* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium ethanolate In ethanol at 20 - 90℃; for 48 h;
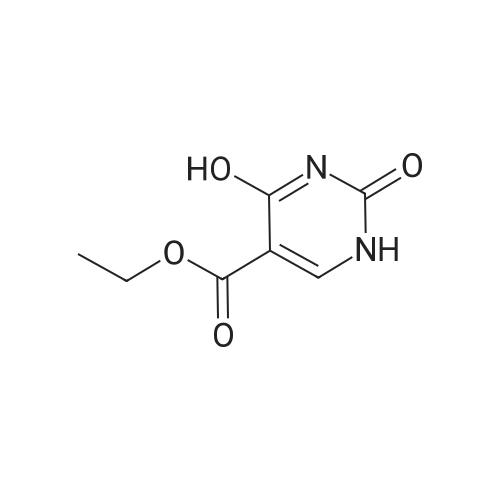
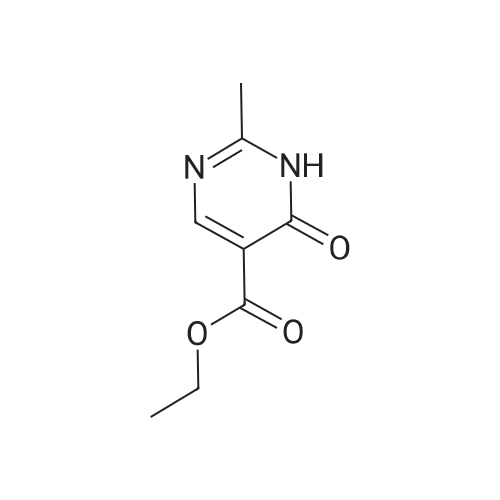
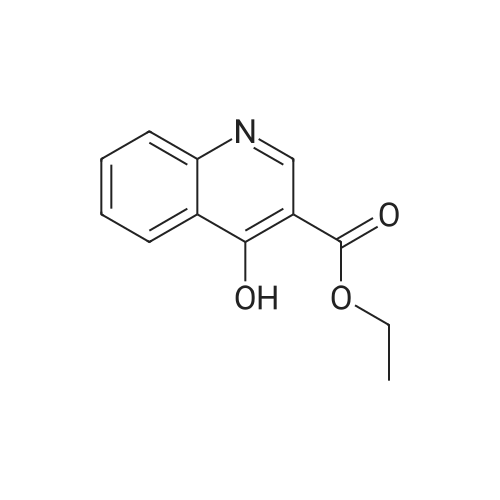
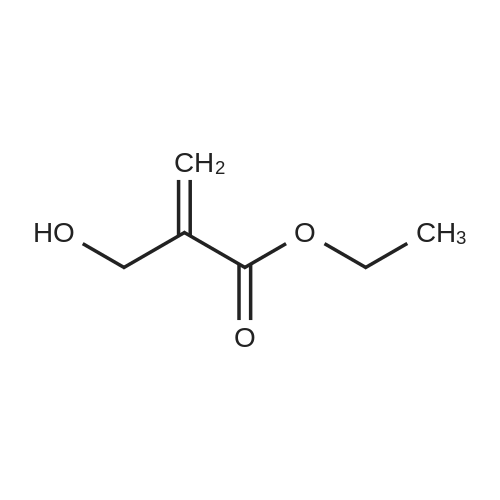
Step 1. Urea (12 g, 200 mmol) was added to EtOH (300 mL) containing NaOC2H5 previously prepared from sodium (5.52 g, 240 mmol, 1.2 eq). Diethyl 2-(ethoxymethylene)malonate (43.2 g, 200 mmol, 1.0 eq) was added and the solution was stirred at 2O0C for 24h, and then stirred at 90 0C for 24h. The alcohol was removed by reduced pressure distillation. Ice- water (100 mL) was added to dissolve the residue. The product was precipitated by adding cold dilute hydrochloric acid. The solid was filtered off to afford ethyl 2,4-dioxo-l,2,3,4-tetrahydropyrimidine-5- carboxylate (8.5 g, 23percent), which was used in next step without further purification.
Reference:
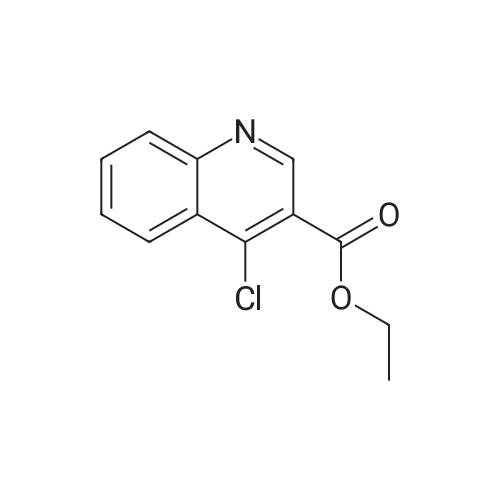
[1] Journal of the American Chemical Society, 1942, vol. 64, p. 794,797
[2] Journal of the American Chemical Society, 1952, vol. 74, p. 4267,4269
3
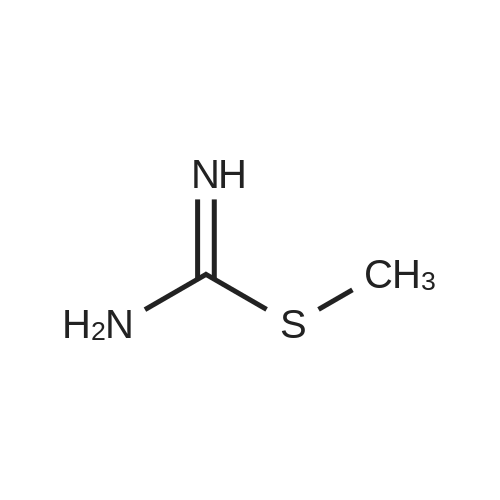
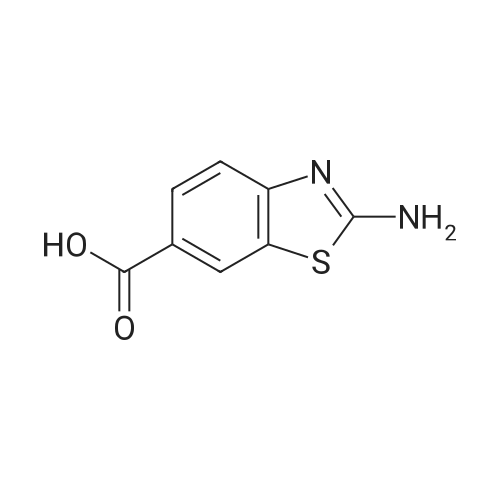
[ 17284-97-8 ]
[ 87-13-8 ]
[ 28593-24-0 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 1980, vol. 17, # 6, p. 1527 - 1529
[2] Journal of Heterocyclic Chemistry, 1995, vol. 32, # 4, p. 1341 - 1350
[3] Arkivoc, 2011, vol. 2011, # 10, p. 298 - 311
4
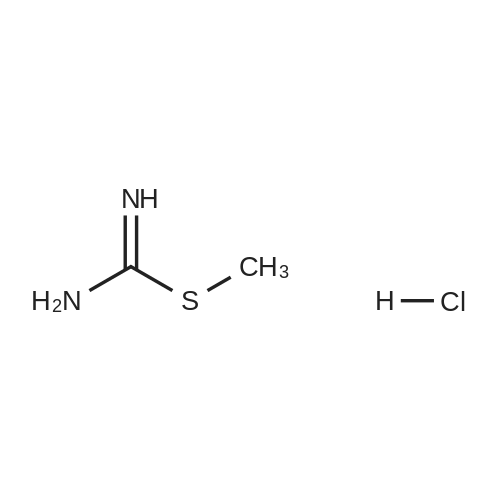
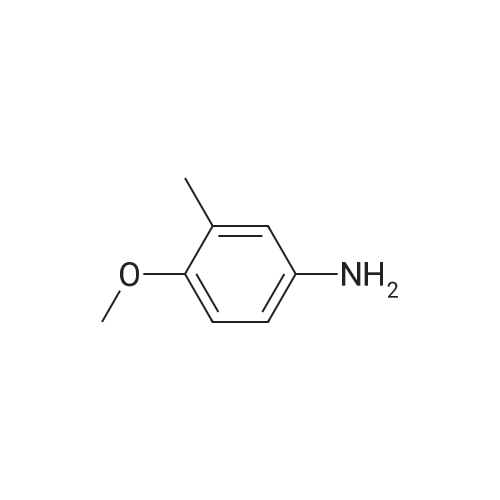
[ 87-13-8 ]
[ 371-40-4 ]
[ 391-78-6 ]
Reference:
[1] Patent: US2009/99220, 2009, A1, . Location in patent: Page/Page column 8-9
[2] Patent: US4560692, 1985, A,
5
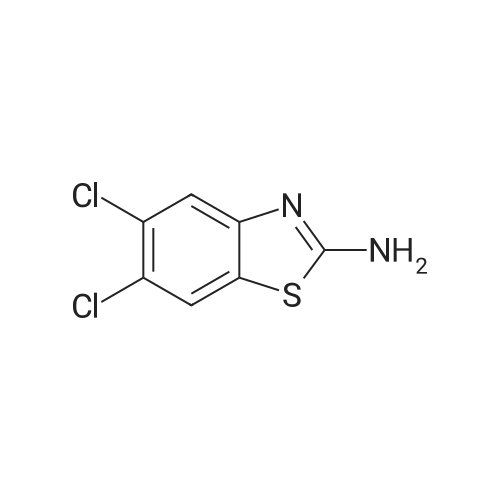
[ 87-13-8 ]
[ 7251-53-8 ]
Yield
Reaction Conditions
Operation in experiment
80%
With sodium ethanolate; hydrazine hydrate In ethanol at 80℃; Cooling with ice
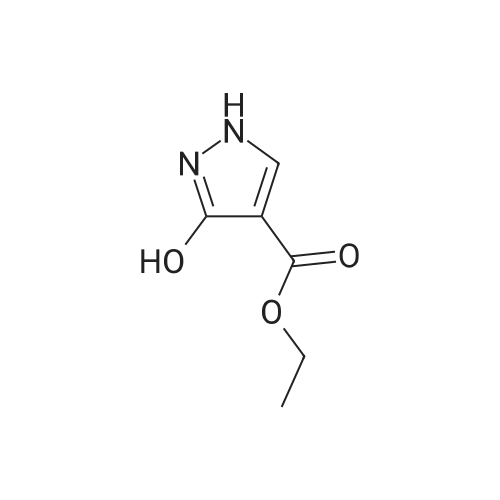
3-Oxo-2,3-dihydro-lH-pyrazole-4-carboxylic acid ethyl ester:[00358] To a solution of sodium ethoxide (20.8 g, 0.31 mol) and diethyl ethoxymethylenemalonate (20 mL, 0.10 mol) in ethanol (400 mL), was added hydrazine monohydrate (10.0 mL, 0.20 mol) with cooling in an ice-cold water bath. The mixture was then heated at 80 0C for 3h. The resulting mixture was diluted with water (200 mL) and neutralized with 1OM HCl solution until pη 6. The mixture was extracted several times with chloroform. The aqueous layer was acidified to pη 2 and extracted again with chloroform. The combined organic layers were dried (Na2SO4) and concentrated to a solid which was washed with methanol and ether and dried to give the product (13.2 g, 80percent) as a solid. 1H NMR (400MHz, DMSO-c/6) δ 7.89 (s, IH), 4.15 (q, 2H, J= 7.12 Hz), 1.23 (t, 3H, J= 7.11 Hz).
76%
Stage #1: With hydrazine In ethanol for 0.333333 h; Stage #2: With sodium hydroxide In ethanol; water at 20℃; for 1.5 h; Cooling with ice
XX. Ethyl 3-oxo-2,3-dihydro-1H-pyrazole-4-carboxylate A solution of diethyl 2-(ethoxymethylene)malonate (40 g, 185 mmol) in absolute ethanol (400 mL) was treated with hydrazine hydrate (8.99 mi, 185 mmol) dropwise. After approximately 20 min, 2N NaOH (25 mL) was added slowly (reaction in an ice-bath), then water (25 mL). The ice-bath was removed and the mixture stirred at rt for 1.5 hrs. The ethanol was removed in vacuo and the aqueous layer diluted with water (25 mL) and partitioned over EtOAc (100 mL). The aqueous layer was collected and cooled in an ice- bath. The pH was adjusted to 5 with 1M HCI, forming a precipitate. The precipitate was filtered, washing with water and dried in vacuo in the presence of CaCI2 to afford ethyl 3-oxo-2,3-dihydro-1H-pyrazole-4- carboxylate (22.31 g, 76percent yield). [MH]* = 157.1
35%
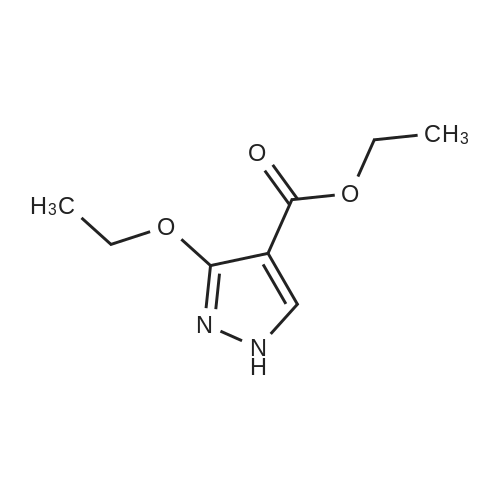
Stage #1: With sodium ethanolate In ethanol at 20℃; for 0.166667 h; Stage #2: With hydrazine hydrate In ethanol at 80℃; for 18 h; Heating Stage #3: With hydrogenchloride In ethanol; water; ethyl acetate at 0 - 20℃; for 1 h;
This compound was synthesized according to the method described in .To a 500-mL recovery flask, a 20percent solution of sodium ethoxide in ethanol (60 mL) and ethyl 2-(ethoxymethylene)malonate (10.5 mL, 524 mmol) were added, and the resulting mixture was stirred at room temperature for 10 minutes. To the obtained mixture, hydrazine monohydrate (5.1 mL, 104 mmol) was added, the resulting mixture was stirred at 80°C for 18 hours with heating, and then the obtained yellow suspension was cooled to 0°C. To the reaction solution vigorously stirred, 1 N hydrochloric acid (180 mL) was slowly added to the mixture at the same temperature to obtain a yellow solution. To the obtained solution, ethyl acetate (150 mL) was added, and the resulting mixture was stirred at room temperature for 1 hour. The organic layer was separated, and then the aqueous layer was extracted with ethyl acetate (100 mL x 2). The combined organic layers were dried over anhydrous sodium sulfate, and the insoluble substance was separated by filtration. The filtrate was concentrated under reduced pressure, and the obtained residue was crystallized by using ethyl acetate and hexane to obtain the title compound (2.82 g, 35percent) as yellow crystals (mixture of tautomers). MS ES M-H = 155
Reference:
[1] Patent: WO2009/89057, 2009, A1, . Location in patent: Page/Page column 56
[2] Patent: WO2016/83816, 2016, A1, . Location in patent: Page/Page column 48
[3] Patent: EP3272750, 2018, A1, . Location in patent: Paragraph 0219; 0220
[4] Yakugaku Zasshi, 1957, vol. 77, p. 800[5] Chem.Abstr., 1957, p. 17893
6
[ 87-13-8 ]
[ 7251-53-8 ]
[ 332066-58-7 ]
Yield
Reaction Conditions
Operation in experiment
41%
With hydrazine hydrochloride In ethanol for 16 h; Reflux
Diethylethoxymethylenemalonate (7) (37.4 g, 0.172 mol) and hydrazine hydrochloride (12.2 g, 0.178 mol) were refluxed in ethanol (500 mL) for 16 hours. The solvent was removed under reduced pressure, the residue was dispersed in water (500 ml) and slowly made basic by the addition of solid sodium hydrogencarbonate. The aqueous phase was extracted with dichloromethane; this organic phase was washed with a 1N solution of sodium hydrogencarbonate three times, dried over sodium sulfate and concentrated to dryness to yield compound 8 as an oil that solidified very slowly (13.22 g, 41 percent). The aqueous phase was cautiously made acid with concentrated hydrochloric acid, saturated with sodium chloride and the resulting precipitate was filtered, washed with water and dried under vacuum while heating at 60 °C to yield compound 9 (10 g, 37 percent) as a white powder.
Reference:
[1] Patent: EP2151434, 2010, A1, . Location in patent: Page/Page column 7; 12
[2] Chemistry - A European Journal, 2010, vol. 16, # 15, p. 4669 - 4677
7
[ 87-13-8 ]
[ 5472-46-8 ]
Reference:
[1] Journal of the Chemical Society, 1937, p. 367[2] Journal of the Chemical Society, 1938, p. 28
8
[ 87-13-8 ]
[ 2134-36-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1645 - 1648
[2] Journal of the Chemical Society, 1937, p. 367[3] Journal of the Chemical Society, 1938, p. 28
[4] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 21, p. 6756 - 6761
[5] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 22, p. 5771 - 5780
9
[ 87-13-8 ]
[ 51940-64-8 ]
Reference:
[1] Patent: WO2011/19405, 2011, A1,
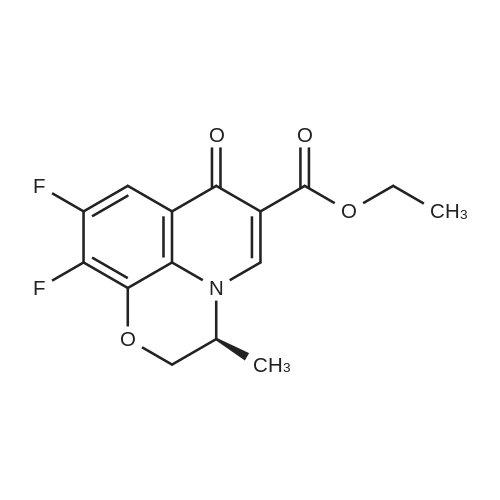
[2] Indian Journal of Heterocyclic Chemistry, 2010, vol. 20, # 2, p. 133 - 136
10
[ 124-42-5 ]
[ 87-13-8 ]
[ 53135-24-3 ]
Yield
Reaction Conditions
Operation in experiment
61%
Stage #1: at 0℃; for 0.333333 h; Stage #2: at 0℃; for 0.5 h; Stage #3: With triethylamine In ethanol for 2 h; Reflux
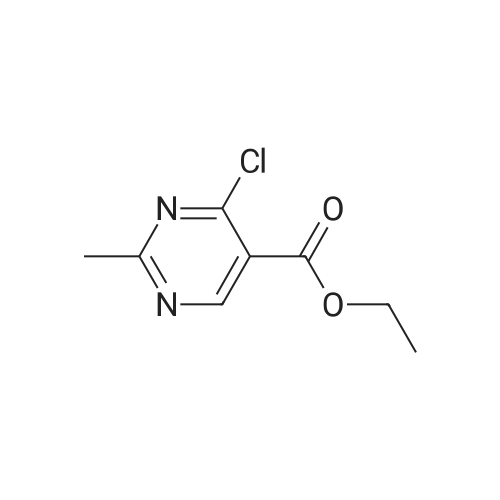
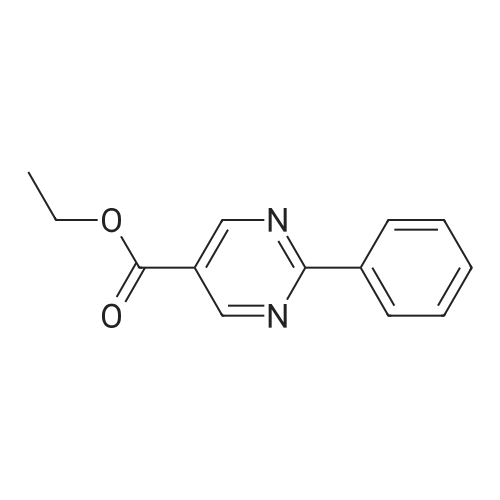
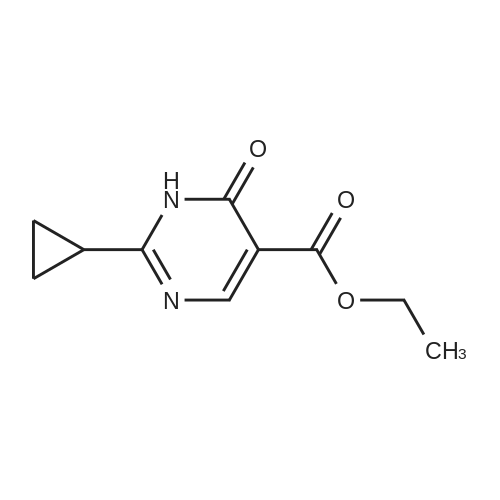
(0294) To a solution of sodium (2.90 g, 126 mmol) in ethanol (150 mL) was added acetoamidine hydrochloride (11.9 g, 126 mmol) at 0° C. The mixture was stirred at 0° C. for 20 minutes, diethyl (ethoxymethylene)malonate (28.6 g, 132 mmol) was added dropwise thereto, the mixture was stirred at 0° C. for 30 minutes, and triethylamine (20 mL, 145 mmol) was added thereto. The mixture was heated under reflux for 2 hours, the reaction mixture was concentrated, water (400 mL) was added thereto, citric acid was added to adjust pH to 4 to 5, and the mixture was extracted with dichloromethane (200 mL) three times. The organic layers were combined, dried over anhydrous sodium sulfate, filtrated, and concentrated. To the resulting concentrated residue was added tert-butyl methyl ether (200 mL), and the precipitate was collected on a filter to give the title compound (14.0 g, 61percent) (0295) 1H-NMR (400 MHz, CDCl3) δ: 1.40 (3H, t, J=7.2 Hz), 2.61 (3H, s), 4.39 (2H, q, J=7.2 Hz), 8.73 (1H, s).
27%
With hydrogenchloride In ethanol; dichloromethane; sodium ethanolate; ethyl acetate
EXAMPLE 30 Ethyl 2-methyl-pyrimidin-6(1H)-one-5-carboxylate STR55 Acetamidine hydrochloride (37.16 g, 0.39 mole) was stirred in sodium ethoxide in ethanol (73 mL of a 21percent solution, 0.20 mole) for 5 minutes. Diethyl ethoxymethylenemalonate (31.5 mL, 0.15 mole) was added, and the reaction mixture was refluxed for 5 hours. The reaction mixture was allowed to cool to room temperature overnight, and diluted with dichloromethane (100 mL). The solution was filtered, washing the solid cake with dichloromethane. The filtrate was concentrated at reduced pressure. The residue was dissolved in dichloromethane (150 mL) and 2.0N HCl (30 mL). The pH of the aqueous layer was 1. The organic layer was washed with water, saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure. The residue was dissolved in hot dichloromethane (50 mL). Ethyl acetate was added (50 mL). The product precipitated. The solution was boiled for 5 minutes, cooled to room temperature, and hexanes were added (50 mL). The resulting crystals were filtered, then washed with ethyl acetate (20 mL) followed by hexanes (50 mL) to yield the title compound (7.22 g, 27percent) as off-white crystals. Rf =0.27 (silica gel, 10percent isopropanol in dichloromethane). The title compound also was prepared by the route described in Example 102.
27%
With hydrogenchloride In ethanol; dichloromethane; sodium ethanolate; ethyl acetate
Example 30 Ethyl 2-methyl-pyrimidin-6(1H)-one-5-carboxylate STR57 Acetamidine hydrochloride (37.16 g, 0.39 mole) was stirred in sodium ethoxide in ethanol (73 mL of a 21percent solution, 0.20 mole) for 5 minutes. Diethyl ethoxymethylenemalonate (31.5 mL, 0.15 mole) was added, and the reaction mixture was refluxed for 5 hours. The reaction mixture was allowed to cool to room temperature overnight, and diluted with dichloromethane (100 mL). The solution was filtered, washing the solid cake with dichloromethane. The filtrate was concentrated at reduced pressure. The residue was dissolved in dichloromethane (150 mL) and 2.0N HCl (30 mL). The pH of the aqueous layer was 1. The organic layer was washed with water, saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure. The residue was dissolved in hot dichloromethane (50 mL). Ethyl acetate was added (50 mL). The product precipitated. The solution was boiled for 5 minutes, cooled to room temperature, and hexanes were added (50 mL). The resulting crystals were filtered, then washed with ethyl acetate (20 mL) followed by hexanes (50 mL) to yield the title compound (7.22 g, 27percent) as off-white crystals. Rf =0.27 (silica gel, 10percent isopropanol in dichloromethane). The title compound also was prepared by the route described in Example 102.
27%
With hydrogenchloride In ethanol; dichloromethane; sodium ethanolate; ethyl acetate
Example 30 Ethyl 2-methyl-pyrimidin-6(1H)-one-5-carboxylate STR52 Acetamidine hydrochloride (37.16 g, 0.39 mole) was stirred in sodium ethoxide in ethanol (73 mL of a 21percent solution, 0.20 mole) for 5 minutes. Diethyl ethoxymethylenemalonate (31.5 mL, 0.15 mole) was added, and the reaction mixture was refluxed for 5 hours. The reaction mixture was allowed to cool to room temperature overnight, and diluted with dichloromethane (100 mL). The solution was filtered, washing the solid cake with dichloromethane. The filtrate was concentrated at reduced pressure. The residue was dissolved in dichloromethane (150 mL) and 2.0N HCl (30 mL). The pH of the aqueous layer was 1. The organic layer was washed with water, saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure. The residue was dissolved in hot dichloromethane (50 mL). Ethyl acetate was added (50 mL). The product precipitated. The solution was boiled for 5 minutes, cooled to room temperature, and hexanes were added (50 mL). The resulting crystals were filtered, then washed with ethyl acetate (20 mL) followed by hexanes (50 mL) to yield the title compound (7.22 g, 27percent) as off-white crystals. Rf =0.27 (silica gel, 10percent isopropanol in dichloromethane). The title compound also was prepared by the route described in Example 102.
27%
With hydrogenchloride In ethanol; dichloromethane; sodium ethanolate; ethyl acetate
Example 30 Ethyl 2-Methyl-pyrimidin-6(1H)-one-5-carboxylate STR50 Acetamidine hydrochloride (37.16 g, 0.39 mole) was stirred in sodium ethoxide in ethanol (73 mL of a 21percent solution, 0.20 mole) for 5 minutes. Diethyl ethoxymethylenemalonate (31.5 mL, 0.15 mole) was added, and the reaction mixture was refluxed for 5 hours. The reaction mixture was allowed to cool to room temperature overnight, and diluted with dichloromethane (100 mL). The solution was filtered, washing the solid cake with dichloromethane. The filtrate was concentrated at reduced pressure. The residue was dissolved in dichloromethane (150 mL) and 2.0N HCl (30 mL). The pH of the aqueous layer was 1. The organic layer was washed with water, saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure. The residue was dissolved in hot dichloromethane (50 mL). Ethyl acetate was added (50 mL). The product precipitated. The solution was boiled for 5 minutes, cooled to room temperature, and hexanes were added (50 mL). The resulting crystals were filtered, then washed with ethyl acetate (20 mL) followed by hexanes (50 mL) to yield the title compound (7.22 g, 27percent) as off-white crystals. Rf =0.27 (silica gel, 10percent isopropanol in dichloromethane). The title compound also was prepared by the route described in Example 102.
27%
With sodium ethanolate In ethanol for 5 h; Heating / reflux
Acetamidine hydrochloride (37.16 g, 0.39 mole) was stirred in sodium ethoxide in ethanol (73 ML of a 21percent solution, 0.20 mole) for 5 minutes.. diethyl ethoxymethylenemalonate (31.5 ML, 0.15 mole) was added, and the reaction mixture was refluxed for 5 hours.. The reaction mixture was allowed to cool to room temperature overnight, and diluted with dichloromethane (100 ML).. The solution was filtered, washing the solid cake with dichloromethane.. The filtrate was concentrated at reduced pressure.. The residue was dissolved in dichloromethane (150 ML) and 2.0N HCl (30 ML).. The PH of the aqueous layer was 1.. The organic layer was washed with water, saturated sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure.. The residue was dissolved in hot dichloromethane (50 ML).. ethyl acetate was added (50 ML).. The product precipitated.. The solution was boiled for 5 minutes, cooled to room temperature, and hexanes were added (50 ML).. The resulting crystals were filtered, then washed with ethyl acetate (20 ML) followed by hexanes (50 ML) to yield the title compound (7.22 g, 27percent) as off-white crystals. Rf=0.27 (silica gel, 10percent isopropanol in dichloromethane).. The title compound also was prepared by the route described in Example 102.
Reference:
[1] Patent: US2016/122319, 2016, A1, . Location in patent: Paragraph 0293-0295
[2] Patent: US6008351, 1999, A,
[3] Patent: US6011158, 2000, A,
[4] Patent: US5656645, 1997, A,
[5] Patent: US5658930, 1997, A,
[6] Patent: US6342504, 2002, B1, . Location in patent: Page column 58
[7] Journal of Organic Chemistry, 1946, vol. 11, p. 741,749
[8] Journal of the Chemical Society, 1937, p. 367[9] Journal of the Chemical Society, 1938, p. 28
11
[ 87-13-8 ]
[ 53135-24-3 ]
Yield
Reaction Conditions
Operation in experiment
61%
Stage #1: With sodium In ethanol at 0℃; for 0.333333 h; Stage #2: With triethylamine In ethanol at 0℃; for 2.5 h; Reflux
Sodium (2.90g, 126mmol) and acetamidine hydrochloride (11.9g, 126mmol) in ethanol solution (150mL), it was added at 0°C. After stirring for 20 minutes at 0°C, Ethoxymethylene diethyl malonate (28.6g, 132mmol) It was added dropwise triethylamine (20mL, 145mmol) and stirred for 30 minutes at 0°C. After heated to reflux for 2 hours, The reaction mixture was concentrated, after addition of water (400 mL), citric acid is added to adjust to pH4 ~ 5, and extracted 3 times with dichloromethane (200 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated. The resulting concentrated residue tert- butyl methyl ether (200 mL) was added, the precipitate By collected by filtration to give the title compound (14.0g, 61percent).
49.5%
With sodium ethanolate In ethanol at 20 - 60℃; for 6.0833 h; Inert atmosphere
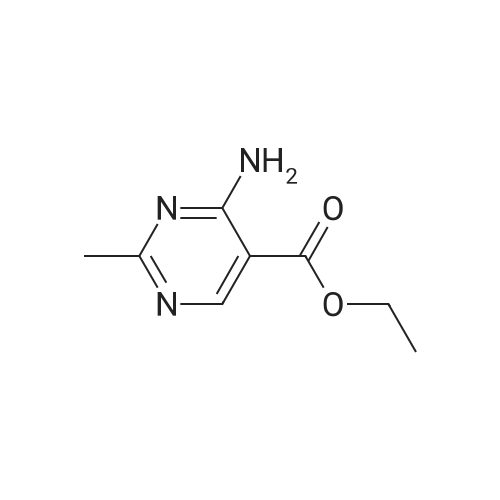
ethyl 2-methyl-6-oxo-1,6-dihydropyrimidine-5-carboxylate diethyl 2-(ethoxymethylene)malonate (9.35 mL, 46.25 mmol) was added dropwise to acetimidamide hydrochloride (4.37 g, 46.25 mmol), and sodium ethoxide (17.27 mL, 46.25 mmol) in ethanol (50 mL) at room temperature over a period of 5 minutes under nitrogen. The resulting solution was stirred at 60° C. for 6 hours. The reaction mixture was evaporated to dryness and redissolved in EtOAc (50 mL).The precipitate was collected by filtration, washed with EtOH (10 mL) and dried under vacuum to afford ethyl 2-methyl-6-oxo-1,6-dihydropyrimidine-5-carboxylate (4.17 g, 49.5percent) as a cream solid, which was used without further purification. 1H NMR (400.13 MHz, DMSO-d6) δ 1.15-1.23 (3H, t), 2.21 (3H, s), 4.09-4.17 (2H, q), 8.31 (1H, s) m/z (ESI+) (M+H)+=183; HPLC tR=0.78 min.
Reference:
[1] Patent: JP2016/108326, 2016, A, . Location in patent: Paragraph 0224; 0225
[2] Patent: US2009/264401, 2009, A1, . Location in patent: Page/Page column 117
12
[ 36896-17-0 ]
[ 87-13-8 ]
[ 53135-24-3 ]
Yield
Reaction Conditions
Operation in experiment
46%
for 26.25 h; Heating / reflux
Acetamidine acetate (37.21 g, 0.31 mole) and diethyl ethoxymethylenemalonate (63 ML, 0.31 mole) were refluxed for 4 h in ethanol (60 ML).. The reaction mixture was allowed to cool for 15 minutes, then acetamidine acetate (37.21 g, 0.31 mole) was added.. The reaction mixture was refluxed for 22 hours, allowed to cool to room temperature, and diluted with water (200 ML) and dichloromethane (200 ML).. The aqueous layer was extracted with 10percent isopropanol/dichloromethane (2*200 ML).. The combined organic extracts were washed with water (50 ML), brine (50 ML), dried over magnesium sulfate, filtered and the solvent was removed.. The residue was recrystallized from chloroform/hexanes in two crops to afford the title compound (24.92 g) in 46percent yield as yellowish crystals. Rf=0.27 (silica gel, 10percent isopropanol in dichloromethane); m.p. 187 to 188° C.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 1997, vol. 7, # 12, p. 1543 - 1548
[2] Patent: US6008351, 1999, A,
[3] Patent: US6011158, 2000, A,
[4] Patent: US5656645, 1997, A,
[5] Patent: US5658930, 1997, A,
[6] Patent: US6342504, 2002, B1, . Location in patent: Page column 92
13
[ 143-37-3 ]
[ 87-13-8 ]
[ 53135-24-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2002, vol. 12, # 3, p. 403 - 406
[2] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 21, p. 6756 - 6761
14
[ 87-13-8 ]
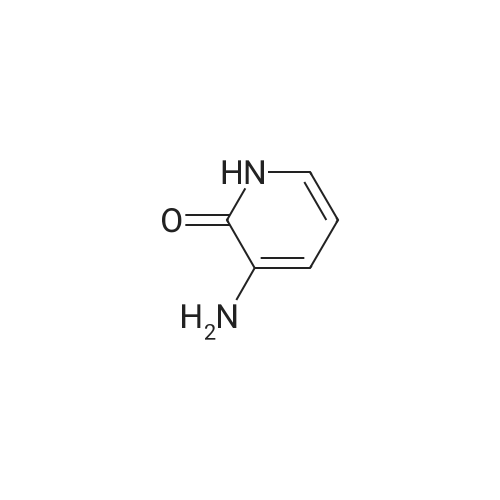
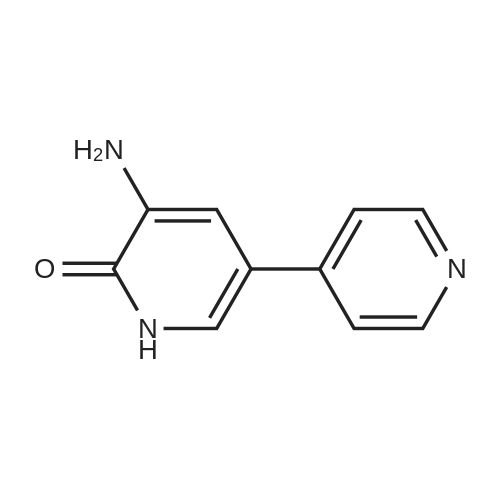
[ 626-43-7 ]
[ 171850-29-6 ]
Reference:
[1] Journal of the American Chemical Society, 2001, vol. 123, # 28, p. 6801 - 6808
Reference:
[1] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1958, vol. 246, p. 1701
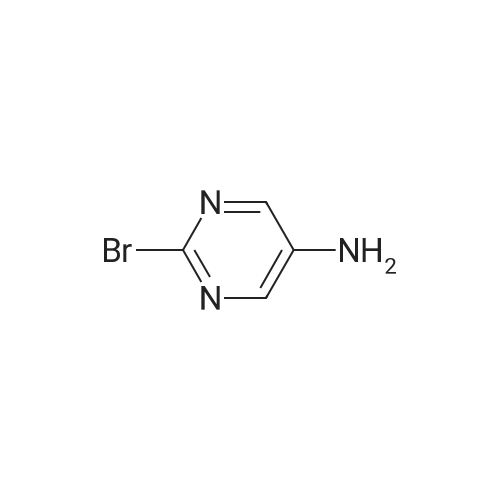
17
[ 87-13-8 ]
[ 5909-24-0 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1645 - 1648
[2] Journal of the American Chemical Society, 1943, vol. 65, p. 350,352
[3] Journal of Medicinal Chemistry, 2014, vol. 57, # 14, p. 6210 - 6225
[4] Patent: WO2015/1567, 2015, A1,
[5] European Journal of Medicinal Chemistry, 2018, vol. 145, p. 673 - 690
[6] Journal of the Chinese Chemical Society, 2018, vol. 65, # 4, p. 445 - 451
[7] Patent: CN108707141, 2018, A,
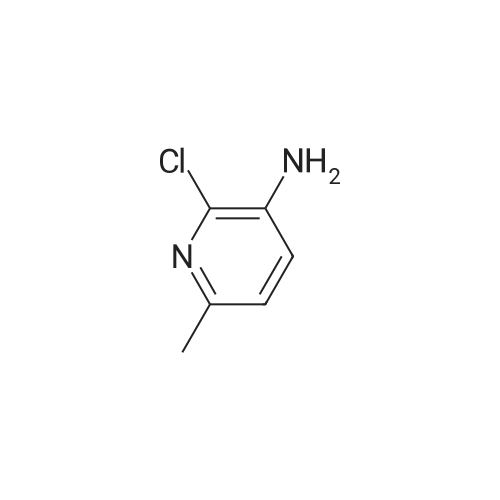
18
[ 109-06-8 ]
[ 87-13-8 ]
[ 88612-71-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 2011, vol. 54, # 13, p. 4773 - 4780
[2] Chemische Berichte, 1955, vol. 88, p. 1831,1837
[3] ACS Medicinal Chemistry Letters, 2010, vol. 1, # 6, p. 263 - 267
[4] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 6, p. 1710 - 1715
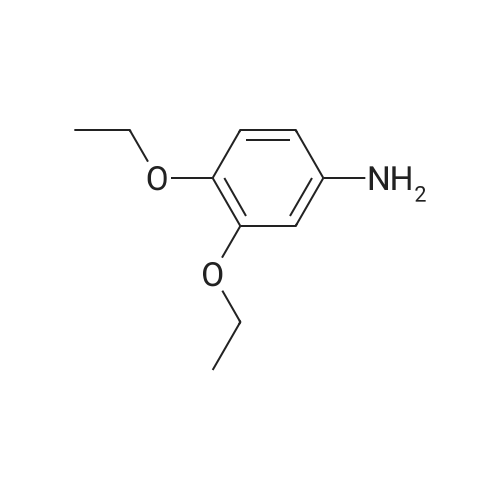
19
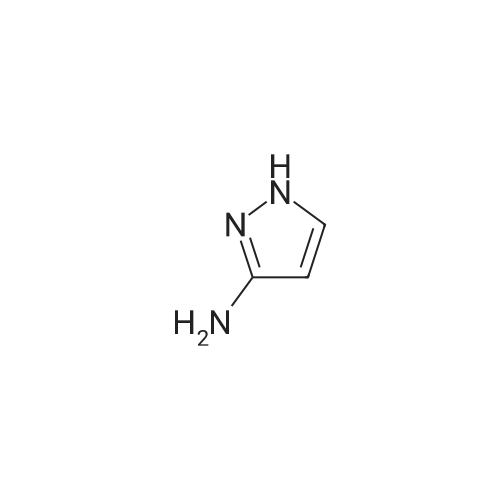
[ 87-13-8 ]
[ 53554-29-3 ]
Yield
Reaction Conditions
Operation in experiment
81%
Stage #1: at 0℃; Inert atmosphere Stage #2: at 0 - 20℃; Inert atmosphere
To a solution of sodium ethoxide (1.36 g, 20.0 mmol) in ethanol (15 mL) was added S-methylisothiourea sulfate (5.57 g, 40.0 mmol) at 0 °C. After a few minutes, diethyl ethoxymethylenemalonate (4.00 mL, 19.8 mmol) was added. After stirring at 0 °C for 3 h, a solution of sodium ethoxide (1.36 g, 20.0 mmol) in ethanol (15 mL) was added. The resulted solution was stirred at room temperature overnight. Ethanol was removed under reduced pressure and the pale yellow residue was dissolved in water. After filtration, the aqueous filtrate was washed with ether and acidified with acetic acid (10 mL). The organic materials were extracted with CHCl3. The combined organic extracts were washed with brine, dried over MgSO4, and concentrated. Recrystallization of the residue with CHCl3 gave compound 10 (3.44 g, 81percent) as colorless needles. Mp 129-130 °C (CHCl3), lit.16 132-133 °C (benzene/petroleum ether). IR (KBr) 3435, 2902, 2805, 1739, 1701, 1529, 1170 cm-1. δH (400 MHz, CDCl3) 1.42 (3H, t, J 6.8 Hz, CH2CH3), 2.60 (3H, s, SCH3), 4.43 (2H, q, J 6.8 Hz, CH2CH3), 8.75 (1H, s, C4-H). δC (150 MHz, DMSO;d6) 13.0, 14.1, 60.2, 111.7, 157.8, 158.9, 163.6, 168.0. LRMS (EI) m/z 214 (M+). HRMS (EI): M+, found 214.0458. C8H10N2O3S requires 214.0412.
Reference:
[1] Tetrahedron, 2011, vol. 67, # 14, p. 2661 - 2669
[2] European Journal of Medicinal Chemistry, 2018, vol. 145, p. 673 - 690
20
[ 2986-19-8 ]
[ 87-13-8 ]
[ 53554-29-3 ]
Reference:
[1] Journal of the American Chemical Society, 1943, vol. 65, p. 350,352
21
[ 53114-57-1 ]
[ 87-13-8 ]
[ 53554-29-3 ]
Reference:
[1] American Chemical Journal, 1907, vol. 37, p. 396
22
[ 62-53-3 ]
[ 87-13-8 ]
[ 26892-90-0 ]
Yield
Reaction Conditions
Operation in experiment
76%
at 100 - 200℃; for 3 h;
Aniline (1.02 mL, 0.011 mol) and diethyl ethoxymethylenemalonate (0.5-2 eq) were weighed into ethanol and heated at 100-200 ° C for three hours.Cool to room temperature, add phenyl ether, heat at 100-200 ° C for 30 min, cool to room temperature,After the reaction was completed, ice water was added, and ethyl acetate (100 mL × 2) was added, and the organic phase was combined, and the organic phase was washed with saturated brine.Dry over anhydrous sodium sulfate, filter,The organic phase was concentrated and the residue was purified by silica gel column chromatography1.6 g (76percent yield);
76%
at 100 - 200℃; for 3 h;
Aniline (1.02 mL, 0.011 mol) and diethyl ethoxymethylenemalonate (0.5-2 eq) were weighed into ethanol and heated at 100-200 ° C for three hours.Cool to room temperature, add phenyl ether, heat at 100-200 ° C for 30 min,Cool to room temperature,After the reaction was completed, ice water was added, and ethyl acetate was extracted (100 mL × 2).The organic phase was combined and the organic phase was washed with brine.Dry over anhydrous sodium sulfate, filter, and concentrate the organic phase.The residue was subjected to silica gel column chromatography to give the productWhite solid 1.6g (76percent yield);
69%
Stage #1: for 3 h; Reflux Stage #2: for 1 h; Reflux
Weigh aniline (1.0 mL) and diethyl ethoxymethylenemalonate (2.2 mL) into an appropriate amount of ethanol, and heat to reflux threeAfter cooling to room temperature, add 40 mL of phenylethyl ether, reflux under heating for 1 hour, and cool to room temperature. After completion of the reaction, add ice water, extract with ethyl acetate (100 mL×2), combine the organic phase, and saturate the organic phase. The mixture was washed with brine, dried over anhydrous sodium sulfate.
66%
at 90 - 100℃; for 3.5 h;
Weighed aniline (0.011 mol) and diethyl ethoxymethylenemalonate (0.012 mol) were added to 20 mL of ethanol and heated at 90 ° C for three hours. Cool to room temperature, add 40 mL of phenyl ether, heat at 100 ° C for 30 min, cool to room temperature.After the reaction was completed, ice water was added, and ethyl acetate was extracted (100 mL×2).The organic phase was combined and the organic phase was washed with brine.Dry over anhydrous sodium sulfate, filter, and concentrate the organic phase.The residue was subjected to silica gel column chromatography to ethyl 4-hydroxy-2-hydro-quinoline-3-carboxylate (66percent yield)
66%
at 90℃; for 3 h;
Weigh aniline (0.011 mol) and diethyl ethoxymethylenemalonate (0.012 mol) into 20 mL of ethanol.Heated at 90 oC for three hours,Cool to room temperature, add 40 mL of phenyl ether and heat at 100 °C for 30 min.After cooling to room temperature, after completion of the reaction, ice water was added, and ethyl acetate was extracted (100 mL×2), the organic phase was combined, and the organic phase was saturated.Washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated.The residue was subjected to silica gel column chromatography toield ethyl 4-hydroxy-2-hydro-quinoline-3-carboxylate (66percent yield)
32%
Stage #1: at 20 - 110℃; for 17 h; Stage #2: at 255℃; for 0.333333 h;
Synthesis of Lipophilic Group Modified Peptide Sequence Based on Nisvastatin; Scheme A; Scheme A; Ethyl 4-hydroxyquinoline-3-carboxylate (Al); [0183] Aniline (2.15g, 23mmol) and diethyl ethoxymethylene malonate (5g, 23mmol) were mixed neat and heated at 110°C for 2h then cooled and allowed to stand at room temperature for 15h. During this time the reaction mixture crystallized. [0184] Dowtherm A (70 mL) was heated to 255°C and the melted crystals were added and the mixture heated at 255°C for 20 min. The mixture was then poured into a stainless steel container cooled to 0°C with an ice bath. Hexanes were added to the cold solution to precipitate the product which was collected by filtration and rinsed with another portion of hexanes. The product was recrystallized from EtOH to give the product as a white solid. (1.6g, 7.3mmol, 32percent, M.P. 309C) that was used without further purification in the next step.
Reference:
[1] Patent: CN108623581, 2018, A, . Location in patent: Page/Page column 6
[2] Patent: CN108623561, 2018, A, . Location in patent: Paragraph 0014
[3] Patent: CN108690024, 2018, A, . Location in patent: Paragraph 0144
[4] Patent: CN108623562, 2018, A, . Location in patent: Paragraph 0067
[5] Patent: CN108623560, 2018, A, . Location in patent: Page/Page column 36
[6] Patent: WO2005/123686, 2005, A1, . Location in patent: Page/Page column 75-76
[7] Journal of Medicinal Chemistry, 1993, vol. 36, # 11, p. 1669 - 1673
[8] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 8, p. 1487 - 1490
[9] European Journal of Medicinal Chemistry, 2010, vol. 45, # 5, p. 1821 - 1827
[10] Patent: US2012/309758, 2012, A1,
[11] Patent: WO2018/107100, 2018, A1,
[12] Patent: US2018/280349, 2018, A1,
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 23, p. 2661 - 2664
[2] Journal of Medicinal Chemistry, 1993, vol. 36, # 11, p. 1669 - 1673
[3] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1448 - 1452
[4] European Journal of Medicinal Chemistry, 2015, vol. 103, p. 1 - 16
[5] Patent: CN108623562, 2018, A,
[6] Patent: CN108690024, 2018, A,
[7] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 12, p. 1205 - 1210
[8] Patent: CN108623560, 2018, A,
[9] Patent: CN108623561, 2018, A,
[10] Patent: CN108623581, 2018, A,
[11] Patent: CN108623588, 2018, A,
25
[ 104-94-9 ]
[ 87-13-8 ]
[ 77156-78-6 ]
Yield
Reaction Conditions
Operation in experiment
78%
With dowtherm A In ethanol for 0.5 h; Heating / reflux
A solution of 4-methoxyaniline (4Og, 0.32 mole) and diethyl ethoxymethylenemalonate (65 mL, 0.32 mole) in Dowtherm A (500 mL) was heated at reflux in a flask fitted with side-arm and condenser, and heating was continued until all the ethanol had distilled off (ca. 0.5 hr). The solution was cooled and pentane was added to give a sticky precipitate. The solvents were decanted off and the residue was treated with more pentane and allowed to stand overnight. The solid was filtered off and washed well with pentane to give the title compound (62.4 g; 78percent, contains traces of Dowtherm A).
78%
for 0.5 h; Heating / reflux
Preparation 10; Preparation of 4-ethenyl-3-fluoro-6-(methyloxy)quinoline; a) 4-Hydroxy-6-methoxy-quinoline-3-carboxylic acid ethyl ester; A solution of 4-methoxyaniline (4Og, 0.32 mole) and diethyl ethoxymethylenemalonate (65 mL, 0.32 mole) in Dowtherm A (500 mL) was heated at reflux in a flask fitted with side-arm and condenser, and heating was continued until all the ethanol had distilled off (ca. 0.5 hr). The solution was cooled and pentane was added to give a sticky precipitate. The solvents were decanted off and the residue was treated with more pentane and allowed to stand overnight. The solid was filtered off and washed well with pentane to give the title compound (62.4 g; 78percent, contains traces of Dowtherm A).
78%
for 0.5 h; Heating / reflux
Preparation 8; Preparation of 4-ethenvl-3-fluoro-6-(methvloxv)quinoline; a) 4-Hydroxy-6-methoxy-quinoline-3-carboxylic acid ethyl ester; A solution of 4-methoxyaniline (40g, 0.32 mole) and diethylethoxymethylenemalonate (65 ml, 0.32 mole) in Dowtherm A (500 ml_) was heated atreflux in a flask fitted with side-arm and condenser, and heating was continued until all theethanol had distilled off (ca. 0.5 hr). The solution was cooled and pentane was added togive a sticky precipitate. The solvents were decanted off and the residue was treated withmore pentane and allowed to stand overnight. The solid was filtered off and washed wellwith pentane to give the title compound (62.4 g; 78percent, contains traces of Dowtherm A).
17%
Stage #1: at 120℃; for 4 h; Inert atmosphere Stage #2: at 260℃; for 8 h; Inert atmosphere
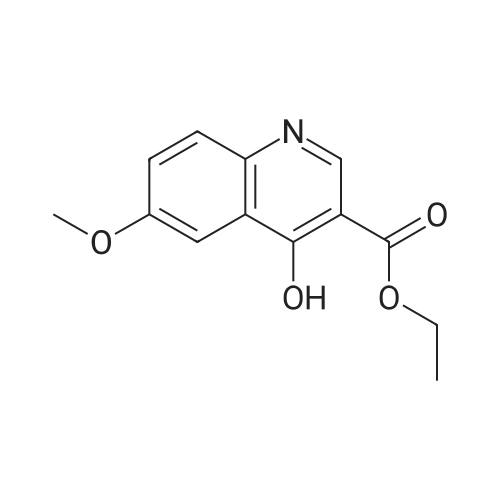
A mixture of 4-methoxyaniline (12.3 g, 100 mmol) and diethyl 2-(ethoxymethylene)malonate (21.6 g, 100 mmol) was stirred at 120 °C under nitrogen for 4 hrs. The solution was cooled to room temperature and Ph20 (100 mL) was added. The reaction mixture was refluxed at 260 °C under nitrogen for 8 hrs. The solution was cooled to room temperature and diluted with hexanes. The resultant precipitate was collected by filtration, washed with 25percent ethyl acetate in hexanes, and dried under vacuum to give ethyl 4-hydroxy-6-methoxyquinoline-3-carboxylate as a pale- yellow solid (4.21 g, 17percent). 1H NMR (400 MHz, DMSO-i¾ δ 1.26 (t, J = 7.0 Hz, 3H), 3.83 (s, 3H), 4.19 (q, J = 7.0 Hz, 2H), 7.32 (dd, J = 3.2, 9.6 Hz, 1H), 7.55 (d, J = 3.2 Hz, 1H), 7.56 (d, J = 9.6 Hz, 1H), 8.47 (s, 1H), 12.27 (s, 1H). MS 248 (MH+).
17%
Stage #1: at 120℃; for 4 h; Inert atmosphere Stage #2: at 260℃; for 8 h; Inert atmosphere
Example 1c ethyl 4-hydroxy-6-methoxyquinoline-3-carboxylate A mixture of 4-methoxyaniline (12.3 g, 100 mmol) and diethyl 2-(ethoxymethylene)malonate (21.6 g, 100 mmol) was stirred at 120° C. under nitrogen for 4 hrs. The solution was cooled to room temperature and Ph2O (100 mL) was added. The reaction mixture was refluxed at 260° C. under nitrogen for 8 hrs. The solution was cooled to room temperature and diluted with hexanes. The resultant precipitate was collected by filtration, washed with 25percent ethyl acetate in hexanes, and dried under vacuum to give ethyl 4-hydroxy-6-methoxyquinoline-3-carboxylate as a pale-yellow solid (4.21 g, 17percent). 1H NMR (400 MHz, DMSO-d6) δ 1.26 (t, J=7.0 Hz, 3H), 3.83 (s, 3H), 4.19 (q, J=7.0 Hz, 2H), 7.32 (dd, J=3.2, 9.6 Hz, 1H), 7.55 (d, J=3.2 Hz, 1H), 7.56 (d, J=9.6 Hz, 1H), 8.47 (s, 1H), 12.27 (s, 1H). MS 248 (MH+).
Reference:
[1] Patent: WO2006/81289, 2006, A2, . Location in patent: Page/Page column 19
[2] Patent: WO2006/2047, 2006, A2, . Location in patent: Page/Page column 74
[3] Patent: WO2006/14580, 2006, A1, . Location in patent: Page/Page column 38
[4] Chemical and Pharmaceutical Bulletin, 2007, vol. 55, # 5, p. 821 - 824
[5] Patent: WO2013/25560, 2013, A1, . Location in patent: Page/Page column 53
[6] Patent: US2015/245642, 2015, A1, . Location in patent: Paragraph 0288; 0289
[7] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 3578 - 3582
[8] Journal of Medicinal Chemistry, 1993, vol. 36, # 11, p. 1669 - 1673
[9] Patent: WO2007/16610, 2007, A2, . Location in patent: Page/Page column 28
Reference:
[1] Farmaco, 1998, vol. 53, # 8-9, p. 579 - 585
[2] Patent: WO2014/57415, 2014, A2,
[3] European Journal of Medicinal Chemistry, 2015, vol. 103, p. 1 - 16
[4] Tetrahedron Letters, 2017, vol. 58, # 8, p. 794 - 796
28
[ 87-13-8 ]
[ 108-46-3 ]
[ 6093-71-6 ]
Reference:
[1] Synthesis, 1982, vol. No. 10, p. 846 - 848
[2] Monatshefte fuer Chemie, 1929, vol. 51, p. 391
[3] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1958, vol. 246, p. 1701
[4] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1958, vol. 246, p. 1701
29
[ 87-13-8 ]
[ 720-01-4 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1645 - 1648
[2] European Journal of Medicinal Chemistry, 2018, vol. 145, p. 673 - 690
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 11, p. 3267 - 3272
[2] RSC Advances, 2014, vol. 4, # 12, p. 6254 - 6260
[3] Journal of Medicinal Chemistry, 2015, vol. 58, # 2, p. 560 - 576
[4] Angewandte Chemie - International Edition, 2017, vol. 56, # 25, p. 7282 - 7287[5] Angew. Chem., 2017, vol. 129, # 25, p. 7388 - 7393,6
32
[ 87-13-8 ]
[ 615-36-1 ]
[ 35975-57-6 ]
Yield
Reaction Conditions
Operation in experiment
3.5 g
at 100℃; for 5 h; Inert atmosphere
A solution of 2-bromoaniline (2.5 g, 14.62 mmol) and ethyl ethoxymethylenemalonate (3.16 g, 14.62 mmol) was heated at 100° C. for 3 h. Then the volatiles were removed by passing a stream of nitrogen and the molten mass was added slowly onto boiling diphenyl ether (10 mL) and the mixture was heated at reflux for 2 h. Then petroleum ether was added to the reaction mixture at rt and the precipitated solid was collected by filtration and dried to afford 3.5 g of the title product. 1H NMR (300 MHz, DMSO d6): δ 11.65 (br s, 1H), 8.45 (s, 1H), 8.17 (d, J=7.8 Hz, 1H), 8.05 (d, J=7.8 Hz, 1H), 7.39-7.34 (t, J=7.8 Hz, 1H), 4.26-4.19 (q, J=7.2, 14.1 Hz, 2H), 1.30-1.26 (t, J=6.9 Hz, 3H).
Reference:
[1] Farmaco, 1998, vol. 53, # 8-9, p. 579 - 585
[2] Journal of Medicinal Chemistry, 1993, vol. 36, # 11, p. 1669 - 1673
[3] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1448 - 1452
[4] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 3578 - 3582
35
[ 106-47-8 ]
[ 87-13-8 ]
[ 21168-41-2 ]
Reference:
[1] Patent: WO2016/196961, 2016, A1,
[2] Journal of Medicinal Chemistry, 2018, vol. 61, # 6, p. 2422 - 2446
36
[ 104007-09-2 ]
[ 87-13-8 ]
[ 27568-04-3 ]
Reference:
[1] Molecules, 2016, vol. 21, # 1,
37
[ 87-13-8 ]
[ 27568-04-3 ]
Reference:
[1] Molecules, 2016, vol. 21, # 1,
38
[ 90-04-0 ]
[ 87-13-8 ]
[ 27568-04-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 8, p. 1487 - 1490
39
[ 87-13-8 ]
[ 26893-12-9 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2015, vol. 103, p. 1 - 16
40
[ 455-14-1 ]
[ 87-13-8 ]
[ 26893-12-9 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 1998, vol. 8, # 19, p. 2629 - 2634
[2] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 8, p. 1487 - 1490
41
[ 87-13-8 ]
[ 23851-84-5 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 11, p. 5283 - 5292
[2] European Journal of Medicinal Chemistry, 2013, vol. 68, p. 422 - 432
[3] European Journal of Medicinal Chemistry, 2014, vol. 74, p. 324 - 332
42
[ 87-13-8 ]
[ 88-17-5 ]
[ 23851-84-5 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 8, p. 1487 - 1490
43
[ 42835-89-2 ]
[ 87-13-8 ]
[ 42835-25-6 ]
Reference:
[1] Patent: US4301288, 1981, A,
[2] Patent: US4301289, 1981, A,
[3] Patent: US4301291, 1981, A,
44
[ 87-13-8 ]
[ 18507-89-6 ]
Reference:
[1] Patent: CN105254560, 2016, A,
[2] Patent: CN107382855, 2017, A,
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 1347 - 1351
[2] Patent: US4081545, 1978, A,
47
[ 87-13-8 ]
[ 41103-17-7 ]
Reference:
[1] Patent: WO2011/99764, 2011, A2,
[2] Journal of Medicinal Chemistry, 2012, vol. 55, # 23, p. 10475 - 10489
48
[ 86119-84-8 ]
[ 122-51-0 ]
[ 105-53-3 ]
[ 87-13-8 ]
Reference:
[1] Patent: US5041619, 1991, A,
49
[ 122-51-0 ]
[ 105-53-3 ]
[ 87-13-8 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 11, p. 3267 - 3272
[2] Polish Journal of Chemistry, 1981, vol. 55, # 6, p. 1393 - 1403
[3] ChemMedChem, 2018, vol. 13, # 10, p. 1004 - 1017
[4] Collection of Czechoslovak Chemical Communications, 1991, vol. 56, # 2, p. 411 - 417
[5] Journal of the Chemical Society, 1948, p. 893
[6] Journal of Organic Chemistry, 1946, vol. 11, p. 196
[7] Journal of Biological Chemistry, 1935, vol. 108, p. 619,624
[8] Acta Chemica Scandinavica (1947-1973), 1973, vol. 27, p. 239 - 250
[9] Magnetic Resonance in Chemistry, 2005, vol. 43, # 2, p. 171 - 173
[10] Patent: US5041619, 1991, A,
[11] Russian Journal of General Chemistry, 2013, vol. 83, # 7, p. 1330 - 1335[12] Zh. Obshch. Khim., 2013, vol. 83, # 7, p. 1330 - 1335
[13] Patent: US2824121, 1954, ,
50
[ 105-53-3 ]
[ 87-13-8 ]
Reference:
[1] Patent: US4058553, 1977, A,
[2] Patent: US4058553, 1977, A,
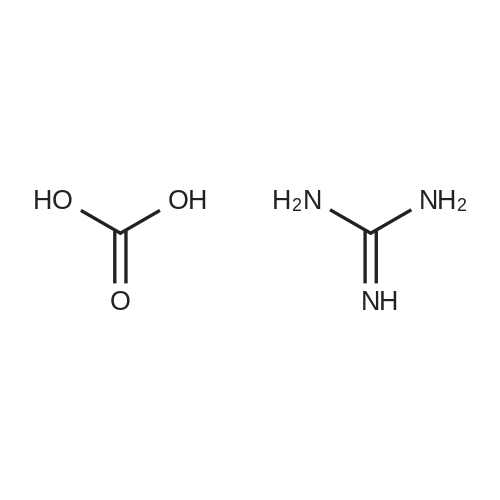
With potassium hydroxide In water at 20 - 35℃; Large scale
In a 20 L flask 1.070 Kg of KOH was dissolved with 10 L of distilled water, stirred to get clear solution and 5 g of silica functionalized magnetic nanoparticles (Fe3O4(at)SiO2) was added. To the above mixture 2 Kg of guanidine carbonate was added and stirring is continued fordissolution. Temperature of the reaction mixture was main-tained around 20 °C. Diethylethoxy methyl melonate (2.4 kg) was added dropwise over 3 h, the temperature of reaction mixture was raised from 20 to 35 °C. A yellow solid was precipitated and reaction was cooled. Then with the help of external magnet Fe3O4SiO2 particles was removed, particles were washed with acetone and dried. The reaction mixture was cooled to 0-5 °C and pale yellow colour thick mixture was filtered and washed with ice cold water and padded well. The product was recrystallized from ethanol/water and dried in oven at 40 °C. The dried product is divided into two parts and recrystallizedin 15 L of 60 percent H2O/40 percent EtOH. The recrystallization was hazy at first but goes clearer in appearance after 1 h of steam heating. This compound is to crystallize and first sign is observedat 39 °C and continued with slow cooling. The process was done for both the parts and dried under vacuum oven. Yield: 95 percent
Reference:
[1] Asian Journal of Chemistry, 2017, vol. 29, # 10, p. 2119 - 2122
[2] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 14, p. 4841 - 4856
[3] Patent: US6194419, 2001, B1,
59
[ 87-13-8 ]
[ 6296-99-7 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 13, p. 3441 - 3444
[2] Journal of the American Chemical Society, 1961, vol. 83, p. 4225 - 4228
[3] Gazzetta Chimica Italiana, 1974, vol. 104, p. 715 - 729
[4] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1981, p. 561 - 570
[5] Australian Journal of Chemistry, 1992, vol. 45, # 10, p. 1571 - 1576
[6] Patent: WO2009/21957, 2009, A2, . Location in patent: Page/Page column 19
[7] Journal of Agricultural and Food Chemistry, 2007, vol. 55, # 3, p. 608 - 613
[8] Patent: US6333336, 2001, B1,
60
[ 76369-62-5 ]
[ 87-13-8 ]
[ 6296-99-7 ]
Reference:
[1] Australian Journal of Chemistry, 2004, vol. 57, # 7, p. 685 - 688
61
[ 87-13-8 ]
[ 6296-99-7 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1897, vol. 297, p. 77
62
[ 62-53-3 ]
[ 87-13-8 ]
[ 54535-22-7 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 120 - 130℃; for 16.5 h;
Aniline (2.733 mL, 29.99 mMol) was stirred in diethyl ethoxymethylenemalonate (6.063 mL, 30.00 mMol) at 120-130°C for 16.5 hours. T.l.c. analysis (ethyl acetate cyclohexane, 1 : 1) showed the presence of one UV-active product (Rf 0.84) and complete consumption of both starting materials. Upon cooling down of the reactionsolution to room temperature, intermediate diethyl 2-((phenylamino)methylene)malonate 35 solidified (as dark yellow crystalline solid,7.899 g, quant.). M.p. 36-37°C; HRMS (EIj: found 263.11531 [M] C14H17N04requires 263.11521; Vmax (thin film): 3265, 3184 (w, NH), 3050 (w, ArC-H), 2981,2936, 2904, 2871 (m, alkyl C-H), 1717 (s, 2 x intramolecularly hydrogen-bondedC=O conjugated with C=C), 1691 (s, C=C-NH), 1655 (s, C=N-), 1623 (s, aryl conjugated C=C), 1255 (s, C-N stretch) cm’; 6H (CD3CN, 500 MHz): 1.31 (3H, t, JCH3,CH2 7.1 Hz, CH3), 1.32 (3H, t, JCH3,CH2 7.2 Hz, CH3), 4.19 (2H, q, JCH2,CH3 7.2 Hz, CH2), 4.25 (2H, q, JCH2,CH3 7.1 Hz, CH2), 7.16 (1H, tt, JparaArH,metaArHs 7.4 Hz, JparaArH,orthoArHs 1.1 Hz, para-ArH), 7.20 (2H, dt, Jortho&-Hs,metaArHs 7.6 Hz,JorthoArHsparaArH 1.0 Hz, 2 x orthoArHs), 7.38 (2H, m, J 7.4 Hz, 2 x metaArHs), 8.48(1H, d, JCH,NH 13.8 Hz, CH-NH), 10.81 (1H, d, JNH,CH 13.6 Hz, CH-NI]); 6c(CD3CN, 125 MHz): 14.1, 14.2 (2 x CH3), 60.3, 60.6(2 x CH2), 93.9 (O=C-C-C=O),117.6(2 x orthoArCs), 125.1 (paraArC), 130.1 (2 x metaArCs), 139.8 (ArCquat-NH),151.9 (NH-CH), 165.6 (C=O), 168.8 (hydrogen bonded C=O).
90%
Reflux
General procedure: To a stirred solution of substituted aniline 8 (50 mmol), diethyl 2-(ethoxymethylene)malonate (55 mmol) in ethanol (EtOH) (250 mL) was refluxed for 3 h. TLC analysis indicated that the reaction was complete. After cooling the reaction mixture solid was separated. The separated solid was filtered and washed with 3percent ethyl acetate:hexane for further purification to afford desired compound as an off-white solid in good yield (89-92percent). 4.1.4.1. Diethyl 2-((phenylamino)methylene)malonate (9a). The compound was prepared according to the general procedure C using compound 8a (4.65 g, 50 mmol), diethyl 2-(ethoxymethylene)malonate (11.87 g, 55 mmol) as an off-white solid (11.83 g, 90percent); m.p: 94-96 C; ESI-MS was found at m/z 264.23 [M+H]+. 1H NMR (300 MHz, CDCl3, TMS):d 8.46 (s, 1H), 7.32e6.84(m, 5H), 6.62 (s, 1H), 4.35 (q,J 7.0 Hz, 4H), 1.32 (t,J 6.8 Hz, 6H).
82%
for 3 h; Reflux
General procedure: A solution of the appropriate aniline (100 mmol), and diethyl ethoxymethylenemalonate (20.4 mL,100 mmol) was heated under reflux for 3 h. The mixture was allowed to cool and then was poured into ice-cold water (100 g). The precipitate was collected by filtration and recrystallized from hexane to give derivatives 8a–c [20,26,27].
62%
at 120℃; for 1 h;
General procedure: A mixture of aryl amines (6 mmol) and EMME (6 mmol)was heated at 120 °C for 1–2 h. The mixture was thenevaporated to dryness to give a residue that was trituratedwith cyclohexane to give a solid that was filtered and driedto afford compound 2e–k. Diethyl 2-((phenylamino)methylene)malonate (2e) Starting from aniline (3 ml); Yield (white powder): 5 g(62 percent); m.p. 44–45 °C; IR (KBr) νmax 1400–1600(aromatic), 1660, 1690 (carbonyl) cm−1; LC-MS (ESI) m/z286.1 (M+Na+).
25 g
at 20 - 55℃; for 3 h;
In a clean round bottom flask charge aniline (10 gm), and di-ethyl (ethoxymethylene) malonate (2.40 gm). The reaction mass was heated to 50 55 C for 3.0 hr and cooled to 25-30 C. To this 80.0 ml of water was added. Again cool to 15-20 C and stir for 2.0 hr . Filter the product and wash with water to 25.0 gm title compound.Purity by HPLC - 98.78 percent.
Reference:
[1] Patent: WO2015/189560, 2015, A1, . Location in patent: Page/Page column 87; 88
[2] Chemistry - A European Journal, 2010, vol. 16, # 9, p. 2938 - 2943
[3] Heterocycles, 2004, vol. 64, p. 177 - 191
[4] Tetrahedron, 1989, vol. 45, # 3, p. 889 - 908
[5] Bioorganic and Medicinal Chemistry Letters, 2006, vol. 16, # 3, p. 537 - 540
[6] Molecular Crystals and Liquid Crystals Science and Technology Section A: Molecular Crystals and Liquid Crystals, 2000, vol. 344, p. 163 - 168
[7] Journal of Medicinal Chemistry, 2006, vol. 49, # 1, p. 70 - 79
[8] Journal of Medicinal Chemistry, 2007, vol. 50, # 22, p. 5471 - 5484
[9] Agricultural and Biological Chemistry, 1981, vol. 45, # 12, p. 2769 - 2774
[10] Synthetic Communications, 2005, vol. 35, # 1, p. 79 - 87
[11] European Journal of Medicinal Chemistry, 2015, vol. 103, p. 1 - 16
[12] European Journal of Medicinal Chemistry, 2018, vol. 150, p. 292 - 306
[13] ACS Medicinal Chemistry Letters, 2018,
[14] Journal of Medicinal Chemistry, 2014, vol. 57, # 12, p. 5405 - 5418
[15] Journal of Medicinal Chemistry, 2006, vol. 49, # 8, p. 2526 - 2533
[16] Molecules, 2014, vol. 19, # 5, p. 6651 - 6670
[17] Medicinal Chemistry Research, 2016, vol. 25, # 9, p. 1861 - 1876
[18] Tetrahedron, 1992, vol. 48, # 29, p. 6135 - 6150
[19] Justus Liebigs Annalen der Chemie, 1897, vol. 297, p. 77
[20] Gazzetta Chimica Italiana, 1974, vol. 104, p. 715 - 729
[21] Justus Liebigs Annalen der Chemie, 1897, vol. 297, p. 77
[22] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1981, p. 561 - 570
[23] Magnetic Resonance in Chemistry, 1996, vol. 34, # 11, p. 972 - 978
[24] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 12, p. 2980 - 2985
[25] Journal of Medicinal Chemistry, 2006, vol. 49, # 21, p. 6351 - 6363
[26] Journal of Medicinal Chemistry, 2006, vol. 49, # 22, p. 6443 - 6450
[27] Chemical Communications, 2005, # 43, p. 5438 - 5440
[28] Patent: US2008/90864, 2008, A1, . Location in patent: Page/Page column 7
[29] Patent: US2008/306048, 2008, A1, . Location in patent: Page/Page column 17
[30] Patent: WO2009/21961, 2009, A1, . Location in patent: Page/Page column 19
[31] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 5, p. 1948 - 1956
[32] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 17, p. 5261 - 5265
[33] Synthetic Communications, 2009, vol. 39, # 24, p. 4375 - 4383
[34] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 2, p. 689 - 693
[35] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1448 - 1452
[36] Patent: US2011/230519, 2011, A1, . Location in patent: Page/Page column 15-16
[37] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 15, p. 5055 - 5058
[38] Patent: US2012/309758, 2012, A1, . Location in patent: Page/Page column 60
[39] European Journal of Medicinal Chemistry, 2013, vol. 67, p. 464 - 468
[40] Patent: WO2013/67410, 2013, A1, . Location in patent: Paragraph 0080
[41] RSC Advances, 2014, vol. 4, # 12, p. 6254 - 6260
[42] Oriental Journal of Chemistry, 2013, vol. 29, # 2, p. 507 - 511
[43] Patent: WO2014/71122, 2014, A1, . Location in patent: Paragraph 00156; 00321; 00322; 00323; 00324
[44] Patent: WO2014/118805, 2014, A1, . Location in patent: Paragraph 0288
[45] ACS Medicinal Chemistry Letters, 2015, vol. 6, # 2, p. 198 - 203
[46] CrystEngComm, 2015, vol. 17, # 4, p. 904 - 915
[47] Patent: WO2015/73231, 2015, A1, . Location in patent: Paragraph 00338-00339
[48] Patent: US2015/231142, 2015, A1, . Location in patent: Paragraph 0350
[49] Journal of Molecular Structure, 2016, vol. 1119, p. 259 - 268
[50] Patent: WO2007/79139, 2007, A2, . Location in patent: Page/Page column 46
[51] Patent: WO2018/64632, 2018, A1, . Location in patent: Paragraph 00211; 00212
[52] Patent: US2017/96397, 2017, A1, . Location in patent: Paragraph 0091
[53] Patent: WO2018/107100, 2018, A1, . Location in patent: Paragraph 00230; 00231
[54] Patent: US2018/280349, 2018, A1, . Location in patent: Paragraph 0094-0095
Reference:
[1] Journal of Chemical Research, Miniprint, 1982, # 6, p. 1726 - 1746
67
[ 87-13-8 ]
[ 77156-85-5 ]
Reference:
[1] Farmaco, 1998, vol. 53, # 8-9, p. 579 - 585
[2] Journal of Medicinal Chemistry, 1993, vol. 36, # 11, p. 1669 - 1673
[3] Journal of Medicinal Chemistry, 2012, vol. 55, # 9, p. 4205 - 4219
[4] Patent: US2014/275548, 2014, A1,
[5] Patent: WO2018/129274, 2018, A1,
68
[ 536-90-3 ]
[ 87-13-8 ]
[ 77156-85-5 ]
Reference:
[1] Patent: WO2016/196961, 2016, A1,
[2] Journal of Medicinal Chemistry, 2018, vol. 61, # 6, p. 2422 - 2446
69
[ 87-13-8 ]
[ 82419-34-9 ]
Reference:
[1] Patent: US4382892, 1983, A,
70
[ 87-13-8 ]
[ 79660-46-1 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1232 - 1244
[2] Journal of Photochemistry and Photobiology A: Chemistry, 2013, vol. 265, p. 41 - 48
[3] Chemical Biology and Drug Design, 2015, vol. 86, # 4, p. 440 - 446
[4] European Journal of Medicinal Chemistry, 2018, vol. 152, p. 377 - 391
71
[ 3862-73-5 ]
[ 87-13-8 ]
[ 79660-46-1 ]
Reference:
[1] Chemical Papers, 2017, vol. 71, # 5, p. 939 - 947
72
[ 106939-42-8 ]
[ 87-13-8 ]
[ 106939-34-8 ]
Yield
Reaction Conditions
Operation in experiment
70%
With ethyl phosphate In diethyl ether
Example 6 Ethyl (S)-(-)-9,10-difluoro-2,3-dihydro-3-methyl-7-oxo-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylate A mixture of 2.66 g of (S)-(-)-7,8-difluoro-3,4-dihydro-3-methyl-2H-[1,4]benzoxazine, as such produced in example 4, and 3.11 g of diethyl ethoxymethylenemalonate was stirred under a nitrogen atmosphere and heated at 140-145°C for 2 hours. After cooling 20 g of ethyl polyphosphate were added and the mixture was again heated at 140-145°C for 1 hour. After cooling the reaction mixture was poured into ice water and extracted three times with chloroform. The collected organic layers were washed with 5percent sodium carbonate solution and water. The organic solution was dried over anhydrous sodium sulfate, filtered and evaporated to dryness. Diethyl ether was added to the residue and the crystalline material was filtered, washed and dried to afford 3.1g of title product (70percent). M.p. 254-255°C.[α] [22/D ] = -64.8° (c=0.25, acetic acid). 1H NMR (CF3COOD, ppm) delta = 1.19 (tr, 3H), 1.44 (d, 3H), 4.38 (m, 4H), 4.84 (m, 1H), 7.75 (dd, 1H), 8.97 (s, 1H).
Reference:
[1] Acta Chemica Scandinavica, Series B: Organic Chemistry and Biochemistry, 1984, vol. 38, # 5 B, p. 359 - 366
74
[ 87-13-8 ]
[ 95-53-4 ]
[ 77156-75-3 ]
Reference:
[1] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2002, vol. 41, # 3, p. 650 - 652
[2] Journal of Molecular Structure, 2013, vol. 1051, p. 299 - 309
[3] Synthetic Communications, 2015, vol. 45, # 20, p. 2386 - 2393
75
[ 87-13-8 ]
[ 77156-75-3 ]
Reference:
[1] Nucleosides and Nucleotides, 1996, vol. 15, # 4, p. 889 - 898
[2] RSC Advances, 2014, vol. 4, # 12, p. 6254 - 6260
[3] Medicinal Chemistry Research, 2016, vol. 25, # 9, p. 1861 - 1876
76
[ 455-14-1 ]
[ 87-13-8 ]
[ 175203-85-7 ]
Reference:
[1] Patent: US6093732, 2000, A,
77
[ 87-13-8 ]
[ 1044770-40-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1645 - 1648
78
[ 54070-74-5 ]
[ 87-13-8 ]
[ 1044770-39-9 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1645 - 1648
79
[ 7732-18-5 ]
[ 87-13-8 ]
[ 343-67-9 ]
Yield
Reaction Conditions
Operation in experiment
50%
With NaOEt In ethanol
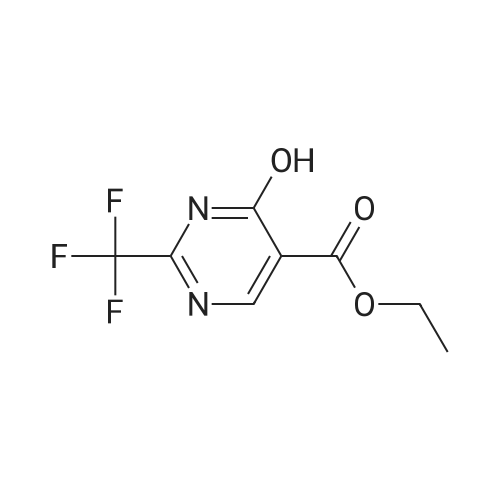
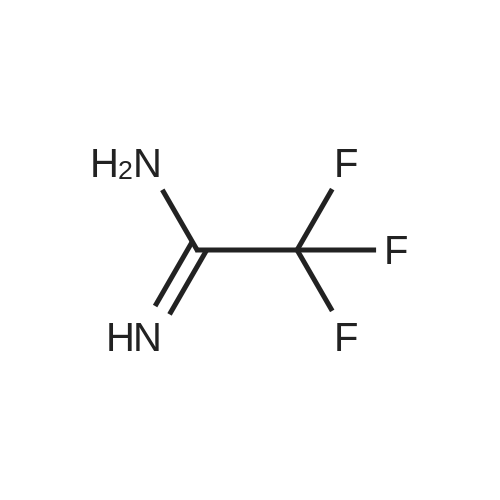
Example 15 ETHYL 2-TRIFLUOROMETHYL-4-HYDROXYPYRIMIDINE-5-CARBOXYLATE A solution of diethyl ethoxymethylenemalonate (35.0 g, 162 mmol), trifluoroacetaridine (18 g, 162 mmol) and NaOEt (11.0 g, 162 mmol) in EtOH (200 mL) was heated at reflux for 6 h. The reaction mixture was concentrated and H2 O (48 naL) was added. The resulting solid was filtered, washed with Et2 O (300 mL) and H2 O (200 mL), and dried to give the title compound (21 g, 50percent yield); m.p.>220° C. (dec.); 1 H NMR (DMSO-d6) δ 8.38, 4.16 (q, 2H), 1.25 (q, 3H).
Reference:
[1] Patent: US5935966, 1999, A,
80
[ 7732-18-5 ]
[ 354-37-0 ]
[ 87-13-8 ]
[ 343-67-9 ]
Yield
Reaction Conditions
Operation in experiment
50%
With NaOEt In ethanol
Example 33 ETHYL 2-TRIFLUOROMETHYL-4-HYDROXYPYRIMIDINE-5-CARBOXYLATE A solution of diethyl ethoxymethylenemalonate (35.0 g, 162 mmol), trifluoroacetamidine (18 g, 162 mmol) and NaOEt (11.0 g, 162 mmol) in EtOH (200 mL) was heated at reflux for 6 h. The reaction mixture was concentrated and H2 O (48 mL) was added. The resulting solid was filtered, washed with Et2 O (300 mL) and H2 O (200 mL), and dried to give the title compound (21 g, 50percent yield); m.p. >220° C. (dec.); 1 H-NMR (DMSO-d6) δ 8.38, 4.16 (q, 2H), 1.25 (q, 3H).
Reference:
[1] Patent: US5811428, 1998, A,
81
[ 354-37-0 ]
[ 87-13-8 ]
[ 343-67-9 ]
Reference:
[1] Journal of medicinal chemistry, 2000, vol. 43, # 21, p. 3995 - 4004
[2] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1645 - 1648
82
[ 87-13-8 ]
[ 478968-48-8 ]
Reference:
[1] Patent: WO2016/128529, 2016, A1,
83
[ 87-13-8 ]
[ 206258-97-1 ]
Reference:
[1] Patent: US2013/210844, 2013, A1,
84
[ 87-13-8 ]
[ 206257-39-8 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 4, p. 1448 - 1452
[2] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 24, p. 7585 - 7596
[3] Patent: WO2016/10869, 2016, A2,
[4] Patent: CN105859684, 2016, A,
[5] Patent: WO2017/11363, 2017, A1,
[6] Molecules, 2018, vol. 23, # 5,
[7] Journal of Medicinal Chemistry, 2018, vol. 61, # 9, p. 3823 - 3841
[8] Bioorganic and Medicinal Chemistry, 2018, vol. 26, # 14, p. 3967 - 3974
[9] Patent: WO2008/119771, 2008, A2,
[10] Patent: WO2006/132739, 2006, A2,
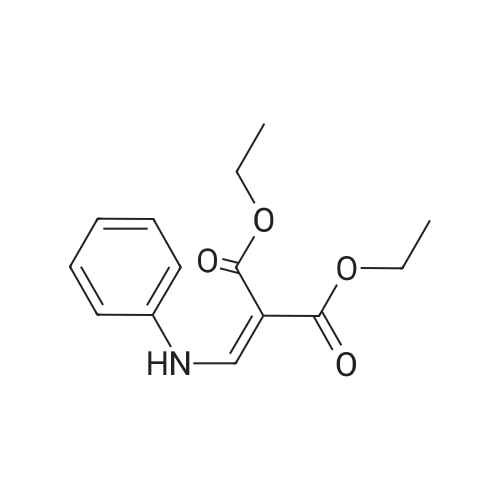
Aniline (2.733 mL, 29.99 mMol) was stirred in diethyl ethoxymethylenemalonate (6.063 mL, 30.00 mMol) at 120-130C for 16.5 hours. T.l.c. analysis (ethyl acetate cyclohexane, 1 : 1) showed the presence of one UV-active product (Rf 0.84) and complete consumption of both starting materials. Upon cooling down of the reactionsolution to room temperature, intermediate diethyl 2-((phenylamino)methylene)malonate 35 solidified (as dark yellow crystalline solid,7.899 g, quant.). M.p. 36-37C; HRMS (EIj: found 263.11531 [M] C14H17N04requires 263.11521; Vmax (thin film): 3265, 3184 (w, NH), 3050 (w, ArC-H), 2981,2936, 2904, 2871 (m, alkyl C-H), 1717 (s, 2 x intramolecularly hydrogen-bondedC=O conjugated with C=C), 1691 (s, C=C-NH), 1655 (s, C=N-), 1623 (s, aryl conjugated C=C), 1255 (s, C-N stretch) cm?; 6H (CD3CN, 500 MHz): 1.31 (3H, t, JCH3,CH2 7.1 Hz, CH3), 1.32 (3H, t, JCH3,CH2 7.2 Hz, CH3), 4.19 (2H, q, JCH2,CH3 7.2 Hz, CH2), 4.25 (2H, q, JCH2,CH3 7.1 Hz, CH2), 7.16 (1H, tt, JparaArH,metaArHs 7.4 Hz, JparaArH,orthoArHs 1.1 Hz, para-ArH), 7.20 (2H, dt, Jortho&-Hs,metaArHs 7.6 Hz,JorthoArHsparaArH 1.0 Hz, 2 x orthoArHs), 7.38 (2H, m, J 7.4 Hz, 2 x metaArHs), 8.48(1H, d, JCH,NH 13.8 Hz, CH-NH), 10.81 (1H, d, JNH,CH 13.6 Hz, CH-NI]); 6c(CD3CN, 125 MHz): 14.1, 14.2 (2 x CH3), 60.3, 60.6(2 x CH2), 93.9 (O=C-C-C=O),117.6(2 x orthoArCs), 125.1 (paraArC), 130.1 (2 x metaArCs), 139.8 (ArCquat-NH),151.9 (NH-CH), 165.6 (C=O), 168.8 (hydrogen bonded C=O).
90%
In ethanol;Reflux;
General procedure: To a stirred solution of substituted aniline 8 (50 mmol), diethyl 2-(ethoxymethylene)malonate (55 mmol) in ethanol (EtOH) (250 mL) was refluxed for 3 h. TLC analysis indicated that the reaction was complete. After cooling the reaction mixture solid was separated. The separated solid was filtered and washed with 3% ethyl acetate:hexane for further purification to afford desired compound as an off-white solid in good yield (89-92%). 4.1.4.1. Diethyl 2-((phenylamino)methylene)malonate (9a). The compound was prepared according to the general procedure C using compound 8a (4.65 g, 50 mmol), diethyl 2-(ethoxymethylene)malonate (11.87 g, 55 mmol) as an off-white solid (11.83 g, 90%); m.p: 94-96 C; ESI-MS was found at m/z 264.23 [M+H]+. 1H NMR (300 MHz, CDCl3, TMS):d 8.46 (s, 1H), 7.32e6.84(m, 5H), 6.62 (s, 1H), 4.35 (q,J 7.0 Hz, 4H), 1.32 (t,J 6.8 Hz, 6H).
82%
In ethanol; for 3h;Reflux;
General procedure: A solution of the appropriate aniline (100 mmol), and diethyl ethoxymethylenemalonate (20.4 mL,100 mmol) was heated under reflux for 3 h. The mixture was allowed to cool and then was poured into ice-cold water (100 g). The precipitate was collected by filtration and recrystallized from hexane to give derivatives 8a-c [20,26,27].
62%
at 120℃; for 1h;
General procedure: A mixture of aryl amines (6 mmol) and EMME (6 mmol)was heated at 120 C for 1-2 h. The mixture was thenevaporated to dryness to give a residue that was trituratedwith cyclohexane to give a solid that was filtered and driedto afford compound 2e-k. Diethyl 2-((phenylamino)methylene)malonate (2e) Starting from aniline (3 ml); Yield (white powder): 5 g(62 %); m.p. 44-45 C; IR (KBr) numax 1400-1600(aromatic), 1660, 1690 (carbonyl) cm-1; LC-MS (ESI) m/z286.1 (M+Na+).
at 140 - 150℃; for 2h;
2-Phenylaminomethylene-malonic acid diethyl ester A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (DMSO-d6) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 110℃; for 3h;
A mixture of aniline (1a) (9.3 g, 0.1 M) and diethyl 2-(ethoxymethylene)malonate (21.6 g, 0.1 M) was heated to 110 C. After 3 hours reaction mixture was cooled and ethanol was evaporated in vacuo to afford off-white solid which was used in next reaction without further purification.The 2-phenylaminomethylene-malonic acid diethyl ester was refluxed in Dowtherm for 15 min to 2 h. The reaction mixture was cooled to 80 C. and solid was collected by filtration and washed with hexane to yield crude ethyl 4-hydroxy-quinoline-3-carboxylate 2a which was used in next step without further purification.
at 150℃; for 0.25h;Microwave irradiation;
2-Phenylaminomethylene-malonic acid diethyl ester: 2-(Ethoxymethylene)-malonic acid diethyl ester (50 mmol) and aniline (50 mmol) are mixed in a flask. The flask is sealed, and the mixture is heated to 1509C in a microwave reactor, stirred for 15 min and then cooled to rt. The crude oil is purified by flash-chromatography (cyclohexane / EtOAc 90 : 10) to yield the title compound in the form of a yellow, viscous oil.
In ethanol;Reflux;
General procedure: The quinolone derivatives 1 were prepared by treating the appropriate aniline (100 mmol) with diethyl ethoxymethylenemalonate (100 mmol) under reflux in ethanol (5 mL) for 2-10 h to obtain the enamine derivatives that were then cyclized in refluxing diphenyl ether for 30 min-6 h [29]. These quinolones (13 mmol) were refluxed in thionyl chloride (20 mL), for 17 h, affording the corresponding chloro-derivatives 2a-g [22]. Reaction of 2a-g (4 mmol) with 2-hydrazinobenzothiazole (8 mmol) in toluene (30 mL), for 3 h, followed by a 2 h reflux in acetic acid gave the corresponding 2-(benzo[d]thiazol-2-yl)-8-substituted-2H-pyrazolo[4,3-c]quinolin-3(5H)-ones 3a-g as solids which were collected by filtration under vacuum, washed with water and subsequent purified by washing with hot ethyl alcohol.
In ethanol; at 30 - 110℃; for 3h;Inert atmosphere;
Procedure for the preparation of ethyl 4-oxo-1,4-dihydroquinoline-3-carboxylate (25) Compound 23 (4.77 g, 47.7 mmol) was added dropwise to compound 22 (10 g, 46.3 mmol) with subsurface N2 flow to drive out ethanol below 30 C. for 0.5 hours. The solution was then heated to 100-110 C. and stirred for 2.5 hours. After cooling the mixture to below 60 C., diphenyl ether was added. The resulting solution was added dropwise to diphenyl ether that had been heated to 228-232 C. for 1.5 hours with subsurface N2 flow to drive out ethanol. The mixture was stirred at 228-232 C. for another 2 hours, cooled to below 100 C. and then heptane was added to precipitate the product. The resulting slurry was stirred at 30 C. for 0.5 hours. The solids were then filtrated, and the cake was washed with heptane and dried in vacuo to give compound 25 as brown solid. 1H NMR (DMSO-d6; 400 MHz) delta 12.25 (s), delta 8.49 (d), delta 8.10 (m), delta 7.64 (m), delta 7.55 (m), delta 7.34 (m), delta 4.16 (q), delta 1.23 (t).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.28 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.29 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (d-DMSO) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
In ethanol; at 30 - 110℃; for 3h;Inert atmosphere;
Compound 23 (4.77 g, 47.7 mmol) was added dropwise to Compound 22 (10 g, 46.3 mmol) with subsurface N2 flow to drive out ethanol below 30 C for 0.5 hours. The solution was then heated to 100-110 C and stirred for 2.5 hours. After cooling the mixture to below 60 C, diphenyl ether was added. The resulting solution was added dropwise to diphenyl ether that had been heated to 228-232 C for 1.5 hours with subsurface N2 flow to drive out ethanol. The mixture was stirred at 228-232 C for another 2 hours, cooled to below 100 C and then heptane was added to precipitate the product. The resulting slurry was stirred at 30 C for 0.5 hours. The solids were then filtered, and the cake was washed with heptane and dried in vacuo to give Compound 25 as a brown solid. 1H NMR (DMSO-d6; 400 MHz) delta 12.25 (s), delta 8.49 (d), delta 8.10 (m), delta 7.64 (m), delta 7.55 (m), delta 7.34 (m), delta 4.16 (q), delta 1.23 (t).
at 125 - 130℃; for 2h;
General procedure: Substituted aniline 1 (0.01 mol) and diethylethoxy methylene malonate 2 (0.01 mol) were mixed and heated at 125-130 0C for 2 h. Ethanol was evaporated off from the resulting mixture ofethyl anilinomethylene malonate 3. The crude solidwas filtered on sintered funnel, dried and recrystallized from n-hexane. The malonate 3 (0.01mol) was refluxed with diphenylether (50 mL) for 1h to give 1,4-dihydro-4-oxoquinoline-3-carboxylicacid ethyl ester 4. After 1 h the solution was cooledand the resulting precipitate was filtered off, washed with n-hexane, and dried. The solid was recrystallized from DMF to give substituted-1,4-dihydro-4-oxoquinoline-3-carboxylic acid ethylester 4.
In ethanol; at 30 - 110℃; for 3h;Inert atmosphere;
[00324] Compound 23 (4.77 g, 47.7 mmol) was added dropwise to compound 22 (10 g, 46.3mmol) with subsurface N2 flow to drive out ethanol below 30 oc for 0.5 hours. The solution wasthen heated to 100-110 oc and stirred for 2.5 hours. After cooling the mixture to below 60 C,diphenyl ether was added. The resulting solution was added dropwise to diphenyl ether that hadbeen heated to 228-232 oc for 1.5 hours with subsurface N2 flow to drive out ethanol. Themixture was stirred at 228-232 oc for another 2 hours, cooled to below 100 oc and then heptanewas added to precipitate the product. The resulting slurry was stirred at 30 oc for 0.5 hours. Thesolids were then filtrated, and the cake was washed with heptane and dried in vacuo to give compound 25 as brown solid. 1H NMR (DMSO-d6; 400 MHz) 8 12.25 (s), 8 8.49 (d), 8 8.10(m), 8 7.64 (m), 8 7.55 (m), 8 7.34 (m), 8 4.16 (q), 8 1.23 (t).
25 g
at 20 - 55℃; for 3h;
In a clean round bottom flask charge aniline (10 gm), and di-ethyl (ethoxymethylene) malonate (2.40 gm). The reaction mass was heated to 50 55 C for 3.0 hr and cooled to 25-30 C. To this 80.0 ml of water was added. Again cool to 15-20 C and stir for 2.0 hr . Filter the product and wash with water to 25.0 gm title compound.Purity by HPLC - 98.78 %.
at 100 - 110℃; for 2.5h;Inert atmosphere;
[00339] Compound 23 (4.77 g, 47.7 mmol) was added dropwise to compound 22 (10 g, 46.3 mmol) with subsurface N2 flow to drive out ethanol below 30 C for 0.5 hours. The solution was then heated to 100-1 10 C and stirred for 2.5 hours. After cooling the mixture to below 60 C, diphenyl ether was added. The resulting solution was added dropwise to diphenyl ether that had been heated to 228-232 C for 1.5 hours with subsurface N2 flow to drive out ethanol. The mixture was stirred at 228-232 C for another 2 hours, cooled to below 100 C and then heptane was added to precipitate the product. The resulting slurry was stirred at 30 C for 0.5 hours. The solids were then filtrated, and the cake was washed with heptane and dried in vacuo to give compound 25 as brown solid. NMR (DMSO-d6; 400 MHz) delta 12.25 (s), delta 8.49 (d), 5 8.10 (m), 5 7.64 (m), delta 7.55 (m), delta 7.34 (m), delta 4.16 (q), delta 1.23 (t).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.28 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.29 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (d-DMSO) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 0C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (DMSO-^) delta 11.00 (d, IH), 8.54 (d, J = 13.6 Hz, IH), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).[0216] 4-Hydroxyquinoline-3-carboxyiic acid ethyl ester
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (DMSO-d6) delta 11.00 (d, 1H), 8.54 (d, J= 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃;
A mixture of aniline (100 g) and Diethyl-2-(ethoxymethylene) malonate (232 g) was heated to 140-150 C. for 2-3 hours. After completion of reaction, the mixture was cooled to 50-55 C. and dried under reduced pressure for 3 hours to obtain 2-Phenylaminomethylene malonic acid diethyl ester as yellow colored solid, which was used in the next step of preparation without purification.
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2- (ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 ^C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification.1H NMR (DMSO-d6) delta 11.00 (d, 1H), 8.54 (d, J = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
Step A: 2-Phenylaminomethylene-malonic acid diethyl ester A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (DMSO-d6) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H) ppm.
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2- (ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C for 2 h. Themixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. ?H NIVIR (DMSO-d6) 11.00 (d, 1H), 8.54 (d, J 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2- (ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. XH NMR (DMSO-ifc) delta 11.00 (d, 1H), 8.54 (d, = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2- (ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140- 150 C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. XH NMR (DMSO-ifc) delta 11.00 (d, 1H), 8.54 (d, = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18- 1.40 (m, 6H).
In ethanol; at 80℃; for 8h;Inert atmosphere;
Compound 1a aniline, ethanol and ethoxymethylenemalonic acid diethyl ester were successively added to a 100 mL single-necked flask, heated to 80 C in an oil bath with electromagnetic stirring and nitrogen/inert gas, and refluxed for 8 h (TLC monitoring reaction Process, developing solvent: V petroleum ether: V ethyl acetate = 6:1), after completion of the reaction, it was cooled, and ethanol was evaporated under reduced pressure to give a black oily liquid 2a.
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2- (ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. XH NMR (DMSO-ifc) delta 11.00 (d, 1H), 8.54 (d, = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
mixture of aniline (25.6 g, 0.275 mol) and diethyl 2- (ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2- phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 'H NMR (DMSO-rfc) d 11.00 (d, 1H), 8.54 (d, J = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
In benzene; at 83℃; for 1.5h;
General procedure: The diethyl 2-((phenylamino)methylene)malonates were synthesized by mixing diethyl (ethoxymethylene) malonate (25mmol) and aniline or p-toluidine (25mmol) in a round bottom flask. 6.5ml benzene was added to it and it was refluxed for 1.5h reaction mixture was concentrated under vacuum and the crude obtained was left to crystallize at room temperature. Solid obtained was washed with hexane and dried [25].
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (DMSO-d6) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 'H NMR (DMSO-rfc) d 11.00 (d, 1H), 8.54 (d, J = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
at 140 - 150℃; for 2h;
A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (DMSO-d6) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
With sodium methylate; In methanol; dichloromethane;
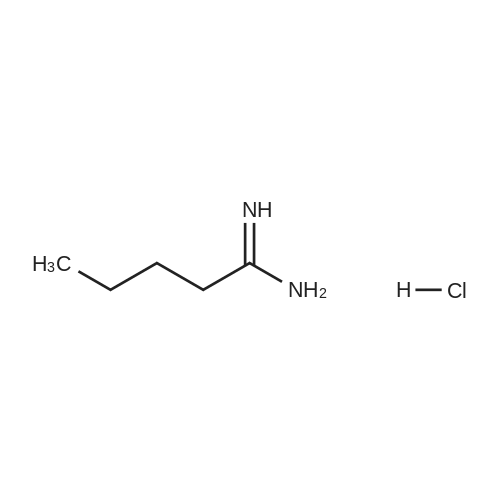
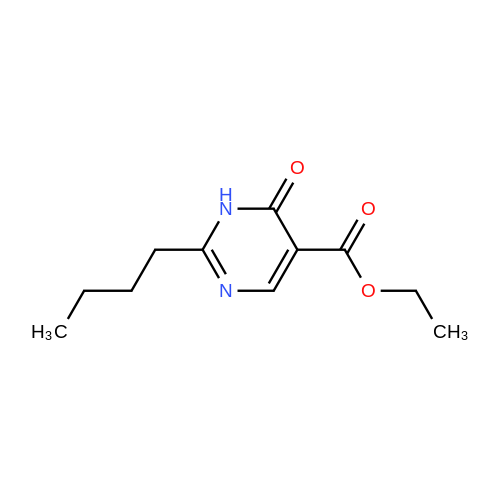
EXAMPLE 4 2-n-Butyl-5-(ethoxycarbonyl)pyrimidin-4(3H)-one To a solution of 2.0 g valeramidine hydrochloride and 750 mg sodium methoxide in 50 mL methanol was added 3.2 g diethyl ethoxymethylenemalonate (Aldrich) at room temperature. After 2 hours, an additional 850 mg sodium methoxide was added and the mixture was allowed to stir overnight. Solvent was removed in vacuo and the remaining material was partitioned between saturated aqueous ammonium chloride solution and ether. The organic layer was removed and the aqueous layer was extracted twice more with ether. The combined organic material was dried over MgSO4, was stripped of solvent in vacuo, and then was Still flash chromatographed in 3% MeOH in CH2 Cl2 to give the title compound as a white solid. 1 H NMR (250 MHz, CDCl3) delta8.73 (s, 1H), 4.37 (4 line m, 2H), 2.77 (3 line m, 2H) 1.80 (m, 2H), 1.41 (m, 2H) 1.38 (3 line m, 3H), 0.96 (3 line m, 3H).
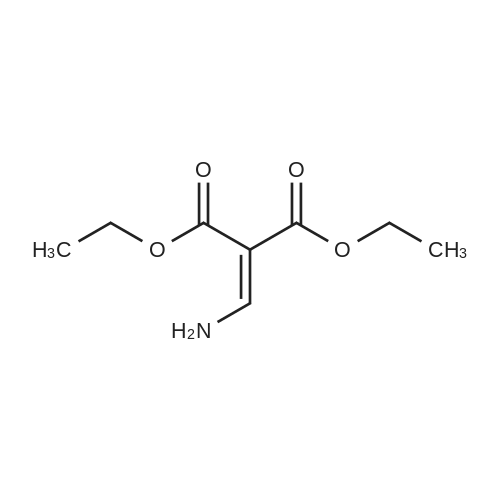
Ammonia was passed through ethanol (200 mL) for 30 minutes. Diethyl 2- (ethoxymethylene) malonate (21.6 g, 0.1 mol, 1.0 eq) was dissolved in an ammonia / ethanol solution and reacted at room temperature for 1 hour. After the reaction was detected by LC-MS and TLC, the reaction was concentrated. The crude product was purified by silica gel column chromatography (100-200 mesh silica gel, PE / EA = 5: 1) to obtain a white solid (18 g, yield: 96%)
With ammonia; In ethanol; at 20℃; for 1.0h;
2-Ethoxymethylene-malonic acid diethyl ester (20 ml, 100 mmol) and a 10% soln. of ammonia in EtOH (37 ml, 220 mmol) are stirred at rt for 1 h. The mixture is evaporated and dried in HV to give 2-aminomethylene-malonic acid diethyl ester that is used without further purification.
With ammonia; In ethanol;
a Diethyl aminomethylenemalonate To diethyl ethoxymethylenemalonate (50.13 g, 0.232 mmol), cooled to -20 C. under nitrogen, was added a 2.0 M solution of ammonia in ethanol (232 ml, 0.464 mmol) and the resulting solution was stirred at room temperature overnight. The solution was then evaporated in vacuo to give a quantitative yield of the title compound as a cream solid; 1H NMR (360 MHz, CDCl3) 5 1.28 (3H, t, J 7.1 Hz), 1.35 (3H, t, J 7.1 Hz), 4.19 (2H, q, J 7.1 Hz), 4.26 (2H, q, J 7.1 Hz), 5.68 (1H, br s), 8.11 (1H, dd), 8.69 (1H, br s).
Example 2: Preparation of diethyl [(l-ethyMH-pyrazol-5-yl)amino]methylidene| propanedioateA mixture of 5-amino-l-ethylpyrazole (5 gm, 0.0448 mole) and diethylethoxy methylenemalonate (10.35 ml, 0.0448 mole) was stirred at 120 C for about 1 hour. The reaction mixture was poured into water and extraction was done with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give viscous oil.Yield: 15 gm (crude) (124%)
100%
at 120℃; for 1h;
Example 2: Preparation of diethyl [(l-ethyl-lH-pyrazol-5-yl)aminolmethylidene| propanedioateA mixture of 5-amino-l-ethylpyrazole (5 gm, 0.0448 mole) and diethylethoxy methylenemalonate (10.35 ml, 0.0448 mole) was stirred at 1200C for about 1 hour. The reaction mixture was poured into water and extraction was done with ethyl acetate. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure to give viscous oil.Yield: 15 gm (crude) (124%)
86%
at 120℃; for 4h;
[l-Ethyl-4-(4-fluoro-2-methyl-phenyl)-lH-pyrazolo[3,4-b]pyridin-5-yl]-methyl-amine a) 2-[(2-Ethyl-2H-pyrazol-3-ylamino)-methylene]-malonic acid diethyl esterTo 2-ethyl-2H-pyrazol-3-ylamine (5.6 g, 0.050 mol) was added 2-ethoxymethylene-malonic acid diethyl ester (10.1 mL, 0.050 mol) and the mixture was heated at 120 C for 4 hours. After cooling down to RT, the crude mixture was purified by column chromatography (Si02,EtO Ac/Heptane 1/1) to give 12.25 g (86 %) of the title compound as a yellow oil. ES-MS m/e: 282.4 (M+H+).
86%
at 120℃; for 4h;
a) 2-[(2-Ethyl-2H-pyrazol-3-ylamino)-methylene]-malonic acid diethyl ester To 2-ethyl-2H-pyrazol-3-ylamine (5.6 g, 0.050 mol) was added 2-ethoxymethylene-malonic acid diethyl ester (10.1 mL, 0.050 mol) and the mixture was heated at 120 C. for 4 hours. After cooling down to RT, the crude mixture was purified by column chromatography (SiO2, EtOAc/Heptane 1/1) to give 12.25 g (86%) of the title compound as a yellow oil. ES-MS m/e: 282.4 (M+H-).
PREPARATION 1 (1-Ethylpyrazol-5-ylamino)methylenemalonate diethyl ester A neat solution of 5-amino-1-ethylpyrazole (20.0 g, 180 mmol) and diethyl ethoxymethylenemalonate (42.8 g, 198 mmol) was heated at 120 C. for 5 h. This material was used directly without further purification. If needed, the product can be distilled at 154-160 C. (0.1 mm Hg) to afford the title compound as a liquid which solidified to afford the title compound as a pale colored solid: mp 50-53 C.
Preparation 1 Preparation of (1-Ethylpyrazol-5-yl-amino)methylenemalonate diethyl ester A neat solution of 5-amino-1-ethylpyrazole (20.0 g, 180 mmol) and diethyl ethoxymethylenemalonate (42.8 g, 198 mmol) was heated at 120 C. for 5 h. This material was used directly without further purification. If needed, the product can be distilled at 154-160 C. (0.1 mm Hg) to afford the title compound as a liquid which solidified to afford the title compound as a pale colored solid: mp 50-53 C.
a. [[(1-Ethyl-5-pyrazolyl)amino]methylene]malonic acid diethyl ester 245 g. 1-Ethyl-5-aminopyrazole (2.2 mol.) and 476 g. ethoxymethylene malonic acid diethyl ester (2.2 mol.) are heated to 120 (bath temperature) for 2 hours with stirring. The ethanol formed by this reaction is removed by means of a water aspirator. Vacuum distillation (b.p. 0.1 154-160) yields 520 g. (84%) of a quick crystallizing oil of [[(1-ethyl-5-pyrazolyl)amino]methylene]malonic acid diethyl ester, m.p. 50-53. The compound is recrystallized from N-hexane, m.p. 55-57. The hydrochloride salt is formed by treating the above product with dilute ethanolic hydrogen chloride solution.
at 120℃; for 1h;
Preparation of Diethyl [(1-ethyl -lH-pyrazol-5-vD amino] methylene. malonateAmixture of 5-amino-l-ethylpyrazole (1 gm, 0.0089 mole) and diethylethoxymethylenemalonate (1.9 ml, 0.0089 mole) was stirred at 1200C for about 1 hour. The reaction mixture was poured into water and extraction was done with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give yellow oil. Yield: 2.5 g. m/z: (M+H-I) 282.0.
at 120℃; for 1h;
Preparation of Diethyl [(1-ethyl -lH-pyrazol-5-yl) amino] methylene} malonateA mixture of 5-amino-l-ethylpyrazole (1 gm, 0.0089 mole) and diethylethoxymethylenemalonate (1.9 ml, 0.0089 mole) was stirred at 1200C for about 1 hour. The reaction mixture was poured into water and extraction was done with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give yellow oil. Yield: 2.5 gm. m/z: (M++.) 282.0.
at 120℃; for 1h;
Example 1 Preparation of Diethyl [(1-ethyl-1H-pyrazol-5-yl)amino]methylene}malonate A mixture of 5-amino-1-ethylpyrazole (1 gm, 0.0089 mole) and diethylethoxymethylenemalonate (1.9 ml, 0.0089 mole) was stirred at 120 C. for about 1 hour. The reaction mixture was poured into water and extraction was done with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give yellow oil. Yield: 2.5 gm. m/z: (M++1) 282.0.
ethyl 4,7-dihydro-2-(4-methylpiperazin-1-yl)-7-oxo-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With acetic acid; at 120℃; for 3h;
General procedure: A solution of aminotriazole 11a-m (3.1mmol), glacial acetic acid (5mL), and diethyl ethoxymethylenemalonate (4.6mmol) was refluxed for 3h. After cooling to room temperature, the resulting precipitate was collected by filtration, washed with cold water and dried. The residue was stirred with cold ethyl ether (20mL) and filtered to afford the desired product. Following general procedure C, compound 12k was isolated as a white solid. Yield 78%, mp=220C dec. 1H NMR (200MHz, DMSO-d6) delta: 9.95 (br s, 1H); 8.41 (s, 1H); 4.17-4.07 (q, J=7Hz, 2H); 3.78-3.45 (m, 4H); 2.76 (s, 3H); 2.52-2.49 (m, 4H); 1.26-1.19 (t, J=7.2Hz, 3H)
1-(4-Methoxybenzyl)-1H-pyrazol-5-amine (3.94 g, 1 9.39 mmol), followed by diethyl 2-(ethoxymethylene)malonate (4 mL, 20 mmol) was added to a 200 mL round bottom flask fitted with a distillation head to remove ethanol. The mixture was heated to 1 30 C for 45 min, then 1 0 mL of diphenyl ether was added and the temperature was raised to 240 C for 2 h. The reaction mixture was then cooled to rt and Et^O ( 1 00 mL) was added. The resulting precipitate was collected by vacuum filtration and dried under vacuum to afford the target compound as a white solid (4 g, 62%).
62%
at 130℃; for 0.75h;
1-(4-Methoxybenzyl)-1H-pyrazol-5-amine (3.94 g, 1 9.39 mmol), followed by diethyl 2-(ethoxymethylene)malonate (4 mL, 20 mmol) was added to a 200 mL round bottom flask fitted with a distillation head to remove ethanol. The mixture was heated to 1 30 C for 45 min, then 1 0 mL of diphenyl ether was added and the temperature was raised to 240 C for 2 h. The reaction mixture was then cooled to rt and Et^O ( 1 00 mL) was added. The resulting precipitate was collected by vacuum filtration and dried under vacuum to afford the target compound as a white solid (4 g, 62%).
62%
In diphenylether; ethanol; at 130 - 240℃; for 2.75h;
Step 2. Ethyl 4-hvdroxy- l -(4-methoxybenzyl )- l H-pyrazolo[3.4-b]pyridine-5- carboxylate. I -(4-Methoxybenzyl )- l H-pyrazol-5-am inc (3.94 g, 1 9 39 mmol), followed by diethyl 2-(ethoxymethylene)malonate (4 mL, 20 mmol) was added to a 200 m L round bottom flask fitted with a disti llation head to remove ethanol. The mixture was heated to 130 C for 45 min, then 10 mL of diphenyl ether was added and the temperature was raised to 240 C for 2 h. The reaction mixture was then cooled to rt and diethyl ether ( 1 00 mL) was added The resulting precipitate was collected by vacuum fi ltration and dried under vacuum to afford the target compound as a white solid (4 g, 62%).
62%
1-(4-Methoxybenzyl)-1H-pyrazol-5-amine (3.94 g, 1 9.39 mmol), followed by diethyl 2-(ethoxymethylene)malonate (4 mL, 20 mmol) was added to a 200 mL round bottom flask fitted with a distillation head to remove ethanol. The mixture was heated to 130 C for 45 min, then 10 mL of diphenyl ether was added and the temperature was raised to 240 C for 2 h. The reaction mixture was then cooled to rt and Et2O ( 1 00 mL) was added. The resulting precipitate was collected by vacuum filtration and dried under vacuum to afford the target compound as a white solid (4 g, 62%).
62%
In diphenylether; at 130 - 240℃; for 2.75h;
[0179] Step 2. Ethyl 4-hydroxy- 1- (4-methoxybenzyl)- 1 H-pyrazolo [3 ,4-bl pyridine-5- carboxylate. 1- (4-Methoxybenzyl)- 1 H-pyrazol-5-amine (3.94 g, 19.39 mmol), followed by diethyl 2-(ethoxymethylene)malonate (4 mL, 20 mmol) was added to a 200 mL round bottom flask fitted with a distillation head to remove ethanol. The mixture was heated to 130 C for 45 mm, then 10 mL of diphenyl ether was added and the temperature was raised to 240 C for2 h. The reaction mixture was then cooled to rt and Et20 (100 mL) was added. The resulting precipitate was collected by vacuum filtration and dried under vacuum to afford the target compound as a white solid (4 g, 62%).
62%
at 130℃; for 0.75h;
[0187] Step 2. Ethyl 4-hydroxy- l-(4-methoxybenzyl)-lH-pyrazolor3,4-blpyridine-5- carboxylate. l-(4-Methoxybenzyl)- lH-pyrazol-5-amine (3.94 g, 19.39 mmol), followed by diethyl 2-(ethoxymethylene)malonate (4 mL, 20 mmol) was added to a 200 mL round bottom flask fitted with a distillation head to remove ethanol. The mixture was heated to 130 C for 45 min, then 10 mL of diphenyl ether was added and the temperature was raised to 240 C for 2 h. The reaction mixture was then cooled to rt and diethyl ether (100 mL) was added. The resulting precipitate was collected by vacuum filtration and dried under vacuum to afford the target compound as a white solid (4 g, 62%
A mixture of <strong>[5369-19-7]3-tert-butylaniline</strong> (5.30 g, 35.5 mmol) and diethyl ethoxymethylenemalonate (10.2 g, 47.30 mmol) were heated at 60 0C for 15 min. then 1 hour at 120 0C. After the generated ethanol was evaporated in vacuo, the crude oil was added dropwise to boiling diphenylether (150 ml) at 200-2500C and the mixture was stirred at 25O0C for 90 min. After cooling to room temperature, the mixture was diluted with hexane (ca. 200 ml) and the precipitate solid was collected to give (4.55 g, 47 percent) of the title compound as a slightly yellow solid. The compound was used for the next step without the determination of NMR, MS for hard solids.
A mixture of <strong>[59702-07-7]1-<strong>[59702-07-7]methylpiperazin-2-one</strong></strong> (183 g, 1.6 mol) and diethyl ethoxymethylenemalonate (346 g, 1.6 mol) in toluene (12 L) was heated at 100 °C overnight. The resultant mixture was concentrated under vacuum. The residue was dissolved in anhydrous THF (8 L), brought to reflux under an atmosphere of nitrogen, and treated with a solution of lithium bis (trimethylsilyl)amide in THF (1 M, 1.05 eq). The reaction mixture was allowed to cool to room temperature and concentrated under vacuum. The residue was partitioned between methylene chloride and dilute aqueous HCI. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with ethyl acetate, cooled to -20 °C, and the solid precipitated was filtered to provide the title compound. 1H NMR (400 MHz, DMSO-d6) 8 8.50 (s, 1H), 7.33 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 4.11 (t, J = 5.5 Hz, 2H), 3.59 (t, J = 5.5 Hz, 2H), 2.92 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H). ES MS M+l = 239
2-Phenylaminomethylene-malonic acid diethyl ester A mixture of aniline (25.6 g, 0.28 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.29 mol) was heated at 140-150 C. for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. 1H NMR (d-DMSO) delta 11.00 (d, 1H), 8.54 (d, J=13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
With potassium carbonate; In ethanol; for 3h;Heating / reflux;
A suspension of <strong>[60481-51-8]3,4-dimethylphenylhydrazine hydrochloride</strong> (2.0 g; 0.012 mol.) and potassium carbonate (3.2 g; 0.023 mol.) in anhydrous ethanol (40.0 mL) was treated dropwise with diethyl(ethoxymethylene)malonate (2.5 g; 0.012 mol.) and the mixture stirred and heated under reflux for 3 h. After cooling the mixture was evaporated and the residue suspended in water and acid;ified to pH 2 followed by extraction with ethyl acetate. After drying and evaporation the resulting intermediate 1-(3,4-dimethyl-phenyl)-3-oxo-2,3-dihydro-1H-pyrazole-4-carboxylic acid; ethyl ester was dissolved in methanol (10.0 mL) and 10percent aqueous sodium hydroxide (10.0 mL) and stirred at room temperature for 3 h and then heated under reflux to effect hydrolysis. The solution was cooled and acid;ified to pH 4 with 3M aqu. hydrochloric acid; and stirred at room temperature for 2 h to effect decarboxylation. The mixture was extracted with ethyl acetate, dried and evaporated to give the title compound (87percent) as a yellow solid. 1H NMR (300 MHz, CDCl3) delta 7.61 (d, J=2.3 Hz, 1H), 7.55 (dd, J=8.2 and 2.3 Hz, 1H), 7.48 (t, J=1.3 Hz, 1H), 1.16 (d, J=8.2 Hz, 1H), 3.50 (d, J=1.3 Hz, 2H)
Intermediate 1 : Ethyl 4-chloro-1-ethyl-lH-pyrazolo [3, 4-b] pyridine-5-carboxylate; A mixture of 5-amino-l-ethyl pyrazol (806g) (e. g. commercially available from Aldrich) and diethyl ethoxymethylenemalonate (1621 ml) (e. g. commercially available from Aldrich) was stirred and heated at 160C under nitrogen, in a 5 litre flask fitted with a Dean-Stark apparatus, for 1. 5 h. Ethanol that distilled out of the reaction mixture (320mol) was collected in the Dean-Stark apparatus. The reaction mixture was stirred and heated at 160C, under nitrogen, for a further 6h, cooled to room temperature and divided into two batches (1200mol + 1000mol :"Batch 1"and "Batch 2"). The first (1200ml) batch ("Batch 1") was divided into two roughly equal portions. Phosphorus oxychloride (1. 85 litres) was added to each portion. The reaction mixtures were then heated at reflux in two 51 flasks for 13h. Excess phosphorus oxychloride was distilled from both flasks under reduced pressure. The residue were cooled to room temperature, then the contents of both flasks were poured slowly onto one portion (10kg) of crushed ice. The mixture was stirred for 15 min and then extracted with diethyl ether (3 x 2. 5 litres). The combined organic were washed with water (2 litres) and brine (2 x 2 litres), then dried over Na2SO4. Evaporation of the solvent afforded the crude Intermediate 1 as a brown oil (865g) which solidified immediately on cooling. An identical procedure was used to prepare a further 710g of crude Intermediate 1 as a solid from"Batch 2"using 3. 1 litres of phosphorus oxychloride ; i. e. a total of 1575g of crude Intermediate 1 was isolated as a solid. This solid (430g) was dissolved in hexane (4. 3 litres, i. e. 10 vols.) by heating to 50C with stirring. Activated charcoal (64. 5g) was added. The mixture was stirred at 50C for 1. 0 h, then filtered through a celite bed. The celite bed was washed with hexane (2 x 430 ml). The combined filtrate and the washing were concentrated to about 950mol and left to stand at 10-15C overnight. The resultant suspension was filtered. The residual solid was washed with chilled hexane (3 x 215 ml slurry wash, plus 2 x 400 ml displacement wash) and dried to give Intermediate 1 (280g) as a pale yellow solid. The combined mother liquor and the washing were concentrated to about 300 ml, then cooled and left to stand at 10-15C overnight to afford an additional 30g of Intermediate 1. LCMS showed MEL+ = 254 ; TRET = 3. 09 min
Step 4: Ethyl 8-hydroxy-2-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-alpha]-pyrazine-7-carboxylate; -A mixture of l-<strong>[59702-07-7]methylpiperazin-2-one</strong> (183 g, 1.6mol) and diethyl ethoxymethylenemalonate (346 g, 1.6 mol) in toluene (12 L) was heated at 100 0C overnight. The resultant mixture was concentrated under vacuum. The residue was dissolved in anhydrous THF (8 L), brought to reflux under an atmosphere of nitrogen, and treated with a solution of lithium bis(trimethylsilyl)amide in THF (I M, 1.05 eq). The reaction mixture was allowed to cool to room temperature and concentrated under vacuum. The residue was partitioned between methylene chloride and dilute aqueous HCl. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with ethyl acetate, cooled to -20 0C, and the solid precipitated was filtered to provide the title compound. lH NMR (400 MHz,DMSO-d6) delta 8.50 (s, IH), 7.33 (s, IH), 4.18 (q, J= 7.1 Hz, 2H), 4.11 (t, J= 5.5 Hz, 2H), 3.59 (t, J= 5.5Hz, 2H), 2.92 (s, 3H), 1.24 (t, J= 7.1 Hz, 3H). ES MS M+l = 239.
A mixture of l-<strong>[59702-07-7]methylpiperazin-2-one</strong> (183 g, 1-6mol) and diethyl ethoxymethylenemalonate (346 g, 1.6 mol) in toluene (12 L) was heated at 100 °C ovemight. The resultant mixture was concentrated under vacuum. The residue was dissolved in anhydrous THF (8 L), brought to reflux under an atmosphere of nitrogen, and treated with a solution of lithium bis (trimethylsilyl) amide in THF (1 M, 1.05 eq). The reaction mixture was allowed to cool to rt and concentrated under vacuum. The residue was partitioned between methylene chloride and dilute aqueous HC1. The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with ethyl acetate, cooled to-20 °C, and the solid precipitated was filtered to provide the title compound. 1H NMR (400 MHz, DMSO-d6) S 8.50 (s, 1H), 7.33 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 4.11 (t, J = 5.5 Hz, 2H), 3.59 (t, J = 5.5 Hz, 2H), 2.92 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H). ES MS M+1 = 239
EXAMPLE 59 8-Chloro-N-((4-chlorophenyl)methyl)-4-hydroxy-5-trifluoromethyl-3-quinolinecarboxamide [] A mixture of 2-chloro-5-trifluoromethylaniline (3.91 g) and diethyl ethoxymethylenemalonate (4.0 mL) is heated at 190°C for 2 h and is then diluted with diphenyl ether (30 mL). The mixture is allowed to cool to rt, filtered, and the white solid is washed with hexane (2 x 10 mL) to afford 1.632 g of the diethyl aminomethylenemalonate. The resulting intermediate (2.63 g) is suspended in diphenyl ether (30 mL) and heated to reflux with a Dean-Stark trap for 3 h. The mixture is allowed to cool to rt and is then poured into hexane (50 mL). The solid is filtered and then washed with hexane (20 mL) and hexane/diethyl ether (1/1, 20 mL) to afford 1.273 g of the quinolinecarboxylate ethyl ester. The ester (400 mg) and 4-chlorobenzylamine (1.52 mL) are heated at 190 °C for 1 h. The resulting mixture is diluted with toluene (4 mL), allowed to cool to rt, and then poured into hexane (50 mL). The solvent is decanted and the remaining oil is crystallized (acetic acid, water) to afford 285 mg of the title compound as a brown solid. Physical characteristics are as follows: Mp 270-1 °C.1H NMR (300 MHz, DMSO) delta 12.33, 10.06, 8.68, 8.10, 7.86, 7.36, 4.52.13C NMR (100 MHz, DMSO) delta 175.05, 164.07, 144.36, 139.12, 138.20, 132.42, 131.85, 129.84, 128.77, 128.44, 126.70 (q), 125.84, 125.20, 122.22, 113.75, 42.00.IR (mull) 3187, 3091, 2925, 2855, 1657, 1604, 1567, 1531, 1462, 1417, 1377, 1366, 1348, 1307, 1272, 1210, 1158, 1139, 1128, 1106, 854, 841, 802, 726 cm-1.Anal. Found: C, 52.12; H, 2.78; N, 6.70; Cl, 16.85.MS (ESI-) for C18H11Cl2F3N2O2m/z 412 (M-H)-.
c) ethyl 8-fluoro-6-(methoxy)-4-oxo-1 ,4-dihydro-3-quinolinecarboxylate A mixture of aniline <strong>[458-52-6]2-fluoro-4-(methoxy)aniline</strong> (22.8 g, 162 mmol) and diethyl [(ethyloxy)methylidene]propanedioate (32.6 mL) were heated to reflux in Dowtherm A under a flow of argon. After 15 minutes (when all ethanol was removed), the mixture was allowed to cool down and was diluted with pentane. A precipitate was formed which was triturated with pentane, filtered and dried under vacuum to afford the product as an oil (33.06 g, 77percent); MS (+ve ion electrospray) m/z 265 (M+H)+.
a 7-Iodo-6-methyl-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid ethyl ester A mixture of <strong>[35944-64-0]3-iodo-4-methylaniline</strong> (10.0 g, 42.9 mmol) and diethyl ethoxy methylenemalonate (12.4 g, 57.2 mmol) was heated at 150 C. for 80 min, and the liberated ethanol was allowed to evaporate. After cooling Dowtherm A (50 mL) was added, then the mixture was heated at 250 C. for 75 min. After cooling to room temperature, heptane (50 mL) was added and the precipitate collected by filtration. The crude material was subsequently triturated in hexane/ethyl acetate (50 mL) and in 70% aqueous ethanol (2*300 mL) to afford the title compound (8.30 g, 54%). White solid, ISP-MS: m/e=358.1 ([M+H]+).
diethyl 2-(((2-bromopyrimidin-5-yl)amino)methylene)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Preparation 3 Diethyl 2-(((2-Bromopyrimidin-5-yl)amino)methylene)malonate (N.2). A mixture of <strong>[56621-91-1]2-bromopyrimidin-5-amine</strong> (Krchnak, V.; Arnold, Z. Coll. Czech. Chem. Commun. 1975, 40, 1396-1402) (3.48 g) and diethyl ethoxymethylenemalonate (5.05 mL) is heated to 135 C. for 1 h. The mixture is allowed to cool affording a solid.
diethyl 2-(((2-chloro-6-methyl-3-pyridinyl)amino)methylene)-malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
17.9 g (95%)
In dichloromethane;
Preparation 42 Diethyl 2-(((2-Chloro-6-methyl-3-pyridinyl)amino)methylene) malonate [BT.2] A mixture of 2-chloro-6-methyl-3-pyridinylamine (8.5 g) and diethyl ethoxy-methylenemalonate (22.0 mL) is heated at 140° C. for 18 hours. The mixture is cooled to room temperature and the resulting solids are recrystallized from a mixture of heptane (400 mL) and CH2Cl2 (2 mL) to obtain 17.9 g (95percent) of the title compound as a pale red solid. Physical characteristics: MS (ESI+) m/z 313 (M+H).
<strong>[39052-12-5]3,4-Diethoxyaniline</strong> (3.7 g) and diethyl ethoxymethylenemalonate (5.3 g) were reacted in the same manner as in Experimental Example 1 to obtain 6,7-diethoxy-3-ethoxycarbonyl-4(1H)-quinolone (4.3 g).
C. Ethyl 7-bromo-6,8-difluoro-4-hydroxyquinoline-3-carboxylate A stirred mixture of 26 g of <strong>[103977-79-3]3-amino-2,6-difluorobromobenzene</strong> and 27 g of diethyl ethoxymethylenemalonate was heated at 150 C. for 1 hour. After cooling, 100 ml of Dowtherm "A" (commercially available high boiling inert solvent mixture of diphenylether and dibenzofuran) was added and the mixture was heated under nitrogen at 260 C. for 1.5 hour. The mixture was cooled to room temperature and 200 ml of hexanes was added. The resulting precipitate was collected by filtration, washed with hexanes and dried to give a cream solid, m.p. 285 C., yield 78% (32.32 g).
The mixture of 4.8 g of this product and 5.3 g of diethyl ethoxymethylenemalonate was heated for 1 hour at from about 140 to 145 C. (bath temperature). After reaction, the ethanol produced was removed by evaporation to provide an oily product. 35 g of ethyl polyphosphate was added thereto and the mixture was stirred for 1 hour at from about 140 to 145 C. (bath temperature). After cooling, the reaction mixture was added to ice-cold water. Precipitate was extracted with 200 ml of chloroform, this procedure was carried out three times, and the extracts were combined and was washed with a 5% potassium hydroxide solution and water. The chloroform layer was dried with sodium sulfate to provide 5.1 g of ethyl 9,10-difluoro-3-methyl-7-oxo-2,3-dihydro-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylate as white powder with m.p. 261 C.
2-(2,2-dicarbethoxyethenylamino)-4,5-dimethylthiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol;
EXAMPLE 11 2-(2,2-Dicarbethoxyethenylamino)-4,5-dimethylthiazole 2-Amino-4,5-dimethylthiazole (2.56 g., 20 mmoles), diethyl ethoxymethylenemalonate (4.8 g., 22 mmoles) and ethanol (5 ml.) were refluxed for 1 hour. The reaction mixture was cooled and the crude product precipitated with hexane.
A mixture of 3-t-butylaniline (67.6 g.) and diethyl ethoxymethylenemalonate was heated on the steam bath for 3 hours, the ethanol formed being collected by distillation. The resulting yellow oil was dissolved in diphenyl ether (200 ml.) and added during 0.5 hour to stirred diphenyl ether (800 ml.) at 250°-260°, the ethanol formed being collected by distillation. The mixture was stirred at 250°-260° for 0.5 hour, then allowed to cool to room temperature and diluted with an equal volume of petroleum ether (b.p. 60°-80°). The precipitate was collected by filtration to give the novel compound ethyl 7-t-butyl-4-hydroxyquinoline-3-carboxylate, m.p. 279°-281°.
To a suspension solution of 6-fluoroquinolin-4-ol (3.67 g, 22.5 mmol) (synthesized in overall yield of 32percent starting from 4-fluoroaniline and diethyl ethoxymethylenemalonate according to a literature procedure (see Price et al., Organic Syntheses, Coll. Vol. 3, p. 272; Vol. 28, p. 38), triphenylphosphine (6.56 g, 25 mmol), and 3-bromo-1-propanol (2.3 mL, 25 mmol) in dry tetrahydrofuran (30 mL) was added diisopropyl azodicarboxylate (4.9 mL, 25 mmol) at 25° C. over 30 minutes under nitrogen and the reaction mixture was left stirring for one hour. Hydrobromic acid (2.8 mL of 48percent aqueous solution, 25 mmol) was added, resulting in precipitation of the titled compound. The product was collected by suction filtration and washed with THF, acetone and ether to give a light yellow crystalline solid (4.17 g, 51percent). 1H NMR (400 MHz, DMSO-d6): 2.45 (2H, m), 3.82 (2H, t, J=6.5 Hz), 4.61 (2H, t, J=5.7 Hz), 7.64 (1H, d, J=6.6 Hz), 8.40 (1H, dt, J=8.5, 2.7 Hz), 7.90 (m, 1H), 8.27 (1H, dd, J=9.4, 4.8 Hz), 9.24 (1H, d, J=6.6 Hz).
With hydrogenchloride; sodium hydroxide; In diphenylether; hexane; water;
(a) 6-Fluoro-4-hydroxyquinoline A mixture of 216 ml of diethyl ethoxymethylenemalonate, 95 ml of 4-fluoroaniline, and 800 ml of diphenyl ether was stirred and slowly heated to 185° with distillation of the ethanol formed. The reaction mixture was then heated to 245° for 1.5 hr. The total amount of ethanol collected was 105 ml. The reaction mixture was allowed to cool to 60° at which time 250 ml of hexane was added. After it had stood overnight, the product was collected, washed with hexane and dried to give 223 g of solid mp 305°-309°. A mixture of the above solid, 600 ml of 3N sodium hydroxide, and 250 ml of water was stirred and heated under reflux for 2 hr. To the hot brown cloudy mixture was slowly added 600 ml of 3N hydrochloric acid to precipitate the product, 6-fluoro-4-hydroxyquinoline-3-carboxylic acid. This product was filtered from the cooled reaction mixture. This damp solid and 600 ml of diphenyl ether was slowly heated to 250° while water was allowed to distill out. The reaction mixture was held at this temperature for 20 min. and then allowed to cool to 100°. Then 500 ml of hexane was added, and the mixture was stirred for 1 hr. The product 6-fluoro-4-hydroxyquinoline, was collected and washed with hexane to give 144 g, mp 209°-212°.
oil [[(1-ethyl-5-pyrazolyl)amino]methylene]malonic acid diethyl ester[ No CAS ]
[ 26823-99-4 ]
Yield
Reaction Conditions
Operation in experiment
a. [[(1-Ethyl-5-pyrazolyl)amino]methylene]malonic acid diethyl ester 245 g. of 1-Ethyl-5-aminopyrazole (2.2 mol.) and 476 g. of ethoxymethylene malonic acid diethyl ester (2.2 mol.) are heated to 120 (bath temperature) for 2 hours with stirring. The ethanol formed by this reaction is removed by means of a water aspirator. Then vacuum distillation (b.p.0.1 154-160) yields 520 g. (84% of theory) of a quickly crystallizing oil [[(1-ethyl-5-pyrazolyl)amino]methylene]malonic acid diethyl ester, m.p. 50-53. The compound is recrystallized from N-hexane, m.p. 55-57. The hydrochloride salt is formed by treating the above product with dilute ethanolic hydrogen chloride solution.
(1-Ethyl-5-byrazolyl)amino]methylene]malonic acid diethyl ester[ No CAS ]
[ 26823-99-4 ]
Yield
Reaction Conditions
Operation in experiment
a. [[(1-Ethyl-5-byrazolyl)amino]methylene]malonic acid diethyl ester 245 g of 1-ethyl-5-aminopyrazole (2.2 mol) and 476 g of ethoxymethylene malonic acid diethyl ester (2.2 mol) are heated to 120 (bath temperature) for 2 hours with stirring. The ethanol formed by this reaction is removed by means of a water aspirator. Vacuum distillation (b.p. 0.1 154-160) yields 520 g (84% of theory) of a quickly crystallizing oil of [[(1-ethyl-5-pyrazolyl)amino]methylene]malonic acid diethyl ester, m.p. 50-53. The compound is recrystallized from N-hexane, and has a melting point of 55-57.
diethyl N-[1,2-dihydro-2-oxo-5-(4-pyridinyl)-3-pyridinyl]aminomethylenemalonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; ethanol;
J-1. Diethyl N-[1,2-dihydro-2-oxo-5-(4-pyridinyl)-3-pyridinyl]aminomethylenemalonate (also named diethyl [1,6-dihydro-6-oxo-(3,4'-bipyridin)-5-ylaminomethylene]propanedioate) -- A mixture containing 9.4 g. of <strong>[60719-84-8]3-amino-5-(4-pyridinyl)-2(1H)-pyridinone</strong>, 10.8 g of diethyl ethoxymethylenemalonate and 100 ml. of ethanol was refluxed with stirring on a steam bath for six and one-half hours. The reaction mixture was filtered through infusorial earth and the filtrate was distilled in vacuo to remove the solvent. The solid residue was recrystallized once from ethanol and then once from methanol using decolorizing charcoal, washed successively with isopropyl alcohol and ether, and then dried to yield 22 g. of diethyl N-[1,2-dihydro-2-oxo-5-(4-pyridinyl)-3-pyridinyl]aminomethylenemalonate, m.p. 218°-222° C.
In methanol; ethanol;
J-1. Diethyl N-[1,2-dihydro-2-oxo-5-(4-pyridinyl)-3-pyridinyl]aminomethylenemalonate (also named diethyl [1,6-dihydro-6-oxo-(3,4'-bipyridin)-5-ylaminomethylene]propanedioate) - A mixture containing 9.4 g. of <strong>[60719-84-8]3-amino-5-(4-pyridinyl)-2(1H)-pyridinone</strong>, 10.8 g. of diethyl ethoxymethylenemalonate and 100 ml. of ethanol was refluxed with stirring on a steam bath for 61/2 hours. The reaction mixture was filtered through infusorial earth and the filtrate was distilled in vacuo to remove the solvent. The solid residue was recrystallized once from ethanol and then once from methanol using decolorizing charcoal, washed successively with isopropyl alcohol and ether, and then dried to yield 22 g. of diethyl N-[1,2-dihydro-2-oxo-5-(4-pyridinyl)-3-pyridinyl]aminomethylenemalonate, m.p. 218°-222° C.
[[[1-[(4-Methoxyphenyl)methyl]-1H-pyrazol-5-yl]amino]methylene]propanedioic acid diethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
at 130℃; for 2.5h;
B. [[[1-[(4-Methoxyphenyl)methyl]-1H-pyrazol-5-yl]amino]methylene]propanedioic acid diethyl ester A mixture of 126 g (0.62 mols) of Part A amine and 140 mL (0.69 mols) of diethyl ethoxymethylenemalonate was heated under reduced pressure with stirring to ~130 under a Dean-Stark apparatus. The reaction mixture was heated for 2.5 h then cooled to room temperature to give 237 g (100%) of compound B as an orange-red oil.
at 120℃; for 5h;
Mixture of 1-(4-methoxy-benzyl)-1H-pyrazol-2-ylamine (1 g, 5.0 mmol) and diethyl ethoxymethylenemalonate (1 mL, 5.0 mmol) is heated at 120 C. for 5 h. It is cooled and then the desired compound is isolated by silica gel column chromatography. MS m/z 374.2 (M+1).
at 120℃; for 1.5h;
A mixture of <strong>[3528-45-8]1-(4-methoxybenzyl)-1H-pyrazol-5-amine</strong> (3 g, 10 mmol) and (ethoxymethylene)propanedioic acid, diethyl ester (3.8 g, 18 mmol) was heated at 120 C. for 1.5 h, then allowed to cool. After cooling, the mixture was concentrated and the resulting residue was purified with CombiFlash eluting with 0-30% EtOAc in hexanes to give an intermediate, LCMS (M+H): 374.1. The intermediate was dissolved in diphenyl ether (5 mL). The resulted solution was heated at 240 C. in microwave reactor for 1 h 20 min. After cooling, the solid crashed out was filtered and washed with hexanes to afford 4.0 g (80%) of the sub-title compound. LCMS calc. for C17H18N3O4 (M+H)+: m/z=328.1. Found: 328.1
at 125 - 130℃; for 2h;Inert atmosphere;
In a round-bottom flask, equipped with a stirrer, thermometer and reflux condenser, mix under nitrogen in the specified order: 1 g (0.0048 mol) of BCD-BTK-13-14 and 1.17 g (0.0051 mol) of diethyl ethoxymethylenemalonate. Heat the mixture for 2 hours at 125-130 . Distill off the solvent using a rotary evaporator. Take the mixture to the next step without additional purification. Yield: 1.88 g (99%).
at 120℃; for 5h;
Acrylonitrile (7.5 g, 141 mmol) is dissolved in THF (75 mL) and cooled at 00C, and hydrazine monohydrate (7.41g, 148 mmol) is added at the same temperature. The reaction mixture is brought to room temperature and stirred for 2 h. p-Anisaldehyde (20.1 g, 148 mmol) is added drop-wise and stirred at room temperature for 1O h. The above reaction mixture is concentrated to give a red oil, followed by drop- wise addition of n-BuONa in 80 mL n-BuOH at 00C. The reaction mixture is brought to room temperature, then heated at 120 0C for 3.5 h. After cooling, the reaction mixture is poured onto 300 mL of cold water and extracted with 2 X 200 mL of Et2O. The ether layer is collected and washed with 3 X 100 mL of IN HCl. The combined acidic layer is collected and neutralized with 50 % NaOH. The mixture is again extracted with 200 mL Of Et2O, washed with water, dried over Na2SO4, and concentrated to give l-(4-Methoxy- benzyl)-lH-pyrazol-2-ylamine as red oil. MS m/z 204.1 (M + 1). A mixture of l-(4-methoxy- benzyl)-lH-pyrazol-2-ylamine (1 g, 5.0 mmol) and diethylethoxymethylene- malonate (1 mL, 5.0 mmol) is heated at 1200C for 5 h, cooled and isolated by silica gel column chromatography. MS m/z 374.2 (M + 1).
General procedure: A solution of aminotriazole 11a?m (3.1mmol), glacial acetic acid (5mL), and diethyl ethoxymethylenemalonate (4.6mmol) was refluxed for 3h. After cooling to room temperature, the resulting precipitate was collected by filtration, washed with cold water and dried. The residue was stirred with cold ethyl ether (20mL) and filtered to afford the desired product. Following general procedure C, compound 12c was isolated as a white solid. Yield 67percent, mp>300°C. 1H NMR (200MHz, DMSO-d6) delta: 14.00 (br s, 1H); 8.64 (s, 1H); 8.14?8.10 (m, 2H); 7.55 (m, 3H); 4.26 (q, J=7.0Hz, 2H); 1.28 (t, J=6.8Hz, 3H)
A mixture of amine (1 equiv.) and diethyl ethoxymethylenemalonate (1.35 equiv.) in 1,2,4-trichlorobenzene is heated at 160 C with a distill collector for 16 hr. Ethanol that forms during the reaction is collected at the top of a short fractionating column. Next, the reaction mixture is allowed to cool and then the solid is filtered off, washed with hexane, dried to give the ethyl ester product.
diethyl 2-(((4-(1H-pyrazol-1-yl)phenyl)amino)methylene)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
In ethanol; for 12h;Reflux;
Synthesis of diethyl 2-((4-(1H-pyrazol-1-yl)phenylamino)methylene) malonate Compound b (4 mmol) and diethyl ethoxymethylidenemalonate (1.5 eq.) were dissolved in ethanol (15 mL), refluxed for 12 hrs, cooled, filtered and dried to give compound c (1.2 g) in 90percent yield. Exact Mass (cal): 329.1376; MS (ESI) m/e(M+1)+: 330.1460.
1.2 g
In ethanol; for 12h;Reflux;
A mixture of 1-phenyl-5-aminopyrazole (0.64g, 4mmol) and diethyl ethoxymethylenemalonate (1.3g, 6mmol) was refluxed in EtOH (15mL) for 12h. After cooling, the reaction mixture was filtered to afford the desired product 33 (1.20g, 90percent yield) 1H NMR (400MHz, DMSO-d6) delta 10.78 (s, 1H), 8.44 (d, J=12.9Hz, 2H), 7.85 (s, 2H), 7.73 (s, 1H), 7.49 (s, 2H), 6.52 (s, 1H), 4.18 (s, 4H), 1.26 (d, J=5.0Hz, 4H). 13C NMR (100MHz, DMSO-d6) delta 151.42, 141.30, 137.93, 136.92, 127.99, 119.95, 118.97, 108.21, 100.00, 93.88, 60.02,14.66. LC/MS (ESI, m/z) [M+H]+: 330.15.
ethyl 5-hydroxy-2-methyl-3-[(pyrazin-2-ylmethyl)amino]-2,3-dihydroisoxazole-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
General procedure: To a mixture of diethyl 2-(ethoxymethylene)malonate (4) (0.6 mmol) and hydroxylamine 5 (0.6 mmol) was added pyridine (0.1 mmol), andthe resulting mixture was stirred at 85 C for 4 h. After cooling the reaction mixture to r.t., amine 2 (0.5 mmol), K2CO3 (0.5 mmol or 1.0mmol) and EtOAc (3 mL) were added. The mixture was stirred at r.t. until the reaction was complete (as detected by TLC monitoring). The insoluble salt (K2CO3) was filtered out and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EtOAc, 10:1 to 4:1) to afford the desired product 3.
diethyl 2-(((2-methylthiazol-5-yl)amino)methylene)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
32%
In ethanol; at 0 - 90℃; for 4h;
2-Methylthiazol-5-amine (4.5 g, 39.41 mmol) was dissolved in EtOH (150 mL). Diethyl 2- (ethoxymethylene)malonate (7.97 mL, 39.41 mmol) was added and the mixture stirred at 90 C for 3 hours. The mixture was cooled to 0 C and stirred for 1 hour, and the resulting solid was filtered, washed with EtOH and dried to give the title compound (3.62 g, 32% yield). ?H NMR (500 MHz, DMSO-d6): 10.74 (d, 1H), 7.94 (d, 1H), 7.53 (s, 1H), 4.19 (q, 2H), 4.11 (q, 2H), 2.58 (s, 3H), 1.24 (m, 6H); MS mlz: 285.06 [M+Hjt
ethyl 2-(3-fluorophenyl)-4-hydroxypyrimidine-5-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium ethanolate; In ethanol; for 2h;Reflux;
To a flask containing commercially available <strong>[75207-72-6](3-fluorobenzenecarboximidoyl) ammonium chloride</strong> (5 g, 28.64 mmol) was added sodium ethoxide in ethanol (21 %) (21 .4 ml_, 57.27 mmol) followed by a solution of diethyl 2-(ethoxy methylene)propanedioate (5.79 ml_, 28.64 mmol) in ethanol (10 ml_). The mixture was heated to reflux for 2 hours and after cooling to room temperature, treated with 6N HCI aq. (50 ml_). The precipitate was filtered and washed with water (2 x 200 ml_). The filtered solid was dissolved in EtOAc (1 litre) and dried over MgS04. The filtrate was re-filtered and the solid washed with water (2 x 25 ml_) before being dissolved in EtOAc (500 ml_), dried over MgS04, combined with the other EtOAc fraction and concentrated in vacuo to a volume of ~ 50m L at which point the formed solid was collected by filtration to afford the titled compound. 1 H NMR (500 MHz, Chloroform -d) d 1 1 .73 (s, 1 H), 9.04 (s, 1 H), 8.26 (d, J = 7.9 (1230) Hz, 1 H), 8.18 (dt, J = 10.1 , 2.2 Hz, 1 H), 7.49 (td, J = 7.9, 5.7 Hz, 1 H), 7.30 - 7.22 (m, 1 H), 4.49 (q, J = 7.1 Hz, 2H), 1 .45 (t, J = 7.1 Hz, 3H).
EXAMPLE 85 1-Ethyl-5,6,8-trifluoro-1,4-dihydro-7-(4-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid A mixture of 65.5 g of <strong>[5580-80-3]2,3,4,5-tetrafluoroaniline</strong> and 86.5 g of diethyl diethylethoxymethylene malonate was stirred and heated at 130 C. for one hour, then allowed to cool and the solid, which was [[(2,3,4,5-tetrafluorophenyl)amino]methylene], diethyl ester, collected. This solid was added to 400 ml of diphenyl ether, heated at 250 C. for one hour, then cooled, diluted with hexane and cooled in ice. The solid was collected, dried and recrystallized from dimethylformamide with charcoal treatment, giving 57.9 g of 5,6,7,8-tetrafluoro-4-hydroxy-3-quinolinecarboxylic acid, ethyl ester.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






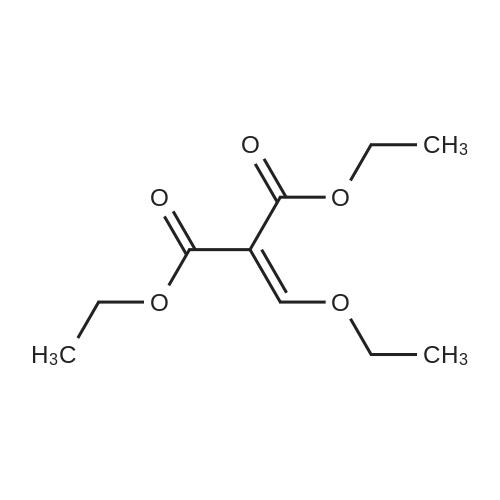

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping