There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
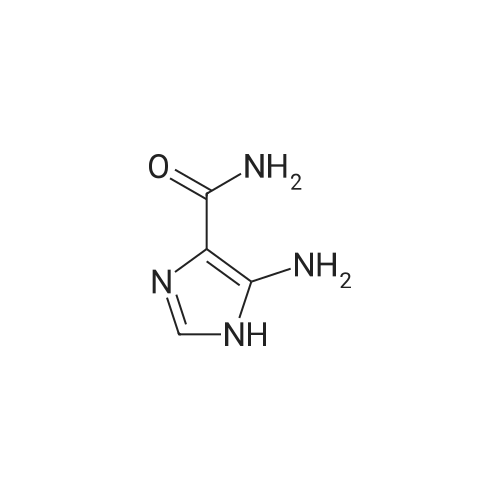
Structure of 85622-93-1 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1993, # 15, p. 1177 - 1178
2
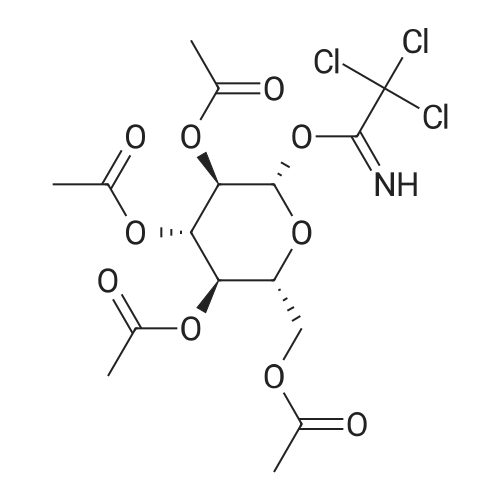
[ 624-83-9 ]
[ 7008-85-7 ]
[ 85622-93-1 ]
Yield
Reaction Conditions
Operation in experiment
76.5%
at 50℃;
EXAMPLE 3: Preparation of TMZ; 15 g of 5-diazo-5 H-imidazole-4 carboxamide was added to the flask containing methyl isocyanate absorbed in dioxane obtained in Example 2. The reaction mass was then heated to 500C and the temperature maintained for 15-18 hours until HPLC showed that the starting material had been consumed. The reaction mixture was then cooled to 300C and 8 ml of acidic water was added.The reaction mixture was stirred for 30 minutes. 60 ml of Ethyl acetate was added. The reaction mixture was stirred for 60 minutes and filtered. The solid obtained was washed with 15 ml of ethyl acetate and suction dried for 15 minutes. The solid was suspended in 45 ml of acetone and stirred for 30 minutes. The solid was filtered and suction dried for 15 minutes. The yield was 18g with a moisture content of 1percent. The corrected yield is 16.5g (78.5percent) with a purity of 99.74percent.
Reference:
[1] Journal of Medicinal Chemistry, 1984, vol. 27, # 2, p. 196 - 201
[2] Patent: WO2010/58430, 2010, A1, . Location in patent: Page/Page column 14
[3] Bioorganic and Medicinal Chemistry Letters, 1996, vol. 6, # 2, p. 185 - 188
[4] Journal of Medicinal Chemistry, 2002, vol. 45, # 25, p. 5448 - 5457
[5] Patent: US2007/225496, 2007, A1, . Location in patent: Page/Page column 4-5
[6] Molecules, 2010, vol. 15, # 12, p. 9427 - 9436
3
[ 188612-53-5 ]
[ 85622-93-1 ]
Yield
Reaction Conditions
Operation in experiment
64%
Stage #1: With acetic acid; lithium bromide In water at 20℃; for 1 h; Stage #2: With sodium nitrite In water at -10 - 20℃; for 6 h; Stage #3: With sodium thiosulfate In water for 0.333333 h;
Glacial acetic acid (25 ml), water (250 ml), LiBr (450 g) were charged and the contents20 were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N-methyl carbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for additional 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to5 °C. The reaction mass was stirred for 1 hr at 0-5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in25 250 ml of water) was added slowly and stirred for 20 minutes. This process yielded an acidic solution containing temozolomide. This acidic solution was extracted using dichloromethane (1000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered to give temozolomide (16.9 g, 64percent yield, 99.4 percent HPLC purity).
47%
Stage #1: With sodium nitrite In water at 0℃; for 0.333333 h; Stage #2: With tartaric acid In water at 0 - 60℃; for 4 h;
500 mL of water and 40 g of carbamoyl-AICA were added in a 2 liter reactor. The reaction mixture was cooled to 0 °C and 15.8 g of sodium nitrite was added. The resulting mixture was stirred for 20 minutes and then a solution of tartaric acid (34 g in 100 mL of water) was added slowly. The reaction mixture was stirred for 2 h at 0°C and then stirred at room temperature for 2 hours. The temperature was adjusted to 60 °C, and the suspension was filtered and the residue washed with hot water. Thefiltrate containing 60percent temozolomide and 40percentazahypoxanthine was acidified to pH 2-3, eluted through a resin with acidified aqueous solution and ethanol. The eluate was clarified through a carbon filter coupled to the column outlet, obtaining a solution of temozolomide with purity greater than 90percent (HPLC) . The obtained solution was concentrated at 60 °C and 25 mbar to between 30 and 40 volumes relative to the theoretical weight of temozolomide to achieve a suspension with fine crystals. The suspension was then cooled to 5-10 °C, stirred for 2 hours and filtered through Btichner funnel. The residue was washed with acidified water, and the collected crystals were dried under vacuum at 50 °C for 4 hours, providing 20g of temozolomide monohydrate as a white solid of 99.98percent purity (HPLC) e 47percent yield. DRXP (2) : 9.385, 11.863; 14.676; 16.826; 18.406; 18.565; 20.322; 28.232 ± 0.2° (Figure 1) IR (cm’) 3421; 3348; 1739; 1670; 1598; 1562; e 1408 (Figure 2).
43%
Stage #1: With acetic acid; lithium chloride In water at 20℃; for 1 h; Stage #2: With sodium nitrite In water at -10 - 20℃; for 6 h; Stage #3: With sodium thiosulfate In water for 0.333333 h;
Glacial acetic acid (25 ml), water (250 ml) and LiCl (225 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N- methylcarbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to 5 °C. The reaction mass was stirred for 1 hr at 0-5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in 250 ml of water) was added slowly and stirred for 20 minutes (solution A). This process yielded an acidic solution containing temozolomide.; Solution A as prepared above in Example 1 is extracted using dichloromethane (5.0 L x5), concentrated to 100 ml stage and filtered to give temozolomide (1 1.5 g, 43percent yield, 99.0 percent HPLC purity).; Solution A as prepared above in Example 1 is extracted using dichloromethane (1000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered to give temozolomide (17.2 g, 65percent yield, 99.3 percent HPLC purity). ; Glacial acetic acid (50 ml), water (500 ml) and LiCl (450 g) were charged and the5 contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N- methylcarbamoyl) imidazole-4-carboxamide (II) (50 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to NaNO2 solution (25 g in 100 ml water) at -10 to 5 °C. The reaction mass was stirred for 1 hr at 0-5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution10 (50 g in 500 ml of water) was added slowly and stirred for 20 minutes . This process yielded an acidic solution containing temozolomide. This acidic solution was extracted using hexane (2000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered to give temozolomide (33.4 g, 63percent yield, 99.2 percent HPLC purity).; Glacial acetic acid (50 ml), water (500 ml) and LiCl (450 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N- methylcarbamoyl) imidazole-4-carboxamide (II) (50 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to sodium nitrite solution (25 g in 100 ml water) at -10 to 5 °C. The reaction mass was stirred for 1 hr at 0- 5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (50 g in 500 ml of water) was added slowly and stirred for 20 minutes. This process yielded an acidic solution containing temozolomide. This acidic solution was extracted using dichloromethane (2000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered the temozolomide.To the above filtered temozolomide, acetone (350 ml) was charged at room temperature, heated to reflux (52-55°C) and maintained for 5 hours, distilled off 50percent of acetone atmospherically at 52-550C, charged acetone (175 ml) and distilled off 50percent of acetone atmospherically at 52-550C, this process was repeated twice and 25percent of aqueous acetone (350 ml, pH of water is acidified to 3.0 using acetic acid) was charged at 520C. Refluxed the contents for 1 hour at 52°C, slowly cooled to room temperature in 1 hour and maintained at room temperature for 2 hours. Chilled the contents to 100C, maintained for 1 hour, filtered the solid, washed with acetone (35 ml) and dried at 45-500C under vacuum to yield temozolomide (33.2 g, 62percent yield, 99.8percent HPLC purity, dichloromethane content : 250 ppm ).
1.5 g
Stage #1: With acetic acid; sodium nitrite In water at -5 - 5℃; for 2 h; Stage #2: With calcium chloride In water at 20℃; for 2 h; Cooling with ice
To a stirred suspension of 5-amino-N 1-methyl-i I-{-imidazole- 1, 4- dicarboxamide (5g. 0.0272 mol) and acetic acid (4.5 mE) was added sodium nitrite (2.5g, 0.0362 moi) in water (5.0 L) at -5 to 0°C at a rate so that temperature does not rise above 0-5°C. The reaction mixture was stirred at 0-5°C for 2h. After consumption of starting material, ice bath was removed and calcium chloride (12.5 g) was added to the reaction mixture and stirred at room temperature for another two hours. The reaction mass was extracted with a 2.5percent solution of dimethyl sulfoxide in dichloromethane (5 X 500 mL), Combined organic layer was dried over sodium sulfate and the solvent was distilled below 40°C. After removing the dichioromethane completely, the dimethyl sulfoxide suspension was cooled to 10-20°C and filtered the solid. The solid was washed with ethyl acetate (15 mE) and dried at room temperature. The obtained solid was siurried in ethyl acetate (40 mE) and heated at 40-45°C for lh. The solid was filtered and dried at room temperature to yield Temozolomide (1 .5g) as an off white solid.11*JMR (DMSO-d6): 5 8.81 (s, 1H), 7,78 (bs, 1H), 7.66 (bs, 1H), 3.86 (s, 3H); Mass (m/z):194.8 (M+);HPLC purity: 99.6 1percent.
The compound TZ-6 (25,0 g, 0,10 mol) was added portionwise to stirred sulfuric acid (d 1.8, 70 mL) at 15–25 °C. The whole mixture was stirred at ambient temperature for 3 h and then poured into 96percent ethanol (800 mL) at 0 °C. The suspension was stirred for 30 min. at 0 °C, the precipitate collected, washed with cold ethanol (50 mL) and dried on air for 16 h. The obtained crude TZ-7 was dissolved in the refluxed mixture of acetone (450 mL), water (150 mL) and acetic acid (38 mL), the solution was filtered, the filtrate refluxed again, cooled to ambient temperature and stirred for 4 h. The precipitate was then collected, washed with acetone/water 3:1 (80 mL) and dried in vacuo for 16 h at 25 °C to afford 10.1 g (52percent) of pharmaceutically pure TZ-7. Elementary analysis: 36.99percent (C), 3.00percent (H), 43.02percent (N). ESI-MS: calcd. for (M+H)+: 194.06, found 194.06.
With 1,8-diazabicyclo[5.4.0]undec-7-ene In acetonitrile
Synthesis 363-Methyl-4-oxo-3,4-dihydro-imidazo[5,1-c/J[1 ,2,3,5]tetrazine-8-carboxylic acid amide(Temozolomide); 1 ,8-Diazabicycloundec-7-ene (DBU) (35 μL, 0.228 mmol, 1.2 eq.) was added dropwise to a suspension of the hydroxymethyl derivative of Temozolomide (3-(hydroxymethyl)-4-oxo- 3,4-dihydroimidazo[5,1-cfl[1,2,3,5]tetrazine-8-carboxamide) (40 mg, 0.19 mmol) and iodomethane (2 M solution in tert-butylmethyl ether, 238 μL, 0.475 mmol, 2.5 eq.) in <n="94"/>acetonitrile (1 ml_). The resulting green suspension became homogeneous and was stirred overnight. The reaction was acidified with 1 N HCI and the yellow solution was extracted four times with ethyl acetate. The combined organic extracts were washed with water and then dried over MgSO4. The solvent was removed under vacuum to give 8 mg of the title compound as a yellow solid (22percent crude). The NMR data of the crude product were consistent with the NMR data of temozolomide. 1H NMR (DMSO d6): 8.22 (s, 1H), 7.81 (S1 1H), 7.68 (s, 1H)1 3.87 (S1 3H). LCMS: 91percent pure; m/z: 217.4 (M+Na+).
Reference:
[1] Chemical Communications, 1997, # 4, p. 363 - 364
10
[ 188612-53-5 ]
[ 85622-93-1 ]
Reference:
[1] Journal of Organic Chemistry, 1997, vol. 62, # 21, p. 7288 - 7294
11
[ 157467-00-0 ]
[ 85622-93-1 ]
Reference:
[1] Journal of the Chemical Society - Series Chemical Communications, 1994, # 14, p. 1687 - 1688
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1995, # 21, p. 2783 - 2789
12
[ 77-78-1 ]
[ 85622-93-1 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 1996, vol. 6, # 2, p. 185 - 188
Reference:
[1] Journal of Organic Chemistry, 1997, vol. 62, # 21, p. 7288 - 7294
15
[ 157466-98-3 ]
[ 85622-93-1 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1995, # 21, p. 2783 - 2789
[2] Journal of the Chemical Society - Series Chemical Communications, 1994, # 14, p. 1687 - 1688
16
[ 157466-97-2 ]
[ 85622-93-1 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1995, # 21, p. 2783 - 2789
[2] Journal of the Chemical Society - Series Chemical Communications, 1994, # 14, p. 1687 - 1688
17
[ 157466-99-4 ]
[ 85622-93-1 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1995, # 21, p. 2783 - 2789
[2] Journal of the Chemical Society - Series Chemical Communications, 1994, # 14, p. 1687 - 1688
18
[ 1025840-09-8 ]
[ 85622-93-1 ]
Reference:
[1] Journal of the Chemical Society. Perkin Transactions 1, 2002, # 16, p. 1877 - 1880
19
[ 157466-96-1 ]
[ 85622-93-1 ]
Reference:
[1] Journal of the Chemical Society - Series Chemical Communications, 1994, # 14, p. 1687 - 1688
20
[ 530-62-1 ]
[ 85622-93-1 ]
Reference:
[1] Patent: WO2018/112589, 2018, A1,
21
[ 85622-93-1 ]
[ 113942-30-6 ]
Yield
Reaction Conditions
Operation in experiment
98.6%
at 15 - 20℃;
Example 1 Preparation of TMZ acid (EP0252682) TMZ (2.577mmol, 0.5g) was mixed with concentrated sulfuric acid (4ml) with agitation. Sodium nitrite (9.4mmol, 0.65g) was dissolved in 2.6ml of water and then dropped into aforementioned mixture on an ice bath at temperature of below 15°C, to stir at room temperature overnight. The resulting mixture continued to be added with 10g of ice and cool for 1 hour in ice-bath. The solid product was colleted by filtration, and dried in vacuo to give 0.493g of TMZ acid. The yield was 98.6percent.
75%
With sulfuric acid; sodium nitrite In water at 0 - 20℃; for 17 h; Inert atmosphere; Darkness
TMZ-carboxylic acid (shown as compound 1 in FIG. 1) was prepared following a previously described procedure. See, e.g., Arrowsmith, J.; Jennings, S. A.; Langnel, D. A. F.; Wheelhouse, R T.; Stevens, M. F. G. Antitumour Imidazotetrazines. Part 39. Synthesis of Bis(Imidazotetrazines) with Saturated Space Groups. J. Chem. Soc., Perkin Trans. 2000, 1, 4432-4438. In a round bottom flask charged with a stir bar and fitted with an addition funnel, TMZ (2.29 grams, 11.8 millimoles) was dissolved in concentrated H2504 (23.6 milliliters), and the resulting yellow solution was cooled at 0° C. under nitrogen. A solution of sodium nitrite (2.51 grams, 36.4 millimoles) in water (23.6 milliliters) was then added drop-wise over 45 minutes, noting the evolution of a brown gas during addition. The mixture was allowed to warm to room temperature and was stirred under nitrogen, protected from light. After stirring for 17 hours, the solution was cooled at 0° C., and the reaction was quenched with ice (61.26 grams). Further stirring at 0° C. resulted in the precipitation of 1 as a fine white solid, which was isolated by vacuum filtration, washed with cold water, and dried under high vacuum to afford 1 in 75percent yield. 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 3.88 (s, 3H), 8.82 (s, 1H), 13.33 (br, 1H). 13C-NMR (125 MHz, DMSO-d6, δ, ppm): 36.32, 127.78, 129.09, 136.48, 139.10, 161.85. HRMS-EI (m/z): [M]+ calculated for C6H5N5O3: 195.0392, found: 195.0395.
Reference:
[1] European Journal of Medicinal Chemistry, 2002, vol. 37, # 4, p. 323 - 332
[2] Patent: EP1798234, 2007, A1, . Location in patent: Page/Page column 11
[3] Patent: US2018/79754, 2018, A1, . Location in patent: Paragraph 0082; 0101-0102
[4] European Journal of Medicinal Chemistry, 2017, vol. 127, p. 691 - 702
[5] Journal of Medicinal Chemistry, 2002, vol. 45, # 25, p. 5458 - 5470
[6] Molecules, 2010, vol. 15, # 12, p. 9427 - 9436
[7] International Journal of Molecular Sciences, 2018, vol. 19, # 2,
EXAMPLE 3: Preparation of TMZ; 15 g of 5-diazo-5 H-imidazole-4 carboxamide was added to the flask containing methyl isocyanate absorbed in dioxane obtained in Example 2. The reaction mass was then heated to 500C and the temperature maintained for 15-18 hours until HPLC showed that the starting material had been consumed. The reaction mixture was then cooled to 300C and 8 ml of acidic water was added.The reaction mixture was stirred for 30 minutes. 60 ml of Ethyl acetate was added. The reaction mixture was stirred for 60 minutes and filtered. The solid obtained was washed with 15 ml of ethyl acetate and suction dried for 15 minutes. The solid was suspended in 45 ml of acetone and stirred for 30 minutes. The solid was filtered and suction dried for 15 minutes. The yield was 18g with a moisture content of 1%. The corrected yield is 16.5g (78.5%) with a purity of 99.74%.
In dimethyl sulfoxide; at 27℃; for 24.5h;
EXAMPLE 3; Preparation of Temozolomide (Formula I); 3 Kg of N'-methyl-N,N-diphenyl urea was charged into a clean and dry reactor equipped with a condenser and a receiver. 3 L of dimethyl sulfoxide and 600 g of 5-diazo-5H-imidazole-4-carboxylic acid amide were charged into the receiver and then stirred for about 10 minutes at 27 C. The reactor containing N'-methyl-N,N-diphenyl urea was heated to 260 C. for a period of 2 hours, and simultaneously vapours comprising methyl isocyanate were collected into the receiver containing 5-diazo-5H-imidazole-4-carboxylic acid amide, maintained at 30 C. After completion of distillation, the reactor containing N'-methyl-N,N-diphenyl urea was cooled to 80 C. and the condenser was detached. The reaction mass in the receiver was stirred for 30 minutes and then stirred for 24 hours in the dark at 27 C. until the starting material was consumed, as confirmed by TLC. After completion of the reaction, 3 L of ethyl acetate was charged and stirred for about 15 minutes at about 27 C. 3 L of n-hexane was charged and the reaction mixture was stirred at 27 C. for 15 minutes. 3 L of ethyl acetate was charged into the reaction mixture and stirred for 1 hour at 27 C. The separated solid was filtered and then the solid was washed with 3 L of ethyl acetate. The final solid was dried at 60 C. for 24 hours under a vacuum of 580 mm Hg to afford 0.577 Kg of the title compound having a purity by HPLC of 99%.
Take a 1L three-neck round bottom flask,Lithium chloride monohydrate (373.03 g, 6175 mmol), glacial acetic acid (29 mL), and water (290 ml) were sequentially added. Stir at room temperature for 30 minutes. Intermediate 3 (29 g, 158.32 mmol,) was added, and after stirring at normal temperature for 30 minutes, the reaction flask was placed in a cooling circulation pump, the temperature was controlled below 0 C, and after cooling for 10 minutes, an aqueous solution of sodium nitrite was added dropwise. (14.5g dissolved in 58ml of water), the temperature is controlled within -10 ~ 5 C, the reaction mixture is within 0 ~ 5 C, stirring for one hour, add I2 (4.00g, 15.8mmol), and then at room temperature After reacting for 2 hours, an aqueous solution of sodium thiosulfate (29 g in 290 ml of water) was added, and after stirring for 20 minutes, the reaction was completed. Each time, it is extracted with 5 L of dichloromethane, extracted 8 to 10 times, and the extract is filtered, and the solution is concentrated by rotary evaporation, and concentrated to about 100 ml of solvent. After suction filtration, a pink temozolomide final product is obtained, melting point: 212 C ( break down).
64%
Glacial acetic acid (25 ml), water (250 ml), LiBr (450 g) were charged and the contents20 were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N-methyl carbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for additional 30 minutes. The reaction mixture was cooled to 0C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to5 C. The reaction mass was stirred for 1 hr at 0-5 C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in25 250 ml of water) was added slowly and stirred for 20 minutes. This process yielded an acidic solution containing temozolomide. This acidic solution was extracted using dichloromethane (1000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered to give temozolomide (16.9 g, 64% yield, 99.4 % HPLC purity).
47%
500 mL of water and 40 g of carbamoyl-AICA were added in a 2 liter reactor. The reaction mixture was cooled to 0 C and 15.8 g of sodium nitrite was added. The resulting mixture was stirred for 20 minutes and then a solution of tartaric acid (34 g in 100 mL of water) was added slowly. The reaction mixture was stirred for 2 h at 0C and then stirred at room temperature for 2 hours. The temperature was adjusted to 60 C, and the suspension was filtered and the residue washed with hot water. Thefiltrate containing 60% temozolomide and 40%azahypoxanthine was acidified to pH 2-3, eluted through a resin with acidified aqueous solution and ethanol. The eluate was clarified through a carbon filter coupled to the column outlet, obtaining a solution of temozolomide with purity greater than 90% (HPLC) . The obtained solution was concentrated at 60 C and 25 mbar to between 30 and 40 volumes relative to the theoretical weight of temozolomide to achieve a suspension with fine crystals. The suspension was then cooled to 5-10 C, stirred for 2 hours and filtered through Btichner funnel. The residue was washed with acidified water, and the collected crystals were dried under vacuum at 50 C for 4 hours, providing 20g of temozolomide monohydrate as a white solid of 99.98% purity (HPLC) e 47% yield. DRXP (2) : 9.385, 11.863; 14.676; 16.826; 18.406; 18.565; 20.322; 28.232 ± 0.2 (Figure 1) IR (cm?) 3421; 3348; 1739; 1670; 1598; 1562; e 1408 (Figure 2).
43 - 65%
Glacial acetic acid (25 ml), water (250 ml) and LiCl (225 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N- methylcarbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to 5 C. The reaction mass was stirred for 1 hr at 0-5 C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in 250 ml of water) was added slowly and stirred for 20 minutes (solution A). This process yielded an acidic solution containing temozolomide.; Solution A as prepared above in Example 1 is extracted using dichloromethane (5.0 L x5), concentrated to 100 ml stage and filtered to give temozolomide (1 1.5 g, 43% yield, 99.0 % HPLC purity).; Solution A as prepared above in Example 1 is extracted using dichloromethane (1000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered to give temozolomide (17.2 g, 65% yield, 99.3 % HPLC purity). ; Glacial acetic acid (50 ml), water (500 ml) and LiCl (450 g) were charged and the5 contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N- methylcarbamoyl) imidazole-4-carboxamide (II) (50 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0C and then added drop wise to NaNO2 solution (25 g in 100 ml water) at -10 to 5 C. The reaction mass was stirred for 1 hr at 0-5 C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution10 (50 g in 500 ml of water) was added slowly and stirred for 20 minutes . This process yielded an acidic solution containing temozolomide. This acidic solution was extracted using hexane (2000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered to give temozolomide (33.4 g, 63% yield, 99.2 % HPLC purity).; Glacial acetic acid (50 ml), water (500 ml) and LiCl (450 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-l-(N- methylcarbamoyl) imidazole-4-carboxamide (II) (50 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0C and then added drop wise to sodium nitrite solution (25 g in 100 ml water) at -10 to 5 C. The reaction mass was stirred for 1 hr at 0- 5 C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (50 g in 500 ml of water) was added slowly and stirred for 20 minutes. This process yielded an acidic solution containing temozolomide. This acidic solution was extracted using dichloromethane (2000 ml) by continuous liquid-liquid extractor, concentrated to 100 ml stage and filtered the temozolomide.To the above filtered temozolomide, acetone (350 ml) was charged at room temperature, heated to reflux (52-55C) and maintained for 5 hours, distilled off 50% of acetone atmospherically at 52-550C, charged acetone (175 ml) and distilled off 50% of acetone atmospherically at 52-550C, this process was repeated twice and 25% of aqueous acetone (350 ml, pH of water is acidified to 3.0 using acetic acid) was charged at 520C. Refluxed the contents for 1 hour at 52C, slowly cooled to room temperature in 1 hour and maintained at room temperature for 2 hours. Chilled the contents to 100C, maintained for 1 hour, filtered the solid, washed with acetone (35 ml) and dried at 45-500C under vacuum to yield temozolomide (33.2 g, 62% yield, 99.8% HPLC purity, dichloromethane content : 250 ppm ).
With acetic acid; sodium nitrite; In water; at -5 - 5℃; for 1.5h;Product distribution / selectivity;
Example 10: Preparation of temozolomideAcetic acid (90 g) was added to a suspension of N-methyl-5-aminoimidazole-l,4-dicarboxamide (100 g) and sodium nitrite (50 g) in water (1000 ml) at -5 to 00C. The reaction mixture was stirred at 0-5 0C for 1.5 hours. After the completion of reaction, calcium chloride (250 g) was added to the reaction mass and heated to 25-30 0C. The reaction mixture was stirred at 25-30 0C for 3 hours and cooled to -5 to 0 0C. The reaction mixture was stirred for 1 hour, resulting solid was filtered and suck dried to give 70 g of wet temozolomide. The filtrate was extracted twice with a mixture of dichloromethane and dimethylsulfoxide (9:1, 500 ml). The resulting organic layer was combined with wet compound (as obtained above) and solvent was distilled off under reduced pressure. Dimethylsulfoxide (450 ml) was added to the resulting reaction mass and heated to 60-70 0C. Charcoal (5 g) was added to the resulting solution and reaction mixture was stirred at 60-70 0C for 10 minutes. The reaction mass was filtered through hyflo bed, filtrate was cooled to 5-10 0C and stirred for 60 minutes. Solid thus precipitated was filtered and suck diied to 78 g of temozolomide.Purification of temozolomide: Temozolomide (75 g) was dissolved in a mixture of acetone (1125 ml) and water (375 ml) at 40-45 0C, activated charcoal (7.5 g) was added to it and the solution was stirred for 10 minutes. The reaction mixture was filtered while hot through hyflo bed. The reaction mixture was slowly cooled to 0-5 0C, stilted for 1 hour, filtered and suck dried for 30 minutes. The resulting product was stirred in acetone and water (1:1, 300ml) at 25-30 0C for 1 hour and cooled to 5-100C. The reaction mixture was further stirred for 30 minutes and filtered. The resulting solid was stirred with acetone (150 ml) for 60 minutes at 25-30 0C, filtered and dried under vacuum at 55-6O0C for 15 hours to give 28 g of temozolomide having purity 99.97% by HPLC.
1.5 g
To a stirred suspension of 5-amino-N 1-methyl-i I-{-imidazole- 1, 4- dicarboxamide (5g. 0.0272 mol) and acetic acid (4.5 mE) was added sodium nitrite (2.5g, 0.0362 moi) in water (5.0 L) at -5 to 0C at a rate so that temperature does not rise above 0-5C. The reaction mixture was stirred at 0-5C for 2h. After consumption of starting material, ice bath was removed and calcium chloride (12.5 g) was added to the reaction mixture and stirred at room temperature for another two hours. The reaction mass was extracted with a 2.5% solution of dimethyl sulfoxide in dichloromethane (5 X 500 mL), Combined organic layer was dried over sodium sulfate and the solvent was distilled below 40C. After removing the dichioromethane completely, the dimethyl sulfoxide suspension was cooled to 10-20C and filtered the solid. The solid was washed with ethyl acetate (15 mE) and dried at room temperature. The obtained solid was siurried in ethyl acetate (40 mE) and heated at 40-45C for lh. The solid was filtered and dried at room temperature to yield Temozolomide (1 .5g) as an off white solid.11*JMR (DMSO-d6): 5 8.81 (s, 1H), 7,78 (bs, 1H), 7.66 (bs, 1H), 3.86 (s, 3H); Mass (m/z):194.8 (M+);HPLC purity: 99.6 1%.
3-(5-{5-[2-(dimethylamino)ethylcarbamoyl]-1-methyl-pyrrol-3-ylcarbamoyl}-1-methylpyrrol-3-yl)-3,4-dihydro-3-methyl-4-oxoimdazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide[ No CAS ]
3-(2-{2-[2-(dimethylamino)ethylcarbamoyl]-1-methylimidazol-4-ylcarbamoyl}-1-methylimidazol-4-yl)-3,4-dihydro-3-methyl-4-oxoimdazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide[ No CAS ]
3-[2-(2-{2-[2-(dimethylamino)ethylcarbamoyl]-1-methylimidazol-4-ylcarbamoyl}-1-methylimidazol-4-ylcarbamoyl)-1-methylimidazol-4-yl]-3,4-dihydro-3-methyl-4-oxoimdazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide[ No CAS ]
7-{D-2-(3-methyl-4-oxo-imidazo[5,1-d]-1,2,3,5-tetrazine-8-carboxyl)amino-2-phenylacetamido}-3-methyl-8-oxo-5-thia-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid[ No CAS ]
8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one hydrochloride[ No CAS ]
[ 85622-93-1 ]
Yield
Reaction Conditions
Operation in experiment
90.3%
With Lewatit Mono Plus MP-64; In acetone; at 60 - 63℃;Purification / work up;
Example 5; A 1.25 cm width column was filled with 10 grams of the ion exchange resin Lewatit Mono Plus MP-64. A solvent mixture comprising 55% water and 45% acetonitrile (20 ml) was added and 30 minutes were allowed for resin swelling. Then, the solution was allowed to drain up to the top of the resin and a solution of the mother liquor of example 1 (50 ml) was passed through the column at a rate of about 1 ml/1 min. The first volume was discarded (10 ml) and then the eluted solution (the eluate) was collected (50 ml) and transferred to a reaction vessel. Temozolomide hydrochloride (3.33 grams, 0.0144 moles) was added and the mixture was heated to 60-63 C. under stirring and filtered at this temperature. Then, the mixture was cooled to 5 C., and stirring was maintained for about 30 minutes to enable crystallization. The crystals thus obtained were collected by filtration and washed with water (2×10 ml) and cold acetone (10 ml). The crystals were dried at 40 C. in vacuum to obtain 2.52 grams (0.013 moles) of Temozolomide base in 90.3% yield. Purity (by HPLC): 99.9%.
88%
With acetic acid; In water; acetone; for 0.166667h;Heating / reflux;Purification / work up;
Example 3 A 500 ml reaction vessel equipped with a magnetic stirrer and a reflux condenser was charged with Temozolomide hydrochloride (7.2 gram, 0.031 mol), acetone (85 ml), acetic acid (1.8 ml) and water (90 ml). The reaction mixture was heated to reflux and stirring was maintained at this temperature for about 10 minutes. The reaction mixture was filtered at elevated temperature and then cooled to 5 C. for about half an hour to enable crystallization. The crystals thus obtained were collected and washed with water (2*20 ml) and cold acetone (20 ml). The crystals were dried at 40 C. in vacuum to obtain 5.3 grams (0.027 mol) of free Temozolomide base (88% yield).
86.7%
With acetic acid; In water; acetonitrile; at 60 - 63℃; for 0.166667h;Purification / work up;
Example 2 A 500 ml reaction vessel equipped with a magnetic stirrer and a reflux condenser was charged with Temozolomide hydrochloride (11.7 grams, 0.0507 mol), acetonitrile (79 ml), acetic acid (2.5 ml) and water (96.5 ml). The reaction mixture was heated to 60-63 C. and stirring was maintained at this temperature for about 10 minutes. The reaction mixture was filtered at this temperature and then cooled to 5 C. for about half an hour to enable crystallization. The crystals thus obtained were collected and washed with water (2*20 ml) and cold acetone (20 ml). The crystals were dried at 40 C. in vacuum to obtain 8.54 grams (0.044 mol) of free Temozolomide base (86.7% yield). Purity (by HPLC): 99.96%.
70%
With acetic acid; In tetrahydrofuran; water; for 0.166667h;Heating / reflux;Purification / work up;
Example 4 A 250 ml reaction vessel equipped with a magnetic stirrer and a reflux condenser was charged with Temozolomide hydrochloride (2.9 grams, 0.0126 mol), THF (23 ml), acetic acid (0.7 ml) and water (28.8 ml). The reaction mixture was heated to reflux and stirring was maintained at this temperature for about 10 minutes. The reaction mixture was filtered at elevated temperature and then cooled to 5 C. for about half an hour to enable crystallization. The crystals thus obtained were collected and washed with water (2*20 ml) and cold acetone (20 ml). The crystals were dried at 40 C. in vacuum to obtain 1.71 grams (0.0088 mol) of free Temozolomide base (70% yield). Purity (by HPLC): 99.8%.
With hydrogenchloride; In water; acetone;pH 2 - 2.2;Purification / work up;
EXAMPLE 2 Preparation of TemozolomideFollowing the method published in J. Org. Chem. 62, 7288-7294 (1997) on page 7293, second example of the first column, a reaction crude is generated starting from 18 g of Carbamoyl AICA, mainly containing Temozolomide and Azaipoxantine in approximately a 1:1 ratio.An amount of 5% HCl sufficient to bring the pH to 2÷2.2 is added to this reaction mixture which is then charged in a 100 ml glass pre-column, charged with 60 ml of XAD 1600 resin, which is connected to a glass column filled with 600 ml of XAD 1600 resin. Both columns are previously washed and conditioned with a solution of water and HCl at a pH of 2÷2.2.The elution is carried out at a flow rate between 0.5 and 2 BV/h, while controlling the eluate fractions by means of HPLC. Once the outflow of azaipoxantine from the column is completed (taking place before the outflow of temozolomide from the column), the pre-column is disconnected and the elution is continued with a mixture of water at pH 2÷2.2 for HCl and ethanol 90:10, while collecting the temozolomide-containing eluate.The solution containing the reunited temozolomide-containing fractions is evaporated to obtain a residue, which is recrystallized by being refluxed after addition of a mixture made up of 240 ml of acetone and 80 ml of acidulated water at pH 2 with HCl. After cooling, the precipitated solid is filtered, washed with the 1:3 mixture of water-acetone, discharged from the filter and dried. 5.8 g of temozolomide are thereby obtained, with HPLC purity >99.9% and single impurity lower than 0.10% (30% yield).
Example 1 3-Methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2.3,5-tetrazin-4(3H)-one (Temozolomide) The imine 3 (700 g, 3.178 mol) and CH2Cl2 (7 L) were placed into a 22 L three-necked flask equipped with a nitrogen inlet, a gas outlet tube, reflux condenser, thermometer, mechanical stirrer, and maintained under a positive pressure of nitrogen. 1,1-Dimethylethyl-isocyanate (442 mL, 3.870 mol) was added to this stirred mixture at 0 C., and after stirring for 10 min a solution of potassium t-butoxide in THF (1.0 M in THF, 3.88 L, 3.88 mol) (as supplied by Aldrich) was added slowly (1 hour). The solution was stirred at 0 C. for 4 hours, when the reaction mixture had become a very thick paste with a deep brown color, and thin layer chromatography (EtOAc/hexanes=1/4) indicated that no more starting material was present. The resulting mixture was quenched with saturated NH4Cl solution (5 L), and the organic layer was separated and washed sequentially with saturated NH4Cl solution (5 L), and brine (5 L). The combined aqueous solution was extracted with CH2Cl2 (1 L). The combined CH2Cl2 solutions were dried over MgSO4 and concentrated under reduced pressure to yield a brown solid. The resulting crude N-(1,1-dimethylethyl)-acetamide derivative was purified by slurrying in hexane (2.5 L) at a concentration of 1-5% at room temperature.
Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60 C. for 20 mm and then cooled to room temperature. H5I06 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg). 1H NMR (400 MHz, DMSO-d6, delta): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).
EXAMPLE 1 Compound A 4[5]-Diazoimidazole-5[4]-carboxamide (500 mg) was suspended in methyl isocyanate (3.0 ml) and stirred in the dark, at ambient temperature, for 21 days. The reaction mixture was then diluted with anhydrous diethyl ether and filtered. The residue was washed quickly with anhydrous methanols then with anhydrous diethyl ether, and dried in air, in the dark, at ambient temperature, to give 8-carbamoyl-3-methyl-[3H]-imidazo[5,1-d]-1,2,3,5-tetrazin-4-one, in the form of a light brown microcrystalline solid (198 mg), m.p. 210 C. (with effervescence and darkening from 160 to 210 C.). [Elemental analysis:- found: C,36.8; H,3.10; 44.2%; C6 H6 N6 O2 requires: C,37.1; H,3.09; N,43.3%].
EXAMPLE 4 Compound A A suspension of 4[5]-diazoimidazole-5[4]-carboxamide (1.37 g) in ethyl acetate (20 ml) was treated with methyl isocyanate (7.0 g) and was stirred in a closed vessel in the dark at room temperature for 3 weeks. The resulting solid was filtered off and washed with diethyl ether to give 8-carbamoyl-3-methyl-[3H]-imidazo[5,1-d]-1,2,3,5-tetrazin-4-one (1.9 g), in the form of a cream-coloured solid, m.p. 212 C. (with effervescence). This material was recrystallized from three different solvent systems to give three different products, each of which had a slightly different IR spectrum. The three products were probably all polymorphs of 8-carbamoyl-3-methyl-[3H]-imidazo[5,1-d]-1,2,3,5-tetrazin-4-one.
A polymorphic form of 8-carbamoyl-3-methyl[-3H]-imidazo[5,1-d]-1,2,3,5-tetrazin-4-one was obtained by dissolving it in acetonitrile, filtering, concentration of the filtrate to dryness, and trituration of the resulting residue with diethyl ether. This material was in the form of an orange-tinged solid, m.p. about 200 C. (with decomposition). [Elemental analysis: C,37.4; H,3.26; N,43.5%]. Its NMR spectrum in dimethylsulphoxide-D6 was identical to that of the abovementioned pale purple solid, but its IR spectrum (KBr disc) showed some differences.
Product distribution / selectivity;
EXAMPLE 4 Purification of Temozolomide (Formula I) 2 Kg of temozolomide was suspended in 4 L of acetone and stirred at 27 C. for 30 minutes. The suspension was cooled to 0 C. and then 4 L of purified water was charged. The obtained mixture was stirred for 30 minutes at 0 C. The suspension was filtered and the solid was suspended in 4 L of acetone. The obtained suspension was cooled to 0 C. and stirred for about 1 hour at 0 C. The suspension was filtered and the obtained solid was suspended in 4 L of acetone and stirred for about 1 hour at 25-30 C., and filtered. The reactor was rinsed with 1 L of acetone and then the wet solid was washed with rinse acetone. The obtained solid was dried at about 60 C. under vacuum to afford 1.52 Kg of the title compound.
36
[ 85622-93-1 ]
[ 1057647-03-6 ]
Yield
Reaction Conditions
Operation in experiment
With water; In acetone; at -5 - 25℃; for 5.5 - 6.5h;
Example 1; <strong>[85622-93-1]Temozolomide</strong> (5gm) was added to a mixture of acetone and water (700ml) in the ratio of 3:1(v/v) and stirred at 20-250C to for about 30 minutes to dissolve the material. Activated carbon(lgm) was added to the solution and the mixture agitated at 20-250C for another 1 hour. The mixture was then filtered and the filtrate stirred at -5 to O0C for 4-5 hours. The solid material <n="15"/>crystallizing out was then filtered and dried at 25-3O0C under vacuum to give crystalline <strong>[85622-93-1]Temozolomide</strong> Monohydrate, having the characteristic X-ray (Powder) diffraction pattern as depicted in Table-I and Fig-1; the characteristic Differential Scanning Calorimetry Thermogram as depicted in Fig-2; and Infra Red spectrum as depicted in Fig-3.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile;
Synthesis 363-Methyl-4-oxo-3,4-dihydro-imidazo[5,1-c/J[1 ,2,3,5]tetrazine-8-carboxylic acid amide(Temozolomide); 1 ,8-Diazabicycloundec-7-ene (DBU) (35 muL, 0.228 mmol, 1.2 eq.) was added dropwise to a suspension of the hydroxymethyl derivative of Temozolomide (3-(hydroxymethyl)-4-oxo- 3,4-dihydroimidazo[5,1-cfl[1,2,3,5]tetrazine-8-carboxamide) (40 mg, 0.19 mmol) and iodomethane (2 M solution in tert-butylmethyl ether, 238 muL, 0.475 mmol, 2.5 eq.) in <n="94"/>acetonitrile (1 ml_). The resulting green suspension became homogeneous and was stirred overnight. The reaction was acidified with 1 N HCI and the yellow solution was extracted four times with ethyl acetate. The combined organic extracts were washed with water and then dried over MgSO4. The solvent was removed under vacuum to give 8 mg of the title compound as a yellow solid (22% crude). The NMR data of the crude product were consistent with the NMR data of temozolomide. 1H NMR (DMSO d6): 8.22 (s, 1H), 7.81 (S1 1H), 7.68 (s, 1H)1 3.87 (S1 3H). LCMS: 91% pure; m/z: 217.4 (M+Na+).
In water; acetone; at -5 - 50℃; for 1.16667h;activated charcoal;Purification / work up;
Method I: <strong>[85622-93-1]Temozolomide</strong> (25g) was dissolved in a mixture of acetone and water (1.5L) in the ratio of 3:1 at 45-500C, activated charcoal (1.25g) was added to the solution. The reaction mixture was stirred for 10 minutes and filtered while hot through hyflo bed. The solution was cooled to -50C and stirred for one hour. The resulting solid was filtered, washed with chilled acetone (50 ml), suck dried for 30 minutes and finally dried under vacuum at 50-600C for 18 hours to obtain 10.8 g of title compound as a off white crystalline powder having purity 99.98 % by HPLC. M.P. 2100C (with effervescence)
In a reactor was added water (358.6 Kg), acetic acid (about 11 Kg), acetone (97.9 Kg), charcoal (2.2 Kg), and crude <strong>[85622-93-1]Temozolomide</strong> (?17 kg). The reaction mixture was heated to around 60 C., followed by filtration in order to remove the charcoal. The filtrate was transferred to another reactor, followed by cooling to allow crystallization. The mixture was stirred at -2-2 C. for at least 1 hour, and then filtered to give the purified <strong>[85622-93-1]Temozolomide</strong> (?12 kg). The purity of the purified <strong>[85622-93-1]Temozolomide</strong> was verified by HPLC (<strong>[85622-93-1]Temozolomide</strong> >99.0%, 2-aza-hypoxanthine (AHX) <0.15%, 5-aminoimidazole-4-carboxamide hydrochloride (AICA) <0.15%, individual unknown <0.10%).
39
[ 85622-93-1 ]
[ 70-11-1 ]
[ 1259488-87-3 ]
Yield
Reaction Conditions
Operation in experiment
1%
at 130℃; for 1h;Inert atmosphere; Sealed tube;
Synthesis 48 3-Methyl-8-(4-phenyloxazol-2-yl)imidazo[5,1-d][1 ,2,3,5]tetrazin-4(3H)-one (WW-037)<strong>[85622-93-1]Temozolomide</strong> (0.200 g; 1.03 mmol) and 2-bromoacetophenone (0.246 g; 1.24 mmol) were stirred in a sealed tube under nitrogen, and heated to 1300C for 1 hour. The mixture was cooled, and then concentrated in vacuo before being applied directly to the head of a chromatography column (SiO2) and purified by column chromatography (DCM:MeOH, 5:1), to give the title compound as a yellow powder (0.003 g; 1 %). deltaH (DMSO-Cf6) 8.97 (1 H, s), 8.88 (1 H, S)1 7.91 (2H, t, J = 7.4), 7.50 (2H, t, J = 7.4), 7.39 (1 H, t, J = 7.4), 3.90 (3H, s).
With 2,4-(4-phenoxyphenyl)-1,3-dithia-2lambda(5),4lambda(5)-diphosphetane 2,4-disulfides; In dichloromethane;Reflux;
(II) Preparation of C8-ThioamidesSynthesis 4 3-methyl-4-oxo-3,4-dihydroimidazo[5, 1 -d][1 ,2,3,5]tetrazine-8-carbothioamideA mixture of <strong>[85622-93-1]temozolomide</strong> (3 g, 15.5 mmol) and Belleau's reagent (4.49 g, 8.5 mmol) was refluxed in DCM (80 mL) overnight. The reaction was quenched with water and the precipitate was filtered and washed with diethyl ether to give the pure title compound as an orange solid. (2.75 g, 84%). deltaH (DMSO-Cf6): 9.92 (1H, s), 9.45 (1 H, s), 8.81 (1H, s), 3.85 (3H, s).
4-oxo-3,4-dihydroimidazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide[ No CAS ]
[ 74-88-4 ]
[ 85622-93-1 ]
Yield
Reaction Conditions
Operation in experiment
Synthesis 3Temozolomide from NorTemozolomideA 60% dispersion of sodium hydride in mineral oil (16.3mg, 0.408mmol, 1.05eq.) was added in one portion to a slurry of 4-oxo-3,4-dihydroimidazo[5,1-d][1 ,2,3,5]tetrazine-8-carboxamide (70mg, 0.389mmol) in dry DMF (4.5mL) at 0C. The resulting suspension was stirred 3 minutes before the addition of methyl iodide (48mu., 0.778mmol, 2 eq.). The mixture became rapidly a green solution and was stirred at 0C for 30 minutes and then at room temperature for 2h. The mixture was concentrated to dryness under high vacuum and the crude product was absorbed on silica. The crude product was purified by flash chromatography using DCM:MeOH 95:5 as eluant and two fractions were collected. The first fraction (46mg, 61%yield) corresponded to the title product. 1H NMR (DMSO d6): 8.82 (s, 1 H, CH), 7.80 (s, 1 H, CONH2), 7.67 (s, 1 H, CONH2), 3.86 (s, 3H, CH3). The 1H NMR data were consistent with the data of an original sample of temozolomide, as shown by an NMR spectrum of a mixture of temozolomide and the product obtained above. The second fraction (18mg) was identified as a 1 :0.70 mixture of the title compound and one of its regioisomers.
In 1,2-dichloro-ethane; at 10℃; for 2.58333h;Inert atmosphere; Reflux;
Example 4; Synthesis of iso-POH Conjugated with Teroosolamide (TMZ) The reaction scheme is die following:Prejpar&fi<m *,| YNo.~ &/& ^mrbmnic id -4-i pn>py me &yGfahex*l«e yim&' thyi ite : Oxalyl chloride (0.26 g, 2,0 mraol) will be added slowly to a mixture of Temo- lamide (Source: OChem Incorporation, Lot No. 07.1 .1 185A; 0.2 g, .1 ,0 mmoi) in 1 ,,2-dichloroethane (13 mL) over a period of 5 mitt while maintaining the temperature at 10 C under Nj. The reaction mixture will be allowed to warm to room temperature and then heated to reflux for 2.5 h. The excess of oxalyl chloride and 1 ,2-dichloroethane will be removed by concentration under vacuum. The resulting residue will be redissoKed in 1 ,2-dichloroethane (20 mL) and the reaction mixture cooled to 5C under N^, A solution of isoperillyl alcohol (0.1.7 g? 1 , 12 mmoi) in .1 ,2-dichloroethane (5 ml.) will be added over a period of 10 min. The reaction mixture will be allowed to warm to room temperature and stirred for 12 h. 1 ,2-Dichloroetbane will he concentrated under vacuum to gi ve a residue which will be triturated with hexanes, The resulting pale yellow solid will be filtered and washed with hexanes.
In 1,2-dichloro-ethane; at 10 - 20℃; for 3.03h;Inert atmosphere; Reflux;
under N2 While maintaining the temperature at 10 , oxalyl chloride(0.13 g, 1.0mmol) was gradually added over a period of 2 minutes to a mixtureof <strong>[85622-93-1]temozolomide</strong> (OChem company, 0. 1 gram, 0.5 mmol) in 1,2-dichloroethane(10mL). The reaction mixture was placed until to room temperature, and fromthen it was heated to reflux for 3 hours. The excess oxalyl chloride and1,2-dichloroethane were removed by concentration in vacuo. Resulting residue was remelted with 1,2-dichloroethane (15 mL), and the reaction mixture was cooledto 10 C under N2.
In 1,2-dichloro-ethane; at 10 - 20℃; for 3h;Reflux; Inert atmosphere;
Oxalyl chloride (0.64 g, 5.15 mmol) was added slowly to a mixture of Temozolamide (Source: OChem Incorporation, Lot 9110918A; 0.5 g, 2.57 mmol) in 1,2-dichloroethane (10 mL) over a period of 2 min while maintaining the temperature at 10 C. under N2. The reaction mixture was allowed to warm to room temperature and then heated to reflux for 3 h. The excess of oxalyl chloride and 1,2-dichloroethane were removed by concentration under vacuum. The resulting residue was redissolved in 1,2-dichlorethane (15 mL) and the reaction mixture was cooled to 10 C. under N2. A solution of Perillyl alcohol (0.086 g, 0.56 mmol) in 1,2-dichloroethane (3 mL) was added over a period of 5 min. The reaction mixture was allowed to warm to room temperature and stirred for 14 h. 1,2-Dichloroethane was concentrated under vacuum to give a residue which was triturated with hexanes. The resulting pale-yellow solid (TMZ-POH or POH-TMZ) was filtered and washed with hexanes. Weight: 0.85 g; Yield: 89%. (0100) 1H-NMR (400 MHz, CDCl3): delta 1.4-2.2 (m, 10H), 4.06 (s, 3H), 4.6-4.8 (m, 4H), 5.88 (br s, 1H), 8.42 (s, 1H), 9.31 (br s, 1H); MS (APCI): No molecular ion peak is observed. m/e: 314 (100%), 286.5 (17%), 136 (12%).
In N,N-dimethyl-formamide; at 20℃; for 72h;Industry scale;
In reactor B was added 5-diazoimidazole-4-carboxiamide (DAICA, 11 Kg), and N,N-Dimethylformamide (26.4 Kg), the mixture was stirred at room temperature, and waiting for the charging of the in-situ prepared methylisocyanate from reactor A.In reactor A was added sodium cyanate (27.5 Kg), and 1,2-Dichlorobenzene (DCB, 110 Kg). The mixture was stirred and heated at ambient pressure to distill out DCB (about 12 Kg). After the distillation, dimethyl sulfate (DMS, 52.8 Kg) was slowly added, maintaining the temperature NLT 160 C. during the addition. The methylisocyanate thus generated was vaporized and then condensed into reactor B. After the charging of methylisocyanate into reactor B, the mixture in reactor B was stirred for about three days at room temperature, the reaction progress was monitored by HPLC (DAICA, NMT 1.0%) for the completion of the reaction.After the reaction, ethyl acetate (124.3 Kg) was charged into reactor B, and the mixture was stirred for at least 30 minutes followed by filtration to give the crude Temozolomide (17 kg).
The compound TZ-6 (25,0 g, 0,10 mol) was added portionwise to stirred sulfuric acid (d 1.8, 70 mL) at 15-25 C. The whole mixture was stirred at ambient temperature for 3 h and then poured into 96% ethanol (800 mL) at 0 C. The suspension was stirred for 30 min. at 0 C, the precipitate collected, washed with cold ethanol (50 mL) and dried on air for 16 h. The obtained crude TZ-7 was dissolved in the refluxed mixture of acetone (450 mL), water (150 mL) and acetic acid (38 mL), the solution was filtered, the filtrate refluxed again, cooled to ambient temperature and stirred for 4 h. The precipitate was then collected, washed with acetone/water 3:1 (80 mL) and dried in vacuo for 16 h at 25 C to afford 10.1 g (52%) of pharmaceutically pure TZ-7. Elementary analysis: 36.99% (C), 3.00% (H), 43.02% (N). ESI-MS: calcd. for (M+H)+: 194.06, found 194.06.
(3-methyl-4-oxo-3,4-dihydroimidazo[5,1-d][1,2,3,5]tetrazine-8-carbonyl)-carbamic acid-4-isopropenylcyclohex-1-enylmethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
Example 7 Synthesis of <strong>[85622-93-1]Temozolomide</strong> POH Carbamate (3-methyl 4-oxo-3,4-dihydroimidazo[5,1-d][1,2,3,5]tetrazine-8-carbonyl)-carbamic acid-4-isopropenyl cyclohex-1-enylmethyl ester) The reaction scheme is the following: Oxalyl chloride (0.13 grams, 1.0 mmol) was added slowly to a mixture of <strong>[85622-93-1]temozolomide</strong> (OChem Incorporation, 0.1 grams, 0.5 mmol) m 1,2-dichloroethane (10 mL) over a period of 2 minutes while maintaining the temperature at 10 C. under N2. The reaction mixture was allowed to warm to room temperature and then heated to reflux for 3 hours. The excess of oxalyl chloride and 1,2-dichloroethane were removed by concentration under vacuum. The resulting residue was re-dissolved in 1,2-dichlorethane (15 mL) and the reaction mixture was cooled to 10 C. under N2.
A solution of isoperillyl alcohol (0.17 g, 1.12 mmol) in 1,2-dichloroethane (5 mL) will be added over a period of 10 min. The reaction mixture will be allowed to warm to room temperature and stirred for 12 h. 1,2-Dichloroethane will be concentrated under vacuum to give a residue which will be triturated with hexanes. The resulting pale yellow solid will be filtered and washed with hexanes. The product iso-POH carbamate may be partially or fully deuterated. For example, one or more of the H atoms may be deuterium.
With tert.-butylhydroperoxide; trifluoroacetic acid; zinc(II) chloride; In water; dimethyl sulfoxide; at 50℃; for 1.08333h;Cooling with ice;
[0090] To a solution of TMZ (3, 25 mg, 0.13 mmol, 1.0 equiv.), ketal sulfinate 1 (120mg, 0.45 mmol, 3.5 equiv.) and ZnC12 (31 mg, 0.22 mmol, 1.75 equiv.) in DMSO (1 mL)was added TFA (0.02 mL, 0.19 mmol, 1.5 equiv.). The reaction mixture was cooled in icebath and TBHP (70% solution in water, 0.07 1 mL, 5.5 equiv.) was added dropwise withvigorous stirring. The stirring was continued at this temperature for 5 minutes. The reaction was warmed to 50C and monitored by HPLC. The reaction stopped after lh and purified by preperative HPLC to give product 3a (7 mg, 28% yield). ?H NMR (400 MHz, CDC13) 7.45 (s, 1H), 7.26 (s, 1H), 6.28 (s, 1H), 4.07 (s, 3H), 2.68-2.52 (m, 4H), 2.16 (s, 3H), 1.98-1.84 (m, 2H). MS (ESI+) mlz: 329, 351 [M+Naj.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +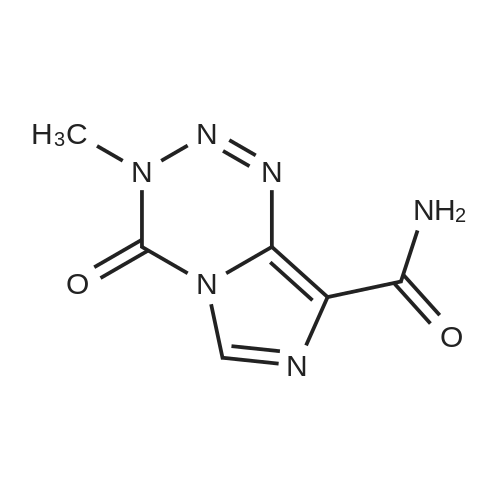

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping