|
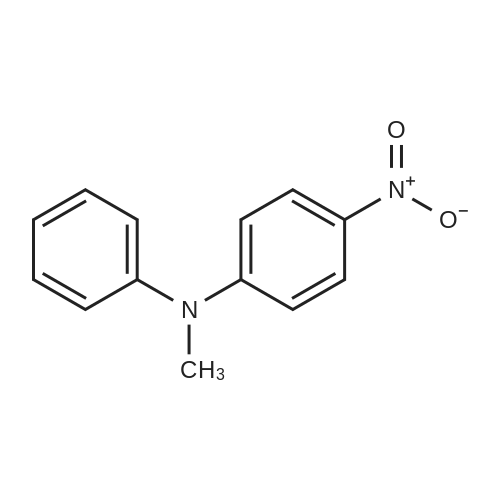
With tetramethyl ammoniumhydroxide; dihydrogen peroxide In water at 66 - 91℃; |
1-15 EXAMPLE 1
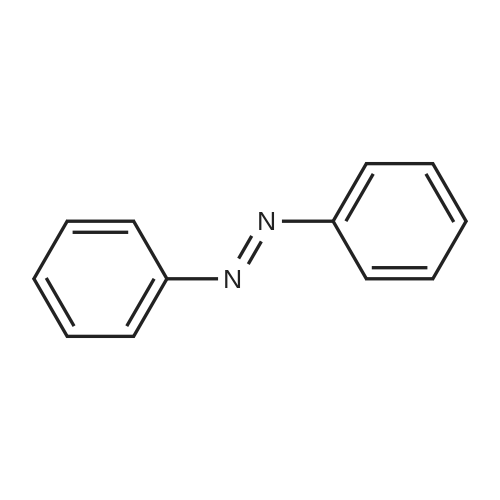
[0045] This example provides reference information, for discussion of the effect of using hydrogen peroxide during the coupling reaction in the other examples. The procedure for Runs 1-3 is similar to that of Example 2, except using plant recycle TMAH (26.8 wt. %) and plant recycle aniline, with base concentration and drying at 62 torr and reaction at 60 torr. The procedure for Runs 4-6 is to charge 145.28 g of fresh aniline (1.56 moles) and 87.36 g of aqueous pre-concentrated fresh TMAH solution (36.0 wt. %, 0.345 moles TMAH) to a 500-mL round bottom flask equipped with a thermocouple, heating mantle, subsurface feed tubes for nitrobenzene and peroxide or water and a Teflon paddle stirrer. With pressure at 70 torr, the mixture is heated to remove 18 mL of water, along with aniline, (?30 minutes) and then nitrobenzene feed (36.93 g, 0.30 moles) is started. Temperature rises from about 66-67 C. to 80 C. during the reaction period, while water and aniline are boiled off. Table 1 gives the times for nitrobenzene feed and reaction hold for all six runs. Water and aniline are boiled off during the hold. Batches for Runs 4-6 are quenched with 20 mL of water after the hold period. The hydrogen peroxide charge is 20.40 g (0.03 moles) of a 5 wt. % aqueous solution concurrent with nitrobenzene. Since water can affect selectivity by protecting TMAH from degradation and by shifting reaction equilibriums, water is fed concurrently with nitrobenzene for direct comparison with peroxide. [0046] The example illustrates that both shorter nitrobenzene feed time and the addition of water can increase selectivity, although water is not very effective for the longer feed time. However, peroxide gave the highest selectivity, 1.9% greater than water addition. More importantly, for a commercial process that involves recycles and waste disposal, aqueous peroxide greatly reduced the levels of two key by-products compared to water alone, viz. azobenzene (by 39%) and phenazine (by 36%). Replicate baseline runs by a slightly different procedure gave selectivities of 92.7% and 92.6%, indicating that the experimental results reported herein are very reproducible. Moreover, the replicates indicate that the small selectivity differences, such as 1.9% higher for peroxide vs. water, are indeed significant. EXAMPLE 2 [0047] Some of the runs in the following examples had relatively low conversions, because the procedure used a fixed nitrobenzene feed time plus hold time rather than holding the batches to reaction completion. This example shows the effect of an extended hold period on selectivity. [0048] The procedure is to charge 432.85 g of plant recycle base (24.4 wt. % TMAH, 1.16 moles) to a 1-L water/glycol jacketed reactor. Begin agitation at 150 rpm and boil off 92 mL water at constant pressure of 65 torr, with the water bath temperature starting at 72 C. and increasing by 1 C. per 10 mL of water removed. Then charge 301.50 g (3.24 moles) of fresh aniline via vacuum. Continue to remove water plus aniline at 65 torr, by raising the bath temperature 1 C. per 9 mL of water removed, while continuously charging 120 mL of aniline from a sidearm pressure-vented dropping funnel. When 72 mL of water has been removed (164 mL total), begin co-feeding 123.11 g of nitrobenzene (1.00 moles) and 27.20 g hydrogen peroxide (10 wt. % aqueous solution, 0.08 moles) subsurface via peristaltic pumps over a period of 80 minutes. Continuously add 60 mL of aniline during the reaction step, while holding pressure at 65 torr and boiling off water plus aniline. Gradually increase bath temperature in 0.5 C. increments to achieve 91 C. in the bath and 80-82 C. in the reactor by the end of the reaction step. Initiate the hold period by reducing pressure to 60 torr and increasing the bath and reactor temperatures by another 1 C. Continue removing water plus aniline during an extended hold. [0049] This example shows that holding a low conversion batch to essentially complete conversion had only a minimum impact on selectivity. In the examples following this one, conversion ranged from 73.4% to 100%. These results show that driving conversion from 89.3% to 99.8% reduced selectivity by only 0.5% and going from 96% to 99.8% conversion reduced selectivity by only 0.2%. Therefore, low conversion for some runs in the following examples does not affect the conclusions. EXAMPLE 3 [0050] A 3-factor, 8-run Design of Experiments (DOE) with pressure, nitrobenzene feed rate, and peroxide as variables was executed. Concentration of peroxide (5 wt. % aqueous solution) and H2O2/NB (0.1 molar ratio) were arbitrarily selected for the four runs using peroxide. To a 500-mL round bottom flask equipped with a heating mantle, thermocouple, subsurface feed tubes for nitrobenzene and peroxide and a Teflon paddle stirrer was charged 130.02 g of recycle base (24.4 wt. %), which was then concentrated to 31 wt. % by boiling off 28 mL of water at the pressure indicated in Table 3. Then 145.28 g of aniline was added and another 16 mL of water was removed, along with aniline (44 mL total water). Then nitrobenzene feed of 36.93 g was started, with continued boil off of water and aniline. When peroxide was used, the 20.40 g of 5 wt. % peroxide solution was co-fed with nitrobenzene at an appropriate feed rate to finish with nitrobenzene. Batches were held as described below, then quenched with 20 mL of water. Reactions were run at 80 C. and either 65 or 95 torr (specified in the design) on a 0.3 mole scale. Hold periods were fixed at 20 minutes for the 110 minute nitrobenzene feed and 45 minutes for the 70 minute nitrobenzene feed, both with continued boil off of water and aniline. [0051] Results in Table 3 show that selectivity is consistently higher when peroxide is used at a low level and the range is much smaller (96.1 to 96.6% with vs. 89.8 to 94.8% without) for variations of reaction pressure and nitrobenzene feed rate. Also, with peroxide more 4-NDPA was made relative to azobenzene, whereas without, nearly equimolar amounts were generated. Less 4-NDPA (by 30-40%) was made with peroxide at the longer nitrobenzene feed time and in all runs, much less azobenzene and phenazine were made with peroxide. Example 1 showed that nitrobenzene feed rate can affect selectivity without peroxide and this example shows that peroxide reduces the effect of both nitrobenzene feed rate and reaction pressure, which is unexpected. EXAMPLE 4 [0052] A refining DOE was completed to assess both 1) amount of peroxide and 2) peroxide concentration for the coupling reaction. The procedure was the same as for Example 3, except for the mole ratios and peroxide concentrations listed in Table 4 and a nitrobenzene feed time of about 70 minutes with a 30 minute hold. Table 4 shows that with a fast nitrobenzene feed rate, selectivity is relatively independent of peroxide concentration, especially at the lower molar ratio. This is surprising, because Example 1 showed that adding water can increase selectivity and the runs with 5 wt. % peroxide had 6.33 times the amount of water as 25 wt. % peroxide. The results also show that selectivity can be affected by the amount of oxidant. A comparison of Runs with equal peroxide concentration in Table 4 shows that the higher mole ratio gave lower selectivity in each case. Again, this is surprising, since twice the amount of water was added at the higher mole ratio. So the benefits of water and peroxide are not additive, as the effect of peroxide predominates. EXAMPLE 5 [0053] This example further illustrates the effect of pressure on selectivity when peroxide is used. The batches were made by a procedure similar to Example 2, with a nitrobenzene feed time of 110 minutes, a hold time of 20 minutes, a different sample of plant recycle base (26.8 wt. %) and plant recycle aniline instead of fresh. The results in Table 5 show that pressure does not have an impact on selectivity when 30 wt. % peroxide is used, just as in Example 3 with 5 wt. % peroxide. This is additional evidence that peroxide mitigates the effect of other reaction variables. EXAMPLE 6 [0054] Example 1 showed that shorter nitrobenzene feed time (80 minutes) alone, or with water, or with aqueous peroxide, can increase selectivity. Example 3 showed that for a fixed peroxide concentration and molar ratio, nitrobenzene feed time (about 75 minutes vs. about 110 minutes) had little effect on selectivity. Example 4 showed that with a short nitrobenzene feed time (about 70 minutes), selectivity is relatively independent of peroxide concentration, especially at the lower mole ratio. [0055] This example explores the effect of peroxide concentration on selectivity for a longer nitrobenzene feed time. A series of batches were made by a procedure similar to that of Example 5. Also, peroxide feed for the 0.064 mole ratio runs was via a piston pump (see Example 14). Peroxide concentration was varied from 5 wt. % to 35 wt. %, with H2O2/NB=0.1 and 0.064. The results in Table 6 show that also for a longer nitrobenzene feed time, selectivity is essentially independent of peroxide concentration. Moreover, phenazine level increases only slightly as significantly less water is charged with the peroxide, which is consistent with Example 4. With a longer nitrobenzene feed time, the rate of removal of water relative to the rate of nitrobenzene charge is greater than for a shorter nitrobenzene feed time. So as less water is charged with the peroxide, the batches become even drier. However, even the lowest water charge in Table 6 has significantly higher selectivity than for water alone with a 110 minute nitrobenzene feed in Table 1. This illustrates that although water and peroxide can both play a role in increasing selectivity, the effect of peroxide is more important. Furthermore, since water can influence the rate of formation of the Meisenheimer complex and the rate of its oxidation by nitrobenzene, it should be possible to improve selectivity with peroxide by tuning H2O2/NB to match the Meisenheimer concentration in the batch. EXAMPLE 7 [0056] A series of coupling reactions were done with a fixed peroxide concentration of 5 wt. % to determine the effect of temperature on selectivity. The procedure was as follows: Charge 130.02 g recycle base (24.4 wt. % TMAH) to 500-mL scale coupler and boil off 28 mL of water. Add 145.28 g aniline and remove another 16 mL of water, along with aniline (44 mL total water). Feed 36.93 g nitrobenzene concurrently with 5 wt. % aqueous peroxide solution at H2O2/NB=0.08 molar, both subsurface, while boiling off water and aniline. Complete the co-feed in 100-110 minutes at the temperatures indicated in Table 7 and at a constant pressure of 65 torr. Hold for 30 minutes, while boiling off water and aniline, and then quench with 20 mL of water. [0057] The results in Table 7 illustrate the effect that the rates of formation and intramolecular oxidation of the Meisenheimer complex have on selectivity with peroxide. As temperature is increased, selectivity reaches a maximum at about 80 C. At lower temperatures, the rate of Meisenheimer formation is too low for the rate of peroxide addition, so that oxidation of aniline to azobenzene by peroxide increases. At higher temperatures, the higher rate of intramolecular Meisenheimer oxidation to p-NDPA reduces the amount of Meisenheimer available for reaction with peroxide, so that again oxidation of aniline to azobenzene by peroxide increases. Also, the selectivity at 70 C. is higher than that obtained without peroxide at otherwise comparable reaction conditions. Therefore, the effective range for this example can be extended down to about 65 C. [0058] Thus, 80 C. is an apparent optimum that is dependent on the reaction procedure. Any procedure change that will change the rate of Meisenheimer formation, such as changing the rate of water removal, will affect selectivity with peroxide. This could shift the optimum selectivity to a different temperature. Moreover, selectivity can be increased at lower and higher temperatures simply by adjusting the molar ratio of H2O2/NB to match the rate of formation or rate of intramolecular oxidation of the Meisenheimer. Thus selectivity is highest at 80 C. for this particular reaction procedure with H2O2/NB=0.08 molar. However, the optimum temperature will vary with other variables, such as water level in the reactor and H2O2/NB mole ratio. Furthermore, varying temperature will require a different mole ratio of H2O2/NB for maximum selectivity. Therefore, the effective ranges given in other examples are not absolute. EXAMPLE 8 [0059] Two sets of coupling reactions were done with fixed peroxide concentrations to determine the effective mole ratio range that would give increased selectivity. The procedure for 5 wt. % peroxide was basically the same as for Example 3. Peroxide and nitrobenzene were fed over 105-110 minutes, with a 20 minute hold period for reaction at 80 C. and 65 torr. The procedure for 30 wt. % was similar to Example 5. [0060] FIG. 1 and Table 8 show that the effective range for 5 wt. % peroxide is about H2O2/NB=0.01-0.20, the preferred range is about H2O2/NB=0.03-0.16 and the most preferred range is about H2O2/NB=0.06-0.12. The optimum molar ratio with 5 wt. % peroxide was H2O2/NB=0.07-0.09 for this procedure, which is about the same as the mole % of 4-NDPA that was made from nitrobenzene. So peroxide reacts in high selectivity to make 4-NDPA with minimum formation of azobenzene. This is a surprising result, due to the very large molar excess of aniline that is available to be oxidized to azobenzene. [0061] The effective mole ratio range for 30 wt. % peroxide is about 0.01-0.25. An optimum cannot be derived from the data, but it appears to be within 0.06-0.21, which is shifted higher than for 5 wt. % peroxide. A more preferred range appears to be within 0.08-0.17. So the effective mole ratio range, preferred range and most preferred range for peroxide are expected to vary with some process variables, such as peroxide concentration, impurity levels in recycle streams, reaction temperature, water removal rate and nitrobenzene feed rate. Therefore, these ranges are not absolute for peroxide, but rather representative. It is envisaged that the effective range could extend to H2O2/NB=0.01-0.4 with recycle base or perhaps even somewhat wider. EXAMPLE 9 [0062] An optimization study was done for fresh base with peroxide to examine the effect of base quality. The procedure was similar to Example 3 for 5 and 20 wt. %, with a 126.89 g charge of 25 wt. % base, and to Example 10 for 35 wt. %. As seen in FIG. 2 and Table 9, fresh base gave flatter and wider optimization curves vs. recycle base. Moreover, the optimum mole ratio and effective range varied with concentration, the maximum selectivity was lower vs. recycle base and selectivity increased after the initial optimum was passed. The upturns are due to the higher water charge as mole ratio increased, which did not occur with 35 wt. % peroxide, for which the least water was added. Selectivity rose because water inhibits oxidation of Meisenheimer by nitrobenzene, so that more is oxidized by peroxide. The results show that at the conditions used for Examples 8 and 9, peroxide is more effective with recycle base. Moreover, the salts in recycle base must moderate the effect of water, since the selectivity upturn did not occur with it. Even so, the selectivity increase with fresh base is substantial. The effective range for 35 wt. % peroxide is about 0.01-0.33 and if water was removed faster with 20 wt. % peroxide, the curve tracks to an effective range of about 0.01-0.46. Since nitrobenzene feed rate can extend well beyond 110 minutes for a commercial process, the effective range could wellEXAMPLE 10 [0063] This example further illustrates the effect |
|
With potassium hydroxide; dihydrogen peroxide; tetramethlyammonium chloride In water at 60℃; for 1h; |
2 COMPARATIVE EXAMPLE 2
[0072] This example examines the effect of peroxide with a strong inorganic base and phase transfer catalyst (PTC), by the following procedure. Aniline (99%, 22.58 g, 240 mmoles), nitrobenzene (99%, 4.97 g, 40 mmoles), hydrogen peroxide (50 wt. % aqueous, molar amount indicated below in FIG. 5), water (water is added such that the total water is kept constant at 2.16 g), potassium hydroxide (86% ground powder, 7.83 g, 120 mmoles) and tetramethylammonium chloride (97%, 4.52 g, 40 mmoles) was charged to a 50-mL round bottom flask equipped with a magnetic stirrer. Peroxide was charged to the reaction mixture before adding KOH & TMACI. Then the flask was quickly stoppered and the reaction was allowed to proceed for 1 hour at 60 C. In this example, azoxybenzene and 2-NDPA were obtained as reaction by-products that were not obtained with TMAH. So these by-products were included in the calculation of selectivity. [0073] FIG. 5 shows that with a strong inorganic base and a phase transfer catalyst, selectivity increases steadily as the mole ratio of peroxide/NB is increased from 0 to unity. However, with a strong organic base, i.e. TMAH, there were optimum mole ratios with both fresh and recycle base. It is unexpected that there is an optimum mole ratio with a strong organic base, but not with a strong inorganic base and PTC that generate the same strong organic base in situ. Moreover, the inorganic system gave 2-NDPA+azoxybenzene levels of 1.1% to 2.4%, whereas none was formed with TMAH. Again, it is surprising that these by-products are formed with a strong inorganic base, but not with a strong organic base. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping