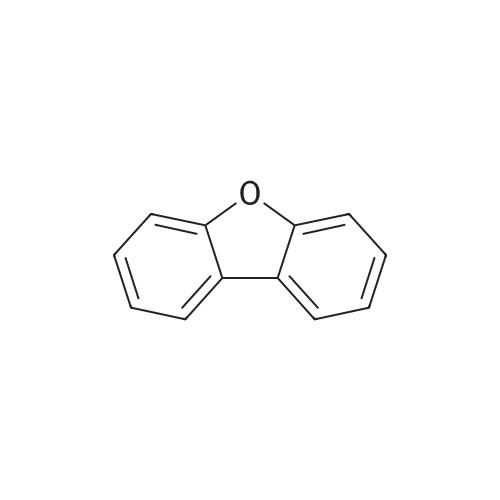
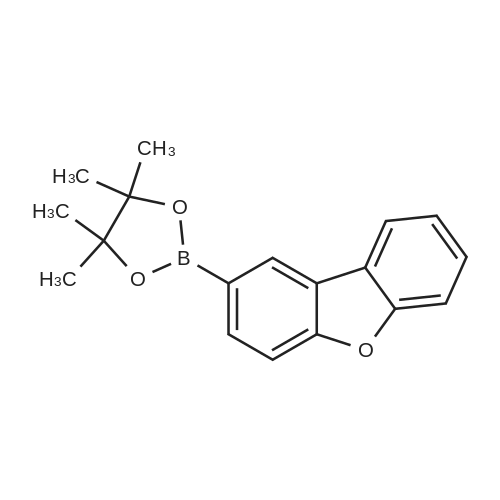
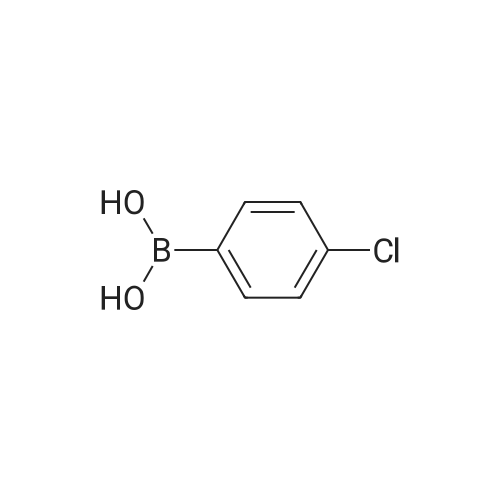
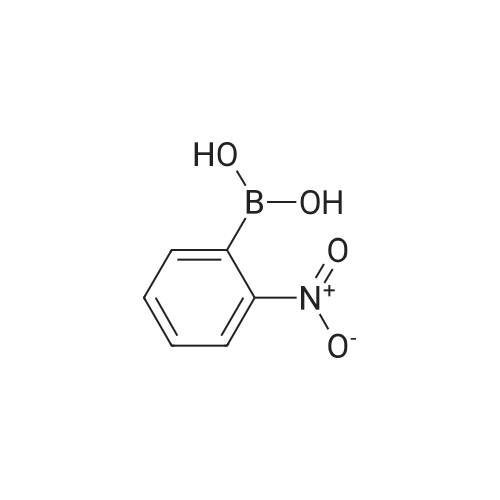
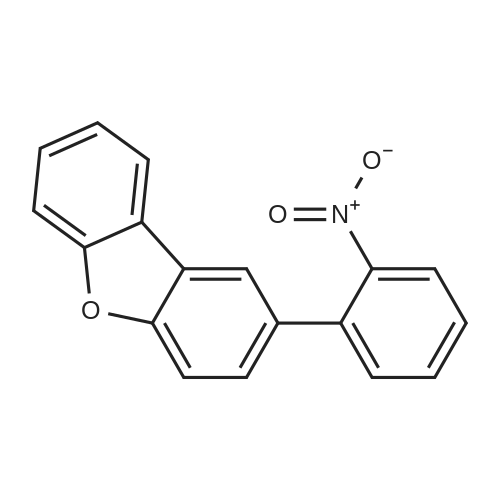
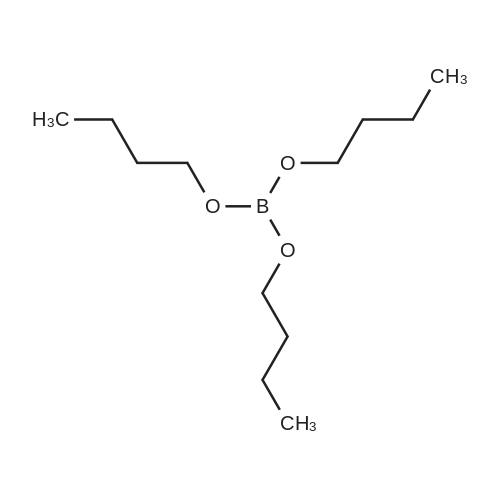
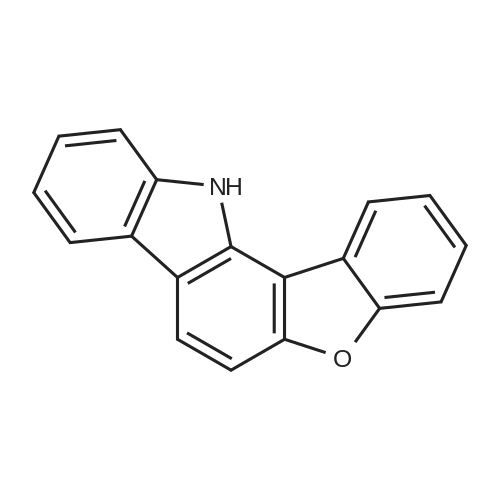
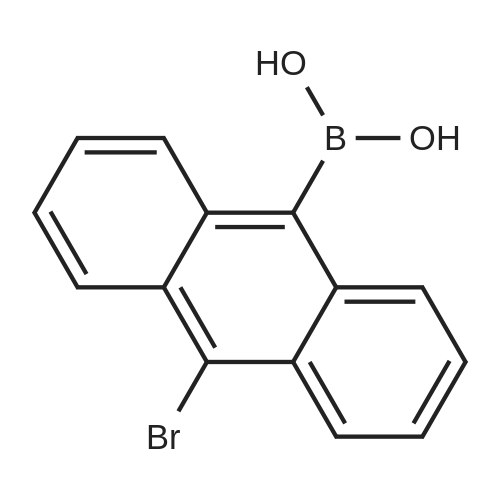
| 80% |
With bromine In chloroform at 20℃; for 24h; |
|
| 77% |
With bromine In dichloromethane |
2.4 Synthesis of SM-1-2(b)
SM-1-1(b) (9.2g, 50 mmol) was dissolved in CH2Cl2 was added dropwise Br2 (2.3ml, 45mmol) slowly and the reaction was stirred for 10 hours. Triisopropyl borate was then added dropwise (1.5 eq.) Lowering the temperature of the reaction back to -78 °C . Stirring at room temperature, diluted with water and it binds the 2N HCl. After completion of reaction, ethyl acetate and water and the organic extract The resulting organic layer was dried over MgSO4, concentrated and recrystallized to give a silicagel column SM-1-2 (b) 9.5g (yield: 77%). |
| 76% |
With hydrogen bromide; dihydrogen peroxide In water monomer; 1,2-dichloro-ethane at 20 - 25℃; for 12.5h; Green chemistry; |
1
In a 500 ml four-necked flask, 33.6 g of dibenzofuran (0.2 mol) was dissolved in 200 ml of dichloroethane, and then 37.1 g of a 48 wt% aqueous solution of hydrobromic acid (0.22 mol) was added thereto, and stirring was started. Maintain a temperature between 20-25 ° C, and drop 23.8 g of a 30 wt% solution of hydrogen peroxide (0.21 mol) in 30 min. After the addition is completed, continue stirring at room temperature for more than 12 h. After the reaction is completed, the lower layer is 100 ml. The aqueous solution of sodium hydrogen sulfite was washed for 1 hour, layered, washed with water, and the lower layer of liquid was added to dichloroethane under reduced pressure to obtain 47.9 g of a white solid, which was recrystallized from ethanol (380 ml) to give a white solid (37.6 g, yield 76%) . |
| 72.1% |
With bromine In dichloromethane at -5 - 20℃; for 22h; Inert atmosphere; |
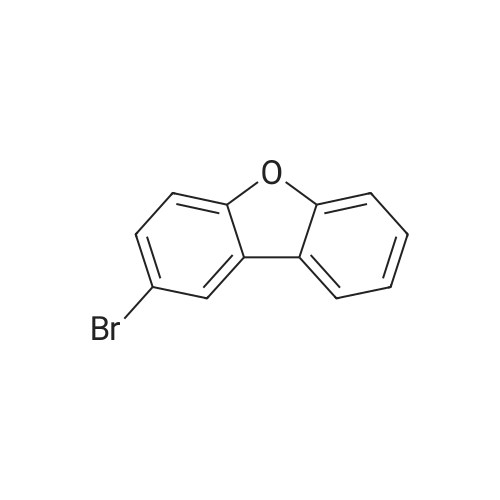
10.1 <1> Synthesis of 2-bromodibenzofuran
Into a 2 L four-necked round bottom flask equipped with a stirrer, a Liebig condenser fitted with a calcium chloride tube, a nitrogen inlet tube, a 500 mL isobaric dropping funnel and a thermometer, 136.8 g (0.813 mol) of dibenzofuran and 900 mL And the mixture was cooled to -5 ° C. or lower in an ice water bath under a nitrogen stream. Subsequently, a solution of 131.3 g (1.62 mol) of bromine diluted with 210 mL of DCM was added dropwise at -5 ° C. or lower over 1.5 hours in a nitrogen stream. After stirring for 0.5 hours in the ice water bath, the ice water bath was removed and the mixture was stirred at room temperature for 20 hours.The obtained reaction solution was transferred to a 2 L separation funnel, the DCM layer was washed in order with 690 mL of water, 210 mL of 20% sodium thiosulfate aqueous solution and 300 mL of water, dried with magnesium sulfate, magnesium sulfate was subjected to suction filtration , And the solvent was distilled off under reduced pressure. Subsequently, the obtained crude crystals were recrystallized from methanol to obtain 145.0 g (yield: 72.1%) of the objective bromide. |
| 70% |
With bromine; glacial acetic acid |
|
| 55% |
With bromine In dichloromethane at 0 - 20℃; for 12.7h; Inert atmosphere; |
1.1 1) Synthesis of Compound (1-a)
(1) Synthesis of Compound (1-a) [0179] [0180] Into a three-necked flask, 269.1 g (1600 mmol) of dibenzofuran and 1280 ml of dichloromethane were charged. The reaction vessel was cooled to 0 °C in a nitrogen atmosphere. After adding 100 ml of a dichloromethane solution of 204.6 g of bromine dropwise into the reaction vessel over 40 min, the contents were stirred at room temperature for 12 h. After the reaction, the reaction vessel was cooled to 0 °C and then 500 ml of water and 100 ml of a 20% aqueous solution of NaHSO4 were added. The reaction solution was extracted with several portions of dichloromethane in a separatory funnel. The extract was washed with 300 ml of a 1 N aqueous solution of sodium hydroxide, dried over anhydrous magnesium sulfate, filtered, and concentrated. The concentrate was dispersed in hexane for washing, to obtain a white solid. [0181] The yield was 136 g and the percent yield was 55%. |
| 55% |
With bromine In dichloromethane at 0 - 20℃; for 12.6667h; Inert atmosphere; |
1.1
Synthesis Example 1 (Synthesis of compound (1)) (1) Synthesis of compound (1-a) [0187] [0188] In a three-neck flask, 269.1g (1600 mmol) of dibenzofuran and 1280 mL of dichloromethane were placed. The reactor was cooled to 0°C in a nitrogen atmosphere. 100 mL of a dichloromethane solution of 204.6g of bromine was added dropwise to the reactor over 40 minutes, and the resulting mixture was stirred at room temperature for 12 hours. After completion of the reaction, the reactor was cooled to 0°C. 500 mL of water was added, and further, 100 mL of an aqueous 20% NaHSO4 solution was added. The sample solution was transferred to a separating funnel, and extracted with dichloromethane several times. The resultant was washed with 300 mL of a 1 N aqueous solution of sodium hydroxide, dried with anhydrous magnesium sulfate, filtrated and concentrated. The resulting product was washed by dispersing in hexane, whereby white solids were obtained. The yield was 136g (55%). |
| 50% |
With bromine; glacial acetic acid at 55℃; |
|
| 31% |
With bromine; glacial acetic acid Inert atmosphere; Cooling; |
A-3.3.i
[Production Example 3] Synthesis of dibenzofuran-2-boronic acid (i) Synthesis of 2-bromodibenzofuran 150 g (892 mmol) of dibenzofuran and 1 L of acetic acid were loaded into a flask. Air in the flask was replaced with nitrogen, and dibenzofuran was dissolved in acetic acid under heat. 188 g (1.18 mol) of bromine were dropped to the solution while the solution was sometimes cooled with water. After that, the mixture was stirred for 20 hours while being cooled with air. The precipitated crystal was separated by filtration, washed with acetic acid and water in the stated order, and dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure. After that, the resultant was repeatedly recrystallized with methanol several times, whereby 66.8 g of 2-bromodibenzofuran were obtained (31% yield). |
| 31% |
With bromine; glacial acetic acid for 20h; Inert atmosphere; Air-cooling; |
3
; Providing 150 g (892 mmol) of dibenzofuran and 1 liter of acetic acid into a flask, the inside of the flask was replaced with the nitrogen gas and the resultant mixture was heated and dissolved. After dropping 188 g (1.18 mol) of bromine while sometimes cooling with water, the mixture was stirred under air-cooling for 20 hours. The precipitated crystal was separated by filtration, washed with acetic acid and water sequentially, and was dried under reduced pressure. After refining the resultant crystal with reduced-pressure distillation, it was subjected to re-crystallization with methanol repeatedly several times to obtain 66.8 g (yield:31%) of 2-bromodibenzofran. Under the atmosphere of argon gas, 400 ml of anhydrous THF was added to 24.7g (100 mmol) of 2-bromodibenzofuran, and while stirring at -40°C, a hexane solution of n-butyllithium with 1.6 M concentration in an amount of 63 ml (100 mmol) was further added. The reacted solution was stirred for 1 hour while warming up to a temperature of 0°C. Cooling the reacted solution down to -78°C again, 50 ml solution of dried THF of trimethyl borate in an amount of 26.0 g (250 mmol) was dropped. The reacted solution was stirred at a room temperature for 5 hours. Adding 100 ml of 1N hydrochloric acid and after stirring the resultant solution for 1 hour, a water layer was removed. After drying an organic layer over magnesium sulfate, the solvent was distillated away under a reduced pressure. The resultant solid was washed with toluene to obtain 15.2 g (yield: 72%) of dibenzofuran-2-boronic acid. The resultant was identified as Intermediate 3 from the analysis in accordance with the FD-MS because the main peak of m/z=212 was shown for C12F9BO3=212. |
| 31% |
With bromine for 20h; Inert atmosphere; Cooling; |
1-3
150 grams (892 mmol) of dibenzofuran and 1 L of acetic acid were loaded into a flask. The air in the flask was replaced with nitrogen, and then the contents were dissolved under heat. 188 grams (1.18 mol) of bromine were dropped to the solution while the flask was sometimes cooled with water. After that, the mixture was stirred for 20 hours under air cooling. The precipitated crystal was separated by filtration, and was then sequentially washed with acetic acid and water. The washed crystal was dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure, and was then repeatedly recrystallized with methanol several times. Thus, 66.8 g of 2-bromodibenzofuran were obtained (in 31% yield) . The resultant was identified as Intermediate 1-3 by FD-MS analysis. |
| 31% |
With bromine; glacial acetic acid for 20h; Inert atmosphere; |
5
; 150 grams (892 mmol) of dibenzofuran and 1 L of acetic acid were added into a flask. The air in the flask was replaced with nitrogen, and then the contents were dissolved under heating. 188 grams (1.18 mol) of bromine were dropped to the solution while the flask was sometimes cooled with water. After that, the mixture was stirred for 20 hours under air cooling. The precipitated crystal was separated by filtration, and was then sequentially washed with acetic acid and water. The washed crystal was dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure, and was then repeatedly recrystallized with methanol several times. Thus, 66.8 g of 2-bromodibenzofuran were obtained (in 31% yield). The resultant was identified as Intermediate 5 by FD-MS analysis. |
| 31% |
With bromine In glacial acetic acid for 20h; Cooling; |
4 Synthesis of Intermediate 4
Intermediate Synthesis Example 4 (Synthesis of Intermediate 4) 150 g of dibenzofuran (892 millimoles) and 1 liter of acetic acid were placed in a flask, which was then replaced with nitrogen and the contents of which were dissolved under heating. After 188 g of bromine (1.18 moles) were dropped while being occasionally cooled with water, the mixture was stirred for 20 hours under cooling with air. The precipitated crystals were filtrated, washed sequentially with acetic acid and water, and dried under reduced pressure. After the crystals obtained were purified by distillation under reduced pressure, recrystallization from methanol was performed several times to obtain 66.8 g of 2-bromodibenzofuran (yield 31 %). The resultant was identified as Intermediate 4 by FD-MS analysis. |
| 30% |
With bromine; glacial acetic acid at 20℃; for 20h; Inert atmosphere; Heating; |
1-2 INTERMEDIATE SYNTHESIS 1-2: Synthesis of intermediate 1-2
In a nitrogen atmosphere, 150 g (0.89 mol) of dibenzofuran was dissolved in 1000 ml of acetic acid under heating. After adding 188 g (1.18 mol) of bromine dropwise, the resultant mixture was stirred at room temperature for 20 h. The precipitated crystals were collected by filtration and successively washed with acetic acid and water. The crude product was recrystallized from methanol several times, to obtain 66.8 g of a white crystal, which was identified as the following intermediate 1-2 by FD-MS analysis (yield: 30%). |
| 30% |
With bromine; glacial acetic acid at 20℃; for 20h; Inert atmosphere; |
1-5 INTERMEDIATE SYNTHESIS 1-5: Synthesis of intermediate 1-5
[0186] In a nitrogen atmosphere, 150 g (0.89 mol) of dibenzofuran was dissolved in 1000 ml of acetic acid underheating. After adding 188 g (1.18 mol) of bromine dropwise, the resultant mixture was stirred at room temperature for20 h. The precipitated crystals were collected by filtration and successively washed with acetic acid and water. The crude product was recrystallized from methanol several times to obtain 66.8 g of white crystal, which was identified asthe following intermediate 1-5 by FD-MS (yield: 30%). |
| 30% |
With bromine; glacial acetic acid at 20℃; for 20h; Inert atmosphere; |
INTERMEDIATE SYNTHESIS 1-4 (Synthesis of intermediate 1-4)
INTERMEDIATE SYNTHESIS 1-4 (Synthesis of intermediate 1-4) [0112] Under a nitrogen atmosphere, 150 g (0.89 mol) of dibenzofuran was dissolved in 1000 ml of acetic acid under heating. After further adding 188 g (1.18 ml) of bromine dropwise, the resultant mixture was stirred at room temperature for 20 h. [0113] The precipitated crystal was collected by filtration and washed successively with acetic acid and water. The recrystallization of the crude product from methanol was repeated several times to obtain 66.8 g of a white crystal, which was identified as the intermediate 1-4 by FD-MS analysis (yield: 30%). |
| 30% |
With bromine; glacial acetic acid at 20℃; for 20h; Inert atmosphere; |
2-4 INTERMEDIATE SYNTHESIS 2-4 (Synthesis of intermediate 2-4)
INTERMEDIATE SYNTHESIS 2-4 (Synthesis of intermediate 2-4)[0111] Under a nitrogen atmosphere, 1000ml of acetic acid was added to 150g (0.89mol) of dibenzofuran, and dissolvedunder heating. After further adding 188 g (1.18 mol) of bromine dropwise, the mixture was stirred at room temperaturefor 20 h. The precipitated crystal was collected by filtration and washed successively with acetic acid and water. Theobtained crude product was recrystallized from methanol several times to obtain 66.8 g of a white crystal, which wasidentified as the intermediate 2-4 by FD-MS analysis (yield: 30%). |
| 23% |
With bromine In glacial acetic acid at 20℃; for 24h; |
|
| 23% |
With bromine; glacial acetic acid at 50℃; for 12h; |
Dibenzofuran 30g (178.4mmol) in a round bottom flask was dissolved in 270g of acetic acid was added bromine 29g (181.5mmol) was dissolved in 6g of acetic acid at 50°C slowly for 4 hours. After stirring the reaction solution at 50°C further for eight hours, and cooling the reactants in distilled water. After the orange solid was dissolved in dichloromethane, washed with a small diyum thio sulphite solution and the organic layer was dried over magnesium sulfite, filtered and the filtrate was concentrated under reduced pressure. The product was recrystallized with dichloromethane / n- hexane to give the 10.1g (yield 23%) of the aimed compound intermediate M-10 as a white solid. |
| 23% |
With bromine; glacial acetic acid at 50℃; for 12h; |
Synthesis of Intermediate M-10
30 g (178.4 mmol) of dibenzofuran was dissolved in 270 g of acetic acid in a round-bottomed flask, and a solution prepared by dissolving 29 g (181.5 mmol) of bromine in 6 g of acetic acid was slowly added thereto at 50° C. for 4 hours. The reaction solution was additionally agitated at 50° C. for 8 hours, cooled down, and added to distilled water. An orange solid was dissolved in dichloromethane, the solution was cleaned with a sodium thiosulfite aqueous solution, an organic layer obtained therefrom was dried with magnesium sulfite and filtered, and the filtered solution was concentrated under a reduced pressure. The concentrated product was recrystallized with dichloromethane/n-hexane, obtaining 10.1 g of a white solid compound, an intermediate M-10 (23% of a yield). |
| 15% |
With bromine; glacial acetic acid at 20℃; for 18h; |
- Synthesis of Intermediate 2
Apparatus set-up: (0271) A 10 L 3 -necked round-bottomed flask, equipped with a mechanical overhead stirrer, reflux condenser, nitrogen inlet and exhaust. (0272) Experimental Procedure: (0273) Dibenzofuran (300 g, 1.783 mol) was taken in acetic acid (2 L) and heated to 45 °C for 30 min to obtain a clear solution. (0274) The mixture was allowed to room temperature and bromine (91.9 mL, 1.783 mol) was slowly added. (0275) The reaction mixture was stirred at room temperature for 18 h. (0276) The reaction mixture was filtered, washed with water ( 1 L) and dried. (0277) The solid obtained was dissolved in ethyl acetate (4 L), washed with sodium thiosulphate solution (1 Kg in 3 L of water), water (1 L), dried over sodium sulphate and concentrated to get 148 g of crude product. (0278) The crude product (148 g) was purified by hot toluene/ methanol crystallization to get 102 g of intermediate 2, taken again in hot ethyl acetate/ hexane and cooled to room temperature to get 76 g of intermediate 2. (0279) Another 65 g of intermediate 2 was prepared from 300 g of dibenzofuran. (0280) Both fractions (76 g + 65 g) were combined and purified again with hot ethyl acetate/ hexane to get 110 g of intermediate 2 with 98.89 % HPLC purity. 1 H-NMR (400 MHz, COCh): δ [ppm] δ 7.36 -7.40 (m, 1H), 7.46 (d, = 8.8 Hz, 1H), 7.49 -7.53 (m, 1H), 7.55 - 7.60 (m, 2H), 7.92 (dd, = 2.0 Hz, 7.6 Hz, 1H), 8.09 (d, = 2.0 Hz, 1H). |
| 13% |
With bromine In glacial acetic acid at 50℃; for 4h; |
11.a
11a) 2-Bromodibenzofuran Bromine (23.8 g, 0.156 mol) in acetic acid (5 g) is added at 50° C. to a solution of dibenzofuran (25 g, 0.149 mol) in acetic acid (230 g). The mixture is then stirred at 50° C. for 4 hours. The reaction mixture is cooled to room temperature and poured into H2O. The orange solid is washed with Na2S2O3 aq. and H2O. The crude product is then purified by recrystallization from toluene/CH2Cl2, wherein the pure product is obtained as a white solid (13% yield). 1H-NMR (CDCl3, ppm): 7.59-7.73 (m, 5H), 7.90 (d, 1H), 8.70 (d, 1H) |
|
With bromine; glacial acetic acid |
|
|
With Carbon tetrachloride; bromine Einwirkung von UV-Licht; |
|
|
With carbon disulfide; bromine |
|
|
With bromine; glacial acetic acid at 0 - 55℃; |
133.1
Example 133: Preparation of (R)-2-(8-bromo-7-(methoxycarbonylamino)dibenzo[b,d]furan-2-sulfonamido)-3-methylbutanoic acidStep 1 : Preparation of 8-bromo-7-nitrodibenzo[b,dlfuran-2-sulfonyl chloride [0282] In a round-bottom flask was added dibenzofuran (15 g) and acetic acid (90 niL). Bromine (6.1 mL) was added via a drop funnel and the mixture was heated overnight at 550C. After the reaction mixture was cooled to O0C, the precipitate was filtered and dried in air to give 2-bromodibenzofuran (11.3 g). |
|
With bromine; glacial acetic acid Inert atmosphere; |
12
Synthesis Example 12 (synthesis of Intermediate 12); Under an argon stream, 150 g of dibenzofuran and 1 L of acetic acid were charged, dibenzofuran was dissolved in acetic acid while heating, and 188 g of bromine was dropped thereinto. The crystal was filtered and washed with acetic acid and water, followed by recrystallization with methanol, whereby 97 g of a bromo compound was obtained. The same reaction as in Synthesis Example 3 was carried out except that the bromo compound was used instead of Intermediate 2, whereby 46 g of white powder was obtained. By an FD-MS analysis, the white powder was identified as Intermediate 12. |
|
With bromine In water monomer; glacial acetic acid |
S.2.1 Step 1:
Step 1: Synthesis of 2-Bromodibenzofuran First, into a four-neck flask equipped with a dropping funnel and a temperature meter were placed 15 g of dibenzofuran and 90 mL of glacial acetic acid, and the inside was refluxed under a nitrogen atmosphere. After that, the flask was heated to 50° C., and 18.8 g of bromine was added dropwise with the dropping funnel for 15 minutes while the temperature of the reaction solution was kept at 60° C. or less. Then, a yellow solid was precipitated after stirring at room temperature for 24 hours. The solution after the reaction was filtered, and washed with 9 mL of acetic acid. Then, washing was performed with water until the yellow solid becomes colorless, and washing was further performed with methanol. The obtained solid was recrystallized from dichloromethane to give 2-bromodibenzofuran, which was the object of the synthesis (a white powder in a yield of 45%). The synthesis scheme of Step 1 is illustrated in the following scheme (y-1). |
|
With bromine; glacial acetic acid for 20h; Inert atmosphere; Cooling; |
10
Under a nitrogen atmosphere, 150 g of dibenzofuran and 1 1 of acetic acid were loaded into a 300-ml three-necked flask, and then the contents were dissolved under heat. 188 Grams of bromine were added dropwise to the solution. After that, the mixture was stirred for 20 hours under air cooling. The precipitated crystal was separated by filtration, and was then sequentially washed with acetic acid and water. The washed crystal was dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure, and was then repeatedly recrystallized with methanol several times. Thus, 66.8 g of 2-bromodibenzofuran were obtained. |
|
With bromine; glacial acetic acid for 20h; Inert atmosphere; Cooling; |
22
Under a nitrogen atmosphere, 1 L of acetic acid were added to 150 g of dibenzofuran, and then the whole was dissolved under heat. 188 g of bromine were dropped to the solution while water cooling occasionally. After that, the mixture was stirred for 20 hours under air cooling. The precipitated crystal was separated by filtration, and was then sequentially washed with acetic acid and water. The washed crystal was dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure, and was then repeatedly recrystallized with methanol several times. Thus, 66.8 g of solid were obtained. The solid was identified as the Intermediate 22 by FD-MS analysis. |
|
With bromine In chloroform at 20℃; for 5.5h; Inert atmosphere; |
3.D
Example 3 [Show Image][Synthesis of Intermediate D] 13.5 g of dibenzofuran, 389 mg of iron chloride (III) and 135 mL of anhydrous chloroform were added under a flow of argon, and the mixture was cooled to a temperature of 140°C. Then, 12.9 g of bromine was dropwise added to the mixture over 30 minutes. The mixture was gradually increased to room temperature and reacted for 5 hours. A saturated sodium sulfite aqueous solution was added to the reaction solution, followed by liquid separation. A crude product obtained by concentration of the organic layer was dissolved with super heat in hexane to precipitate crystals, followed by filtration. This recrystallization treatment was repeated four times, and activated carbon treatment was carried out. The resulting solid was dried under reduced pressure to obtain 8.2 g of a white solid. The white solid was identified as Intermediate D by the FD-MS analysis. |
|
With bromine; glacial acetic acid for 20h; Inert atmosphere; |
4
Under a nitrogen atmosphere, 1 L of acetic acid was added to 150 g of dibenzofuran, and then the contents were dissolved under heat. 188 g of bromine were dropped to the solutionwhile the solution was occasionally water cooled. After that, the mixture was stirred for 20 hours under air cooling. The precipitated crystal was separated by filtration, and was then sequentially washed with acetic acid and water. The washed crystal was dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure, and was then repeatedly recrystallized with methanol several times. Thus, 66.8 g of a solid was obtained. The solid was identified as an intemediate-4 by FD-MS analysis. |
|
With bromine In glacial acetic acid at 20℃; for 20h; Inert atmosphere; |
13
Under nitrogen atmosphere, 1000 ml of acetic acid was added to 150 g (0.89 mol) of dibenzofuran, followed by heating and dissolution. Furthermore, 188 g (1.18 mol) of bromine was added thereto dropwise, and then the resulting mixture was stirred at room temperature for 20 hours. The precipitated solids were filtered out, and washed with acetic acid and water sequentially. The recrystallization of the crude product from methanol was repeated several times to obtain 66.8 g of white solids. The solids were identified as Intermediate 13 by FD-MS analysis. |
|
With bromine; glacial acetic acid Inert atmosphere; Cooling; |
4
Synthesis Example 4 (synthesis of intermediate 4)> Under a nitrogen atmosphere, 1,000 mL of acetic acid were added to 150 g of dibenzofuran, and then the contents were dissolved under heat. 188 g of bromine were dropped to the solution while the solution was occasionally cooled with water. After that, the mixture was stirred for 20 hours under air cooling. The precipitated crystal was separated by filtration, sequentially washed with acetic acid and water, and dried under reduced pressure. The resultant crystal was purified by distillation under reduced pressure, and was then repeatedly recrystallized with methanol several times. Thus, 66.8 g of a solid were obtained. The solid was identified as the intermediate 4 by FD-MS analysis. |
|
With bromine; glacial acetic acid at 20℃; |
|
|
With bromine; glacial acetic acid at 20℃; |
|
|
With N-Bromosuccinimide In acetonitrile at 80℃; for 72h; Inert atmosphere; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping