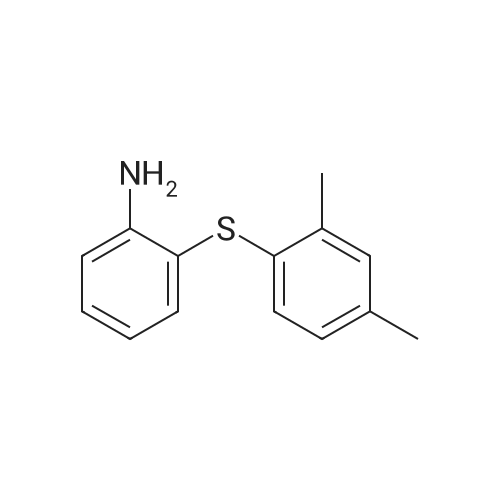
N-(2-(2,4-dimethylphenylthio)phenyl)furan-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With 1,1'-carbonyldiimidazole; In N,N-dimethyl-formamide; for 1h;
General procedure: To a mixture of <strong>[1019453-85-0]2-(2,4-dimethylphenylthio)benzenamine</strong> (5, 8.7 mmol) and acid derivative (10 mmol) inDMF (10 mL) at 0-5 C, was added dropwise coupling agentsolution CDI (17.5 mmol in 5 mL DMF) The reaction mass,allowed to warm at room temperature and stirred for an 1 h(TLC check with hexane/ethyl acetate 9:1). Quenched reaction mass with water (15 mL) and filtered to afford desiredproduct (6i-o).
With 1-hydroxy-7-aza-benzotriazole; N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In dichloromethane; at 20℃; for 12h;
Step 2: To a solution of compound 21a (1 eq.) in dichloromethane, compound 11a (1.5 eq.HOAT (1.5 eq.), HATU (2 eq.), DIPEA (6 eq.), stirred at room temperature for 12 hours.The solvent is then sparged and isolated by direct column chromatography to afford intermediate 21b.
With 1-hydroxy-7-aza-benzotriazole; N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In dichloromethane; at 20℃; for 12h;
Step 1: To a solution of compound 23a (1 eq.) in dichloromethane, compound 11a (1.5 eq.HOAT (1.5 eq.), HATU (2 eq.), DIPEA (6 eq.), stirred at room temperature for 12 hours.The solvent is then sparged off and directly chromatographed to afford intermediate 23b.
With 1-hydroxy-7-aza-benzotriazole; N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In dichloromethane; at 20℃; for 12h;
Step 1: To a solution of compound 24a (1 equivalent) in dichloromethane, compound 11a (1.5 eq.) was added sequentially.HOAT (1.5 eq.), HATU (2 eq.), DIPEA (6 eq.), stirred at room temperature for 12 hours.The solvent is then sparged off and directly isolated by column chromatography to afford intermediate 24b.
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl acetamide;
General procedure: To a solution of corresponding acids (RCOOH, 0.63 mmol), HOBt (170 mg, 1.26 mmol), EDCI (242 mg, 1.26 mmol), and DIPEA (0.208 mL, 1.26 mmol) were added to DMA (60 mL), and then 4 (186 mg, 1.26mmol) were added, the mixture was stirred overnight. The mixture was dissolved in water, then the aqueous solution was extracted with ethyl acetate (50 mL 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by column chromatography to give N-(4-nitrophenethyl)amide (5) in a good yield. And to a solution of 5 in MeOH was added 10% Pd/C under H2 at reflux overnight, then the mixture was dissolved in water, then the aqueous solution was extracted with ethyl acetate (50 mL 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by column chromatography to give N-(4-aminophenethyl)amide (6) in a good yield. Then the mixture of 3 and 6 was added NaCNBH3 in MeOH and the solution was stirred under reflux overnight. After reaction completed, the solvent was removed under reduced pressure and the residue was dissolved in water, the aqueous solution was extracted with ethyl acetate (50 mL 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by column chromatography to afford the corresponding compounds (7-26) in good yields.
With sodium carbonate; at 240℃; under 30003 Torr; for 24h;Sealed tube; Inert atmosphere;
10g of furoic acid,8 ml of methanol and 1 g of sodium carbonate were sequentially introduced into the autoclave.Sealed, nitrogen purged 3 times, carbon dioxide to 4MPa,The reaction temperature is 240 C, and the reaction time is 24 h.The dialkyl 2,5-furandicarboxylate can be obtained.Sodium carbonate was removed by filtration, and excess methanol was distilled from the filtrate.In this embodiment, the conversion of furfural is 100%.The yield of dialkyl 2,5-furandicarboxylate was 91%.
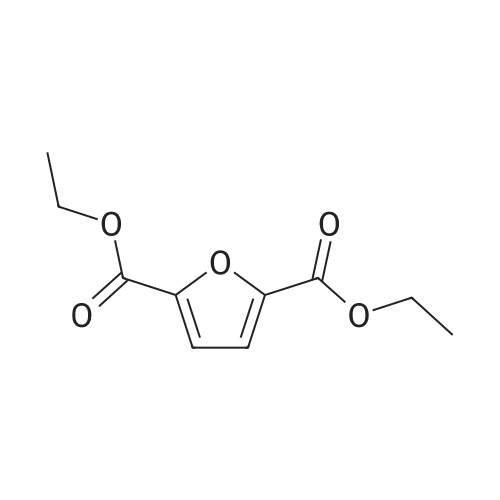
With potassium carbonate; at 240℃; under 7500.75 Torr; for 6h;Sealed tube; Inert atmosphere;
10 g of furoic acid, 11 ml of ethanol and 3 g of potassium carbonate were sequentially introduced into the autoclave, sealed, and purged with nitrogen three times.Charging carbon dioxide to 1 MPa,When the reaction temperature is 240 C and the reaction time is 6 h, a dialkyl 2,5-furandicarboxylate can be obtained.Potassium carbonate was removed by filtration, and excess ethanol was distilled from the filtrate.In the present example, the conversion of furfural was 100%, and the yield of dialkyl 2,5-furandicarboxylate was 93%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping