* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the American Chemical Society, 1932, vol. 54, p. 3413,3417
[2] Nagasaki med. J., 1958, vol. 33, p. 825,826[3] Chem.Abstr., 1958, p. 18591
5
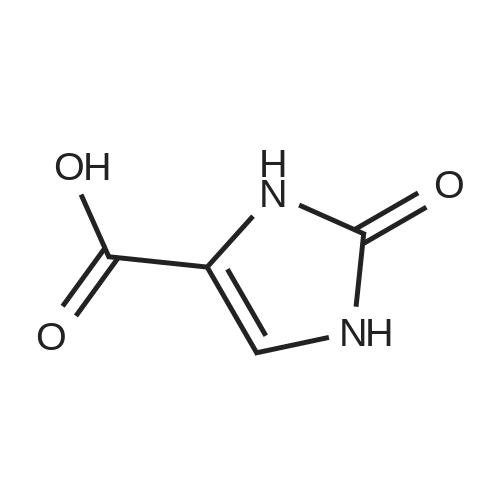
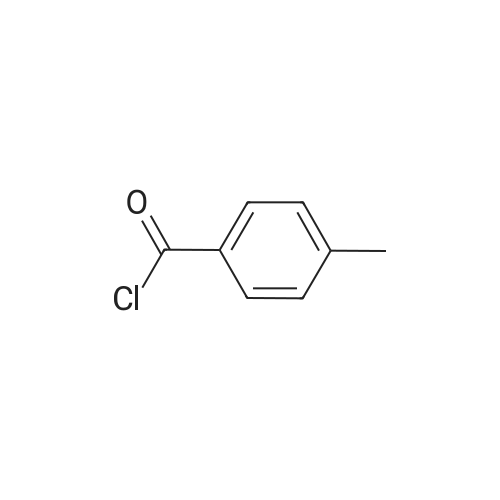
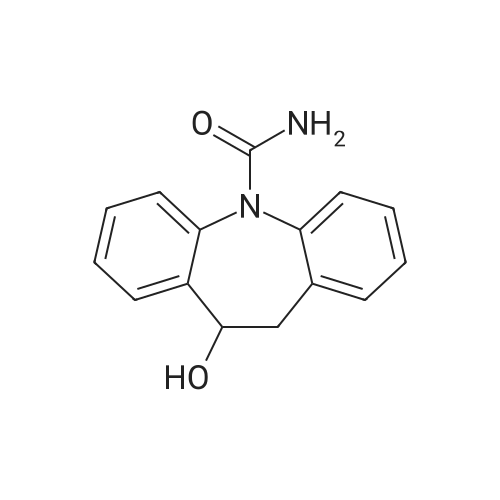
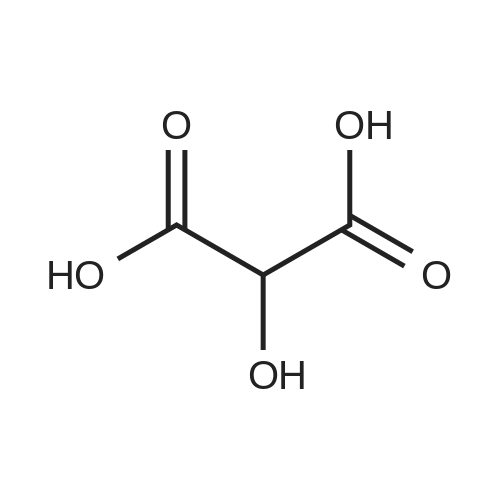
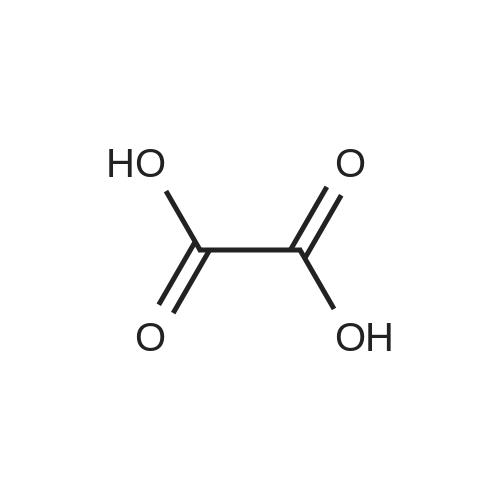
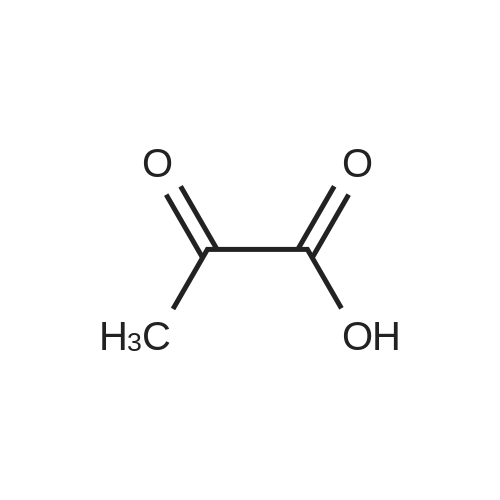
[ 87-69-4 ]
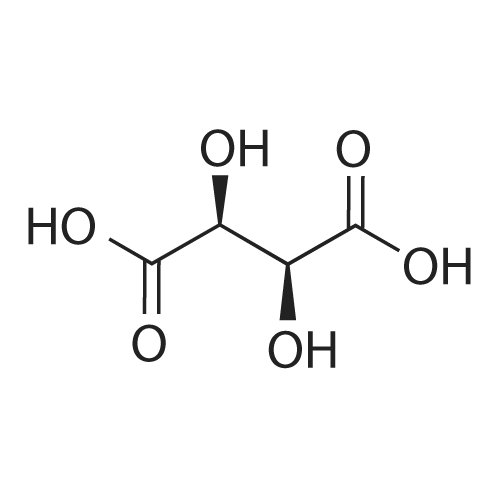
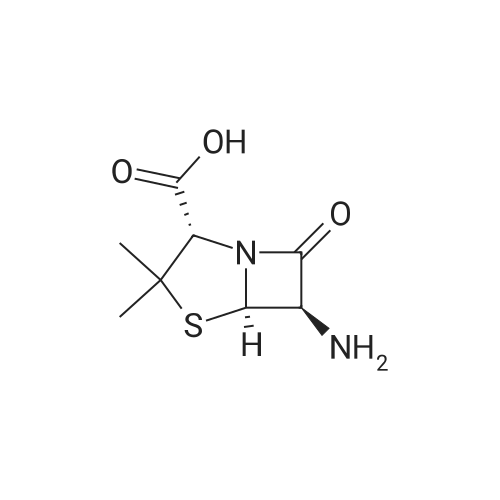
[ 4760-34-3 ]
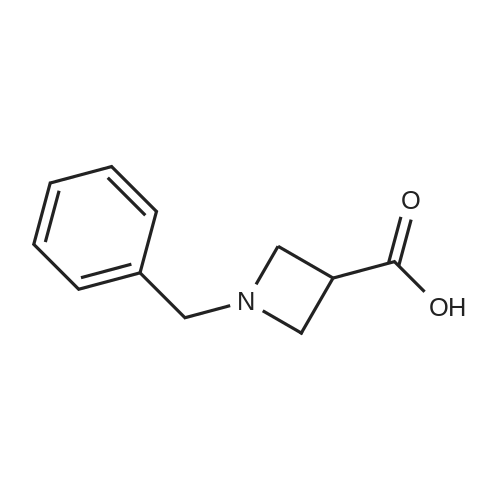
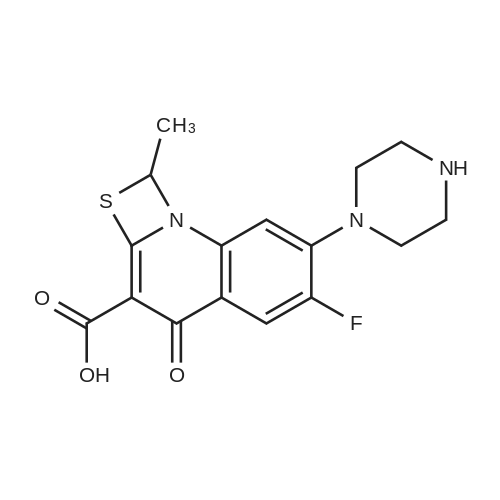
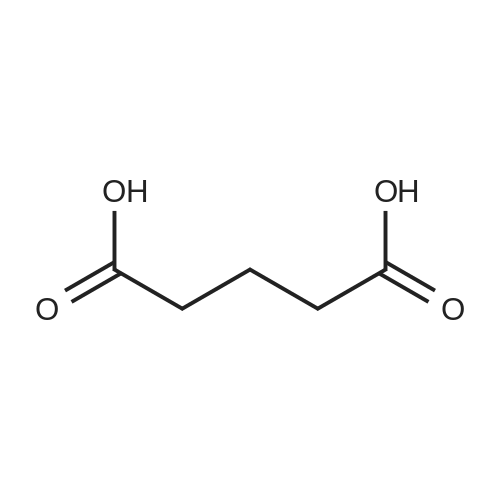
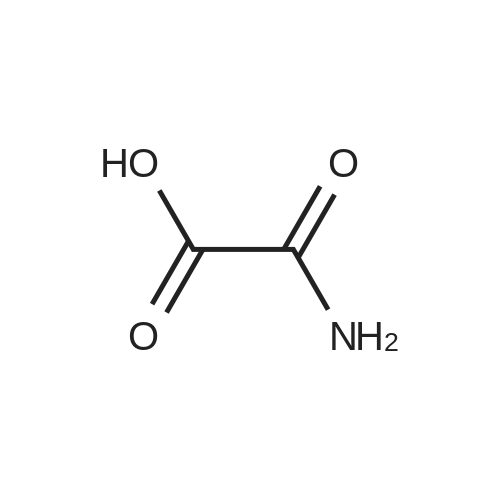
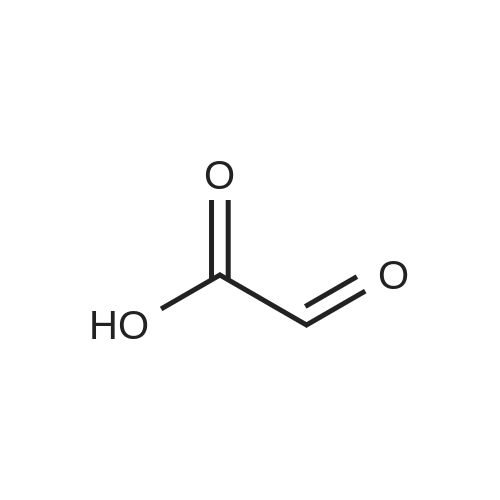
[ 3012-80-4 ]
Reference:
[1] Liebigs Annalen der Chemie, 1980, # 4, p. 542 - 556
6
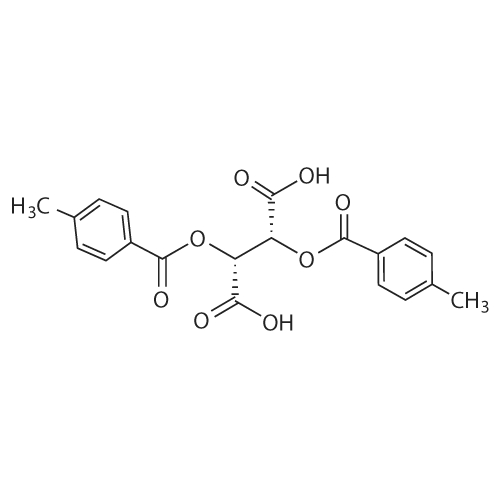
[ 26096-30-0 ]
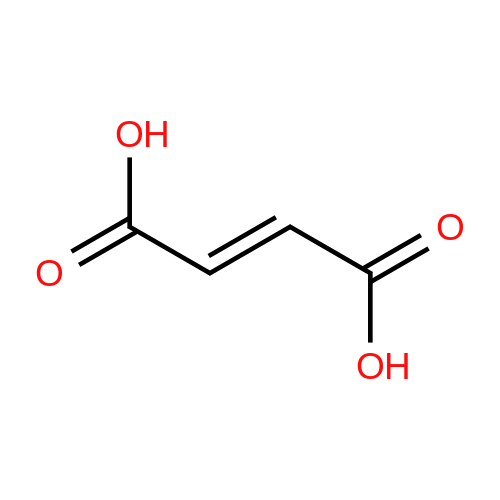
[ 87-69-4 ]
[ 94985-27-0 ]
[ 36476-78-5 ]
Reference:
[1] Patent: US4812581, 1989, A,
7
[ 87-69-4 ]
[ 98-88-4 ]
[ 2743-38-6 ]
Yield
Reaction Conditions
Operation in experiment
97.2%
With copper(II) sulfate In toluene for 4 h; Autoclave; Large scale
To the 3000 L autoclave, 450 kg of T-tartaric acid and 600 L of toluene were added. 2 kg of copper sulfate was added with stirring. 880 kg of benzoyl chloride was added dropwise and the reaction was continued for 4 hours. The reaction was continued for 4 hours, Dibenzoyl tartaric acid anhydride 1211.7kg, put it into 5000L reactor, add water and toluene 1211.7kg, heated to reflux, keep 3 hours, cooling to room temperature discharge, rejection of L-dibenzoyl tartaric acid 1059.8kg.The L-dibenzoyl tartaric acid obtained in this example was subjected to TEM test. The test structure was shown in Fig. 5, and it was found that the purity of the product L-dibenzoyl tartaric acid was 99.09percent and the total yield was 97.2percent.
Reference:
[1] Patent: CN104496806, 2016, B, . Location in patent: Paragraph 0099-0102
[2] Journal of Chemical Research, Miniprint, 1984, # 5, p. 1518 - 1530
(1)Into 500mL three-necked glass flask L - (+) - tartaric acid 30g,Then take the amount of anhydrous ethanol 150mL added to three-necked flask,Turn on stirringControl the internal temperature at 0 ~ 30 ,95.4 g of thionyl chloride was added dropwise,Time is 1.5h,Dropping the process of exothermic,After the addition was completed, the temperature was raised to 50 ° C,Reaction 2h,Then vacuum distillation,Remove ethanol,Get L - (+) - diethyl tartrate crude;(2)To the L - (+) - diethyl tartrate crude was added potassium bicarbonate 3.3g,Warmed to 20 ° C,Stirring reaction 3h,Filter to remove the solid,39.6 g of L - (+) - tartaric acid diethyl ester was obtained as a colorless or light yellow oily liquid,The molar yield was 96.2percentAfter testing,Product purity is 99.4percent.
94%
at 25℃; for 12 h; Cooling with ice
(1) under the ice water bath, to tartaric acid L - (substituted (I - 1 - 1), 100 g, 0.67 µM) anhydrous ethanol (600 ml) is dropped in the adds the chlorination sulfoxide (107 ml), at room temperature (about 25 °C) reaction 12 h, at low temperature the solvent is removed under reduced pressure, dissolved in EtOAc, saturated sodium bicarbonate washing to neutral, dried, concentrated under reduced pressure to remove the solvent, and concentration to obtain a colorless oil of, substituted (I - 1 - 2) compound of formula (138 g, yield 94percent).
88%
for 10 h; Reflux
A mixture of (2R,3R)-(+)-tartaric acid 1 (22.0 g, 150 mmol) and a catalytic amount of H2SO4 in EtOH (100 mL) was stirred at reflux for 10 h. After completion of the reaction, the mixture was concentrated in vacuo. A silica gel mesh was prepared from the residue and submitted to flash chromatography (silica gel: EtOAc/hexane as eluent) to provide 2 (26.4 g, 88percent). 1H-NMR (CDCl3, δ=7.26 ppm, 400 MHz,): 1.27 (t, J = 7.2 Hz, 6H), 3.36 (d, J = 6.6 Hz, 2H), 4.26 (q, J = 7.2 Hz, 4H), 4.5 (d, J = 6Hz, 2H).
Reference:
[1] Tetrahedron Letters, 2013, vol. 54, # 36, p. 4851 - 4853
[2] RSC Advances, 2015, vol. 5, # 15, p. 11687 - 11696
[3] Patent: CN107337603, 2017, A, . Location in patent: Paragraph 0025; 0026; 0027; 0028; 0029; 0030; 0031-0040
[4] Journal of Organic Chemistry, 2012, vol. 77, # 19, p. 8465 - 8479,15
[5] Patent: CN106966943, 2017, A, . Location in patent: Paragraph 0090; 0091
[6] Organic Letters, 2005, vol. 7, # 22, p. 5047 - 5050
[7] Tetrahedron, 2007, vol. 63, # 35, p. 8645 - 8657
[8] Tetrahedron Asymmetry, 2001, vol. 12, # 22, p. 3081 - 3088
[9] Journal of Enzyme Inhibition and Medicinal Chemistry, 2016, vol. 31, # 2, p. 219 - 228
[10] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1993, # 7, p. 805 - 812
[11] RSC Advances, 2013, vol. 3, # 6, p. 1976 - 1986
[12] Bulletin of the Korean Chemical Society, 2016, vol. 37, # 12, p. 1910 - 1911
[13] Ann.Chim.applic., 1935, vol. 25, p. 321[14] Chem. Zentralbl., 1935, vol. 106, # II, p. 3084
[15] Annales de Chimie (Cachan, France), 1943, vol. <11>18, p. 155[16] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1943, vol. 216, p. 64
[17] Monatshefte fuer Chemie, 1949, vol. 80, p. 256,259
[18] Chemische Berichte, 1936, vol. 69, p. 2279
[19] Ciencia, 1947, vol. 8, p. 175[20] Chem.Abstr., 1949, p. 127
[21] Collection of Czechoslovak Chemical Communications, 1977, vol. 42, p. 3069 - 3078
[22] Canadian Journal of Chemistry, 1963, vol. 41, p. 393 - 398
[23] Helvetica Chimica Acta, 1977, vol. 60, p. 301 - 325
[24] Tetrahedron Asymmetry, 2006, vol. 17, # 14, p. 2101 - 2107
[25] Tetrahedron, 1984, vol. 40, # 22, p. 4617 - 4623
[26] Tetrahedron, 2007, vol. 63, # 27, p. 6346 - 6357
[27] Chemistry Letters, 2007, vol. 36, # 12, p. 1436 - 1437
[28] Tetrahedron Asymmetry, 2011, vol. 22, # 3, p. 257 - 263
[29] RSC Advances, 2013, vol. 3, # 43, p. 20298 - 20307
12
[ 64-17-5 ]
[ 87-69-4 ]
[ 87-91-2 ]
Reference:
[1] Journal of the Chemical Society, 1898, vol. 73, p. 302
[2] Journal of the Chemical Society, 1922, vol. 121, p. 536,539
[3] Yakugaku Zasshi, 1927, p. 150[4] Chem. Zentralbl., 1928, vol. 99, # I, p. 1643
[5] Chemische Berichte, 1895, vol. 28, p. 3255
[6] Bulletin des Societes Chimiques Belges, 1920, vol. 29, p. 61,66[7] Chem. Zentralbl., 1920, vol. 91, # I, p. 817
[8] Chemische Berichte, 1895, vol. 28, p. 3255
[9] Journal of the American Chemical Society, 1921, vol. 43, p. 369
[10] Anales de la Real Sociedad Espanola de Fisica y Quimica, vol. 23, p. 415[11] Chem. Zentralbl., 1926, vol. 97, # I, p. 877
[12] Journal of the Chemical Society, 1922, vol. 121, p. 536,539
[13] Journal of the Chemical Society, 1910, vol. 79, p. 168
[14] Journal of the Chemical Society, 1929, p. 1897
The temperature of the solution was raised to 40 ° C and 150 g of L-tartaric acid was added. The mixture was stirred at 40 ° C for 1h and then at 20 ° C 20h, precipitation of a large number of solid, filtration, filter cake with cold isopropyl alcohol (100ml) wash, 40 ° C vacuum drying 8h, you can get white powderL-tartaric acid salt of the compound X in the form of 302. 6g.
Reference:
[1] Patent: CN106279095, 2017, A, . Location in patent: Paragraph 0050
Add p-anisic acid (77 g, 0.55 mol) to 100 mL SOCl2 and stir overnight at room temperature. Evaporate the excess SOCl2 to give p-anisoyl chloride. Add (2R,3R)-(+)-tartaric acid (25 g, 166 mmol) and stir the mixture and heat at 170°C for an hour. Allow the mixture to cool to 100°C and add 200 mL toluene. Cool the mixture to room temperature and add another 100 mL toluene. Collect the precipitate, rinse with toluene and dry. Reflux the crude product in a mixture of 300 mL acetone and 20 mL water for two hours. Then add 200 mL water and evaporate the acetone. Add another 200 mL water and collect the precipitate, rinse with water and dry. Reflux the product in 200 mL toluene for 15 minutes and collect the precipitate while the mixture is hot. Rinse the precipitate with 50 mL warm toluene and dry to give (2R,3R)-(-)-di-(p-anisoyl)tartaric acid.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide hemi-tartarate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; water; at 20 - 60℃;Product distribution / selectivity;
About 40-50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-lH-indol-3- yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base monohydrate was suspended in 1 mL of a solvent as listed in Table 17. A stoichiometric amount of tartaric acid was subsequently added to the suspension. The mixture was stirred at either 600C or ambient temperature (where a clear solution formed, stirring continued at 4C). Solids were collected by filtration and analyzed by XRPD, TGA and in some instances 1H-NMR.Table 17[0087] The salt forming reaction of the free base with tartaric acid required heating to elevated temperatures. A highly crystalline, anhydrous salt that decomposed above 200C was isolated as a hemi-tartarate and was labeled as form A. Form B was isolated once in isopropyl alcohol and water at 60C and, although very similar in structure with A, significant differences were seen in its XRPD pattern.
In water; isopropyl alcohol; at 60℃;
About 40-50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-lH-indol-3- yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base monohydrate was suspended in 1 mL of a solvent as listed in Table 17. A stoichiometric amount of tartaric acid was subsequently added to the suspension. The mixture was stirred at either 600C or ambient temperature (where a clear solution formed, stirring continued at 4C). Solids were collected by filtration and analyzed by XRPD, TGA and in some instances 1H-NMR.Table 17[0087] The salt forming reaction of the free base with tartaric acid required heating to elevated temperatures. A highly crystalline, anhydrous salt that decomposed above 200C was isolated as a hemi-tartarate and was labeled as form A. Form B was isolated once in isopropyl alcohol and water at 60C and, although very similar in structure with A, significant differences were seen in its XRPD pattern.
In ethanol; at 60℃;Product distribution / selectivity;
About 40-50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-lH-indol-3- yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base monohydrate was suspended in 1 mL of a solvent as listed in Table 17. A stoichiometric amount of tartaric acid was subsequently added to the suspension. The mixture was stirred at either 600C or ambient temperature (where a clear solution formed, stirring continued at 4C). Solids were collected by filtration and analyzed by XRPD, TGA and in some instances 1H-NMR.Table 17[0087] The salt forming reaction of the free base with tartaric acid required heating to elevated temperatures. A highly crystalline, anhydrous salt that decomposed above 200C was isolated as a hemi-tartarate and was labeled as form A. Form B was isolated once in isopropyl alcohol and water at 60C and, although very similar in structure with A, significant differences were seen in its XRPD pattern.
In ethanol; water; at 4 - 60℃;Product distribution / selectivity;
EXAMPLE 17; Formation of Hemi-Tartarate Salt; About 40 to 50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base monohydrate was suspended in 1 ml of a solvent as listed in Table 17. A stoichiometric amount of tartaric acid was subsequently added to the suspension. The mixture was stirred at either 60 C. or ambient temperature (where a clear solution formed, stirring continued at 4 C.). Solids were collected by filtration and analyzed by XRPD, TGA and in some instances 1H-NMR.; The salt forming reaction of the free base with tartaric acid required heating to elevated temperatures. A highly crystalline anhydrous salt that decomposed above 200 C. was isolated as a hemi-tartarate and was labeled as form A. Form B was isolated once in isopropyl alcohol and water at 60 C. and, although very similar in structure with A, significant differences were seen in its XRPD pattern.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide hemi-L-tartarate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96.6%
In ethanol; at 60℃; for 2h;
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3 -[4- [[[2-(2-methyl- IH- indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 50 mL of absolute ethanol were charged in a 250 mL 3-neck flask equipped with a magnetic stirrer and an addition funnel. The mixture was heated to 60C, and to the hot suspension were added dropwise 0.83 g (5.5 mmol, 10% excess) of L-tartaric acid dissolved in 15 mL absolute ethanol. Initially, large yellow agglomerates formed that prevented adequate stirring, but overtime these were converted to free flowing and stirrable yellow powder. Stirring continued at 60C for 2 hours. The mixture was subsequently cooled to room temperature and placed in an ice bath for approximately 30 minutes. The yellow powder was recovered by filtration and washed once by cold absolute ethanol (10 mL). It was dried overnight under vacuum to yield 4.1 g of the L-tartarate (hemi-tartarate) salt of N-hydroxy-3-[4-[[[2-(2-methyl-lH-indol-3- yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (96.6%).
96.6%
In ethanol; at 60℃;Product distribution / selectivity;
EXAMPLE 27; Formation of L-Tartarate Salt; 3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 50 ml of absolute ethanol were charged in a 250 ml 3-neck flask equipped with a magnetic stirrer and an addition funnel. The mixture was heated to 60 C., and to the hot suspension were added dropwise 0.83 g (5.5mmol, 10% excess) of 1-tartaric acid dissolved in 15 ml absolute ethanol. Initially, large yellow agglomerates formed that prevented adequate stirring, but overtime these were converted to free flowing and stirrable yellow powder. Stirring continued at 60 C. for 2 hours. The mixture was subsequently cooled to room temperature and placed in an ice bath for approximately 30 min. The yellow powder was recovered by filtration and washed once by cold absolute ethanol (10 ml). It was dried overnight under vacuum to yield 4.1 g of the 1-tartarate (hemi-tartarate) salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (96.6%).
20.00 g (38.86 mmol) of telmisartan was suspended in 200 ml of methanol, 6.6 g (38.86 mmol) of tartaric acid was added, and the mixture was refluxed for 2 hours.After cooling the reaction solution to room temperature, the crystalline solid was filtered, washed with 50 ml of methanol, and dried under vacuum at 50 for 24 hours to obtain 22.1 g of telmisartan stannate.The yield was 83.2%.
In methanol; for 3h;Heating / reflux;
Example 9 ; <strong>[144701-48-4]Telmisartan</strong> tartrateI g (2 mmol) of telmisartan is suspended in 10 mL of methanol and then 0.30 g (2 mmol) of tartaric acid is added. The suspension is heated under reflux temperature for 3 h and then cooled to room temperature, filtered, washed with water and dried at 350C in a vacuum drier. 1.2 g telmisartan tartrate is isolated.T: 230-2350CIR: 1697, 1446, 1266, 1130, 758, 740XRD: crystalline, figure 18
To the 1200 L reactor was added [60C] racemic 2-methylpiperazine (100 kg) from a drum. H20 (240 L) was added, and the solution was cooled to [13C.] To the 1200 L receiver was added L-tartaric acid (150 kg). [H20] (140 L) was added, and the slurry was stirred for 1 h 35 min until dissolution of the solids was complete. The L-tartaric acid solution was transferred to the 1200 L reactor over 2 h while maintaining a temperature of [10-22C] in the 1200 L reactor followed by a [H20] rinse (20 L). Ethanol (163 kg) was added to the 1200 L reactor, and the solution was cooled to [2C.] The resulting slurry was stirred for 2 h at [2C,] and filtered through a 36"Nutsche filter sending the filtrate into the 1200 L receiver. The 1200 L reactor and 36"Nutsche filter were washed with H20 (200 L), and the solids were dried to yield 214 kg of 12% ee [(171%] based on the title compound). These solids were recharged to a clean 1200 L receiver and H20 (630 L) was added to the 1200 L receiver, which was heated to [85C] until all the solids had dissolved. The solution was filtered through an in-line filter (C) into the 1200 L reactor, cooled to [5C,] and stirred for 2 h. The resulting slurry was filtered through a clean 36"Nutsche filter sending the filtrate into the 1200 L receiver. The 1200 L reactor and 36"Nutsche filter were washed with [H20] (200 L), and the solids were dried to yield 104 kg of 93% ee (83% based on the title compound). These solids were recharged to a clean 1200 L receiver and [H20] (254 L) was added to the 1200 L receiver, which was heated to [85C] until all the solids had dissolved. The solution was filtered through an in-line filter (C) into the 1200 L reactor, cooled to [5C,] and stirred for 2 h. The resulting slurry was filtered through a clean 36"Nutsche filter sending the filtrate into the 1200 L receiver. The 1200 L reactor and 36"Nutsche filter were washed with H20 (200 L), and the solids were dried to yield 92 kg of 99% ee (74% based on the title compound). [D1] does not describe the preparation of the title compound but makes reference [TO J.] Med. Chem. 1990, 33, 1645-1656 (D2). The yield of the title compound according to D2, starting from racemic 2-methylpiperazine was 35%. M. Pt. 255.0-257. [0C.] 'H NMR (400 MHz, [D20)] : [6] 4.79 [(D20,] reference), 4.36 (2H, s), 3.73-3. 64 (4H, [M),] 3.43 [(1H,] td J = 13.7, 3.0 Hz), 3.34 [(1H,] td, J = 12.7, 3.1 Hz), 3.17 [(1H,] dd, J = 14.2 12.8 Hz), 1.41 (3H, d, J = 6.1 Hz), 0.00 (TMS, reference). [13C] NMR (100 MHz, [D20)] : [6] 178.46 (s), 73.91 (d), 49.02 (d), 49.00 [(MEOH,] reference), 45.82 (t), 40.56 (t), 40.10 (t), 15.42 (q). IR (diffuse reflectance) 3426 (s), 3011 (s), 2999 (s), 2888 (s), 2785 (s, b), 2740 (s, b), 2703 (s, b), 2649 (s, b), 2483 (s, b), 2483 (s, b), 2361 (s), 2354,2340, 2248,1638 (s), cm HRMS (FAB) [CALCD FOR C5HL2N2 +HI 101.] 1079, found [101.] 1080. [[A]] [25D] = [24] (c 1.00, water). Anal. Calcd for [C4H606.] [C5HL2N2C,] 43.20 ; H, 7.25 ; N, 11.19. Found: C, 41.25 ; H, 7.45 ; N, 10.71.
In water; N,N-dimethyl-formamide; at 20℃; for 2 - 4.5h;Resolution of racemate;Product distribution / selectivity;
Example 1(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate dimethyl formamide solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide water mixture (15 % water in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield = 7.8 gm.%Yield: 82.62%.Melting point; 1370COptical Purity by Chiral HPLC: 99.00%. Example 2(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate dimethyl formamide solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide water mixture (20 % water in DMF). To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product. Yield = 9.0 gm.%Yield: 95.3% EPO <DP n="16"/>Optical Purity by Chiral HPLC: 98.86%Example 3(S)-<strong>[88150-42-9]Amlodipine</strong>-L--hemitartrate dimethyl formamide solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 220 ml dimethyl formamide water mixture (10 % water in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield = 9.0 gm.%Yield: 95.3%Optical Purity by Chiral HPLC: 98.69%. Example 4(S)-<strong>[88150-42-9]Amlodipine</strong>-L--hemitartrate dimethyl formamide solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide water mixture (15 % water in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47g, 0.01 moles) in 20 ml DMF + water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 1.5 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield = 8.0 gm. %Yield: 84.74%Melting point; 137COptical Purity by Chiral HPLC: 99.94%
52.9%
In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 4.5h;Resolution of racemate;Product distribution / selectivity;
Example 6(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate dimethyl formamide solvate (RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide dichloromethane mixture (15 % dichloromethane in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + dichloromethane mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield: 5.0gms (w/w)%Yield: 52.9%.Melting point; 137C Optical Purity by Chiral HPLC: 99.38%
42.37%
In methanol; N,N-dimethyl-formamide; at 20℃; for 4.5h;Resolution of racemate;Product distribution / selectivity;
Example 5(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate dimethyl formamide solvate (RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide methanol mixture (15 % methanol in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + methanol mixture (proportion as referred above) EPO <DP n="17"/>was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone(10 ml) and dried to give the title product.Yield: 4.0 gms (w/w) % Yield: 42.37%..Melting point; 137COptical Purity by Chiral HPLC: 99.70%.
31.77%
In hexane; N,N-dimethyl-formamide; at 20℃; for 4.5h;Resolution of racemate;Product distribution / selectivity;
Example 7(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate dimethyl formamide solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide hexane mixture (15 % hexane in DMF). To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + hexane mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product. Yield: 3.0 gms.%Yield: 31.77%.Melting point; 137COptical Purity by Chiral HPLC: 99.97%.
21.18%
In ethyl acetate; N,N-dimethyl-formamide; at 20℃; for 4.5h;Resolution of racemate;Product distribution / selectivity;
Example 8(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate dimethyl forinamide solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide ethyl acetate mixture (15 % ethyl acetate in DMF). To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid(1.47g, 0.01 moles) in 20 ml DMF + ethyl acetate mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product. Yield: 2.0 gms%Yield: 21.18%.Melting point; 137COptical Purity by Chiral HPLC: 99.37%.
The solution of L- (+)-tartaric acid (1.872 g, 0.51 mole equivalents) in dimethyl sulfoxide (25 ml) was added to the solution of (R, [S)-AMLODIPINE] (10 g, 24.46 [MMOLE)] in dimethyl sulfoxide (25 [ML)] under stirring. Precipitation was observed within 5 minutes after the addition, and the resulting slurry was stirred overnight at room temperature. The resulting solid was filtered off. [CH2C12] (50 ml) was added to the obtained filtrate, which was then stirred at room temperature for 40 hours. The resulting slurry was cooled to 5 [C,] stirred for 2 hours, and then filtered. The resulting solid was dried overnight at 50 [C IN] vacuo to give a solid (5.48 [G)] having the [FOLLOWING'H-NMR] data. Fig. 1 shows the'H-NMR chart of the solid, which means that the solid is [(S)- (-)-AMLODIPINE-HEMI-L-TARTRATE-1/4-DMSO-SOLVATE. ] [1H-NMR] (CD30D) : 7.04-7. 41 (m, 4H), 5.40 (s, 1H), 4.72 (gq, 2H), 4.36 (s, [1H),] 4.02 (m, 2H), 3.77 (m, 2H), 3.57 (s, 3H), 3.28 (m, 2H), 2.65 (s, DMSO), 2.31 (s, 3H), 1.15 (t, 3H) (2)
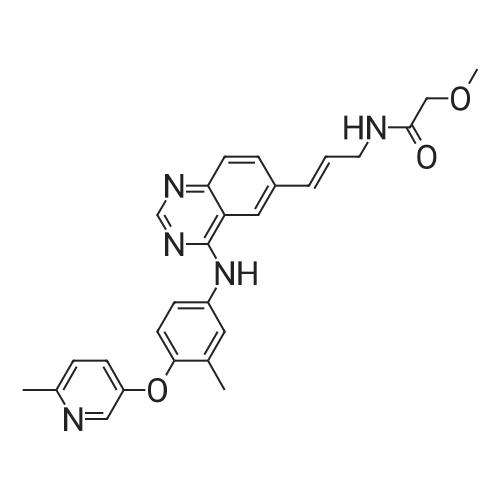
E-2-methoxy-N-(3-{4-[3-methyl-4-(6-methyl-pyridin-3-yloxy)-phenylamino]-quinazolin-6-yl}-allyl)-acetamide tartrate[ No CAS ]
E-2-methoxy-N-(3-{4-[3-methyl-4-(6-methyl-pyridin-3-yloxy)-phenylamino]-quinazolin-6-yl}-allyl)-acetamide hemitartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; dichloromethane; water; at 20℃; for 0.5h;
Several racemic E-2-METHOXY-N- (3- {4- [3-METHYL-4- (6-METHYL-PYRIDIN-3-YLOXY)- phenylamino]-quinazolin-6-yl)-allyl)-acetamide tartrates were produced. The synthesis of the monotartrate hemihydrate and hemitartrate hemihydrate were produced started with the production of amorphous material. This material was synthesized by dissolving 3 grams E-2- METHOXY-N- (3- {4- [3-METHYL-4- (6-METHYL-PYRIDIN-3-YLOXY)-PHENYLAMINO]-QUINAZOLIN-6-YL)-ALLYL)- acetamide free base in 20: 3 (v/v) ethanol (ETOH)/DICHLOROMETHANE (-50 mL). A solution of D, L-tartaric acid was prepared by dissolving 2 grams of D, L-tartaric acid in 10 mL water. The two solutions were combined and stirred at room temperature FOR-30 minutes. The solvent was reduced to yield the amorphous material.
To stirred solution of 10.50 gm (0.0256 mole) of RS amlodipine in 30 ml DMSO was added a solution of 1.93 gm (0.128 moles, 0.5 eq.) of L(+) tartaric acid in 30 ml DMSO. The solid starts separating from clear solution within 5-10 mins. This was stirred for 3 hrs and solid was filtered off, washed with acetone and dried to give 6.66 gm (46.2%) R(+) amlodipine hemi L(+) tartarate mono DMSO solvate. mp. 160162 C., 95.2% d.e. by chiral HPLC[J. Chrom. B. 693,367, (1997), J. Luksa, Dj. Josic, B. Podobric, B. Furlan, M. Kremser.]EXAMPLE 2 R(+)<strong>[88150-42-9]Amlodipine</strong> HemiL(+) Tartarate Mono DMSO Solvate from RS <strong>[88150-42-9]Amlodipine</strong> To a stirred solution of 100 gm (0.245 moles) of RS amlodipine in 300 ml DMSO was added a solution of 9.2 gm (0.06 moles, 0.25 eq) of L(+) tartaric acid in 300 ml DMSO. The solid starts separating from clear solution within 5-10 mins. This was stirred for 3 hrs and solid was filtered off, washed with acetone and dried to give 52.3 gm (36.2%) R(+) amlodipine hemi L(+) tartarate mono DMSO solvate. mp. 160.162 C., 98.2% d.e. by chiral HPLC. EXAMPLE 3 R(+)<strong>[88150-42-9]Amlodipine</strong> HemiL(+)Tartarate Mono DMSO Solvate from RS <strong>[88150-42-9]Amlodipine</strong> To a stirred solution of 100 gm (0.245 moles) of RS amlodipine in 150 ml DMSO was added a solution of 9.2 gm (0.06 moles, 0.25 eq) of L (+) tartaric acid in 100 ml DMSO. The solid starts separating from clear solution within 5-10 mins. This was stirred for 3 hrs and solid was filtered off, washed with acetone and dried to give 58.6 gm (40.5%) R (+) amlodipine hemi L(+) tartarate mono DMSO solvate. mp. 160-162 C., 96.8% d.e. by chiral HPLC.
(4-R)-2-[(2-aminoethyl)oxy]methyl}-4-(2-chlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate hemi L(+) tartarate mono DMSO solvate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
36.2 - 46.2%
for 3h;Product distribution / selectivity;
To stirred solution of 10.50 gm (0. 0256mole)of RS amlodipine in 30ml DMSO wasadded a solution of 1. 93gm (0.128 moles, 0.5 eq. ) of L (+) tartaric acid in 30 ml DMSO. The solid starts separating from clear solution within 5-10 mins. This was stirred for 3 hrs and solid was filtered off, washed with acetone and dried to give6. 66 gm(46. 2%) R (+) amlodipine hemi L (+) tartarate mono DMSO solvate, mp.160-162 C, 95.2% d. e. by chiralHPLC [J. Chrom. B. 693,367, (1997), J. Luksa, Dj. Josic, B. Podobric, B. Furlan, M. Kremser.]; Example 2: R (+) <strong>[88150-42-9]Amlodipine</strong> hemiL (+) tartarate mono DMSO solvate from RS amlodipine; To a stirred solution of100gm (0.245 moles) of RS amlodipine in 300ml DMSO was added a solution of 9.2 gm (0.06 moles, 0.25 eq)of L (+) tartaric acid in 300 ml DMSO. The solid starts separating from clear solution within5-10 mins. This was stirred for 3 hrs and solid was filtered off, washed with acetone and dried to give 52.3gm (36.2%) R (+) amlodipine hemi L (+) tartarate mono DMSO solvate. mp.160-162 C, 98.2%d.e. by chiralHPLC.; Example 3: R (+) <strong>[88150-42-9]Amlodipine</strong> hemiL (+) tartarate mono DMSO solvate from RS amlodipine; To a stirred solution of100gm (0.245 moles) of RS amlodipine in 150ml DMSO was added a solution of 9. 2gm (0.06 moles, 0.25 eq)of L (+) tartaric acid in 100 ml DMSO. The solid starts separating from clear solution within 5-10 mins. This was stirred for 3 hrs and solid was filtered off, washed with acetone and dried to give 58.6gm (40.5%) R (+) amlodipine hemi L (+) tartarate mono DMSO solvate. mp.160-162 C, 96.8 % d. e. by chiralHPLC.
In still some embodiments, decitabine salts were prepared from weak acids (3.0<pKa<5). For example, decitabine salts of (+)-L-tartaric, citric, L-lactic, succinic, acetic, hexanoic, butyric, or propionic acid (11-18, respectively, depicted above) were prepared by the following procedure: Decitabine (1.0 g, 4.4 mmol) was suspended in methanol (50 mL) in a round bottom flask (50 mL) and flushed and closed with nitrogen before adding acid (liquid acid: 0.4-4.4 mL; solid acid: 2-5 g) and each was heated in a sonicator at 30-55 C. until complete dissolution. If after 30 minutes complete dissolution hadn't been achieved, more methanol (5 mL) was added every 10 minutes. The solution was allowed to cool to 23 C. and then stored at 0 C. for NLT 12 hrs. The first crop of crystalline product was filtered and dried in vacuo for NLT 12 hr.
(d) R-<strong>[106133-20-4]Tamsulosin</strong> Tartrate Salt (R)-<strong>[106133-20-4]Tamsulosin</strong> free base (3.5 gm, 8.57 mmol) was dissolved in ethyl acetate (150 mL) at reflux temperature. (+)-Tartaric acid (0.65 gm, 4.33 mmol) was dissolved in ethyl acetate (100 mL) separately at reflux temperature and added slowly to the solution of tamsulosin at 40 45 C. The mixture was stirred at reflux temperature for 20 minutes, then cooled to room temperature and stirred for 2 hrs. The precipitate was filtered under vacuum, washed with ethyl acetate (15 mL), the off white sold collected and dried in hot air oven at 60 C. to afford 3.3 gm of <strong>[106133-20-4](R)-tamsulosin</strong>(+)-tartrate salt of melting range 179.5-182.7 C., chemical purity by HPLC: 99.93% and 100% of <strong>[106133-20-4](R)-tamsulosin</strong>(+)tartrate salt (chiral HPLC). DSC: 179-180.70 (sharp); XRD: 14.9, 11.5, 22.2, 25.3, 20.4, 19.3, 22.9, 20.1, 25.9 & 10.2 (2 theta values in decreasing order of intensity).
A sample of 1 g of this material was treated with 1.5 g of (2R, 3R) -tartaric acid in MeOH/ether to obtain 2.3 g of the tartrate salt [a] p = +10. 6 (c=1, H20)'. ' [a] 22 D = +11. 1 (c=9.57, H20), Sleevi et al. J. Med. Chem. , (1991), Vol 34, n4, 1314- 1328).
R(+) amlodipine-hemi L(+)-tartarate mono DMSO solvate[ No CAS ]
S(-) amlodipine-hemi L(+)-tartarate mono DMSO solvate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46.15%; 44.4%
EXAMPLE -1 <strong>[88150-42-9]Amlodipine</strong> hemi L tartarate-mono-DMSO solvate mp 160-162§C [alpha]t = +24.32 (c=1, R(+) <strong>[88150-42-9]Amlodipine</strong>-hemi-L-tartarate mono DMSO solvate and S(-) <strong>[88150-42-9]Amlodipine</strong>-hemi-L tartarate mono DMSO solvate from (RS) <strong>[88150-42-9]Amlodipine</strong>. To a stirred solution of 10.50 gm (0.0256 mole), of RS Amiodipine in 30 ml of DMSO was added a solution of 1.93 (0.128) mole (0.5 equiv) of L(+) Tartaric acid in 30 ml DMSO. The solid starts separating from clear solution within 5-10 min. This was stirred for 3 hrs. and the solid was filtered off, washed with acetone and dried to give 6.66 gm, 46.15% R(+) MeOH). The filtrate was seeded with S(-) amiodipine hemi L(+) tartarate salt. and left overnight the solid was filtered off and washed with 10ml acetone and dried to give 6.41 gm, 44.4% S(-) amlodipine-hemi L(+)-tartarate mono DMSO solvate.mp 169.5-171.5§C = -14.1 (c=1, MeOH) 90% de by chiral HPLC. (J.Chrom., B 693, 367 (1997) J. Luksa, Dj. Josic, B. Podobinc, B. Furlan, M. Kremser]
methyl N-[(RS)-methylbenzyl]azetidine-2-(RS)-carboxylate[ No CAS ]
tartaric acid salt of methyl N-[(S)-methylbenzyl]azetidine-2-(S)-carboxylate[ No CAS ]
[ 87-69-4 ]
L-tartaric acid salt of methyl N-[(S)-methylbenzyl]azetidine-2-(S)-carboxylate[ No CAS ]
[ 2133-34-8 ]
Yield
Reaction Conditions
Operation in experiment
In methanol; toluene;
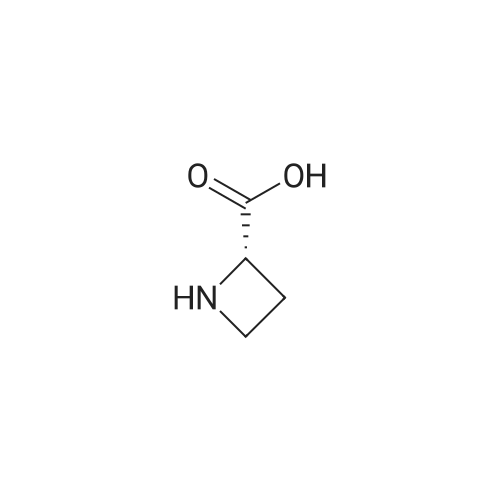
Example 12 200 g of 49.2% methyl N-[(RS)-methylbenzyl]azetidine-2-(RS)-carboxylate solution in toluene was added dropwise to a solution of L-tartaric acid (6.84 g) in methanol (10.26 g) at 60 C. 0.005 g of tartaric acid salt of methyl N-[(S)-methylbenzyl]azetidine-2-(S)-carboxylate was added as seed crystals to the mixture and was maintained at the temperature for 0.5 hour. To the resulting slurry was added 30 g of 50% methyl N-[(RS)-methylbenzyl]azetidine-2-(RS)-carboxylate in toluene solution was added dropwise at 60 C. over 1 hour. The slurry was cooled to 0 C. and crystals were collected by filtration. The obtained crystals were washed with cold methanol and dried in vacuo to give L-tartaric acid salt of methyl N-[(S)-methylbenzyl]azetidine-2-(S)-carboxylate (16.5 g, yield: 15.7%, 99.7% d.e.). The obtained crystals were treated in the following Example 14, 15 or 16 to yield (S)-azetidine-2-carboxylic acid. (>99% e.e).
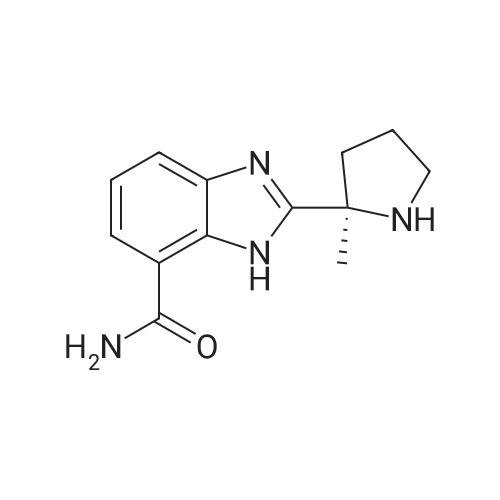
2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
EXAMPLE 3B; <strong>[912444-00-9]2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide</strong> A solution of Example 3A (500 mg, 1.32 mmol) in methanol (10 ml) was treated with 10% Pd/C (150 mg) under hydrogen for overnight (balloon). Solid material was filtered off and the filtrate was concentrated. The residual solid was further purified by HPLC (Zorbax C-18, CH3CN/H2O/0.1%TFA) and was converted to bis-HCl salt to provide Example 4 as white solid (254 mg). Co-crystallization of the free base with 1 equivalent of L-tartaric acid in methanol gave a single crystal that was suitable for X-ray study. The X-ray structure with L-tartaric acid was assigned the R-configuration. MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): delta 2.00 (s, 3 H), 2.10-2.19 (m, 1 H), 2.30-2.39 (m, 1 H), 2.45-2.51 (m, 1 H), 2.61-2.66 (m, 1 H), 3.64-3.73 (m, 2 H), 7.40 (t, J=7.95 Hz, 1 H), 7.77 (d, J=8.11 Hz, 1 H), 7.80 (d, J=7.49 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72; N, 17.66. Found: C, 49.10; H, 5.52; N, 17.61.
In water; N,N-dimethyl-formamide; at 20℃; for 8.5 - 9.5h;Resolution of racemate;
Example 13 Preparation of (R)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate DMF solvate.L-tartaric acid (1.47gms; 0.01 moles) was added to the mother liquor obtained fromExample 1. and stirred at ambient temperature for 240-300 minutes, till complete precipitation of the product The title compound was then filtered and dried.Yield: 6.4 gms. %Yield; 67.7%.
With diisobutylaluminium hydride; In water; ethyl acetate; toluene;
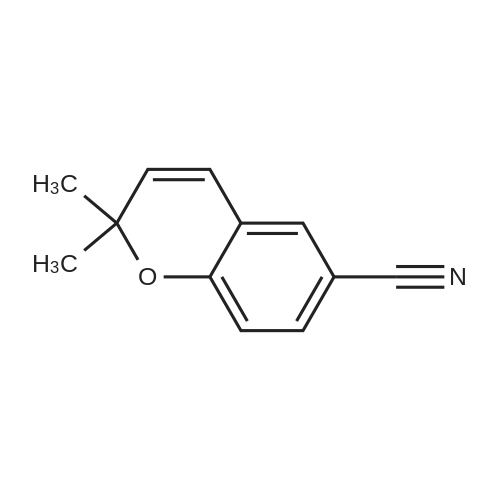
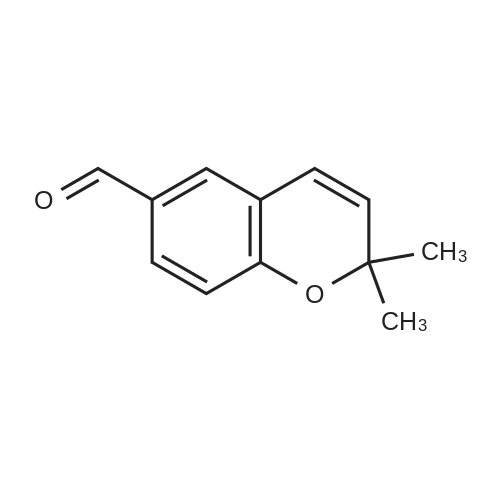
PREPARATION 8 <strong>[33143-29-2]2,2-Dimethyl-2H-1-benzopyran-6-carbonitrile</strong> (10 g) was dissolved in toluene (54 ml), and the solution was cooled to -78 C. under a nitrogen atmosphere, to this solution was added diisobutylaluminum hydride (1M solution in toluene, 81 ml) over 30 minutes, and the mixture was stirred at -78 C. for 10 minutes. The reaction mixture was quenched at -78 C. with ethyl acetate (54 ml) followed by 1M tartaric acid solution in water (100 ml). The cooling bath was removed and the mixture was vigorously stirred for 1 hour. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, and concentrated to give 2,2-dimethyl-2H-1-benzopyran-6-carbaldehyde (9.9 g) as a pale yellow oil, which was used without further purification. IR (Film): 1690, 1640 cm-1. NMR (CDCl3, delta): 1.47 (6H, s), 5.70 (1H, d, J=10Hz), 6.38 (1H, d, J=10Hz), 6.86 (1H, d, J=8Hz), 7.52 (1H, d, J=2Hz), 7.64 (1H, dd, J=2, 8Hz), 9.82 (1H, s).
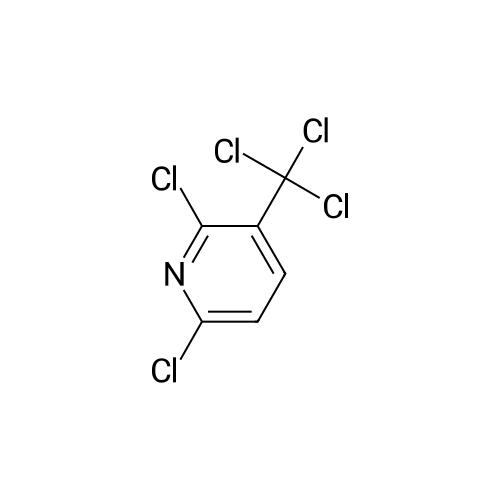
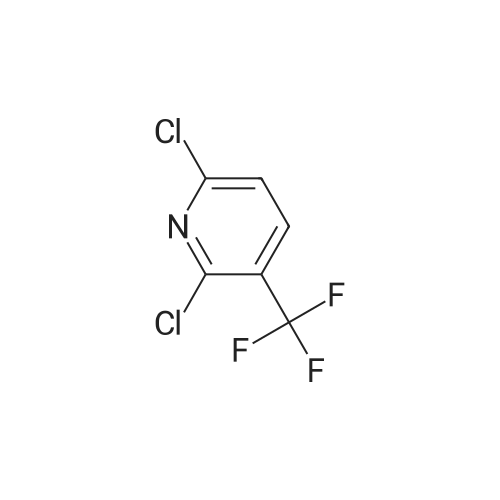
2,6-Dichloro-3-trifluoromethylpyridine (Example 14) 266 g (1 mol) of 2,6-dichloro-3-trichloromethylpyridine, obtained by chlorinating 2,6-dichloro-3-chloromethylpyridine with Cl2 under irradiation with UV light, and 5 g of antimony trifluoride are mixed and are heated under gentle reflux in a three-necked flask with an upright condenser (internal temperature of the melt 240C). Portions of 10 g each of antimony trifluoride are added at intervals of 10 minutes each, up to a total quantity of 191 g (1.065 mols). The reaction mixture is then heated under reflux for a further 15 minutes and then the reflux condenser is replaced by a fractionating column and the resulting trifluoromethyl compound is distilled off at normal pressure. The distillate is diluted with 2 l of diethyl ether and the ethereal solution is washed with a solution of 1 kg of tartaric acid in 4 l of water. After distilling off the diethyl ether, 162 g of crude 2,6-dichloro-3-trifluoromethylpyridine are obtained, which, on distillation at 68 - 71C/12 mm Hg, gives 138.4 g (64% of theory) of pure 2,6-dichloro-3-trifluoromethylpyridine.
Example 29 (3S,4S)-1-Benzyl-3,4-dihydroxypyrrolidine (BDHP). BDHP was obtained according to the method described in Ber., 1986, 199, 3326 and in Chem. Phar. Bull., 1991, 39, 2219. A mixture of 15 g of L-tartaric acid and 1.9 mL of benzylamine in xylene was heated at reflux for 7 h to afford, after crystallization, 11 g (52percent) of an N-benzylimide intermediate: mp.=199-201° C. (lit. 196-98° C., Can. J. Chem. 1968, 46, 3091); [alpha]D23 =+138 (c=1, MeOH) (lit. [alpha]D23 =+138 (c=1, MeOH), Chem. Phar. Bull., 1991, 39, 2219).
With diisobutylaluminium hydride; In tetrahydrofuran; dichloromethane;
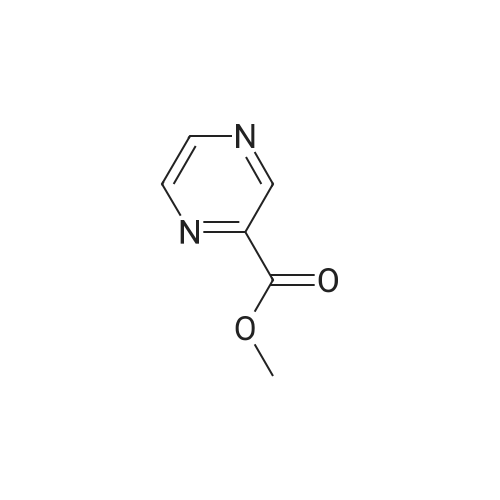
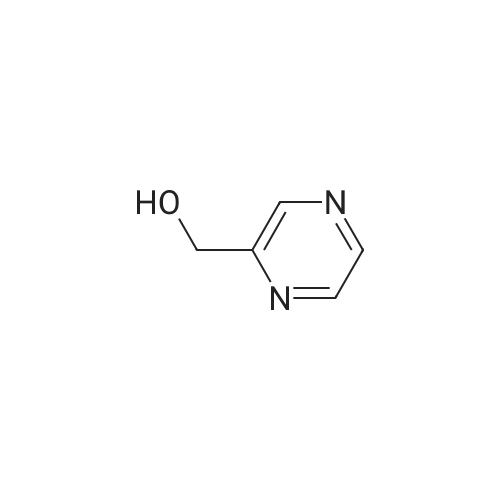
a 2-Hydroxymethylpyrazine To methyl 2-pyrazinecarboxylate (1.80 g) in THF (60 ml) was added diisobutylaluminium hydride (1 M solution in THF; 39 ml) at -78 C. with stirring. The solution was allowed to warm to room temperature, and stirred for 24 h. The reaction was quenched with solid tartaric acid, then aqueous sodium potassium tartrate, and stirred for 30 min at room temperature. Saturated aqueous sodium hydrogen carbonate was added until the pH of the solution was >7. The solution was washed with ethyl acetate (3*200 ml), and the organic layers combined, washed with saturated sodium chloride solution (1*200 ml), dried (magnesium sulfate) and concentrated in vacuo. The residue was purified by flash chromatography (silica gel, eluent =5% methanol in dichloromethane) to yield 2-hydroxymethylpyrazine as a dark brown oil (0.16 g). 1H NMR (250 MHz, CDCs) delta3.42 (1 H, br s), 4.85 (2 H, s), 8.55 (2 H, m), 8.68 (1 H, s); MS (ES+) m/e 111 [MH+].
di-(R-(+)-N-propargyl-1-aminoindan) L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol;Heating / reflux;Product distribution / selectivity;
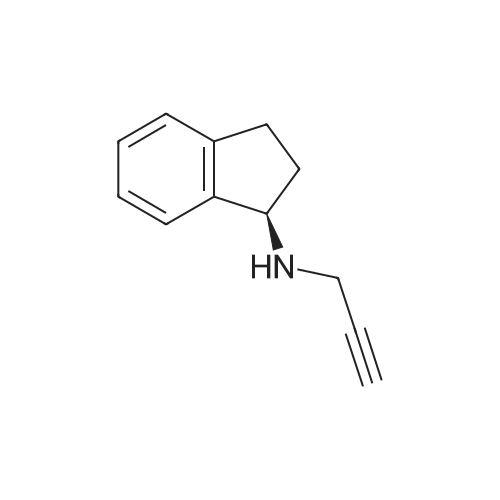
An example was designed to produce crystals as described in U.S. Pat. No. 6,630,514, example 6A. A first solution of 5.0 g of L-tartaric Acid in 55 ml methanol was prepared and heated to reflux. Then, a solution of 5.7 g of (R)-PAI in 55 ml methanol was added to the first solution while heating and stirring. 322 ml of t-butylmethyl ether was added to the resulting mixture under reflux conditions over 20 minutes. Crystallization of <strong>[136236-51-6]rasagiline</strong> tartrate took place during the addition. The batch was cooled to 17 C. and filtered. The solid product was washed on a filter with t-butylmethyl and dried under vacuum. Dry solid product (7.6 g white crystalline powder) was sampled and analyzed. Analysis: m.p. 176.4-178.4 C., Purity by TLC-one spot (PAI) R-PAI tartrate final product was attained.
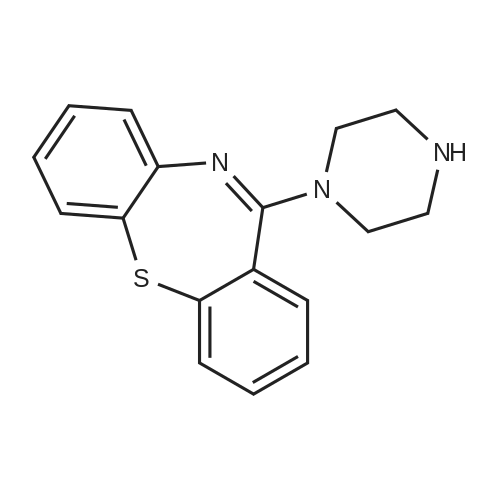
11-piperazin-1-yldibenzo[b,f][1,4]thiazepine L-tartaric acid salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93.8%
In acetonitrile;Heating / reflux;
In 5 mL of acetonitrile was dissolved 500 mg (1.693 mmol) of 11-piperazin-l- yldibenzo[b,fj[l,4]thiazepine. Ih 5 mL of acetonitrile was dissolved 254 mg (1.693 mmol) of L- tartaric acid with heating. The solutions were combined and a slightly gummy solid precipitated immediately. The mixture was diluted with an additional 5 mL of acetonitrile. The solid did not dissolve, but triturated into a free-flowing solid. The mixture did not change overnight. The solids were collected, washed with acetonitrile (5 mL), and dried under vacuum at 40 0C resulting in 707 mg crystalline solid (93.8%). mp 170-175 0C (dec). 1H NMR (DMSOd6) was consistent with the title salt.Polarized light microscopy showed that the solid was composed of irregularly shaped crystalline particles. DSC revealed two broad endotherms at 118.3 and 175.1 0C (Figure 1). The lower temperature endothermic event is likely due to water loss and the higher temperature endothermic event may be due to a melt followed by decomposition. TGA revealed two weight loss transitions (Figure 1). The lower temperature weight loss transition coincided with the lower temperature DSC event and was measured to correspond to approximately 1 mole <n="13"/>equivalent of water. The higher weight loss transition coincided with the higher DSC event. Dynamic vapor sorption (DVS) studies indicated that the salt was hygroscopic (Figure 2). Moisture gain was reversible with little hysteresis and amounts to about 1 mole equivalent at 90% RH which was in addition to the moisture already contained in the salt.
(S)-amlodipine-L-hemitartrate dimethylformamide solvate[ No CAS ]
[ 103129-81-3 ]
Yield
Reaction Conditions
Operation in experiment
82.62 - 95.3%
In water; at 20℃; for 2 - 4.5h;Product distribution / selectivity;
EXAMPLE 1 (S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate (RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide water mixture (15% water in DMF). To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product. Yield=7.8 gm. % Yield: 82.62%. Melting point; 137 C. Optical Purity by Chiral HPLC: 99.00%. EXAMPLE 2(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide water mixture (20% water in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield=9.0 gm.% Yield: 95.3%Optical Purity by Chiral HPLC: 98.86%EXAMPLE 3(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 220 ml dimethyl formamide water mixture (10% water in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield=9.0 gm.% Yield: 95.3%Optical Purity by Chiral HPLC: 98.69%.EXAMPLE 4(S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide water mixture (15% water in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+water mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 1.5 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield=8.0 gm.% Yield: 84.74%Melting point; 137 C.Optical Purity by Chiral HPLC: 99.94%
52.9%
In dichloromethane; at 20℃; for 4.5h;Product distribution / selectivity;
EXAMPLE 6; (S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate; (RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide dichloromethane mixture (15% dichloromethane in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+dichloromethane mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield: 5.0 gms (w/w)% Yield: 52.9%.Melting point; 137 C.Optical Purity by Chiral HPLC: 99.38%
42.37%
In methanol; at 20℃; for 4.5h;Product distribution / selectivity;
EXAMPLE 5; (S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate(RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide methanol mixture (15% methanol in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+methanol mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield: 4.0 gms (w/w)% Yield: 42.37%.Melting point; 137 C.Optical Purity by Chiral HPLC: 99.70%.
31.77%
In hexane; at 20℃; for 4.5h;Product distribution / selectivity;
EXAMPLE 7; (S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate; (RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide hexane mixture (15% hexane in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+hexane mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield: 3.0 gms.% Yield: 31.77%.Melting point; 137 C.Optical Purity by Chiral HPLC: 99.97%.
21.18%
In ethyl acetate; at 20℃; for 4.5h;Product distribution / selectivity;
EXAMPLE 8; (S)-<strong>[88150-42-9]Amlodipine</strong>-L-hemitartrate Dimethyl Formamide Solvate; (RS)-<strong>[88150-42-9]Amlodipine</strong> (16 gm, 0.04 moles) was dissolved in 140 ml dimethyl formamide ethyl acetate mixture (15% ethyl acetate in DMF).To this mixture, a solution of L-tartaric acid, prepared by dissolving tartaric acid (1.47 g, 0.01 moles) in 20 ml DMF+ethyl acetate mixture (proportion as referred above) was added drop wise in 30 min. The mixture was stirred for 4 hr at room temperature after addition. The slurry was filtered and the residual solid was washed with acetone (10 ml) and dried to give the title product.Yield: 2.0 gms% Yield: 21.18%.Melting point; 137 C.Optical Purity by Chiral HPLC: 99.37%.
(S)-amlodipine-L-hemitartrate dimethylformamide solvate[ No CAS ]
(R)-amlodipine-L-hemitartrate dimethylformamide solvate[ No CAS ]
[ 103129-81-3 ]
Yield
Reaction Conditions
Operation in experiment
96.25%
at 20℃; for 4.5h;Product distribution / selectivity;
The effect of the co-solvent in the dimethyl formamide mixture on the resolution of racemic amlodipine-L-hemitartrate salt has been described in Table-I for better understanding.5. Note: Without the co-solvent, the optical purity of (S)- amlodipine-L-hemitartrate DMF solvate is only 76%, with yield of 96.25% (w/w).
Example 8 Preparation of 1-cyclopropyl-7-[(S,S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid -L-(+)-tartarate (IIa) (Insitu preparation without isolation of Moxifloxacin base) 750 ml of N, N-dimethylformamide, 50 g of 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic acid (IV) and 25.70 ml of [S,S]-2,8-diazabicyclo [4.3.0] nonane (V) were stirred at 65 - 70 C for 6-8 hours. Completion of the reaction was monitored by HPLC. The reaction mass was cooled to 25-30 C, 50 g of L-(+)-tartaric acid was added, and the reaction mass was stirred at 75 - 80 C for 3-4 hours. The reaction mass was cooled to 25-30C and stirred for 10 12 hours. The solid was filtered off with suction, washed with 150 ml of N, N-dimethylformamide and dried under vacuum at 75C, affording 84.5 g of the title compound. Yield: 90.32 % Purity: 99.80 % (HPLC) Chiral impurity (R,R isomer content): 0.06 %
750mlDMF <strong>[151096-09-2]moxifloxacin</strong> was added 50g, 18 7g L- (+)-tartaric The reactionwas stirred, 75~80 C under3 hours, cooled to 25~30 C after the reaction was stirred for 10~12 hours.Suction filtered and the solid washed with lOOmlDMF, 75 C and dried in vacuoto yield <strong>[151096-09-2]moxifloxacin</strong> tartaric 67. 3g (98. 08%).
90.2%
In N,N-dimethyl-formamide; at 25 - 80℃; for 13 - 16h;Product distribution / selectivity;
Example 2; Preparation of 1-cyclopropyl-7-[(S,S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid -L-(+)-tartarate (IIa); 750 ml of N, N'-Dimethylformamide, 50 g of 1-cyclopropyl-7-[(S,S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (obtained from Example 1) and 18.7 g of L-(+)-tartaric acid were stirred at 75 - 80 C for 3-4 hours. The reaction mass was cooled to 25 - 30C and stirred for 10 12 hours. The solid was filtered off with suction, washed with 100 ml of N, N dimethylformamide and dried under vacuum at 75C, affording 62.0 g of the title compound. Yield: 90.2 % Purity: 99.80 % (HPLC) Chiral impurity (R,R isomer content): 0.04 % 1H NMR (400 MHz, DMSO) delta 0.80-1.30(br,m, 2H), 0.80-1.30(br,m, 2H), 1.50-1.73 (br, 2H), 1.50-1.73 (br, 2H), 2.7(m,2H), 3.02(m, 1H), 3.52(m, 2H), 3.56(s,3H), 3.9 (m, 2H), 3.97 (m,1H), 4.12 (m, 1H), 7.66(d, 1H), 8.65(s, 1H) 13C NMR(400 MHz, DMSO) delta 21.80(CH2), 19.62(CH2), 8.22(CH2), 9.631(CH2), 35.25(CH), 39.51(CH), 39.92(CH), 40.65(CH), 43.04(CH2), 55.06(CH), 56.65(CH2), 56.70(CH2), 61.42(CH3), 106.49(CH), 106.2, 116.85, 136.92, 137.0, 140.25, 150.18(CH), 151.35 ,165.8, 174.43, 175.89, 175.92. IR 3410 (N-H/O-H stretching), 2927, 2862 (aliphatic C-H stretching), 1716 (-C=O, stretching), 1623,1510( Ar C=C, stretching), 1443,1344 (aliphatic CH, bending), 1192 (C-F, stretching), 1045 (C-N, stretching), 805 (Aromatic C-H, bending)cm -1
(i) With Tartaric AcidA 30% CAPTISOL formulation was prepared by adding 2.7 ml water to a vial containing 0.9 g CAPTISOL. The mixture was then mixed on a vortexer to give 3 ml of a clear solution.In order to prepare a formulation of 25 mg/ml solution of compound 12, 1 ml of the 30% CAPTISOL solution was added to a vial containing 25 mg of compound 12 and 8.6 mg tartaric acid and the resulting mixture was mixed on a vortexer or sonicated at 30 C. for 15 to 20 minutes to give a clear yellowish solution. The resulting solution is stable at room temperature.In order to prepare a formulation of 30 mg/ml solution of compound 12, 1 ml of the 30% CAPTISOL solution was added to a vial containing 30 mg of compound 12 and 10.4 mg tartaric acid (1.0 eq) at room temperature.In order to prepare a formulation of 40 mg/ml solution of compound 12, 1 ml of the 30% CAPTISOL solution was added to a vial containing 40 mg of compound 12 and 17.9 mg tartaric acid (1.3 eq) at 36 C.In order to prepare a formulation of 50 mg/ml compound 12 in CAPTISOL, 1 ml of 30% CAPTISOL was added to 50 mg compound 12, 22.5 mg tartaric acid (1.3 eq) at 37 C.In order to prepare a formulation of 60 mg/ml compound 12 in CAPTISOL, the 30% CAPTISOL was added to a vial containing 60 mg compound 12 and 26.9 mg tartaric acid (1.3 eq) at 36 C. The solution was diluted in 1× water and 2×D5W. The diluted solution is stable at room temperature for >12 h.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide hemi-L-tartarate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96.6%
In ethanol; at 0 - 60℃;Cooling with ice;Product distribution / selectivity;
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 50 mL of absolute ethanol were charged in a 250 mL 3-neck flask equipped with a magnetic stirrer and an addition funnel. The mixture was heated to 60 C., and to the hot suspension were added dropwise 0.83 g (5.5 mmol, 10% excess) of L-tartaric acid dissolved in 15 mL absolute ethanol. Initially, large yellow agglomerates formed that prevented adequate stirring, but overtime these were converted to free flowing and stirrable yellow powder. Stirring continued at 60 C. for 2 hours. The mixture was subsequently cooled to room temperature and placed in an ice bath for approximately 30 minutes. The yellow powder was recovered by filtration and washed once by cold absolute ethanol (10 mL). It was dried overnight under vacuum to yield 4.1 g of the L-tartarate (hemi-tartarate) salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (96.6%).
In dichloromethane; at 25 - 30℃;Reflux;Product distribution / selectivity;
<strong>[93413-62-8]O-desmethylvenlafaxine</strong> (40 g), dichloromethane (400 mL) and L-tartaric acid (22.8 g) were charged into a round bottom flask and the mixture was stirred for about 10 minutes at 25-300C. The mixture was heated to reflux and stirred for about 90 minutes. The mixture was cooled to 25-300C. The formed solid was filtered, washed with dichloromethane (40 ml_) and dried at about 600C under vacuum for about 3 hours to afford the title compound (yield 62.2 g, HPLC purity 99.9%). The product is crystalline Form B, characterized by a powder X-ray diffraction pattern substantially in accordance with Fig. 4.
In methanol; for 1h;Reflux;Product distribution / selectivity;
Example 8 Desvenlafaxine tartrate (60) 0.53 g (2 mmol) of desvenlafaxine was suspended in 13 ml of methanol and 0.30 g (2 mmol) of tartaric acid was added. The suspension was heated at reflux temperature for 1 h. The solution was filtered; filtrate was cooled to room temperature and evaporated to solid residue to obtain 0.80 g of desvenlafaxine tartrate. This product was suspended in diethyl ether for 1h and the suspension was filtered and the product was dried and desvenlafaxine tartrate was isolated. Mp = 95 - 107C. IR: 2937, 1730, 1616, 1517, 1448, 1402, 1265, 1135, 842, 679
(1R)-N-(prop-2-ynyl)-2,3-dihydro-1H-inden-1-amine hemihydrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
In isopropyl alcohol; at 40℃; for 2.16667h;
Preparation of <strong>[136236-51-6]rasagiline</strong> hemitartrate In 726 ml of 2-propanol 40.9 g of L ( + ) -tartaric acid (anhydrous) were dissolved under heating to 40C to obtain Solution 1. Separately Solution 2 was prepared by dissolving 110.3 g of <strong>[136236-51-6]rasagiline</strong> free base in 168 ml of 2-propanol under stirring and heating to 40 C. Solution 2 was slowly added under rigorous stirring to Solution 1 within 130 minutes. The obtained suspension was then cooled to 15-20C within 1 hour and further stirred at this temperature for 1 hour. The product was filtered, washed with 70 ml of pre-cooled 2-propanol (having a temperature of 5-10 C) and dried in vacuum drier at 50C and a pressure under 50 mbar . 132.6 of crystalline product was obtained with an assay of 100% and a molar yield of 88.0% (enatiomeric purity 100.00 area%, chromatographic purity 100.00 area%) .
72%
In methanol; butanone; for 1.16667h;Reflux;
Rasagiline free base (5.0 g) and methanol (75 mL) were charged in a flask followed by addition of L-(+)-tartaric acid (1.95 g) and mixture was stirred for 10 minutes. To the mixture, methyl ethyl ketone (25 mL) was added and mixture was heated to reflux temperature followed by stirring at the same for 1 hour. The reaction mixture was cooled to room temperature and maintained at the same for 1 hour. The solid obtained is isolated by filtration and washed with methyl ethyl ketone (10 mL) followed by drying at 60C under vacuum to afford title compound in 72% yield. HPLC purity is 99.98%.
In isopropyl alcohol; at 40℃; for 2h;Product distribution / selectivity;
Example 51; Preparation of Rasagiline Mixture of Tartrate Form I and Hemitartrate Form IRasagiline base (1.61 g) was dissolved in 2-propanol (6.9 mL). L-Tartaric acid (1.41 g) was added and the mixture was stirred for 2 h at 40 C. The mixture was allowed to cool to ambient temperature and stirred for 24 hours at this temperature. The mixture was filtered and dried under vacuum at 40 C. Yield 2.04 g.
4-[2-(dimethylamino)-1-(1-hydroxycyclohexyl)ethyl]phenol L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; at 20℃;Reflux;
Example 14[00155] A 100 ml flask equipped with a magnetic stirrer was charged with ODV base (3 g, 11.39 mmol) and EtOH 95% (9ml), the suspension being stirred at reflux. L-Tartaric acid (2.Og 12.57 mmol) was added and a clear solution was obtained. After stirring overnight at room temperature, the mixture was cooled to -1O0C. Toluene (6 ml) was added and a clear solution appeared. The mixture was evaporated to dryness to get amorphous ODV Tartarate. X-ray powder diffraction pattern is depicted in Figure 14.
Example 7; Preparation of CffH-VRivastigmine Tartrate; To a solution of lOOg of (S)-[I -(3 -hydroxyphenyl) ethyl] dimethylamine (VIII) in 500ml of acetone, pulverized potassium carbonate (125.2g) was added. The reaction mixture was heated to 40-450C for 1 hour. A solution of iV-ethyl-iV-methylcarbamoyl chloride (77.5 g) in acetone (300ml) was added to the reaction mixture and the temperature of the reaction mixture was raised to 55-600C followed by stirring at the same temperature for 8-10 hours. After the completion of the reaction, the reaction mixture was cooled and filtered. The solvent was then removed under reduced pressure, water (500ml) and toluene (300ml) were added followed by pH adjustment to 1 -2 using concentrated hydrochloric acid. After separating the organic layer, ethyl acetate (500ml) was added to the aqueous layer and pH was adjusted to 9-11 with liquid ammonia. The organic layer was separated and concentrated under reduced pressure to obtain oil. To this oily mass, acetone (750ml) and activated charcoal (5g) were added and stirred for 30 minutes. The reaction mixture was then filtered first through hyflo bed and then through micron filter. (L)-(+)-Tartaric acid was added to the filtrate and the reaction mixture was stirred for 4-5 hours and filtered. The solid thus obtained was washed with acetone and dried to obtain the title compound as solid
Example 7 Preparation of (S)-(-)-Rivastigmine Tartrate To a solution of 100 g of (S)-[1-(3-hydroxyphenyl)ethyl]dimethylamine (VIII) in 500 ml of acetone, pulverized potassium carbonate (125.2 g) was added. The reaction mixture was heated to 40-45 C. for 1 hour. A solution of N-ethyl-N-methylcarbamoyl chloride (77.5 g) in acetone (300 ml) was added to the reaction mixture and the temperature of the reaction mixture was raised to 55-60 C. followed by stirring at the same temperature for 8-10 hours. After the completion of the reaction, the reaction mixture was cooled and filtered. The solvent was then removed under reduced pressure, water (500 ml) and toluene (300 ml) were added followed by pH adjustment to 1-2 using concentrated hydrochloric acid. After separating the organic layer, ethyl acetate (500 ml) was added to the aqueous layer and pH was adjusted to 9-11 with liquid ammonia. The organic layer was separated and concentrated under reduced pressure to obtain oil. To this oily mass, acetone (750 ml) and activated charcoal (5 g) were added and stirred for 30 minutes. The reaction mixture was then filtered first through hyflo bed and then through micron filter. (L)-(+)-Tartaric acid was added to the filtrate and the reaction mixture was stirred for 4-5 hours and filtered. The solid thus obtained was washed with acetone and dried to obtain the title compound as solid
(1R)-N-prop-2-ynyl-2,3-dihydro-1H-inden-1-amine L-(+)-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; isopropyl alcohol;Reflux;Product distribution / selectivity;
Rasagiline free base (8.0 g), L-(+)-tartahc acid (2.10 g), methanol (56 mL) and isopropyl alcohol (40 mL) are placed in a flask and the mixture is stirred for 5- 15 minutes. The mixture is heated to reflux and maintained for 60-90 minutes under constant stirring. The mixture is then cooled to 25-35C and stirred for 60- 90 minutes. The solid obtained is filtered under vacuum, washed with isopropyl alcohol (8 mL), and dried for 6-8 hours at 60-700C to afford the title compound. HPLC purity is 99.94%
In methanol; isopropyl alcohol;Reflux;
Rasagiline free base (8.0 g), L-(+)-tartaric acid (2.10 g), methanol (56 mL) and isopropyl alcohol (40 mL) are placed in a flask and the mixture is stirred for 5-15 minutes. The mixture is heated to reflux and maintained for 60-90 minutes under constant stirring. The mixture is then cooled to 25-35 C. and stirred for 60-90 minutes. The solid obtained is filtered under vacuum, washed with isopropyl alcohol (8 mL), and dried for 6-8 hours at 60-70 C. to afford the title compound.HPLC purity is 99.94%
L-(+)-Tartaric acid (20gm, 0.13mol) is stirred with acetic anhydride (51.49gm, 0.5mol) and a catalytic quantity of sulphuric acid (96 %) at about 25-3O0C. The reaction is exothermic and the temperature raise to about 60-650C. Thereafter, the reaction mixture is heated to reflux and stirred at reflux temperature for 10 min. The reaction mass is concentrated at about 65-700C under reduced pressure and the remaining residue mass is co-evaporated with toluene (26mL).The residue is dissolved in methylene chloride (277ml) and racemic 10,l l-Dihydro-10- hydroxy-5H-dibenz[b,f]azepine-5-carboxamide (28.2gm, O.l lmol), pyridine (9.57gm, 0.12mol) and 4-dimethylaminopyridine (0.54gm, 0.004mol) are added to the solution. Thereafter, the reaction mixture is stirred at about 25-300C for forty minutes and then water (197mL) is added. The reaction mass is stirred at about 15-200C for about 12 hours. The precipitated solid is filtered, washed with water (2 x 28mL) and dried at 40- 45C under reduced pressure to afford the intermediate, diacetyl tartarate half-ester (26.1gm).
Example 14Preparation of (R)-<strong>[88150-42-9]Amlodipine</strong>-D-hemitartrate DMF solvate.D-tartaric acid (1.47gms; 0.01 moles) was added to the mother liquor obtained fromExample 1. and stirred at ambient temperature for 240-300 minutes, till complete precipitation of the product. The title compound was then filtered and dried.Yield: 6.4 gms.%Yield; 67.7%.
(R)-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In dimethyl sulfoxide; at 20℃; for 20h;
Racemic <strong>[112984-60-8]ulifloxacin</strong> (105 g) was dissolved in DMSO (1500 mL). L-tartrate (27 g) solution in DMSO (405 mL) was added to the racemic ulifoxacin solution with agitation. Cloudiness and precipitation appear. After 20 hours of agitation at an ambient temperature, the mixture was filtered and dried under vacuum to yield 82 g of solid. The solid was recrystallized in DMSO to yield 34 g of (R)- 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a] quinoline-3-carboxylic acid-L-tartrate salt. The salt was dispersed in water and the dispersion was neutralized with 2% NaOH aqueous solution to a pH value of 7 to 8. The precipitate was filtered and dried to yield 22 g of (R) -6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid. It has specific rotation [Show Image] (c=0.15, 0.1mol/L NaOH), optical purity e.e>95%.
In dimethyl sulfoxide; at 20℃; for 20h;Resolution of racemate;
Example 2 Preparation of (R)-(+)-uliflourxacin; 105 g of racemic uliflourxacin was dissolved in 1,500 mL of DMSO. 27 g of L-tartaric acid was dissolved in 405 mL dimethyl sulfoxide dropwise while stirring to allow that the solution became turbid and the precipitation occurred. The solution was stirred at room temperature for 20 hours and then filtered. The collected solid was dried under vacuum to obtain 82 g solid which was recrystallized in dimethyl sulfoxide to obtain 34 g of dextrouliflourxacin-L-tartarte. Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After filtration and drying, 22 g of (R)-uliflourxacin was obtained, having a chemical name (R)-(+)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline -3-carboxylic acid. Specific rotation [alpha]20D= +132.4 (c=0.5, 0.1 mol/L methanesulfonic acid), optical purity e.e. 96%.
(R)-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In dimethyl sulfoxide; at 20℃; for 20h;
Example 2; Preparation of (R)-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid (Compound 3); Racemic <strong>[112984-60-8]ulifloxacin</strong> (105 g) was dissolved in DMSO (1500 mL). L-tartrate (27 g) solution in DMSO (405 mL) was added to the racemic ulifoxacin solution with agitation. Cloudiness and precipitation appeared. After 20 hours of agitation at an ambient temperature, the mixture was filtered and dried under vacuum to yield 82 g of solid. The solid was recrystallized in DMSO to yield 34 g of (R)-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid-L-tartrate salt. The salt was dispersed in water and the dispersion was neutralized with 2% NaOH aqueous solution to a pH value of 7 to 8. The precipitate was filtered and dried to yield 22 g of (R)-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid. It had specific rotation [alpha]D20=+139.2 (c=0.15, 0.1 mol/L NaOH), optical purity e.e>95%.
(S)-3-[(1-dimethylamino)ethyl]phenyl-N-ethyl-N-methyl-carbamate L-(+)-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a solution of 100 g of S- [1 -(3 -Hydroxyphenyl)ethyl] dimethyl amine (XI) obtained in Example 1 1 in 500 ml of acetone, pulverized potassium carbonate (125.2 g) was added. The reaction mixture was heated to 55-60C for 1 hour. A solution of N-ethyl- N-methyl carbamoyl chloride (77.5 g) was added to the reaction mixture followed by stirring at the same temperature for 8-10 hours. After the completion of the reaction, the reaction mixture was cooled and filtered. The solvent was then removed under reduced pressure, water (500 ml) and toluene (500 ml) were added followed by pH adjustment to 1-2 using concentrated hydrochloric acid. After separating the organic layer, toluene (500 ml) was added to the aqueous layer and pH was adjusted to 9-11 with 10% sodium hydroxide solution. The organic layer was separated and concentrated under reduced pressure to obtain oil. To this oily mass, acetone (750 ml) and activated charcoal (5 g) were added and stirred for 30 minutes. The reaction mixture was then first filtered through hyflo bed and then through micron filter. L-(+ -tartaric acid was added to the filtrate and the reaction mixture was stirred for 4-5 hours and filtered. The solid thus obtained was washed with acetone and dried to obtain the title compound as solid.
[3'-([N-(tert-butoxycarbonyl)glycyl](methyl)amino}methyl)biphenyl-4-yl]methylmethanesulfonate[ No CAS ]
N-methyl-N-({4'-[(4-methyl-1,4-diazepan-1-yl)methyl]biphenyl-3-yl}methyl)glycinamide L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a solution of [3'-([N-(tert-butoxycarbonyl)glycyl](methyl)amino}methyl)biphenyl-4-yl]methylmethanesulfonate (163 mg) in DMF (1.6 ml) was added 1-methyl-1,4-diazepane (80 mg), followed by stirring at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and then purified by silica gel column chromatography (CHCl3/MeOH). The product was dissolved in MeOH (1.6 ml), and 4 M hydrogen chloride/EtOAc (0.8 ml) was added thereto, followed by stirring at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and then a saturated aqueous sodium hydrogen carbonate solution was added thereto, followed by extraction with CHCl3. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (CHCl3/MeOH). The product was dissolved in EtOH (2 ml), and L-tartaric acid (9 mg) was added thereto, followed by stirring at room temperature for 1 hour. The reaction mixture was concentrated under reduced pressure, and then EtOAc was added thereto. The precipitated solid was collected by filtration to obtain N-methyl-N-({4'-[(4-methyl-1,4-diazepan-1-yl)methyl]biphenyl-3-yl}methyl)glycinamide L-tartrate (16 mg).
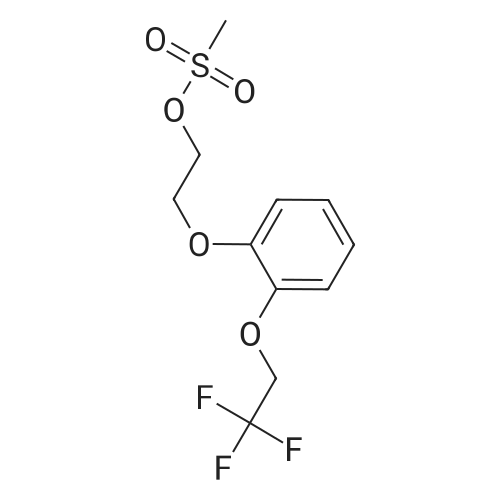
Example 1: Preparation of tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l- yl}propyl benzoate (IV) Method A: To the solution of 3-{7-cyano-5-[(2R)-2-aminopropyl]-2,3-dihydro-lH- indol-l-yl} propyl benzoate (II) (1 mole) in acetonitrile were added sodium carbonate (41.3 grams) and 2-(2-(2,2,2-trifluoroethoxy)phenoxy)ethyl methane sulfonate of formula (III) (79.6 grams) and heated to reflux temperature till reaction completion. After completion, reaction mass cooled to room temperature, water and ethylacetate added over it, stirred and layer separated. The obtained organic layer was washed with brine solution. To the organic layer, L (+) tartaric acid (29.2 grams) was added at room temperature and stirred. The solid obtained was filtered the solid and dried under vacuum at 50 - 55 C; Yield: 110 grams.
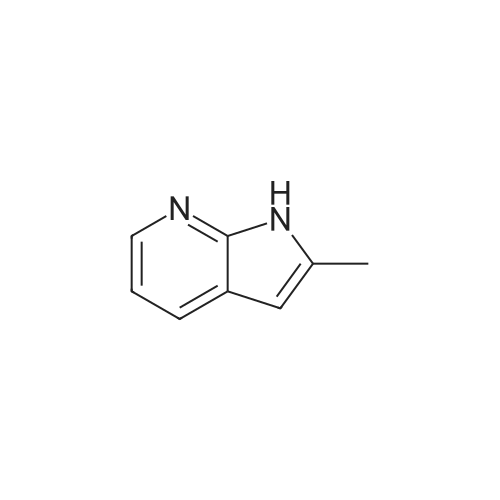
2-methyl-1H-pyrrolo[2,3-b]pyridin-7-ium L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
843 g
In ethanol; water; at 65℃; for 0.333333h;Large scale;
L-tartaric acid (505 g, 3.37 mol) was dissolved in EtOH (3.4 L) and H2O (140 mL) then heated to 65C. <strong>[23612-48-8]2-Methyl-1H-pyrrolo[2,3-b]pyridine</strong> (S4 crude, 1.5 kilos) was added over 20 minutes, then reaction diluted with 1:1 EtOH/iPrOAc (2 litres) and a further <strong>[23612-48-8]<strong>[23612-48-8]2-methyl-1H-pyrrolo[2,3-b]pyridin</strong>e</strong>(S4 crude, 1.5 kilos) added. The resulting yellow suspension was cooled to 0C, stirred for 2 hours thenfiltered to give <strong>[23612-48-8]2-methyl-1H-pyrrolo[2,3-b]pyridin</strong>-7-ium-L-tartrate (843 g, 90%yield), which was taken on directly to the next step.
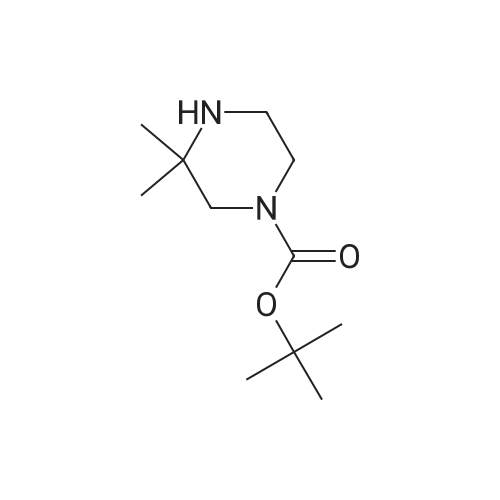
tert-butyl 3,3-dimethylpiperazine-1-carboxylate hemi-D,L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
27.1 kg
In ethanol; at 20 - 52℃;Inert atmosphere;
2,2-dimethylpiperazine (1 1 .5 kg, 101 mol) was dissolved in ethanol (48.5 L) and the solution was cooled to approximately 9C. Di-tert-butyl dicarbonate (21 .9 kg, 100 mol) was dissolved in ethanol (41 .7 L). The solution of di-tertbutyl dicarbonate was added to the solution of dimethylpiperazine over a period of 2 h 30 min, keeping the temperature of the reaction below 15C. Ethanol (12.4 L) was added and the solution was stirred overnight at a temperature between 12-25C. The reaction was warmed to reflux and 75 L were distilled off. Ethanol (76 L) was added to the reaction and the solution was heated to 52C and transferred to a suspension of D,L-tartaric acid (7.5 kg, 50.0 mol) in ethanol (25.2 L), and warmed to 51 C. Ethanol (25.3 L ) was added and the reaction was kept at 20C overnight. The precipitate was filtered off and washed with ethanol (28.1 L). The solid was dried in a vacuum oven at 50C overnight to yield compound (XVIII) (27.1 kg, 93%) with 99% purity according to GC analysis.
Step 5: (S)-4-((1-(5-(2-methoxyquinolin-1-ium-3-yl)-JH-imidazol-2-yl)- 7-(methylamino)- 7-oxoheptyl)carbamoyl)quinuclidin-1-ium mono L-tartrate salt (A5) A solution of <strong>[40117-63-3]4-carboxyquinuclidin-1-ium chlorhydrate</strong> (1.3 eq.) in DMF (0.2 M) was treated with TBTU (1.3 eq.) and NMM (2.6 eq.). The reaction mixture was stirred at room temperature for 10 minutes and then added to a solution of A4 in DMF (0.2 M). The reaction was stirred at RT for 2 h and subsequently was purified by RP-HPLC (Acetonitrile/H20 + 0.1 % TFA). Theproduct was obtained as TFA salt which was partitioned between DCM and sat. aq. NaHCO3. The organic phase was separated, dried over Na2504 and concentrated under reduced pressure. The resulting syrup was dissolved in acetonitrile/H20 (2:3) and treated with L-tartaric acid (1 eq.). The resulting solution was lyophilized to obtain the title compound. 1H-NMR (400 MHz, 300 K, DMSO-d6) oe 8.73 (br s, 1H), 7.94 (t, 2H, J 9.6 Hz), 7.76 (d, 1H, J 8.0 Hz), 7.67 (br s,1H), 7.60 (m, 2H), 7.42 (t, 1H, J8.0 Hz), 5.02 (m, 1H), 4.13 (s, 3H), 3.93 (s, 2H), 3.18 (t, 6H, J7.2 Hz), 2.54 (d, 3H, J4.4 Hz), 2.05-1.91 (m, 8H), 1.51-1.23 (m, 8H). MS (ESj C29H38N603:519 (M+H).
Step 5: (S)-4-((1-(5-(6-methoxynaphthalen-2-yl)-JH-imidazol-3-ium-2-yl)- 7-(methylamino)- 7-oxoheptyl)carbamoyl)quinuclidin-1-ium mono L-tartrate salt (E5) A solution of <strong>[40117-63-3]4-carboxyquinuclidin-1-ium chlorhydrate</strong> (1.3 eq.) in DMF (0.2 M) was treated with TBTU (1.3 eq.) and NMM (2.6 eq.). The reaction mixture was stirred at room temperature for 10 mm and then added to a solution of E4 in DMF (0.2 M). The reaction was stirred at RT for 2 h, then filtered and purified by RP-HPLC (CH3CN/H20 + 0.1 % TFA). The product was obtained as TFA salt which was partitioned between EtOAc and sat. aq. NaHCO3. The organicphase was separated, dried (Na2504) and concentrated under reduced pressure. The resulting pale yellow solid was dissolved in acetonitrile/H20 (1:1) and treated with L-tartaric acid (1 eq.). The resulting solution was lyophilized to obtain the title compound. (400 MHz, 300K, DMSOd 6) oe: 11.5 (br s, 1H), 8.14 (br s, 1H), 7.95-7.73 (m, 4H), 7.66 (m, 1H), 7.54 (m, 1H), 7.27 (d, 1H, J2 Hz), 7.12 (dd, 1H, J8.8 and 2.4 Hz), 4.99 (m, 1H), 3.89 (s, 2H), 3.86 (s, 3H), 3.16 (m,6H), 2.54 (d, J4.4 Hz), 3H), 2.02 (t, 2H, J7.4 Hz), 2.01 (m, 6H), 1.83 (m, 2H), 1.48 (m, 2H),1.35-1.15 (m, 4H). MS (ESj C30H39N503: 518 (M+H).
Step 8: (S)-4- ((1- (5- (2-methoxyquinolin-1-ium-3-yl)oxazol-2-yl)- 7- (methylamino)- 7-oxoheptyl)carbamoyl)quinuclidin-1-ium mono L-tartrate salt (F8)A solution of <strong>[40117-63-3]4-carboxyquinuclidin-1-ium chlorhydrate</strong> (1.3 eq.) in DMF (0.1 M) was treated with HOBt (1.3 eq.), EDC HC1 (1.3 eq.) and DIPEA (1.3 eq.). The reaction mixture was stirred at room temperature for 10 minutes and then added to F7. The reaction was stirred for at RT for 48 h, filtered and directly purified by RP-HPLC (Acetonitrile/H20 + 0.1 % TFA). The product was obtained as TFA salt which was partitioned between DCM and sat. aq. NaHCO3. Theorganic phase was separated dried over Na2504 and concentrated under reduced pressure. The resulting solid was dissolved in acetonitrile/H20 (1:1) and treated with L-tartaric acid (1 eq.). The resulting solution was lyophilized to obtain the title compound. MS (ESj C29H37N504: 520 (M+H).
9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine tartarate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1 g
In acetonitrile;Reflux;
To a solution of 9-[(R)-2-[[(S)-[[(S)-l-(isopropoxycarbonyl) ethyl] amino] phenoxy phosphinyl] methoxy] propyl] adenine (lgm, 0.0021 mol) in acetonitrile (20 gms), L- tartaric acid (0.28 gms, 0.0019 mol) was added and the reaction mixture was heated to reflux to dissolve the solids. The reaction mass was filtered in hot condition, filtrate cooled to 5°C and maintained for 16 hours. The product was isolated by filtration, rinsed with acetonitrile (6 gms), and dried to afford 1.0 g of Tartarate as a white crystalline powder. HPLC purity: 99.42percent The XRPD is set forth in Figure 07.
(R)-2-(2-aminothiazol-4-yl)-4’-[2-[(2-hydroxy-2-phenylethyl)amino]ethyl]acetanilide monotartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
In 2-methyltetrahydrofuran; at 20 - 25℃; for 1h;
Example 9: Preparation of Mirabegron Monotartrate SaltTo Mirabegron free base (3.0 g, 0.01 moles) was added 2-methyltetrahydrofuran (30 mL) and DL-tartaric acid (1.1 g, 0.01 moles). Themixture was stirred at 20-25 C for 1 hour and the obtained suspension was filtered, washed with 2-methyltetrahydrofuran (1 x 6 mL) and dried to afford Mirabegron monotartrate salt (3.4 g, 83% yield). A PXRD diffractogram of this sample is shown in Figure 5. 1H NMR (300 MHz, DMSO-d6): O 10.0 (s, 1H), 8.8-7.8(bs, IH), 7.6-7.5 (m, 2H), 7.5-7.3 (m, 5H), 7.3-7.1 (m, 2H), 6.9 (s, 2H), 6.3 (s, IH), 4.8-4.6 (m, 1H), 4.1Hz (s, 2H), 3.4 (s, 2H), 3.2-2.8 (m, 6H).
(3R)-3-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70%
In ethanol; at 50℃; for 1h;
200 mg (0.653 mmol) of (3R)-3-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-l- yl]propanenitrile was dissolved in 8 mL of ethanol by heating to 50C applying a continuous agitation. 98 mg (0.653 mmol) of L-tartaric acid was dissolved in 2 mL of ethanol at room temperature. The ethanol solution of L-tartaric acid was drop-wise added to the solution of (3R)-3- cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazoi-l-yl]propanenitrile at 50C, agitated for additional 60 minutes at 50C and then cooled back to room temperature. The suspensions was stirred at room temperature overnight and then filtered and dried with vacuum suction. Product: 211 mg; yield: 70% HPLC purity: 90.0% The sligthly yellowish powder obtained was analyzed by FTIR spectroscopy. Similarly, the same result was obtained using any of the recrystallization solvents listed in the Table 10.
9-[(R)-2-[[(S)-[[(S)-1-(isopropoxycarbonyl)ethyl]amino](phenoxy)phosphinyl]methoxy]propyl]adenine L-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
520 mg
In ethyl acetate;Reflux;
The TAF (476mg, 1.0mmol) and tartaric acid (150mg, 1.0mmol) was suspended in ethyl acetate (10.0 ml of) the solution was heated at reflux until the solids dissolve, hot filtration of the undissolved particles, an appropriate amount of n-hexane was added until cloudiness clarification and still appears after heating slowly cooled to room temperature, and overnight at this temperature, product was isolated by filtration, washed with cold ethyl acetate and n-hexane (V / V = 1: 1) mixed solution was washed, and dried in vacuo to give 520mg of a white solid powder.
9-[(R)-2-[[(S)-[[(S)-1-(isopropanoyl)ethyl]amino]phenoxyphosphinyl]methoxy]propyl]adenine hemitartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
97%
With ethanol; In butanone; at 75 - 80℃;
To a reactor equipped with a stirrer was added 9 - [(R) -2 - [[(S) - [[((S) -1- (isopropoxycarbonyl) ethyl] amino] phenoxyphosphinyl] Methoxy] propyl] adenine (5.00 g), L-tartaric acid (1.56 g), ethanol (30 ml) and butanone (10 mL). The mixture was heated to 75 ° C to 80 ° C to dissolve the solid. The filtrate was cooled to 0 ° C to 5 ° C over 4 hours. The temperature was maintained at 1-18 hours, and the resulting slurry was filtered and washed with 0.2 mL of butanone (0 ° C to 5 ° C). The solid was dried under vacuum at 50 & lt; 0 & gt; C to give 6.35 g of tenofovir &Yield: 97percent, HPLC purity 99.79percent (area normalization),
200 mg
In ethyl acetate;Reflux;
The TAF (476mg, 1.0mmol) and tartaric acid (75.0mg, 0.5mmol) was suspended in ethyl acetate (5.0ml) solution was heated to reflux, hot filtered undissolved particles, an appropriate amount of n-hexane was added until cloudy after reheated to clarify and still slowly cooled to -5 ~ 0 , and overnight at this temperature, the product was isolated by filtration, washed with cold ethyl acetate and n-hexane (V / V = 1: 1) was washed with a mixed solution, and dried in vacuo to give 200mg of a white solid powder.
N-[2-hydroxy-2-(2,3-dihydrobenzo[β][1,4]dioxin-6-yl)-1-pyrrolidin-1-ylmethylethyl]-3-(4-methoxyphenoxy)propionamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65.7%
compound 163 was prepared according to the above scheme 1A. 3- (4-methoxyphenoxy) propanoic acid (Example1D1,34.47g, 169mmol, 96% purity by HPLC), DCC (34.78g, 169mmol ) and N-hydroxysuccinimide (19.33,169mmol) combining as dry powder, wasadded methylene chloride (500 mL). The suspension mechanically overnight at ambient temperature and stirred under a nitrogen atmosphere. HPLCanalysis showed complete conversion of acid to NHS ester (N-hydroxy succinyl ester). Mixture to Step 5 of the amine (50 g, 169 mmol) was addedand stirring continued for 2.5 hours. HPLC showed conversion and loss of both amines of NHS ester and step 5 to product. The reaction mixture wasvacuum filtered through a Buchner funnel to remove DCC urea. The solid urea was washed with 500mL of methylene chloride. The organic layerswere combined, placed in a separatory funnel and treated with 1.0 M NaOH in 500 mL. The layers were separated, again placed in a separatoryfunnel and the organic layer was set, and treated with. 6% HCl solution (adjusted to pH = .03-0.34, a solution of 100 mL). Two clear layers areformed. The resulting two-phase solution was poured into Erlenmeyer flask, was carefully neutralized to pH7.2-7.4 with a saturated solution of sodiumbicarbonate (solution of about 200mL). The organic layer was separated from the aqueous layer, dried over sodium sulfate and evaporated to give ayellow oil of 83.6 g (theoretical yield: 77.03g). In isopropyl alcohol (500 mL), heated by dissolving the oil was transferred to a 1L round bottom flaskequipped with a mechanical stirrer and heating mantel. The solution was heated to 50 , you set the machine agitator speed 53-64rpm. Tartaric acid(25.33g, 168mmol) was dissolved in deionized water (50 mL), it was added at 50 C. To the stirred solution. When the solution is changed totransparent from milky white, adding a seed crystal to the mixture, crystallization began immediately (temperature jumps to 56 ). After 20 minutes,the mixture was set to cool to a temperature 35 C. (The cooling takes 1.15 hours). Remove the heating and the solution was stirred for 12 hours.The resulting dark slurry was filtered through a Buchner funnel. Use of ice-cold isopropyl alcohol (100 mL), and any solid remaining in the flask waswashed on the funnel. The material was transferred to a drying tray, 3 days under vacuum, heated to 48 (material weight after two days is 76g, theweight after three days was 69.3g). The solid place was analyzed by LC indicates a purity of 98.1% (AUC), residual solvent analysis showed that thematerial has a isopropyl alcohol 3472ppm, DSC (differential scanning calorimeter), the dissolution point 134. 89 showed . It was collected all whitesolid 69.3g (overall yield 65.7%).
3-(7-cyano-5-[(2R)-2-(2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino)propyl]-2,3-dihydro-1H-indol-1-yl)propylbenzoate (2R,3R)-tartrate salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
79.3%
40 g of 3-5-[(2R) -2-aminopropyl] -7-cyano-2,3-dihydro-1H-indol-1-yl propylbenzoate (2R, 3R) -tartrate,Acetonitrile 320 ml,20.6 g of anhydrous sodium carbonate, 26.9 g of 2- [2- (2,2,2-trifluoroethoxy) phenoxy] ethyl methanesulfonate was added and stirred under reflux for 24 hours.The reaction solution was cooled, filtered, concentrated under reduced pressure, and 400 ml of ethyl acetate and 320 ml of purified water were added to the concentrate, followed by stirring, separating the layers, and concentrating the organic layer under reduced pressure.To the concentrate, 240 ml of ethanol and 10.5 g of L-(+)-tartaric acid were added, stirred at room temperature for 5 hours, filtered and dried under reduced pressure to make 3-7-cyano-5-[(2R) -2- (2- [ 2- (2,2,2-trifluoroethoxy) phenoxy] ethyl amino) propyl] -2,3-dihydro-1H-indol-1-yl propylbenzoate (2R, 3R) -tartrate Obtained 45.1 g. (Yield 79.3%, ee value = 99.97% or more).
4.6 g
To the Butyl acetate solution of N- alkylated indoline derivative obtained in the reaction step, 35mL of butyl acetate and tartaric acid 1.3 g were added and stirred, and then heated to around 80 C andstirred. After confirming that the tartaric acid was dissolved, then cooled toabout 20 C and stirred for 2 hours. The precipitated solid was collected by filtration, and the resulting wet material was dried under reduced pressure in the vicinity of 40 , to obtain N- alkylated indoline derivative tartrate salt4.6g (purity 96.1%, dialkyl body content 0. 38%) (64.7% yield from the indoline derivative tartrate salt) as a white crystalline.
(S)-dexmedetomidine-L-(+)-tartaric acid salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77.1%
In ethanol; isopropyl alcohol; at 20℃;Reflux;
L- (+) - tartaric acid (9 g, 60 mmol)Was added to a solution of metomidol (12 g, 60 mmol) in ethanol (250 ml).The suspension was heated to reflux until complete dissolution,Followed by stirring at room temperature overnight,Filtration gave a white solid (9 g).The resulting solid was heated to reflux in isopropanol (200 ml)Followed by stirring at room temperature overnight,Filtration gave (13.5 g).The obtained solid was purified once more in the same manner,To obtain a solid 8. lg, purity 99.9%, yield 77.1%.
(3β)-17-(1H-benzimidazol-1-yl)androst-5,16-dien-3-ol tartarate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetone; at 25℃; for 12h;
10.5 mg of the compound of the formula (I) was suspended in 0.5 mL of acetone, 5.8 mg of tartaric acid was added and the mixture was incubated at room temperature (25 ± 2 ° C)The reaction was stirred for 12 hours and the solid was collected.
(1R,2S)-1-(6-bromo-2-methoxy-3-quinolyl)-4-dimethylamino-2-(1-naphthyl)-1-phenylbutan-2-ol tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
34%
In acetonitrile;
( 1 R,2S)- 1 - (6-Bromo-2-methoxy-3 -quinolyl)-4-dimethylamino-2-( 1 -naphthyl)- 1 -phenyl-butan- 2-ol in the amount of 100 mg (0.18 mmol) was suspended in 4 ml of acetonitrile. 27 mg (0.18 mmol) of tartaric acid was added to this suspension. A white solid substance was separated during slow evaporation of the solvent at the room temperature. Yield 43 mg (34%). Melting point 103C (DSC). XRPD: Fig. 10
The temperature of the solution was raised to 40 C and 150 g of L-tartaric acid was added. The mixture was stirred at 40 C for 1h and then at 20 C 20h, precipitation of a large number of solid, filtration, filter cake with cold isopropyl alcohol (100ml) wash, 40 C vacuum drying 8h, you can get white powderL-tartaric acid salt of the compound X in the form of 302. 6g.
To the reaction flask was added AZD9291 (5 g, 10 mmol, 1.0 eq), 50 mL of acetone was added and dissolved in an aqueous solution (5 mL) of tartaric acid (1.8 g, 12 mmol, 1.2 eq) under stirring to precipitate a pale yellow solid , Filter, filter cake with a small amount of acetone leaching, drying, 5g (7.7mmol) light yellow granular solid, molar yield of 77%.The test: purity 99.9%;
(3R)-3-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile L-tartaric acid salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
79%
In ethanol; at 50℃;
1g (3.264 mmol) of <strong>[941678-49-5]ruxolitinib</strong> is suspended in 8 mL of acetonitrile at 50C applying a continuous stirring. The solution of 0.54 g of L-tartaric acid (3.6 mmol)in 2 mL ethyl-acetate is drop-wise added to the suspension of <strong>[941678-49-5]ruxolitinib</strong> at 50C, while continuously stirred, forming a clear solution. The solution is further stirred at 50C for additional 1 hour, while precipitation occurred.The suspension formed was cooled back to room temperature and stirred overnight at that temperature. The solid precipitated is collected by filtration and dried at laboratory condition Product: 1.18 g (2.587 mmol) off-white crystalline solid Yield: 79% HPLC: 99.4% XRPD pattern was measured (Figure 24) and showed that the compound is in a crystalline state that was designated as Crystal modification 5 of <strong>[941678-49-5]ruxolitinib</strong> L-tartaric acid salt.1H-NMR spectrum was measured (Figure 29) and showed that the compound confirms the structure with an API: L-tartaric acid stoichiometry of 1:1.5
Crystalline free base of 4-[3 -chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxy-quinoline-6-carboxamide (form B) in the quantity of 198 mg (4.64 i04 mol) together with73.84 mg (4.87.1 mol) of L-(+)-tartaric acid was dissolved in 3.76 ml of methanol in a hot state. The solution was cooled down to the room temperature under continuous stirring and the mixture was stirred at the laboratory temperature for 12 h. Then, the solvent was left to evaporate in a vacuum drier at the room temperature. UHPLC purity 99.9%. X-ray powderpattern in Fig. 2. Melting point in accordance with DSC 169C.
20 g (100 mmol) of 4- [1- (2,3-dimethylphenyl) ethyl] -1H-imidazole and 9 g (70 mmol) of L - (+) - tartaric acid were added to 80 ml of absolute ethanol,Heated to 65 stirring,After the reaction was dissolved to continue stirring for two hours,Then cooled to 10 C,Stir overnightAt this time a large number of white solid,Suction filtration,The filter cake was washed with absolute ethanol,Get <strong>[113775-47-6]dexmedetomidine</strong> tartrate 16.6g,Yield 47.3%The ee value is 99.24%.
The free amine product of the previous step 1 was dissolved in 300 ml of absolute ethanol,(L) -tartaric acid ethanol solution (28ml, 0.028mol) was added within 5 minutes. After the addition was completed, the reaction mixture was refluxed for 2 hours. The mixture was slowly cooled down and the crystals were lyophilized to be completely crystallized and filtered. The cake was washed with anhydrous ethanol 100ml x 2) and dried to give 5.9g light yellow solid in 60% yield.
110 mL of acetonitrile was added to Compound 7 (25 g, 0.13 mol), heated to reflux,L-tartaric acid (15.2 g, 0.104 mol) was added portionwise and after refluxing for 2 hours after addition,Slowly reduced to 10 C (within 1.5 hours) and stirred at this temperature overnight. After filtering off the solids,The solid was added with ethanol 85mL, heated to reflux and stirred for 1 hour,After cooling to room temperature crystallization 8 hours, filtered off the solid,And dried under reduced pressure (45 C) to give 16.4g as an off-white solid,The obtained solid was added with 50 mL of ethyl acetate, and 30 mL of 0.1 M sodium hydroxide solution was added thereto,After stirring for 20 minutes, the mixture was extracted twice with ethyl acetate (2 * 50 mL)The organic phase was washed once with saturated citric acid (100 mL), the organic phase was dried over anhydrous sodium sulfate,The organic phase was evaporated under pressure to give a pale yellow oil 8.7g, dichloromethane was added 60mL,Di-tert-butyl dicarbonate(10.8 g, 0.05 mol). Triethylamine (5.70 g, 0.057 mol) and DMAP (0.460 g, 3.80 mmol) were added and the mixture was reacted at room temperature for 8 hours. The reaction mixture was washed with saturated bicarbonate , Dried over anhydrous sodium sulphate and the organic phase was autoclaved to give 12.1 g of light yellow solid.
5 g of 4-amino-5-chloro-2,1,3-benzothiadiazole and 6.07 g of <strong>[64205-92-1]imidazoline-2-sulfonic acid</strong> were weighed out and added to 25 ml of sec-butanol, and 12.13 g of tartaric acid was added. The temperature was raised to 95 to 100°C, and the reaction was stirred for 8 hours. Add 25 mg of activated carbon, stir for about 30 minutes, and filter while still hot. The filtrate was cooled to 20-25°C and stirred for 2 hours. The mixture was filtered, the cake was added to 60 ml of saturated ethanolic hydrogen chloride solution, stirred for 4 hours, and filtered. The filter cake was washed with anhydrous ethanol and dried. The filter cake was added 65 ml of isopropanol, refluxed to complete dissolution, natural cooling and crystallization, filtration, the filter cake was washed with isopropanol, and dried to obtain solid tizanidine hydrochloride 6.82g, the yield of 87.3percent, the purity of 99.99percent .
General procedure: Stage I (0144) Table 11 illustrates the selected counter ions for the salt screening of beta-GPA. Salt screening experiments were designed in 1:1.1 equivalence (eq) for beta-GPA to counter ion. [table-us-00011-en] TABLE 11 List of selected counterions beta- Counterion Counterion GPA sequence molecular Sample ID (mg) Counterion wt 2162-42-1 to 4 30 Hydrochloric 1 36.46 acid (36-38%)* 2162-42-5 to 8 30 Hydrobromic 2 80.91 acid (48%)* 2162-42-9 to 12 30 Sulfuric acid 3 98.08 (95-98%)* 2162-42-13 to 16 30 Phosphoric acid 4 98.00 (85%)* 2162-42-17 to 20 30 Methane sulfonic 5 96.11 acid (98%)* 2162-42-21 to 24 30 Maleic acid 6 116.07 2162-42-25 to 28 30 Fumaric acid 7 116.07 2162-42-29 to 32 30 Tartaric acid 8 150.09 2162-42-33 to 36 30 Ethanesulfonic 9 110.13 acid 2162-42-37 to 40 30 Ethanedisulfonic 10 190.20 acid 2162-42-41 to 44 30 Citric acid 11 192.12 2162-42-45 to 48 30 Malic acid 12 134.09 2162-42-49 to 52 30 Lactic acid 13 90.08 2162-42-53 to 56 30 Aspartic acid 14 133.1 2162-42-57 to 60 30 Succinic acid 15 118.09 2162-42-61 to 64 30 Sodium 16 40.00 hydroxide 2162-42-65 to 68 30 Potassium 17 56.11 hydroxide 2162-42-69 to 72 30 Oxalic acid 18 90.03 2162-45-1 to 4 30 Magnesium 19 58.32 hydroxide 76 salt screening experiments of beta-GPA with 19 different counter ions were set up with 30 mg of beta-GPA. Sets of four vials for each counterion were set up with four different solvents (0.3 mL): ethanol:water (9:1), isopropanol, acetone:water (9:1) and acetonitrile. (0146) Appropriate amounts of beta-GPA and the counterion were dissolved in the respective solvents and heated to 70-75 C. until dissolved. An additional 0.1 mL of water was added to the samples containing isopropanol, acetone:water (9:1) and acetonitrile. To samples containing L-aspartic acid, around 1.5 mL of water was required to dissolve the solids. After a clear solution was obtained, the samples were left for stirring at room temperature. Solids were observed in the following samples: 2163-42-4, 25, 26, 27, 28, 45 and 53 through 75. The solids were filtered and analyzed by XRPD immediately as wet sample. The samples that did not yield solids were placed in the oven at 50 C. for drying. The following samples resulted in solids after overnight drying: 2162-42-2, 1, 2, 3 and 21 through 24. The experiments with L-aspartic acid, sodium hydroxide, potassium hydroxide, and magnesium hydroxide resulted in the precipitation of either beta-GPA or the counterion. All the experimental observations were recorded after every step and are listed in Table 12. [table-us-00012-en] TABLE 12 Results of Salt screening Sample ID Counterion Solvent After 24 hours After Drying XRPD 2162-42-1 Hydrochloric EtOH:H2O (9:1) Clear Solution White Solid Pattern 1A 2162-42-2 Acid IPA Clear Solution White Solid 2162-42-3 Acetone:H2O (9:1) Clear Solution White Solid 2162-42-4 MeCN White Solid N/A 2162-42-5 Hydrobromic EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-6 Acid IPA Clear Solution Gel N/A 2162-42-7 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-8 MeCN Clear Solution Gel N/A 2162-42-9 Sulfuric Acid EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-10 IPA Clear Solution Gel N/A 2162-42-11 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-12 MeCN Clear Solution Gel N/A 2162-42-13 Phosphoric Acid EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-14 IPA Clear Solution Gel N/A 2162-42-15 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-16 MeCN Clear Solution Gel N/A 2162-42-17 Methanesulfonic EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-18 Acid IPA Clear Solution Gel N/A 2162-42-19 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-20 MeCN Clear Solution Gel N/A 2162-42-21 Maleic Acid EtOH:H2O (9:1) Clear Solution White Solid Pattern 6A 2162-42-22 IPA Clear Solution White Solid 2162-42-23 Acetone:H2O (9:1) Clear Solution White Solid 2162-42-24 MeCN Clear Solution White Solid 2162-42-25 Fumaric Acid EtOH:H2O (9:1) White Solid N/A Pattern 7A 2162-42-26 IPA White Solid N/A 2162-42-27 Acetone:H2O (9:1) White Solid N/A 2162-42-28 MeCN White Solid N/A 2162-42-29 L-Tartaric Acid EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-30 IPA Clear Solution Gel N/A 2162-42-31 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-32 MeCN Clear Solution Gel N/A 2162-42-33 Ethanesulfonic EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-34 Acid IPA Clear Solution Gel N/A 2162-42-35 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-36 MeCN Clear Solution Gel N/A 2162-42-37 Ethanedisulfonic EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-38 Acid IPA Clear Solution Gel N/A 2162-42-39 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-40 MeCN Clear Solution Gel N/A 2162-42-41 Citric Acid EtOH:H2O (9:1) Clear Solution Gel N/A 2162-42-42 IPA Clear Solution Gel N/A 2162-42-43 Acetone:H2O (9:1) Clear Solution Gel N/A 2162-42-44 MeCN Clear Solution Gel N/A 2162-42-45 L-Malic Acid EtOH:H2O (9:1) White Solid N/A Pattern 12A 2162-42-46 IPA Clear Solution Gel N/A 2162-42-47 Acetone:H2O (9:1) Clear Solution Gel N/...
8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one L-(+)-tartrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
3.82 g
In tetrahydrofuran; water; at 40 - 50℃;
4g of <strong>[283173-50-2]rucaparib</strong> base (12.36 mmol), prepared according to the process described in Example 5, was dissolved in 60 mL of THF/H20 mixture (2: 1) while heating at 40-50°C. To the clear filtered solution of <strong>[283173-50-2]rucaparib</strong> base, 3,72 g (2 eq) of L-(+)-tartaric acid dissolved in 15 mL of water, was added while stirring at 40-50°C. Clear reaction mixture was then cooled down and crystallization occurred while stirring at 20-25°C. Obtained suspension was additionally stirred for 1 hour at 0-5°C. Crystals were filtered off under vacuum, washed with 2*20 mL H20 and analyzed by XRPD as Form I of <strong>[283173-50-2]rucaparib</strong> L-(+)-tartarate. Wet crystals were dried in vacuum oven at 50°C for 4 hours to obtain 3.82 g of material that was analyzed by XRPD as Form II of <strong>[283173-50-2]rucaparib</strong> L-(+)-tartrate.
(2R,3R)-2,3-dihydroxybutanedioic acid (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In water; at 20℃;
A sample of the Amorphous Freebase of <strong>[1310726-60-3]upadacitinib</strong> (28.2 mg) was transferred to an amber vial. Water (200 .iL) and L-tartaric acid (34.5 mg (approximately 3 equivalents)) were added to the vial. The suspension was vortexed under ambient conditions until all the solids dissolved. The solution in the vial was magnetically stirred at 0 C. The following day, the solids were isolated from the solution and left at ambient temperature for a short period of time prior to characterization. Conversion to the <strong>[1310726-60-3]upadacitinib</strong> Tartrate Hydrate (tetrahydrate) was confirmed by PXRD analysis.
3-(7-cyano-5-[(2R)-2-(2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino)propyl]-2,3-dihydro-1H-indol-1-yl)propylbenzoate (2R,3R)-tartrate salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
717 g
General procedure: To a solution of SI-II (531 g, 1.46 mol) as the free base in tert-butanol (2.5 L) was added IM-A (528 g, 1.68 mol) and sodium carbonate (92.9 g, 0.876 mol), all in a round bottom flask (5 L). The reaction mixture was heated under reflux until TLC (dichloromethane/methanol, 20/1) showed the reaction to be complete. After cooling, the mixture was extracted with ethyl acetate twice (2 L 2). The combined organic layer was washed with brine, dried over sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure and iso-propanol (4 L) was added. To the solution was added an aqueous solution of L-tartaric acid (329 g, 2.19 mol) with stirring for 2 h at room temperature.The precipitate was collected and washed with iso-propanol/water (100 ml/100 ml) twice and recrystallized from iso-propanol/water (2.6 L/1.3 L) to give SI-IV tartrate(717 g, 67.2%, HPLC 98.5%) as a white solid. SI-III (free base): 1H NMR(400 MHz, CDCl3): d 1.01 (d, 3H), 2.07 (t, 2H), 2.40 (m, 1H), 2.56 (m, 2H), 2.63 (m,2H), 2.97 (m, 2H), 3.49 (t, 2H), 3.67 (t, 2H), 4.03 (m, 2H), 4.30 (m, 2H), 4.39 (t, 2H),6.81-6.96 (m, 6H), 7.36 (m, 2H), 7.47 (m, 1H), 7.91 (m, 2H), 8.01 (d, 2H). The motherliquor was purified by column chromatograph to give the IM-C as a yellow oil.15 IMC:1H NMR (400 MHz, CDCl3): d 1.03 (d, 3H), 2.13-2.38 (m, 5H), 2.82 (t, 2H), 3.03(m, 4H), 3.51 (t, 2H), 3.72 (t, 2H), 4.00 (m, 4H), 4.35 (q, 4H), 4.48 (t, 2H), 6.88-7.04(m, 12H), 7.45 (m, 2H), 7.55 (m, 1H), 8.08 (d, 2H). These NMR data agreed with theliterature values.
With sodium hydroxide; In methanol; water; at 90℃; for 2h;
Dissolve 2.0 g of tartaric acid (purchased from Tishila (Shanghai) Chemical Industry Development Co., Ltd.) in 50 mL of methanol, add the equivalent of NaOH, stir at room temperature until a white solid precipitates, filter with suction and wash with 30 mL of methanol three times to obtain sodium tartrate . Sodium tartrate is dissolved in 50mL of water to prepare an aqueous solution of sodium tartrate (A); 5.6g of <strong>[60372-77-2]lauroylarginine ethyl ester hydrochloride</strong> is dissolved in 40mL of water and heated to 90 C until the lauroylarginine ethyl salt The acid salt was completely dissolved to prepare an aqueous solution of lauryl arginine ethyl ester hydrochloride (B); the sodium tartrate aqueous solution (A) was slowly added to the aqueous solution of lauryl arginine ethyl ester hydrochloride at 90 C ( In B), the mixture is continuously stirred and reacted for 2 hours, cooled to room temperature, filtered, and the precipitate is sufficiently washed with pure water, and the precipitate is dried under vacuum at 60 C. to obtain 6.3 g of a tartaric acid ion-pair compound.
The cocrystal was obtained by liquid-assisted grinding of amixture of CBZ (122 mg) and dl-TA (78 mg) at 30 kHz for15 min, with the addition of methanol (20 ml) to the mixture.DSC (Q1000, TA Instruments, 5 Cmin1) reveals the meltingof the material at Tm = 168 C.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping