| 85% |
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium; diisopropylamine; In tetrahydrofuran; hexane; water; at -95 - 0℃; for 0.5h;pH 7.0;Aqueous potassium phosphate buffer solution; |
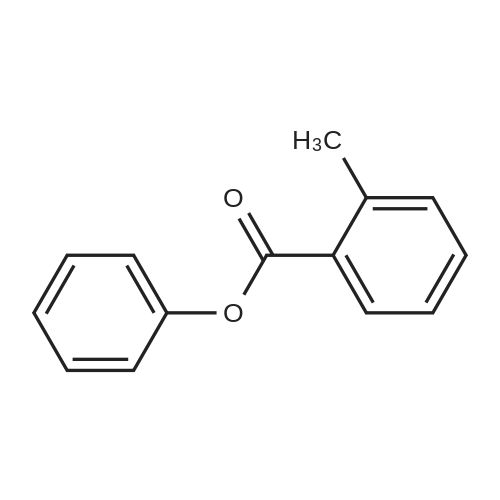
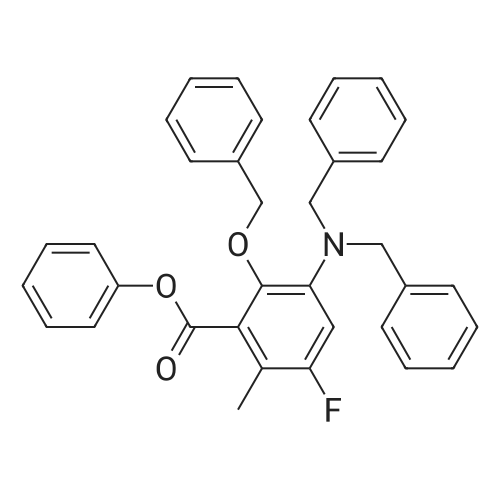
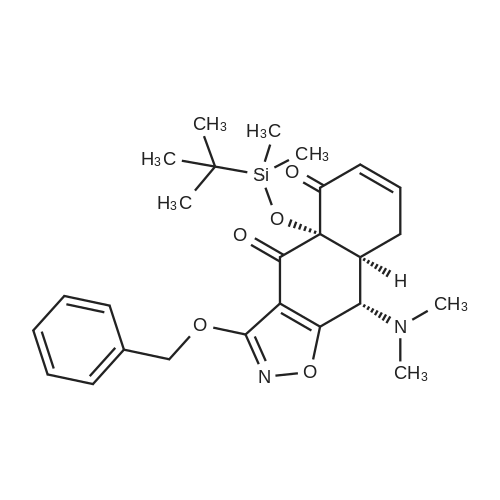
N,N-dimethylformamide (20 muL) was added was added to a solution of the carboxylic acid JDB1-113-SM (500 mg, 3.67 mmol, 1.0 equiv) and oxalyl chloride (367 mul, 4.22 mmol, 1.15 equiv) in dichloromethane (20 mL) at 23 C. Vigorous gas evolution was observed. After stirring for 80 min at 23 C, phenol (863 mg, 9.18 mmol, 2.5 equiv), pyridine (890 muL, 11.0 mmol, 3.0 equiv), and dimethylaminopyridine (3 mg) were added in sequence. The resulting solution was stirred for 90 min at 23 C, whereupon an aqueous potassium phosphate buffer solution (pH 7.05,0.2 M, 5.0 mL) was added. The resulting mixture was partitioned between water (30 mL) and ethyl acetate (50 mL). The aqueous phase was extracted with an additional 50-mL portion of ethyl acetate. The organic layers were combined and washed with an aqueous sodium hydroxide solution (50 mL, 1M), brine (50 mL), and then dried over anhydrous sodium sulfate. The dried solution was decanted and concentrated, affording a colorless oil (850 mg). The product was purified by flash column chromatography (25: 75 ethyl acetate-hexanes), providing the ester JDB1-113 as a colorless oil (774 mg, 99%). [00211] Rf 0.43 (3: 7 ethyl acetate-hexanes); 'H NMR (300 MHz, CDCl3) No. 8.18 (d, 1H, J = 8.1 Hz, ArH), 7.49-7.20 (m, 8H, ArH, OArH), 2.69 (s, 3H, ArCH3); ¹3C NMR (100 MHz, CDC13) No. 165.8, 150.9, 141.3, 132.7,132.0, 131.2, 129.5, 128.5, 125.9, 125.8, 121.8, 22.0; FTIR (neat film), cm-1 3046 (w), 2923 (w), 1739 (s), 1594 (m), 1487 (m), 1287 (m), 1241 (s), 1189 (s), 1159 (m), 1041 (s), 733 (s) ; HRMS (ES) m/z calcd for (C14H12O2+NH4) + 230.1181, found 230.1187. [00212] A solution of n-butyllithium in hexanes (1.47 M, 38.0 muL, 0.0565 mmol, 8.26 equiv) was added to a solution of diisopropylamine (7.4 muL, 0.057 mmol, 8.3 equiv) in tetrahydrofuran (0.50 mL) at -78 C. The reaction mixture was briefly (10 min) transferred to an ice bath, with stirring, then was cooled to -78 C. Hexamethylphosphoramide (13.9 muL, 0.113 mmol, 16.5 equiv) was added to the mixture prepared above at -78 C. The resulting mixture was stirred for 5 minutes whereupon a colorless solution was formed. The resulting solution was added dropwise via cannula to a solution of the ester JDB1-113 (10.0 mg, 0.0471 mmol, 6.88 equiv), and the enone DRS6 (3.3 mg, 0.00684 mmol, 1.00 equiv) in tetrahydrofuran (0.50 mL) at -95 C dropwise via cannula. The light red mixture was allowed to warm to -70 C over 30 min and was then partitioned between an aqueous potassium phosphate buffer solution (pH 7.0,0.2 M, 5.0 mL) and dichloromethane (20 mL). The organic phase was separated and the aqueous phase was further extracted with an additional 20-mL portion of dichloromethane. The organic phases were combined and dried over anhydrous sodium sulfate. The dried solution was decanted and concentrated, affording a yellow solid. The product was purified by preparatory HPLC on a Coulter Ultrasphere ODS column (10 mum, 250 x 10 mm, 3.5 mL/min, Solvent A: water, Solvent B: methanol, UV detection at 350 nm) using an injection volume of 500 muL methanol and a linear gradient elution of 85-100% B over 30 min. The peak at 25- 30 min was collected and concentrated to give enol JDBI-87 as a white solid (3.5 mg, 85%). [00213] Rf 0.46 (3:7 acetate-hexanes) ; ¹H NMR (500 MHz, CD2C12) No. 15.53 (s, 1H, enol), 7.94 (d, 1H, J= 7.9 Hz, ArH), 7.54 - 7.28 (m, 8H, ArH, OCH2ArH), 5.37-5.34 (m, 2H, OCH2Ph), 4.05 (d, 1H, J= 10.7 Hz, CHN(CH3)2), 3.24- 3.18 (m, 1H, CHCH2CHCHN(CH3)2), 2.99 (dd, 1H, J= 15.5, 5.6 Hz, CHH'CHCH2CHCHN(CH3)2), 2.88 (dd, 1H, J= 15.5, 15.5 Hz, CHH'CHCH2CHCHN (CH3)2), 2.61 (dd, 1H, J= 4.4, 10.7 Hz, CHCHN(CH3)2), 2.54- 2.44 (m, 7H, N(CH3)2, CHH'CHCHN(CH3)2), 2.14 (d, 1H, J= 14.3 Hz, CHH'CHCHN(CH3)2), 0.86 (s, 9H, TBS), 0.25 (s, 3H, TBS), 0.12 (s, 3H, TBS) ; ¹3C NMR (100 MHz, CD2Cl2) No. 187.8, 183.0, 182.8, 182.4, 167.7, 141.7, 135.4,133.4, 130.9, 129.0, 128.9, 128.9,128.1, 127.5,126.5, 108.5, 106.8, 82.1, 72.8, 61.5, 58.5, 46.9, 41.9, 38.6, 29.0, 25.9, 23.1, 19.1, -2.6,-3.7; HRMS (ES) m/z calcd for (C34H40N3O6Si+H) + 601.2734, found 601.2730. [00214] Hydrofluoric acid (1.1 mL, 48% aqueous) was added to a polypropylene reaction vessel containing a solution of the enol JDB1-114 (15.1 mg, 0.0251 mmol, 1.0 equiv) in acetonitrile (10 mL) at 23 C. The resulting mixture was stirred vigorously at 23 C for 12 hr, then was poured into water (50 mL) containing K2HP04 (4.7 g). The resulting mixture was extracted with ethyl acetate (3 x 25 mL). The organic phases were combined and dried over anhydrous sodium sulfate. The dried solution was filtered and the filtrate was concentrated, furnishing the intermediate alcohol as a yellow solid (12.2 mg, 99%). Pd black was added in one portion to a solution of the residue in methanol-dioxane (1:1, 3.0 mL). An atmosphere of hydrogen was introduced by briefly evacuating the flask, then flushing with pure hydrogen (1 atm). The mixture was stirred at 23 C for 20 min. Within 5 min, the color changed from light yellow to... |
| 85% |
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium; diisopropylamine; In tetrahydrofuran; hexane; at -95 - -70℃; for 0.5h; |
(0242) A solution of n-butyllithium in hexanes (1.47 M, 38.0 muL, 0.0565 mmol, 8.26 equiv) was added to a solution of diisopropylamine (7.4 muL, 0.057 mmol, 8.3 equiv) in tetrahydrofuran (0.50 mL) at -78 C. The reaction mixture was briefly (10 min) transferred to an ice bath, with stirring, then was cooled to -78 C. Hexamethylphosphoramide (13.9 muL, 0.113 mmol, 16.5 equiv) was added to the mixture prepared above at -78 C. The resulting mixture was stirred for 5 minutes whereupon a colorless solution was formed. The resulting solution was added dropwise via cannula to a solution of the ester JDB1-113 (10.0 mg, 0.0471 mmol, 6.88 equiv), and the enone DRS6 (3.3 mg, 0.00684 mmol, 1.00 equiv) in tetrahydrofuran (0.50 mL) at -95 C dropwise via cannula. The light red mixture was allowed to warm to -70 C over 30 min and was then partitioned between an aqueous potassium phosphate buffer solution (pH 7.0, 0.2 M, 5.0 mL) and dichloromethane (20 mL). The organic phase was separated and the aqueous phase was further extracted with an additional 20-mL portion of dichloromethane. The organic phases were combined and dried over anhydrous sodium sulfate. The dried solution was decanted and concentrated, affording a yellow solid. The product was purified by preparatory HPLC on a Coulter Ultrasphere ODS column (10 mum, 250 × 10 mm, 3.5 mL/min, Solvent A: water, Solvent B: methanol, UV detection at 350 nm) using an injection volume of 500 muL methanol and a linear gradient elution of 85-100% B over 30 min. The peak at 25-30 min was collected and concentrated to give enol JDB1-87 as a white solid (3.5 mg, 85%). Rf 0.46 (3:7 ethyl acetate-hexanes); 1H NMR (500 MHz, CD2Cl2) delta 15.53 (s, 1H, enol), 7.94 (d, 1H, J = 7.9 Hz, ArH), 7.54 - 7.28 (m, 8H, ArH, OCH2ArH), 5.37-5.34 (m, 2H, OCH2Ph), 4.05 (d, 1H, J = 10.7 Hz, CHN(CH3)2), 3.24-3.18 (m, 1H, CHCH2CHCHN(CH3)2), 2.99 (dd, 1H, J = 15.5, 5.6 Hz, CHH'CHCH2CHCHN(CH3)2), 2.88 (dd, 1H, J = 15.5, 15.5 Hz, CHH'CHCH2CHCHN(CH3)2), 2.61 (dd, 1H, J = 4.4, 10.7 Hz, CHCHN(CH3)2), 2.54-2.44 (m, 7H, N(CH3)2, CHH'CHCHN(CH3)2), 2.14 (d, 1H, J = 14.3 Hz, CHH'CHCHN(CH3)2), 0.86 (s, 9H, TBS), 0.25 (s, 3H, TBS), 0.12 (s, 3H, TBS); 13C NMR (100 MHz, CD2Cl2) delta 187.8, 183.0, 182.8, 182.4, 167.7, 141.7, 135.4, 133.4, 130.9, 129.0, 128.9, 128.9, 128.1, 127.5, 126.5, 108.5, 106.8, 82.1, 72.8, 61.5, 58.5, 46.9, 41.9, 38.6, 29.0, 25.9, 23.1, 19.1, -2.6, -3.7; HRMS (ES) m/z calcd for (C34H40N3O6Si+H)+ 601.2734, found 601.2730. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping