| 23% |
With 2,6-dimethylpyridine; dmap; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 3.5h; |
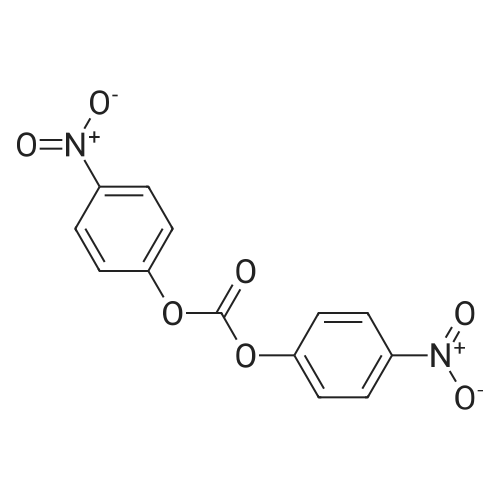
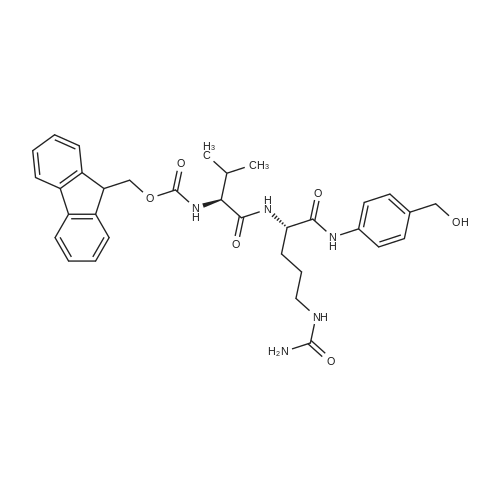
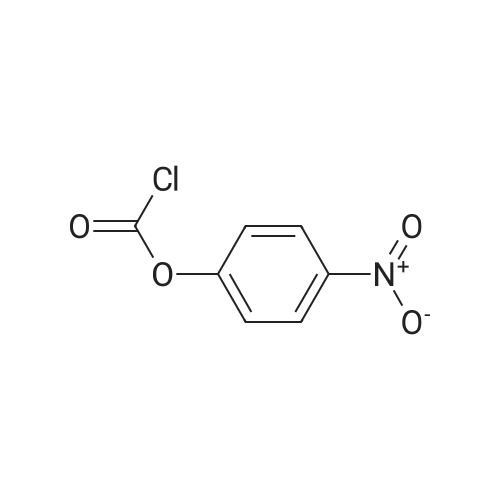
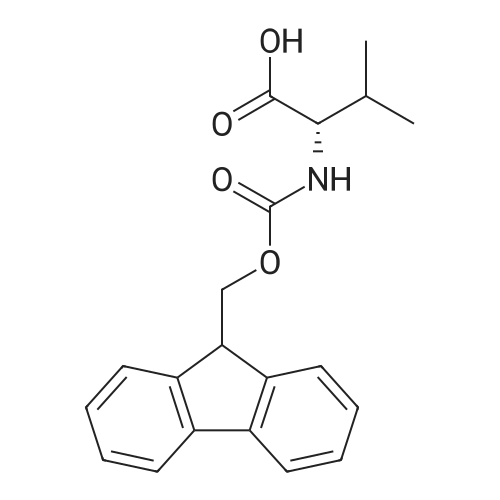
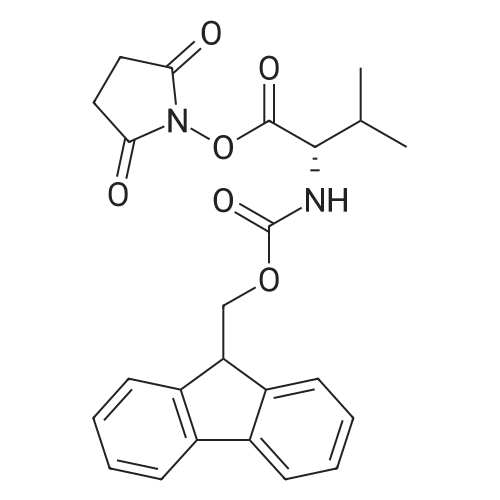
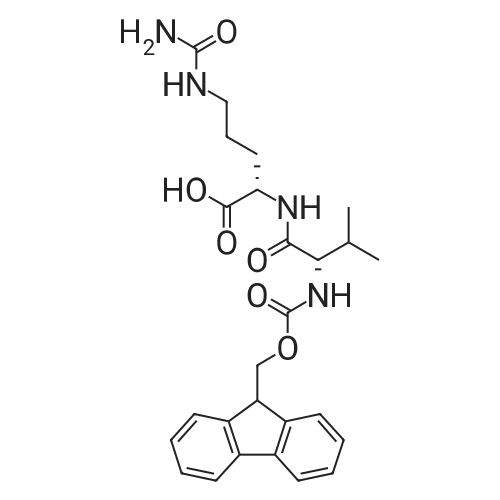
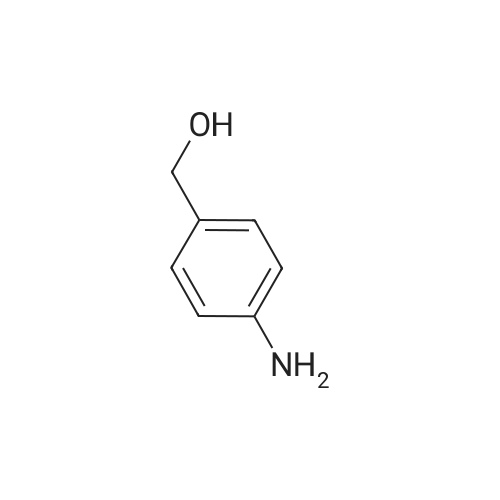
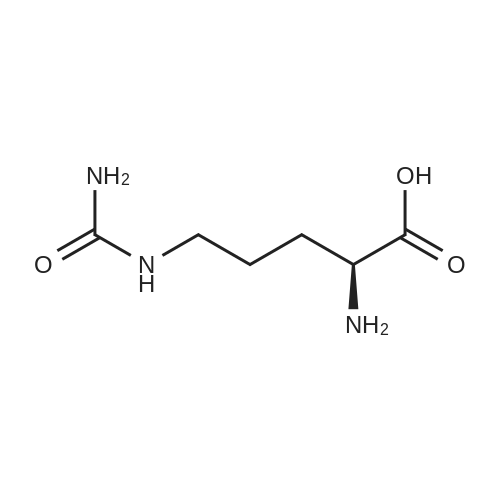
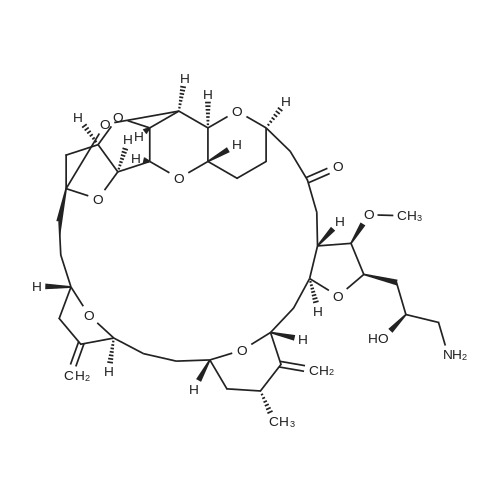
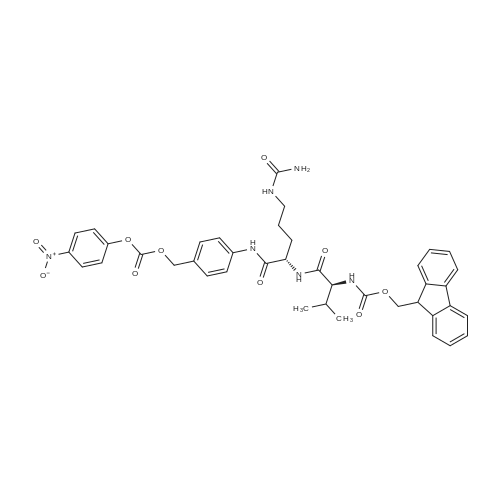
Example A67 Preparation of Ar-{7-[(2,5-dioxopyrrolidin-l-yl)oxy]-7-oxoheptanoyl}-L-valyl-N-[4-([(2- [(3Lambda,55,7Lambda,8Lambda)-7-{(1£,3^-5-[(25,35,5Lambda,^ dimethyltetrahydro-2H-pyran-2-yl]-3-methylpenta-l,3-dien-l-yl}-8-hydroxy-l,6- dioxaspiro[2.5]oct-5-yl]acetyl}hydrazinyl)carbonyl]oxy}methyl)phenyl]-A^-carbamoyl-L- ornithinamide (B190). Step 1. Synthesis of N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-valyl-N-[4-( [(2- [(3R,5S,7R,8R)-7-{(l£,3£)-5-[(2S,3S,5R,6R)-5-[(2Z,4S)-4-(acetyloxy)pent-2-enoyl]amino}-3,6- dimethyltetrahydro-2H-pyran-2-yl]-3-methylpenta-l,3-dien-l -yl}-8-hydroxy-l,6-dioxaspiro[2.5]oct-5- yl]acetyl}hydrazinyl)carbonyl]oxy}methyl)phenyl]-N5-carbamoyl-L-ornithinamide (B191). To a solution of B6 (19.4 mg, 0.035 mmol, 1 eq.) in NN-dimethylformamide (0.5 mL) at rt was added NN-diisopropylethylamine (24.7 muL, 0.14 mmol, 4 eq.), 2,6-lutidine (16.3 mu^, 0.14 mmol, 4 eq.), 4- NN-dimethylamino pyridine (4.3 mg, 0.035 mmol, 1 eq.) and N-[(9H-fluoren-9-ylmethoxy)carbonyl]- L-valyl-N5-carbamoyl-N-[4-([(4-nitrophenoxy)carbonyl]oxy}methyl)phenyl]-L-ornithinamide (40.6 mg, 0.053 mmol, 1.5 eq.), and the reaction was stirred for 2.5 h. More N-[(9H-fluoren-9- ylmethoxy)carbonyl]-L-valyl-N5-carbamoyl-N-[4-( [(4-nitrophenoxy)carbonyl]oxy}methyl)phenyl]-L- ornithinamide (13.5 mg, 0.018 mmol, 0.5 eq.) was added, and the reaction was stirred for another 1 h. The reaction was purified by reverse phase chromatography (Method A) to give B191 as a white solid. Yield: 9.4 mg, 0.0081 mmol, 23%. LCMS (Protocol D): m/z 1177.8 [M+H] , retention time = 0.90 minutes. Step 2. Synthesis of L-valyl-N-[4-( [(2-[(3R,5S,7R,8R)-7- {(l£',3£')-5-[(2S,3S,5R,6R)-5- [(2Z,4S)-4-(acetyloxy)pent-2-enoyl]amino}-3,6-dimethyltetrahydro-2H-pyran-2-yl]-3-methylpenta- 1 ,3 -dien- 1 -yl } - 8 -hydroxy- 1 ,6-dioxaspiro [2.5 ] oct-5 - yl]acetyl}hydrazinyl)carbonyl]oxy}methyl)phenyl]-N5-carbamoyl-L-ornithinamide acetate salt (B192). The title compound was prepared in 56% yield from 9.4 mg (0.008 mmol, 1.0 eq) of B191 and 13.6 mg (0.16 mmol, 20.0 eq) of piperidine using the procedure described for preparation of compound B47. LCMS (Protocol D): m/z 955.8 [M+H]+, retention time = 0.65 minutes. Step 3. Synthesis of N-{7-[(2,5-dioxopyrrolidin-l-yl)oxy]-7-oxoheptanoyl}-L-valyl-N-[4- ([(2-[(3R,5S,7R,8R)-7-{(l£,3£)-5-[(2S,3S,5R,6R)-5- [(2Z,4S)-4-(acetyloxy)pent-2-enoyl]amino}- 3,6-dimethyltetrahydro-2H-pyran-2-yl] -3 -methylpenta- 1 ,3-dien- 1 -yl} -8-hydroxy- 1 ,6- dioxaspiro [2.5] oct-5 -yl] acetyl} hydrazinyl)carbonyl] oxy } methyl)phenyl] -N5-carbamoyl-L- ornithinamide (B190). To a solution of B192 (4.5 mg, 0.004 mmol, 1 eq.) in NN- dimethylformamide (0.3 mL) at rt was added NN-diisopropylethylamine (3.5 muL, 0.02 mmol, 5 eq.) followed by l,l'-[(l,7-dioxoheptane-l,7-diyl)bis(oxy)]dipyrrolidine-2,5-dione (prepared as in J.Am.Chem.Soc. 2006, 128, 2802, 8.9 mg, 0.025 mmol) 6.2 eq.), and the reaction was allowed to stir for 35 min. The reaction was purified by reverse phase chromatography (Method A) to give B190 as a white solid. Yield: 1.65 mg, 0.0014 mmol, 34%. LCMS (Protocol D): m/z 1194.80 [M+H]+, retention time = 0.75 minutes. lH NMR (500 MHz, CD3CN) delta 9.06 (s, 1 H), 8.17 (s, 1 H), 7.71-7.63 (m, 2 H), 7.35-7.25 (m, 2 H), 7.19 (d, J= 7.6 Hz, 1 H), 6.73 (d, J= 6.6 Hz, 1 H), 6.47 (d, J= 8.8 Hz, 1 H), 6.41- 6.30 (m, 2 H), 5.96-5.85 (m, 2 H), 5.67-5.50 (m, 2 H), 5.33-5.24 (m, 1 H), 5.08-4.99 (m, 2 H), 4.74 (s, 1 H), 4.57-4.48 (m, 1 H), 4.39-4.25 (m, 2 H), 4.15-4.08 (m, 1 H), 3.82-3.75 (m, 1 H), 3.67-3.59 (m, 1 H), 3.55-3.47 (m, 1 H), 3.35-3.20 (m, 2 H), 3.12-2.99 (m, 2 H), 2.82-2.73 (m, 5 H), 2.66-2.52 (m, 6 H), 2.46-2.38 (m, 2 H), 2.36-2.20 (m, 4 H), 1.98 (s, 3 H), 1.77-1.57 (m, 11 H), 1.53-1.36 (m, 6 H), 1.30 (d, J= 6.6 Hz, 3 H), 1.06 (d, J= 6.6 Hz, 3 H), 1.00-0.91 (m, 9 H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping