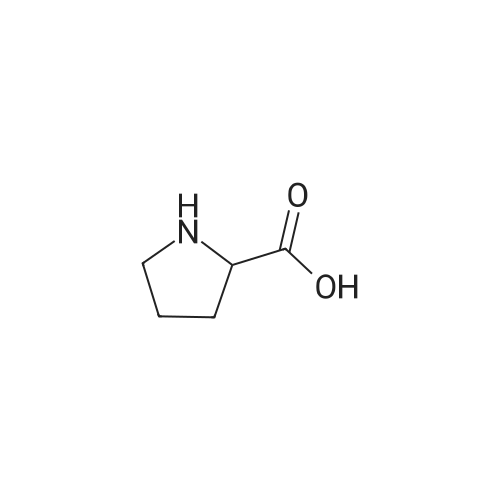
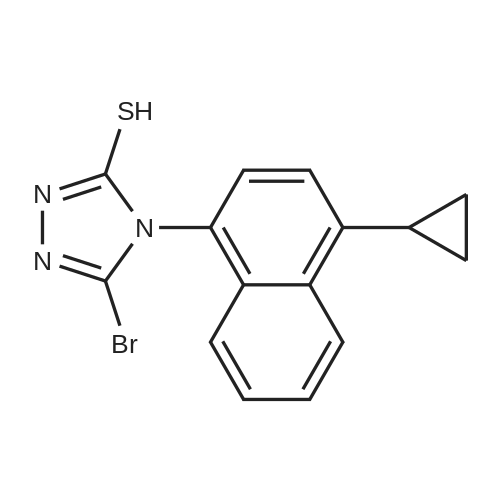
| 92% |
With lithium hydroxide In tetrahydrofuran; ethanol at 0 - 20℃; for 0.75h; |
|
| 90% |
With methanol; sodium hydroxide at 16℃; for 0.333333h; |
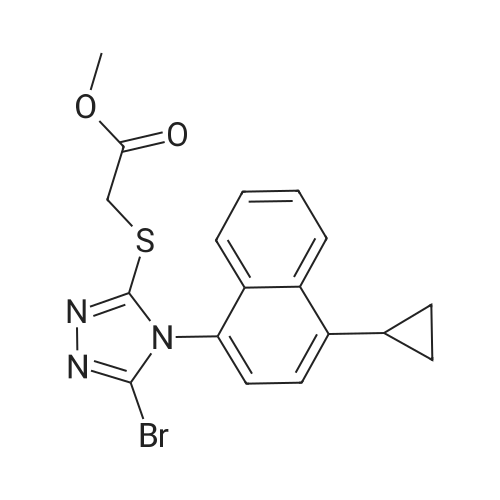
13 Example 13. Preparation of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate
Example 13. Preparation of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate [078] Methyl 2-((5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-yl)thio)acetate (7.0 g), methanol (28 mL) and 1 mole sodium hydroxide solution (20 mL) were placed in a flask, the mixture was stirred at room temperature (16°C) for 20 min, then was added water (50 mL), and 0.5 mol/L of dilute hydrochloric acid was added to the reaction mixture till pH value was 2.5, after the addiction was complete, the mixture was extracted with CH2CI2, the organic phase was combined and CH2CI2 was removed under reduce pressure to obtain yellow solid 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate 6.0 g, yield 90%, LC-Ms: m/z (ESI): 404,406(M+H)+, 1H MR (400 MHz,CDCl3): δ 8.58 (d, J=8.0 Hz, 1 H), 7.71 (m, 1 H), 7.62 (m, 1 H), 7.40 (s, 2 H), 7.24 (d, J=8.4 Hz, 1 H), 4.00 (dd, J=16.0, 25.6 Hz, 2 H), 2.46 (m, 1 H), 1.21 (m, 2 H), 0.9 l(m, 2 H). |
| 90% |
With water; lithium oxide In tetrahydrofuran for 0.75h; Cooling with ice; |
1.6 6) The compound 2 - ((5-bromo-4- (4-cyclopropyl-naphthalen-1-yl) -4H-1,2,4- triazol-3-yl) thio) acetic acid(Lesinuard, I) Synthesis
The intermediate compound VIII (2.7mmol, 1.14g) was dissolved in 10mL of tetrahydrofuran, the ice bath was continued stirring dropwise under stirring an aqueous solution of lithium oxide 45min. Was added 2M hydrochloric pH = 7, was added 20mL of water was distilled off four-fifths of the solvent, continue adjusting pH = 2 ~ 3, the solid was filtered off, and then the precipitated solid was filtered and washed with water at 55-60 of Lesinuard dried in vacuo at a temperature of crude product, then recrystallized, filtered and dried to give the pure product Lesinuard0.99g, 90.0% yield. |
| 89.1% |
Stage #1: 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid methyl ester With water; sodium hydroxide at 20℃; for 24h;
Stage #2: With hydrogenchloride In water |
1.5 (5) Synthesis of Les-09
418 mg of 2-(5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4-hydro-1,2,4-triazol-3-ylsulfanyl)acetic acid AThe ester was placed in a 100 mL flask and 10 mL of a 2 mol/L sodium hydroxide aqueous solution was added. The reaction was carried out at room temperature for 2 h, and 24% hydrochloric acid was added.The pH of the mixture is 3, then it is extracted with 20 mL of ethyl acetate, 10 mL of n-heptane is added, crystallized, filtered, and dried to obtain the product 2-(5-Bromo-4-(4-cyclopropyl-1-naphthyl)-4-hydro-1,2,4-triazol-3-ylsulfanyl)acetic acid 360 mg, 89.1% yield; pureThe degree is 99.6%. |
| 89% |
With sodium hydroxide In tetrahydrofuran |
|
|
With water; sodium hydroxide In toluene at 18 - 25℃; |
5 Compound 5. Example 5: Synthesis of Compound 1 from Compound 11
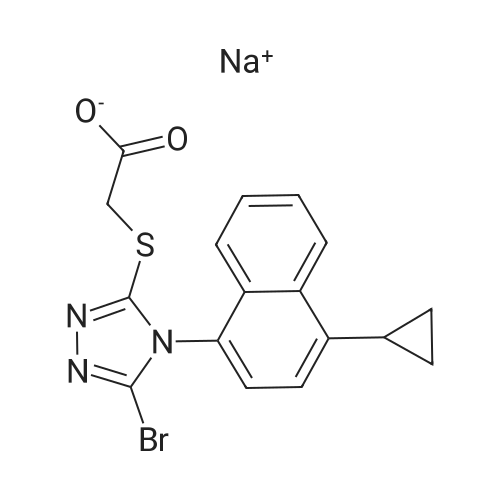
[0340] Compound 11 and methyl bromoacetate were dissolved in DMF and stirred at a temperature between 14 and 22 °C to give Compound 12. The product was isolated by cooling the reaction mixture to a temperature of 10-15 °C followed by an adjustment of the pH with aqueous sodium bicarbonate. The resulting solid, Compound 12, was filtered and was washed with water first, then with cold (0-5 °C) ethyl acetate. [0341] Compound 12 was mixed with copper (II) bromide and potassium nitrite in acetonitrile and stirred at a temperature between 14 and 20 °C until reaction completion. After addition of aqueous sodium hydroxide and citric acid to the reaction mixture, the product, Compound 3, was extracted using toluene and the organic layer was washed several times with aqueous solutions of ammonium hydroxide and sodium citrate to remove copper salts. [0342] Crude Compound 3 in solution underwent base-mediated hydrolysis with the addition of an aqueous sodium hydroxide solution. The biphasic mixture was stirred at 18-25 °C until completion of the ester hydrolysis then the pH of the aqueous layer was adjusted to between 8 and 9 using an aqueous solution of hydrobromic acid. After separation of the two phases, ethyl acetate was added to the aqueous layer, and the pH was adjusted to between 5.15 and 5.35 to extract the product, Compound 1 , into the organic layer. This process was repeated several times until all of Compound 1 was extracted. The combined organic layers were heated at 30-35 °C then circulated through a bed of activated carbon and then through a filter with porosity lower than 0.5 micron. An aqueous solution of sodium bicarbonate was added to extract Compound 1 into the aqueous layer, the layer was concentrated under reduced pressure at a temperature below 40 °C and acetic acid was added while maintaining the temperature between 40 and 60 °C. [0343] The mixture was heated to a temperature between 73 and 77 °C and crude free acid of Compound 1 crystallized, at which point water was added. The crude free acid was filtered at 7- 13 °C, washed with a cold mixture of acetic acid and water, and dried under reduced pressure at a temperature below 50 °C. [0344] The sodium salt (Compound 4) was formed by addition of an equimolar aqueous solution of sodium hydroxide to a suspension of crude Compound 1 in water stirred at 18-25 °C. Compound 4 crystallized upon cooling of the aqueous mixture, was filtered and washed with cold water, then was dissolved in warm water and filtered through a filter with porosity no higher than 1 micron. Ethyl acetate was added, the biphasic mixture was heated to 30-35 °C and an aqueous solution of hydrobromic acid was added. Compound 1 was extracted into the organic layer after addition of ethyl acetate. The organic layer containing the product was washed with water then concentrated under reduced pressure. Compound 1 crystallized from the solution. Variations in the amount of n-heptane are added to complete the crystallization. The crystalline Compound 1 was filtered and washed with a mixture of ethyl acetate and n-heptane and dried under reduced pressure while maintaining the temperature of the drier below 50 °C. |
|
Multi-step reaction with 2 steps
1: sodium hydroxide / toluene; water / 18 - 25 °C / Large scale
2: hydrogen bromide / water; ethyl acetate / 30 - 35 °C / pH 2 - 4 / Large scale |
|
|
With water; sodium hydroxide In toluene |
|
|
Multi-step reaction with 2 steps
1: sodium hydroxide / tetrahydrofuran; water / 2 h
2: hydrogenchloride / water |
|
| 5.8 g |
Alkaline conditions; |
3.E E) Preparation of Lesinurad
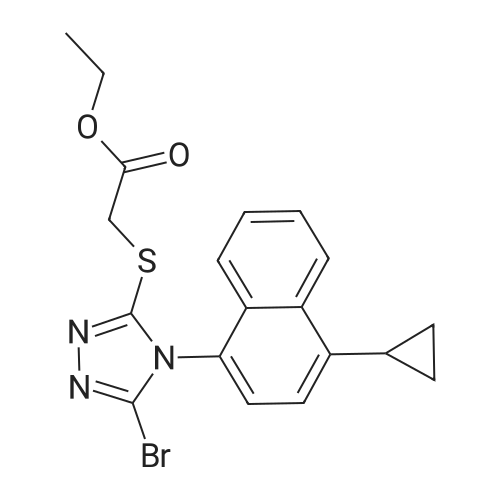
5 - [(Methoxycarbonyl) methylthio] -4- (4-cyclopropylnaphthalen-1-yl) -4H- 1, 2,4- triazole-3-carboxylic acid (6.0 g, 16 mmol) was dissolved in chloroform (20 mL), potassium carbonate (2.4 g, 17 mmol) was added and the reaction mixture was stirred at 25 ° C for 10 min, slowly Into phosphorus tribromide (4.7g, 17mmol), the reaction mixture was stirred at 90 ° C for 12 hours, TLC point plate to determine the completion of the reaction, Concentration to dryness gave the crude intermediate 2 - [4- (4-cyclopropylnaphthalen-1-yl) -5-bromo-4H-1,2,4-triazol-3-yl] thio}Methyl acetate, and then under known ester hydrolysis reaction under alkaline conditions, the reaction is completed, concentrated to dryness by rotary evaporation, slowly added Ethanol (10 mL), crystallized by cooling to -10 ° C for 3 hours, filtered and recrystallized from isopropanol to give a solidorol, an off-white solid(5.8g), yield 92.1%, the reaction of this step with Example 1. |
| 26.1 g |
With sodium carbonate; sodium hydroxide at 20℃; |
1.10
(10) 2-[[5-Bromo-4-(4-cyclopropyl-1-naphthalene)-4H-1,2,4-triazol-3-yl]thio] which is produced in the step (9) In 1330.1 g of methyl acetate, 100 ml of a mixed solution of an inorganic alkali sodium hydroxide and sodium carbonate was added, and at a normal temperature, a hydrolysis reaction occurred to generate a hydrolysis reaction.2-[[5-Bromo-4-(4-cyclopropyl-1-naphthalene)-4H-1,2,4-triazol-3-yl]thio]acetic acid 14 26.1 g, which is the present invention Resinad. |
| 123.0 g |
With water; sodium hydroxide for 2h; |
The crude product of Resinard in the embodiment of the present invention is synthesized by referring to CN104736522A.
In a 5L three-necked flask, add 160.0g of Intermediate 4 and 1.28L of tetrahydrofuran, start stirring, and raise the temperature to 3542.0.Stir until dissolved; lower the temperature to 2732, add 148.0g NBS, and react for 3.04.0h;Reduce the temperature to 5, add 800ml of toluene and 800ml of purified water, stir and separate the liquid, and use 800ml of 3% sodium bisulfite solution for the organic phase,Wash with 800ml of 5% sodium bicarbonate solution, let stand for liquid separation, collect the organic phase, add 580ml of 1mol/L sodium hydroxide to the organic phase, react for 2h,Separate the liquid, collect the aqueous phase, concentrate, cool the crystal, filter, and wash with water to obtain a wet product; add 720ml of purified water to the wet product,Heat to 35, stir to dissolve, add 1.6L of ethyl acetate, adjust the system pH=24 with 24% hydrobromic acid, stir for 30min,Separate the liquid, wash the organic phase once with 320ml of purified water, concentrate the organic phase until the solid precipitates,Keep warm at 3842, continue to stir and crystallize for 34h, then slowly reduce the temperature to 510, stir for 2h,Filtration, washing, and vacuum drying under reduced pressure to obtain 123.0 g of crude Recinald. The purity is 95.12%, |
| 3.3 kg |
With water; sodium hydroxide In tetrahydrofuran at 25 - 35℃; for 2h; Large scale; |
3 Synthesis of Crude Lesinurad
Under the condition of 20°C-30°C, add 2.5kg of formula III compound and DMF10L into the reaction kettle and stir, add DBU (18.5kg) into the reaction system and continue stirring;Continue to add 14.5kg of methyl thioglycolate,After the addition, continue to keep the temperature at 20°C-30°C and react for 2 hours.Continue to slowly add 3.5 kg of methyl chloroacetate into the reaction system, and continue to heat and stir the reaction for 1 hour to stop the reaction;Slowly add 25L of ethyl acetate to the system, keep warm and stir, continue to slowly add 10L of 1N hydrochloric acid to the system, stir for 10-20 minutes, and let stand for layering;Collect the organic phase, and evaporate the organic phase under reduced pressure to almost no fraction.Continue to add 18L of tetrahydrofuran to the concentrate to dissolve.Control the temperature at 25-35°C, drop the pre-prepared sodium hydroxide solution (9.3kg sodium hydroxide dissolved in 20L purified water) into it; after the addition, keep it warm and react for 2 hours,After the completion of the reaction, the reaction system was distilled under reduced pressure until the system was almost free of fractions. The pH of the reaction system was adjusted to 6-7 with hydrochloric acid, extracted twice with ethyl acetate, and the two ethyl acetate layers were combined to remove ethyl acetate.Get 3.3 kg of the crude Lesinurad for use (theoretical 2.57 kg). |
|
With lithium hydroxide In tetrahydrofuran; ethanol at 20℃; |
|
|
Multi-step reaction with 2 steps
1: sodium hydroxide / toluene; water / 10 - 15 °C
2: hydrogen bromide / water / pH 4 - 5 |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping