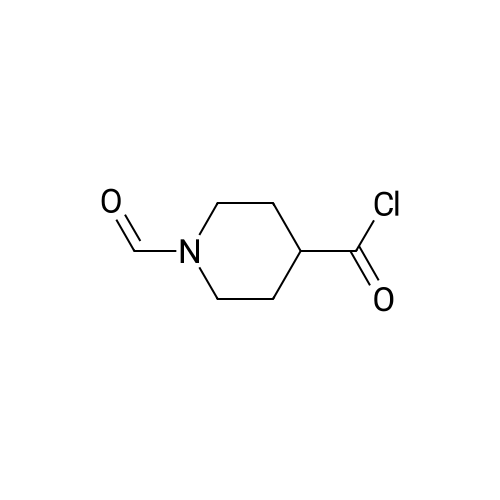
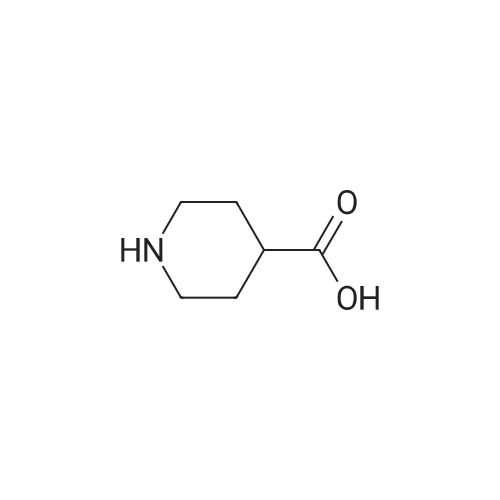
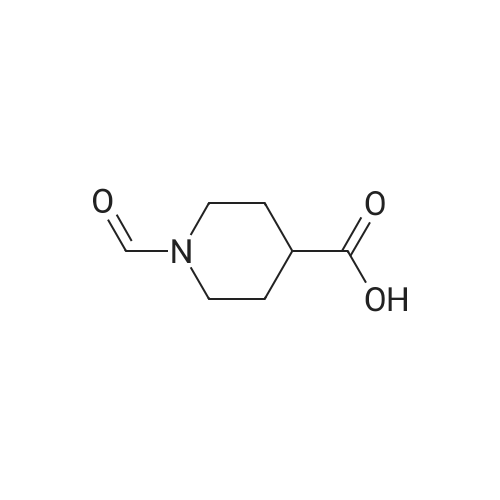
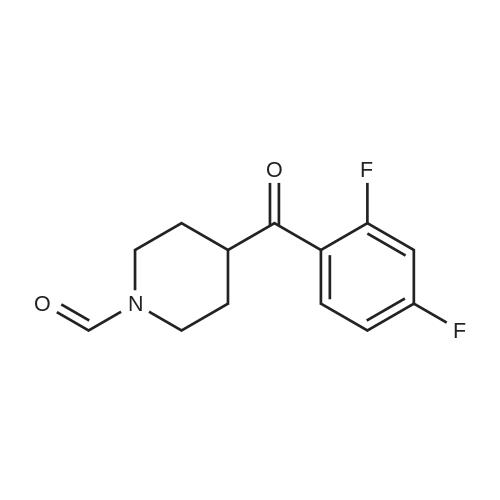
| 60% |
With triethylamine; In methanol;Autoclave; Inert atmosphere; Reflux; |
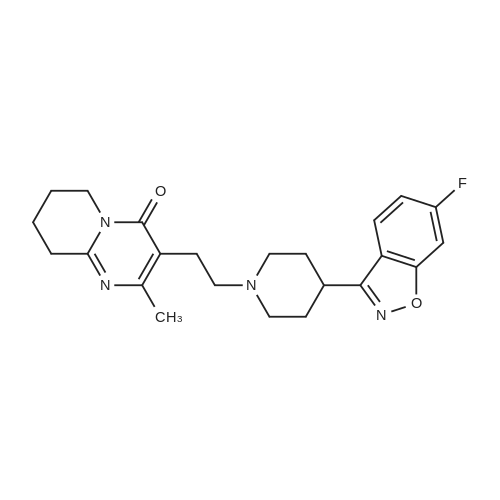
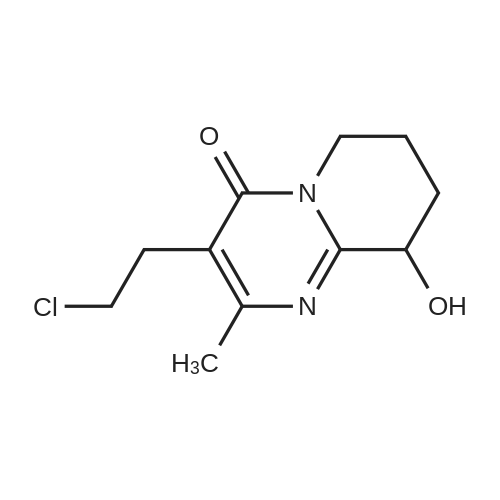
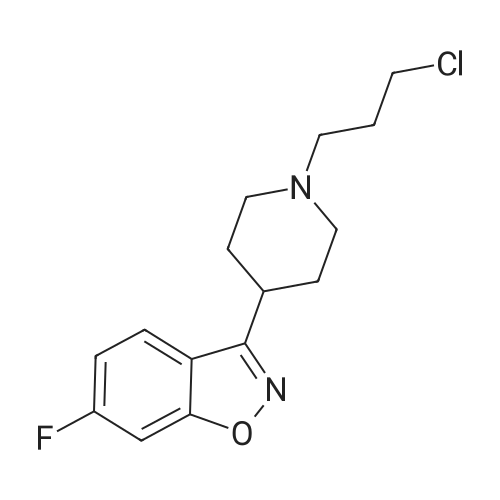
Example (1): Process for the preparation of Paliperidone baseMethanol was taken in an autoclave (closed system), nitrogen was purged for 1 hour. To the methanol solution, 6-fluoro-3-piperidin-4-yl- benzo[d]isoxazole hydrochloride (100 g), 9-hydroxy-3-(2-chloroethyl)-2- methyl-6,7,8,9-tetrahydro-4H-pyrido(l,2-a)-pyrimidine-4-one (108.2 g) and triethylamine (117.9 g) were added and heated to reflux. The nitrogen blanketing was removed and the reaction mass was stirred at reflux temperature under closed condition. After completion of the reaction the reaction mixture was cooled to 15-35 C, the obtained solid was filtered, washed with methanol and dried to yield Paliperidone base (96- 106 g).The above table clearly indicates that the yield of the present invention is very high with respect to the prior art methods. Preparation of Paliperidone under nitrogen atmosphere as disclosed in prior art in an atmospheric pressure i.e. only inert atmosphere yielded Paliperidone with less than 1% of Diketo impurity. But in the present invention, the reaction is carried out in the pressurized vessel i.e in an autoclave under closed condition. In this process, the formation of Diketo impurity was reduced less than 0.2%, preferably less than 0.1%. The probable reason for the reduction in the formation of Diketo impurity by the process of the present invention is performing the reaction in inert condition and closed condition.The following table provides the level of Diketo impurity obtained by the prior art process and process of the present invention.The above table clearly indicates that the level of Diketo impurity is less, approximately less than 0.1% when the reaction was performed under closed condition, on the other hand it was 0.4-1.0 % by the prior art process . |
| 34.33 - 34.74% |
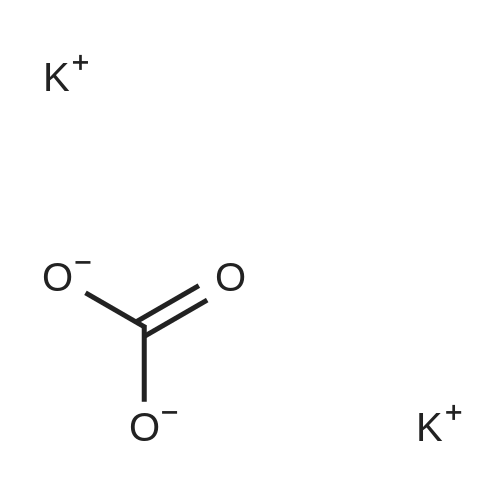
With potassium carbonate; potassium iodide; In N,N-dimethyl-formamide; at 25 - 65℃; for 19h; |
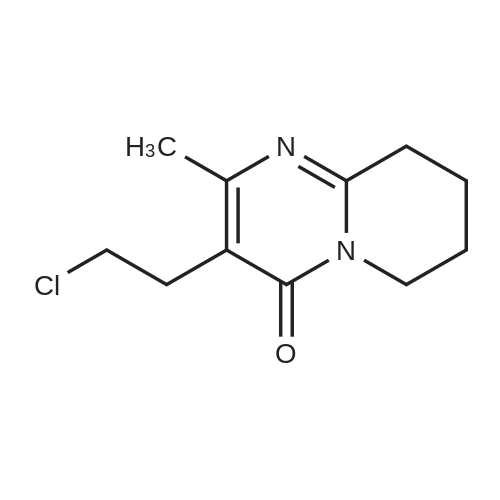
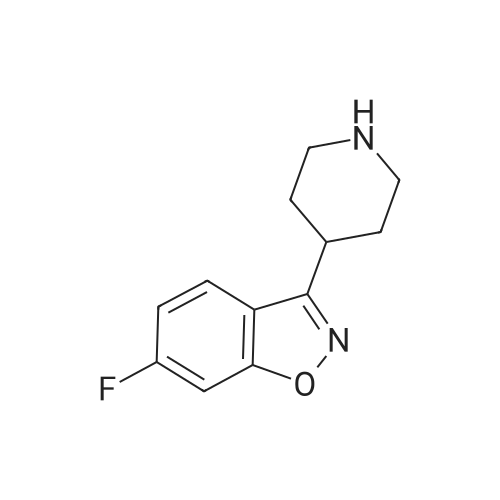
Into a clean 2L, three-necked RB flask equipped with shaft, condenser, and thermo socket was charged 600 ml of DMF, lOOg (0.404 moles) of 3-(2-Chloroethyl)-6,7,8,9- tetrahydro-9-hydroxy-4H-pyrido[l52-a]pyrimidin-4-one of formula- VI obtained from Example 5, 67.19g (0.404 moles) of potassium iodide, 111.91g (0.808moles) of potassium, carbonate and 103.68g (0.404 moles) of 6- fluoro-3-(4- piperidinyl)-l,2- benzisoxazole HCl (formula-VII). The reaction mass was heated to 60-650C and maintained for 18h. Reaction mass was cooled to 25-300C, stirred for Ih and filtered under vacuum. The wet cake was washed with 200ml of DMF. The wet cake was leached with 2 x 500ml of water and 1 x 500ml of methanol. The wet material was dried in the oven at 70-750C for Ih to yield 114g of technical grade paliperidone. <n="17"/>Above technical grade paliperidone was recrystallized from methanol via charcoal treatment to get 6Og (34.74% yield) of pure paliperidone. Purity by HPLC is > 99.8%.S Alternatively technical grade paliperidone was purified by the following chemical method.Above technical grade paliperidone (114g) was dissolved in 6.84L of methanol at 65- 7O0C. Charcoal (20 g) was added to the resultant solution and filtered under vacuum using a filter aid. The solvent was distilled of from the filtrate at 60-650C using0 rotavapor under vacuum to get a cream colored solid. Methanol (575 ml) was added to the solid and stirred for 45min at 25-300C. The reaction mass was filtered and dried in an oven at 70-750C to get 90 g of paliperidone.Into a clean and dry IL, three-necked RB flask equipped with shaft, thermo socket,5 addition funnel and stopper was charged 57.0 ml of cone. HCl and 570 ml of water.The reaction mass was stirred for 5min and charged the above solid (9Og). The reaction mass was stirred for 5-10min at 25-300C to get yellow colored clear liquid. pH of the reaction mass was adjusted to 6.0 to 6.5 by dropwise addition of dilute(concentrated aq ammonia solution was diluted with equal volume of water) aq.0 ammonia solution. The reaction mass was stirred for 30min and filtered under vacuum. The wet solid was dried in the oven at 70-750C to get 88g of paliperidoneHCl.Above solid was transferred taken into a IL RB flask containing 570 ml of methanol.5 The suspension was stirred for 30min at 25-3O0C, filtered under vacuum and dried in the oven at 70-750C for Ih to get 85 g pure paliperidone HCl. HPLC purity is > 99.80%.Above paliperidone HCl salt (85.0 g) and 2280 ml of water were suspended in a RB0 flask at 25-300C. pH was adjusted to 8.5 to 9.0 by adding aq. potassium carbonate solution (prepared from 57 g of potassium carbonate and 570 ml of water). The resultant cream-colored suspension was stirred for 30 min at 25-300C and filtered under vacuum. The wet cake was washed with 1500ml of water followed by 300ml of methanol. The wet cake was triturated with 450 ml of methanol for 30 min and <n="18"/>filtered. Paliperidone was dried in vacuum oven at 65-70C for 4h to afford 59 g (34.33%) of pure paliperidone. Purity by HPLC is > 99.8%. 1H-NMR (CDCl3): 1.75 (m, 2H, aliphatic-H), 1.95 (m, IH, aliphatic-H), 2.15 (m, 5H, aliphatic-H), 2.24 -2.40 (m, 5H, aliphatic-H), 2.56 (t, 2H, -CH2-, J = 6.84 Hz), 2.76 (t, 2H, -CH2-, J = 7.82 Hz), 3.09 (m, IH, aliphatic-H), 3.18 (d, 2H, aliphatic-H, J =11.72 Hz), 3.92 (m, 2H, aliphatic-H), 4.15 (s, IH, -OH, D2O exchangeable), 4.50 (dd, IH, aliphatic-H J = 3.91 Hz), 7.07 (m, IH, aromatic-H), 7.23 (m, IH aromatic-H), 7.70 (dd, IH, aromatic-H, J= 2.93 Hz). |
| 32.8% |
With diisopropylamine; In methanol; at 60℃; for 14h; |
Methanol 100ml, 6-fluoro-3-piperidin-4-yl-1,2-benzoisoxazole hydrochloride 10.27g (0.04mole), 3-(2-chloroethyl)-2-methyl-9-benzyloxy-6,7,8,9-tetrahydro-pyrido[1,2-a]pyrimid in-4-one 9.17g (0.04mole), diisopropylamine 10ml were added to a reaction bottle and reacted at 60C for 14 hours. The solvent was distilled out at a reduced pressure, water and chloroform (q.s.) were added, the pH was adjusted to 8 with 10% NaOH, the obtained chloroform extract was washed with water for three times, and dried over anhydrous magnesium sulfate. The solvent was distilled out at a reduced pressure to obtain a viscous product at which time 100mL isopropanol was added to make it disolve, and stood overnight. The precipitated crystal was filtered out to obtain a crude product 9.0g. Colum chromatography refinement: a colum chromatography on silica gel using chlorform-methanol (100:2) as eluant was conducted to obtain the refined product, the compound [formula (II)] 5.6g (yield: 32.8%), mp: 174.7-178.3C. |
| 21.6% |
With diisopropylamine; In methanol; at 60℃; |
12.5 g of CMHTP was added thereto, followed by stirring overnight at 60 C.The reaction mixture was concentrated and the oil residue was dissolved in trichloromethane and washed with water. The organic layer was dried, filtered and concentrated by evaporation.The residue was purified twice by column chromatography on silica gel and then a mixture of trichloromethane and ammonia saturated methanol (95: 5 by volume) was used as eluent,Followed by elution using a mixture of trichloromethane and ammonia-saturated methanol (95: 5 by volume).The pure fractions were collected, concentrated and then crystallized with 2-propanone.After cooling, the precipitate was filtered off, washed with a mixture of 2-propanol and 2,2'-oxybispropane and recrystallized with 2-propanone.The crystal recovery was filtered and dried to obtain 3.6 g of paliperidone (Yield: 21.6%). |
|
With potassium carbonate; potassium iodide; In N,N-dimethyl-formamide; at 60 - 65℃; for 18h;Product distribution / selectivity; |
Charged 3-[2-chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[ 1 ,2- a]pyrimidin-4-one (5Og), N,N-dimethyl formamide (300ml), potassium iodide (33.66g), potassium carbonate (56.06g) and 6-fluoro-3-(4-piperidinyl)-l,2- benzisoxazole monohydrochloride (52.03g) were charged in IL 3NRB flask, reaction mass was stirred for 18h at 60-650C. after maintenance reaction mass was cooled to room temperature, filtered the solid and washed with N,N-dimethyl formamide(100ml). Resulting solid was leached twice with demineralized water (2x500ml) followed by methanol (250ml), dried at 70-750C for Ih to yield paliperidone crude product; 3-[2-Chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[l,2- a]pyrimidin-4-one (10Og), N,N-dimethyl formamide (600ml), potassium iodide(67.08g), potassium carbonate (111.7Og) and 6-fluoro-3-(4-piperidinyl)-l,2- benzisoxazole monohydrochloride (103.68g) were charged in 2L 3NRB flask, reaction mass was stirred for 18h at 60-650C. after maintenance reaction mass was <n="9"/>cooled to room temperature, filtered the solid and washed with N,N-dimethyl formamide (200ml). Resulting solid was leached twice with demineralized water (2x500ml) followed by methanol (500ml), dried at 70-750C for Ih to yield paliperidone crude product. |
|
With triethylamine; In methanol; for 30 - 32h;Heating / reflux; |
Example (1):General procedure for preparing crude Paliperidone base:To a methanol (50OmL) solution, nitrogen was purged for 30 minutes to remove the nascent oxygen. 6-Fluoro-3-piperidin-4-yl-benzo [d] isoxazole Hydrochloride (5Og), 9-Hydroxy-3-(2-chloro ethyl)-2-methyl-6, 7.8,9-tetrahydro-4H-pyrido ( 1.2-a) pyrimidine-4-one (52g) and triethylamine (55) were added and stirred at reflux for 30- 32hours. The reaction mixture was cooled to 25-35C and filtered off to yield paliperidone base. |
|
With sodium hydrogencarbonate; potassium iodide; In water; acetonitrile;Heating / reflux; |
To a suitable reactor is charged the compound of formula III (60 g, 1 eq), 6F-3-4-Piperidinyl-1,2-Benzisoxazole HCl (63.4 g, 1.05 eq), sodium bicarbonate (51.4 g, 2.5 eq), potassium iodide (4.8 g, 0.12 eq), water (60 mL, 1 vol) and acetonitrile (540 mL, 9 vol). The slurry is heated to reflux and agitated for several hours. After the reaction is completed, the resulting solids are filtered and washed with water. The wet cake is dried under reduced pressure to provide paliperidone (90.4 g). |
|
|
79 g of 6-Fluoro-3-(4- piperidinyl)-l,2-benzisoxazole hydrochloride, 375 ml of methanol, and 78.2 g of triethyl amine were charged in a reaction vessel at 25-300C. The reaction mixture was stirred for 5 minutes. 75 g of the compound of formula II and 375 ml methanol were added to the above mass. The reaction mixture was heated to 60-630C and then maintained at 60-630C to achieve the desired conversion. The reaction mixture was then cooled to 40-450C. Methanol was distilled off under reduced pressure up to two volumes. 375 ml of water was added to the reaction mixture and stirred for 20-30 minutes at 25-300C. The <n="15"/>solid was filtered and washed twice with 150 ml water followed by 2x150 ml acetone to obtain 87 g crude paliperidone. (Purity of compound of formula I >97%). |
|
|
To a solution of 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole HCl (formula-6) (20 grams) in methanol (100 ml) added sodium carbonate (12.7g) and heated the mixture to 50-55C for 20-30 min. the reaction mixture was cooled to 35-40C and filtered. The filtrate was taken in a round bottomed flask and 3-(2-chloroethyl)-9-hydroxy-2- methyl-6,7,8,9-tetrahydro-4H-pyrido[l,2-a]pyrimidin-4-one (formula-7) (18.9 grams) was added to it followed by diisopropyl ethyl amine (15.5 grams). The reaction mixture was refluxed for 20-24 hours. The solvent was distilled off under reduced pressure; dichloromethane (600ml) was added to the reaction mixture and stirred for 15-20 min. The reaction mixture was washed with aqueous sodium hydroxide solution (3x100 ml) followed by water at 20-25C. The organic layer was separated and the solvent was distilled off under reduced pressure. Methanol (100 ml) was added to the reaction mixture and refluxed for 30-45 min. The reaction mixture was cooled to 20- 25C, stirred for one hour, filtered and washed with methanol to provide the title compound as a solid. Yield: 22 grams. |
|
|
A mixture of 3-(2-chloroethyl)-9-hydroxy-2-methyl-6,7,8,9-tetrahydro-4H- pyrido[l,2-a]pyrimidin-4-one (formula-8) (170.1 grams), 6-fluoro-3-(4-piperidinyl)-l,2- benzoisoxazole monohydrochloride (formula-6) (150 grams), sodium carbonate (113.3 grams), and potassium iodide (9.7 grams) in acetonitrile (1.5 L) was heated to 850C. The reaction mixture was heated at 85C for 6 hrs. The reaction mixture was then cooled to - 10C and stirred for 1.5 hrs. The solid obtained was filtered, washed with cold acetonitrile. The wet solid was taken in water, and acetic acid was added to the mixture to adjust the pH to 4.0. The reaction mixture was treated with acidic carbon and filtered through hyflow bed. The filtrate was treated with sodium hydrosulphite (hydrose). The pH of the reaction mixture was adjusted to 12 by adding aqueous sodium hydroxide solution and stirred for 45 min. The solid obtained was filtered, washed with water and dried to provide the title compound. Yield: 224 grams. |
|
With sodium carbonate; In 1-ethyl-3-methylimidazol-3-ium ethyl sulfate; at 80℃;Product distribution / selectivity; |
In a flask there were mixed 1.00 g (3.89 mmole) of 6-fluoro-3-(4-piperidinyl)-l,2- benzisoxazole hydrochloride and 0.95 g (3.89 mmole) of 3-(2-chloroethyl)-9- hydroxy-2-methyl-6,7,8,9-tetrahydro-4H-rhoyrido[l,2-a]rhoyrimidin-4-one, 0.83 g (7.79 mmole) of sodium carbonate and 10 ml of ionic solvent l-ethyl-3-methyl-imidazolium ethylsulfate. The suspension was heated to 80 0C or 90 0C and left stirring at this temperature overnight. HPLC showed that the conversion was 88%.It was cooled to room temperature and co-solvent (water and dichloromethane, acetone, ethanol, methanol, water) was added for precipitation of the product, it was filtered and rinsed with this solvent, well sucked off. |
|
With sodium carbonate; In acetonitrile; at 25 - 65℃; for 31.17 - 46.17h;Product distribution / selectivity; |
3-(2-chloroethyl-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[l,2-a]pyrimidin-4-one (60 gm, 0.165 mole) was added to acetonitrile (300 ml) at 25-35C and stirred for 10 mins at 25-35C. 6-Fluoro-3-piperidin-4-yl-benzo[d]isoxazole.HCl (55.9 gm,0.218 mole) and sodium carbonate anhydrous (78.8 gm, 0.743 mole) was added. Resultant mass was stirred for 10 min at 25-35C. The temperature was slowly raised to 60- 65Cand maintained at 60-65C for 30-45 hrs. Subsequently, mass was cooled and maintained at 25-35C for 1 hr. The solid is filtered and treated with water (600 ml) for 1 hr. The solid was again filtered, washed with water and IPA and dried at 40-45C to give 85 gm of paliperidone. |
|
With sodium carbonate; In acetonitrile; at 25 - 65℃; for 31.17 - 46.17h; |
3-(2-chloroethyl-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[l,2-a]pyrimidin-4-one (60 gm, 0.165 mole) was added to acetonitrile (300 ml) at 25-350C and stirred for 10 mins at 25-35C. 6-Fluoro-3-piperidin-4-yl-benzo[d]isoxazole.HCl (55.9 gm,0.218 mole) and sodium carbonate anhydrous (78.8 gm, 0.743 mole) was added to the reaction mass. The reaction mass was then stirred for 10 min at 25-35C and temperature was raised to 60-65C. The reaction mass was then maintained at 60-65C for 30-45 hrs, slowly cooled and maintained at 25-35C for 1 hr. The resultant solid was filtered and treated with water (600 ml) for 1 hr. The solid was again filtered, washed with water and IPA and dried at 40-45C to give 85 gm of Form II of paliperidone. |
|
With diisopropylamine; In methanol; at 60℃;Product distribution / selectivity; |
3-(2-chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[l,2- a]pyrimidine-4-one (5.0 gm,0.021 moles), 6-fluoro-3-(4-piperidinyl)-l,2-benzisoxazole mono hydrochloride (4 gm,0.018 molar), diisopropylamine (4 gms,0.039 moles) and methanol (48 ml) were stirred overnight at 60C. The reaction mass was evaporated and the residue was suspended in dichloromethane (50 ml) and washed with water. The solvent was evaporated and the residue treated with acetone to get paliperidone crude (5.0 gms). The crude paliperidone (4 gm) was charcoalised in isopropyl alcohol at 75- 80 C, the filtrate cooled and stirred for 1 hr. The reaction mass was again cooled to 0- 5C and stirred for 2 hrs. Slurry was filtered and washed with chilled isopropyl alcohol. The wet cake was dried at 35-40C under vacuum (3.3 gm) to obtain polymorphic Form I of paliperidone |
|
|
Example- 4 Preparation of 3-[2-[4-(6-FIuOrO-I,, 2-benzsoxazoI-3-yl) piperidin-1-yl] ethyl]-9-hydroxy -a-methyl-ThetaJ.beta.theta-tetrahydro^H-pyrido [1,2-a] -pyrmidin- 4-one (Formula-1)100 gm of purified . 6-Fluoro-3-piperidin-4-yl-1 , 2-benzisoxazole hydrochloride (Formula-2), and 143.4 gm potassium carbonate, 6.5 gm potassium iodide in 900 ml of acetonitrile was charged at 300C. The mixture was heated upto 55-600C and stirred for 1 hr. The previously prepared solution of 3-(2-chloroethyl)-9-hydroxy-2-methyl-6,7,8,9-tetrahydro- 4H-pyrido[1 ,2-a]pyrimidin-4-one (Formula-3) dissolved into 600 ml acetonitrile and charged drop wise into the reaction mass at about 600C followed by stirring for 46 h. The acetonitrile was distilled at 500C- 550C under reduced pressure. The crude product was obtained and slurried in water with stirring for 1 hr at 250C-SO0C. If required water wash is repeated. The solid product was dried in vacuum oven at 60 - 650C to provide crude Paliperidone (purity >;98%, dimer compound <;0.1 %) |
|
|
Example-1 Preparation of Paliperidone100 gm of 6-fluoro-3-piperidin-4-yl-1 , 2-benzisoxazole hydrochloride, 125.8 gm diiso- propylethyl amine, potassium iodide and methanol were stirred at room temperature. The reaction mixture heated with stirring. 100 gm of 3-(2-chloroethyl)-9-hydroxy-2-methy|-6,7,8,9- tetrahydro-4H-pyrido[1 ,2-a]pyrimidi?-4-one was added with stirring. The reaction mass was cooled to room temperature and stirred for an hour. The reaction mass was filtered and washed with methanol. The wet cake was stirred with water at 50-550C and washed with water. The product was dried under vacuum to give crude paliperidone (120-125 gm) (HPLC Purity >; 97 %; Water content <; 1 %) |
| 96.5%Chromat. |
triethylamine; In methanol;Inert atmosphere; Reflux; |
Example 1 - Synthesis of 3-[2-[4-(6-fluoro-1,2-benzoisoxazol-3-yl)-1-piperidinyl]ethyl-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-a]-pyrimidin-4-one hydrochloride (Paliperidone hydrochloride) 6-Fluoro-3-(4-piperidinyl)-1,2-benzoisoxazole hydrochloride (300 g, 1.17 mols), 3-(chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (343 g, 1.40 mols) and triethylamine (272 g, 2.69 mols) are suspended in methanol (1.5 1) in a 3000 ml reactor under nitrogen atmosphere, and the reaction mixture is heated at reflux temperature for 18-20 h. Conversion to the product is checked by HPLC titre, obtaining a yield of 96.5% in solution. The mixture is concentrated to a residue. The so obtained product has an XRPD spectrum as shown in Figure 5, and a DSC thermogram as shown in Figure 6, which are characteristic of paliperidone crystalline Form I. The mixture is taken up with demineralised water (1.5 1) and 36.5% hydrochloric acid (113 g, 1.13 mols), obtaining a solution with a pH of 3-4. A solid then reprecipitates, and the mixture is cooled to 0C and filtered. The solid is washed with demineralised water cooled to 0-5C (2 x 150 ml), and then with acetone cooled to 0-5C (3 x 200 ml). The solid is dried in oven under reduced pressure at a temperature of 50C for 16-18h. 451 g of paliperidone hydrochloride is obtained, with a potentiometric titre of 99.9%, an argentimetric titre of 7.8%, 99.2% HPLC purity, and a total yield of 90%. The product has an XRPD spectrum as shown in Figure 1, and a DSC thermogram as shown in Figure 2. |
| 96.5%Chromat. |
With triethylamine; In methanol;Inert atmosphere; Reflux; |
Example 1; Synthesis of 3-[2-[4-(6-fluoro-1,2-benzoisoxazol-3-yl)-1-piperidinyl]ethyl-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-a]-pyrimidin-4-one hydrochloride (Paliperidone hydrochloride)6-Fluoro-3-(4-piperidinyl)-1,2-benzoisoxazole hydrochloride (300 g, 1.17 mols), 3-(chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (343 g, 1.40 mols) and triethylamine (272 g, 2.69 mols) are suspended in methanol (1.5 l) in a 3000 ml reactor under nitrogen atmosphere, and the reaction mixture is heated at reflux temperature for 18-20 h. Conversion to the product is checked by HPLC titre, obtaining a yield of 96.5% in solution. The mixture is concentrated to a residue. The so obtained product has an XRPD spectrum as shown in FIG. 5, and a DSC thermogram as shown in FIG. 6, which are characteristic of paliperidone crystalline Form I.The mixture is taken up with demineralised water (1.5 l) and 36.5% hydrochloric acid (113 g, 1.13 mols), obtaining a solution with a pH of 3-4. A solid then reprecipitates, and the mixture is cooled to 0 C. and filtered. The solid is washed with demineralised water cooled to 0-5 C. (2×150 ml), and then with acetone cooled to 0-5 C. (3×200 ml). The solid is dried in oven under reduced pressure at a temperature of 50 C. for 16-18 h. 451 g of paliperidone hydrochloride is obtained, with a potentiometric titre of 99.9%, an argentimetric titre of 7.8%, 99.2% HPLC purity, and a total yield of 90%. The product has an XRPD spectrum as shown in FIG. 1, and a DSC thermogram as shown in FIG. 2. |
|
With triethylamine; In methanol;Reflux; |
To a methanol (500 mL) solution, nitrogen was purged for 30 minutes to remove the nascent oxygen. 6-Fluoro-3-piperidin-4-yl-benzo[d]isoxazole Hydrochloride (50 g), 9-Hydroxy-3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido (1,2-a) pyrimidine-4-one (52 g) and triethylamine (55) were added and stirred at reflux for 30-32 hours. The reaction mixture was cooled to 25-35 C, and filtered off to yield paliperidone base. Yield: 75 g. |
|
With water; diisopropylamine; In methanol; at 20 - 67℃;pH 10.1;Inert atmosphere;Product distribution / selectivity; |
Example 1 Synthesis of crude paliperidone 20.00 g of 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazol hydrochloride; 19.82 g of 3-(2-chloroethyl)-9-hydroxy-2-methyl-6,7,8,9-tetrahydro-4h-pyrido[1,2-a]pyrimidin-4-one; from 23.62 g to 27.54 g (33 mL to 38,4 mL) of dry diisopropylamine; 0.70 mL of water and 100 mL to 140 mL of methanol were charged to a 500 ml three-necked flask at room temperature, pH of suspension is 10,1. The reaction was carried out in inert atmosphere (N2) and protected from light. The suspension was heated to 65 C to 67 C in 90 minutes until a clear solution was obtained, After one hour the reaction product began to precipitate from the reaction mixture. The reaction mixture was left at 65 - 67 C for 30 hours, cooled to 30 C in 60 minutes, stirred for 30 minutes at 30 C and filtered. The product was washed with 40 mL of methanol. 27,5 g of paliperidone with a water content of 0,5 % were isolated, Purity was 98.6 area %, level of impurity (IV) was 0.12 area %. |
|
With diisopropylamine; In methanol; water; at 20 - 67℃; for 31.5h;pH 10.1;Inert atmosphere; Darkness;Product distribution / selectivity; |
Example 1Synthesis of crude paliperidone20.00 g of 6-fluoro-3-(4-piperidinyl)-l,2-benzisoxazol hydrochloride; 19.82 g of 3-(2- chloroethyl)-9-hydroxy-2-methyl-6,7,8,9-tetrahydro-4h-pyrido[l,2-a]pyrimidin-4-one; from 23.62 g to 27.54 g (33 mL to 38.4 mL) of dry diisopropylamine; 0.70 mL of water and 100 mL to 140 mL of methanol were charged to a 500 mL three-necked flask at room temperature. pH of suspension is 10,1. The reaction was carried out in inert atmosphere (N2) and protected from light. The suspension was heated to 65 0C to 67 0C in 90 minutes until a clear solution was obtained. After one hour the reaction product began to precipitate from the reaction mixture. The reaction mixture was left at 65 - 67 C for 30 hours, cooled to 30 0C in 60 minutes, stirred for 30 minutes at 30 0C and filtered. The product was washed with 40 mL of methanol. 27.5 g of paliperidone with a water content of 0.5 % were isolated. Purity was 98.6 area %, level of impurity (IV) was 0.12 area %. |
|
With triethylamine; In methanol;Industry scale; Reflux;Product distribution / selectivity; |
Example 28.0L of methanol, 1.04 Kg (4.28 moles) of 3-(2-chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-4H- pyrido[l,2-a]pyrimidin-4-one, 1.10 kg (10.89 moles) of triethyl amine and 1.0 kg (3.89 moles) of 6-fluoro-3(4-piperidinyl)-l,2-benzisoxazole hydrochloride were charged. The reaction mass was heated to reflux. The completion of reaction was monitored by HPLC. The reaction mass was cooled to 25-30C and further chilled to 0-5C, stirred for 30 min. and centrifuged. The cake obtained was washed with 0.5 L of chilled methanol. Weight of wet product containing paliperidone and inorganic salts was 1.0 kg, HPLC purity of paliperidone: 98.88%, keto- paliperidone impurity: 0.05%. The wet product obtained was further used for purification process. |
|
With triethylamine; In methanol; at 23 - 65℃; |
EXAMPLE-4PREPARATION OF PALIPERIDONE6-Fluoro-3-piperidino-l ,2-benzisoxazol hydrochloride (36.46 g) was suspended into methanol (300 ml), followed by addition of triethylamine (47.5 g, 65.5 ml) and 3-(2- chloroethyl)-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[l ,2-a]pyrimidin-4-one (30 g) at 23-30C. The content was heated to 63-65C and maintained till completion of reaction. After completion of reaction, the reaction mass was cooled, filtered and dried to give title compound.Yield: 45 gChromatographic Purity (By HPLC): 99.29%, Diketo compound: 0.21% |
|
With dmap; N-ethyl-N,N-diisopropylamine; In methanol; at 20 - 70℃;Inert atmosphere;Product distribution / selectivity; |
Example 4; Preparation of Crude Paliperidone Using N,N-Dimethyl Amino Pyridine (DMAP)A 100 ml Flask equipped with a mechanical stirrer, reflux condenser was charged with CMHTP (7.15 gm), FBIP.HCl (5 gm), diisopropyl ethyl amine (6.5 gm), 4-N,N-dimethyl amino pyridine (0.125 gm) and methanol (50 ml) and stirred at room temperature. The reaction mixture was then refluxed at 60-70 C. for 8 to 10 hr. After completion of the reaction the reaction mixture was cooled to 0 C. and the product obtained was filtered and suck dried. The wet product was dried under vacuum at 50-55 C.Crude Paliperidone: 99.7%N-Oxide Impurity: 0.06%Carboxylate impurity: Not detected |
|
With triethylamine; In methanol; at 60 - 63℃;Product distribution / selectivity; |
79 g of 6-Fluoro-3,4-(piperidinyl)-l,2-benzisoxazole hydrochloride, 750 ml of methanol, and 78.2 g of triethyl amine were charged in a reaction vessel at 25-30C. 75 g of the compound of formula II was added to the above mass. The reaction mixture was heated to 60-63C and then maintained at 60-63 C to achieve desired conversion. The reaction mixture was then cooled to 40-45C. Methanol was distilled off under reduced pressure to obtain a thick mass. 375 ml of methylene chloride was added to the reaction mass followed by 375 ml of water. The reaction mixture was stirred for 10-15 minutes and then filtered to obtain a clear solution. The layers were separated, and the aqueous layer was extracted twice with (2x190 ml) methylene chloride.Organic layers were combined and washed thrice with (3x190 ml) water. The organic layers were subjected to distillation under vacuum at 35C to remove methylene chloride. 75 ml of acetone was added to the thick mass and distilled to strip off methylene chloride. 750 ml of acetone was charged to the reaction mass, which was then heated to achieve reflux. The reflux was maintained for 30 minutes and then cooled to 0-5C and maintained for 45-60 minutes. The reaction mass was filtered, and the solid was washed twice with chilled (2x75 ml) acetone. The solid was dried at 70C to obtain crude paliperidone. Dry wt. = 60 g (Purity of compound of formula I = 99.07%) |
|
|
300 ml of acetonitrile was charged followed by 20.0 g of 3-(2-chloroethyl)-9-hydroxy-2- methyl-6,7,8,9-tetrahydro-4H-pyrido-[1 ,2-a]-pyrimidin-4-one and 21.0 g of 6-fluoro-3-(4- piperidinyl)-1 ,2-benzisoxazole hydrochloride at room temperature. The reaction mass was stirred at room temperature for 5 minutes and 20.0 g of potassium carbonate and 0.7 g of potassium iodide were charged. Further, 0.2 g of sodium borohydride was charged and the temperature was raised to 65+/-2C. The reaction mass was maintained at 65+/-2C for 25 hours. After reaction completion, the reaction mass was slowly cooled to room temperature. The solids were filtered, washed with 50 ml of acetonitirle and then dissolved in 800 ml of methylene chloride at room temperature. The contents were heated to 30-35C, maintained for 10 minutes, filtered and washed with 20 ml of methylene chloride. The clear filtrate was distilled completely under vacuum below 35C and replaced with 50 ml of acetone. Further, 300 ml of acetone was charged, heated to 45C and stirred for 30 minutes. The reaction mass was cooled to room temperature and stirred for 1 hour. The product was filtered, washed with 20 ml of acetone and dried under vacuum at 50-55C to yield paliperidone (30 g, yield : 150% w/w, efficiency : 85. 1 %, keto impurity by HPLC : 0.01%, total impurity level by HPLC: 1.0 %) |
| 22 g |
|
Example-2 Preparation of Paliperidone To a solution of 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole HCl (formula-6) (20 grams) in methanol (100 ml) added sodium carbonate (12.7 g) and heated the mixture to 50-55 C. for 20-30 min. the reaction mixture was cooled to 35-40 C. and filtered. The filtrate was taken in a round bottomed flask and <strong>[130049-82-0]3-(2-chloroethyl)-9-hydroxy-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one</strong> (formula-7) (18.9 grams) was added to it followed by diisopropyl ethyl amine (15.5 grams). The reaction mixture was refluxed for 20-24 hours. The solvent was distilled off under reduced pressure; dichloromethane (600 ml) was added to the reaction mixture and stirred for 15-20 min. The reaction mixture was washed with aqueous sodium hydroxide solution (3*100 ml) followed by water at 20-25 C. The organic layer was separated and the solvent was distilled off under reduced pressure. Methanol (100 ml) was added to the reaction mixture and refluxed for 30-45 min. The reaction mixture was cooled to 20-25 C., stirred for one hour, filtered and washed with methanol to provide the title compound as a solid. Yield: 22 grams. |
| 30 g |
With sodium tetrahydroborate; potassium carbonate; potassium iodide; In acetonitrile; at 20 - 65℃; for 25h; |
300 ml of acetonitrile was charged followed by 20.0 g of 3-(2-chloroethyl)-9-hydroxy-2-methyl-6,7,8,9-tetrahydro-4H-pyrido-[1,2-a]-pyrimidin-4-one and 21.0 g of 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride at room temperature. The reaction mass was stirred at room temperature for 5 minutes and 20.0 g of potassium carbonate and 0.7 g of potassium iodide were charged. Further, 0.2 g of sodium borohydride was charged and the temperature was raised to 65±2 C. The reaction mass was maintained at 65±2 C. for 25 hours. After reaction completion, the reaction mass was slowly cooled to room temperature. The solids were filtered, washed with 50 ml of acetonitrile and then dissolved in 800 ml of methylene chloride at room temperature. The contents were heated to 30-35 C., maintained for 10 minutes, filtered and washed with 20 ml of methylene chloride. The clear filtrate was distilled completely under vacuum below 35 C. and replaced with 50 ml of acetone. Further, 300 ml of acetone was charged, heated to 45 C. and stirred for 30 minutes. The reaction mass was cooled to room temperature and stirred for 1 hour. The product was filtered, washed with 20 ml of acetone and dried under vacuum at 50-55 C. to yield paliperidone (30 g, yield: 150% w/w, efficiency: 85.11%, keto impurity by HPLC: 0.01%, total impurity level by HPLC: 1.0%) |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping