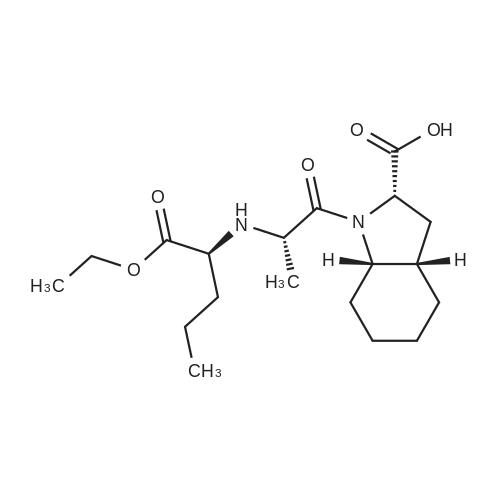
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
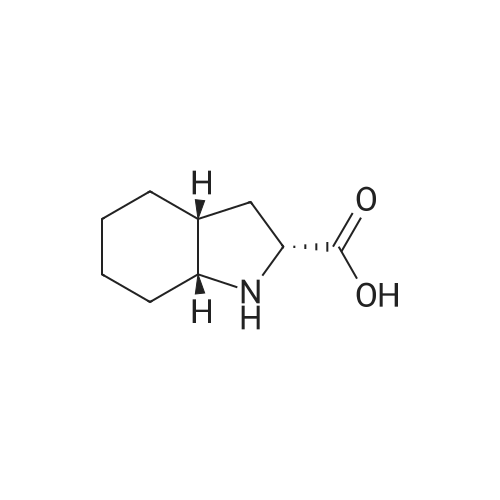
Introduce 200 g of the compound obtained in the previous Step, dissolved in acetic acid, and then 5 g of Pt/C 10percent into a hydrogenator. Hydrogenate under a pressure of 5 bars at ambient temperature until the theoretical amount of hydrogen has been absorbed. Remove the catalyst by filtration and then cool to a temperature of from 0 to 5° C. and collect the resulting solid by filtration. Wash the filter cake and dry it to constant weight. (2S, 3aS, 7aS)-Perhydroindole-2-carboxylic acid is obtained in that manner in a yield of 87percent and with an enantiomeric purity of 99percent.
With 8 % Pd/C; hydrogen In methanol at 55 - 65℃; Autoclave
In a 1 L autoclave,(S) -indoline-2-carboxylic acid (0.3 mol, 49.0 g) and methanol (600 mL)Then, 4.9 g of 8percent wet palladium carbon wetted with methanol was added,Mechanical agitation. Through the nitrogen replacement air 3 times,And then hydrogen gas replacement nitrogen 3 times,Replace the kettle when the pressure is 0.5Mpa. after finishing,Continue to hydrogen to 5.0Mpa, set the heating temperature at 60 ,Maintaining the temperature of the reaction system at 55 to 65 ° C,TLC (developing solvent: chloroform: methanol: triethylamine = 6: 4: 1, ninhydrin)Tracking to the raw point of the reaction is complete. The hydrogen was vented and the hydrogen was replaced with nitrogen three times.Discharge, filter, take the filtrate under reduced pressure,A crude solid was obtained as a white solid. Finally, recrystallization from ethanol,A white solid was obtained in 37.3 g yield, 73.5percent yield,Purity 98.6percent (HPLC),
Stage #1: With chloro-trimethyl-silane; triethylamine In dichloromethane at 20 - 25℃; for 2 h; Stage #2: With triethylamine In dichloromethane at -15℃; for 2 h;
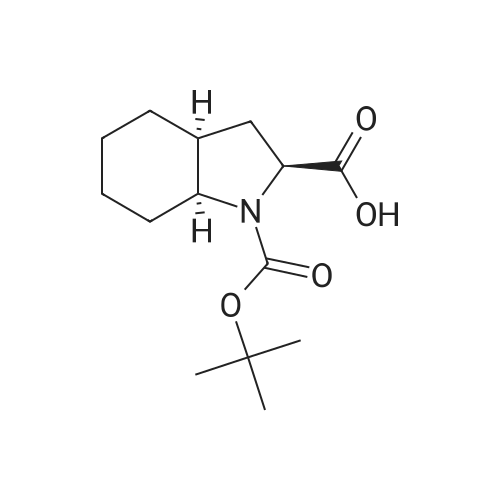
Example 3 Preparation of perindopril erbumin from isopropyl acetate ((2S,3aS,7aS)-l-((2S)-2-(((lS)-l- (ethoxycarbonyl)butyl)amino)-1-oxopropyl)octahydro-1H- indol-2-carboxylic acid 2-methylpropane-2-amine salt) ; Trimethylchlorosilane (2.92 ml) and triethyl amine (3.2 ml) were added to (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (3.72 g) in dichloromethane (40 ml) at 20 - 25°C and it was stirred at 20 - 25°C for two hours. After two hours triethyl amine (2.77 ml) was added, the suspension was cooled to-15°C, a suspension of N-((S)-l-carbethoxybutyl)- L-alanyl chloride hydrochloride (5.5 g) in dichloromethane (80 ml), which had been cooled to-15°C, was poured thereto and the stirring was continued at-15°C for two hours. The reaction solution was heated to 0°C, the precipitated triethylamine hydrochloride was filtered off and washed with dichloromethane (10 ml), water (33 ml), wherein NaOH (0.8 g) had been dissolved, was added to the filtrate, and the pH was adjusted to 4.2 with a 20percent NaOH solution. The organic phase was separated and the aqueous layer was washed with dichloromethane (2 x 20 ml). The combined dichloromethane layers were evaporated, the residue was dissolved in isopropyl acetate (125 ml), the undissolved part was filtered off and t-butylamine (2.2 ml) was added to the filtrate. The precipitated crystals were dissolved at the boiling point of the solution, the clear solution was cooled to 10 - 20°C and the stirring was continued for two hours. After two hours the precipitated crystals were filtered off, washed with isopropyl acetate (15 ml) and dried at 35 - 40°C in an air drier. Perindopril erbumin (7.65 g; 86percent) in an a form having a purity over 99percent was obtained with individual impurities present in not more than 0.1percent each.
80%
Stage #1: With chloro-trimethyl-silane; triethylamine In dichloromethane at 20 - 25℃; for 2 h; Stage #2: With triethylamine In dichloromethane at -15℃; for 2 h;
Example 2 Preparation of perindopril erbumin ((2S,3aS,7aS)-1-((2S)-2- (((lS)-l-(ethoxycarbonyl)butyl)amino)-l- oxopropyl) octahydro-1H-indol-2-carboxylic acid 2- methylpropane-2-amine salt) ; Trimethylchlorosilane (2.86 ml) and triethyl amine (3.08 ml) were added to (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (3.72 g) in dichloromethane (60 ml) at 20 - 25°C and stirred at 20 - 25°C for two hours. After two hours triethyl amine (2.8 ml) was added, the suspension was cooled to -15°C, a suspension of N-((S)-l-carbethoxybutyl)- L-alanyl chloride hydrochloride (5.5 g) in dichloromethane (60 ml), which has been cooled to-15°C, was poured thereto and the stirring was continued at-15°C for two hours. The reaction solution was heated to 0°C, brine (25 ml) with dissolved NaOH (0.8 g) was added, and the pH was adjusted to 4.2 with a 20percent NaOH solution. The organic phase was separated and the aqueous layer was once more washed with dichloromethane (20 ml). The combined dichloromethane layers were evaporated, the residue was dissolved in ethyl acetate (100 ml), the undissolved part was filtered off and t-butylamine (2.2 ml) was added to the filtrate. The precipitated crystals were dissolved at the boiling point of the solution, the clear solution was cooled to 10 - 20°C and the stirring was continued for two hours. After two hours the precipitated crystals were filtered off, washed ,with ethyl acetate (12 ml) and dried at 35 - 40°C in an air drier. Perindopril. erbumin (7.1 g; 80percent) in an a form having a purity over 99percent was obtained with individual impurities present in not more than 0.1percent each.
78%
Stage #1: With chloro-trimethyl-silane; triethylamine In dichloromethane at 20 - 25℃; for 2 h; Stage #2: With triethylamine In dichloromethane at -15℃; for 2 h;
Example Preparation of perindopril erbumin from N,N- dimethylformamide ((2S,3aS,7aS)-l-((2S)-2-(((lS)-l- (ethoxycarbonyl)butyl)amino)-l-oxopropyl)octahydro-lH- indol-2-carboxylic acid 2-methylpropane-2-amine salt) ; Trimethylchlorosilane (2.92 ml) and triethyl amine (3.2 ml) were added to (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (3.77 g) in dichloromethane (40 ml) at 20 - 25°C and stirred at 20 - 25°C for two hours. After two hours triethyl amine (2.77 ml) was added, the suspension was cooled to-15°C, a suspension of N-((S)-l-carbethoxybutyl)- L-alanyl chloride hydrochloride (5.5 g) in dichloromethane (40 ml), which had been cooled to-15°C, was poured thereto and the stirring was continued at-15°C for two hours. The reaction solution was heated to 0°C, the precipitated triethylamine hydrochloride was filtered off and washed with dichloromethane (10 ml), water (33 ml), wherein NaOH (0.8 g) had been dissolved, was added to the filtrate, and the pH was adjusted to 4.2 with a 20percent NaOH solution. The organic phase was separated and the aqueous layer was washed with dichloromethane (2 x 20 ml). The combined dichloromethane layers were evaporated, the residue was dissolved in N, N-dimethylformamide (90 ml),. the undissolved part was filtered off and t-butylamine ,(2.2 ml) was added to the filtrate. The precipitated crystals were dissolved at 80°C, the clear solution was cooled to 10 - 20°C and the stirring was continued for two hours. After two hours the precipitated crystals were filtered off, washed with N,N- dimethylformamide (9 ml) and dried at 35 - 40°C in an air drier. Perindopril erbumin (6.9 g; 78percent) in an a form having a purity over 99percent was obtained with individual impurities present in not more than 0.1percent each.
Stage #1: With sodium hydroxide In tetrahydrofuran; water for 0.166667 h; Stage #2: at 20℃; for 12 h;
L-octahydroindole-2-carboxylic acid (1.69 g, 10 mmol) was added to the reaction flask,Add 30 mL of THF and 30 mL of distilled water to stir,A 7 mL aqueous solution of NaOH (0.68 g, 17 mmol) was added dropwise to the ice bath,Plus,The reaction was stirred for 10 min.(Boc) 2O (2.84 g, 13 mmol)Rose to room temperature for 12h.TLC detection reaction is completed, diluted with water, with methyl tert-butyl ether extraction impurity removal,The water phase with citric acid PH to acidic,Ethyl acetate was extracted three times, the organic phases were combined, washed with saturated brine, dried over Na2SO4,Filtration and concentration gave 3.21 g of compound 1 in 100percent yield.
98%
With triethylamine In tetrahydrofuran; water at 20℃;
Triethylamine (8.25 mL, 59 mmol) was added to (S)-octahydroindole-2- carboxylic acid (10.0 g, 59 mmol) in a 1/1 volume/volume mixture of tetrahydrofuran/water (200 ML total), followed by addition of di-tert-butyl dicarbonate (14.0 g, 65 mmol). The reaction mixture was stirred overnight at room temperature, and then concentrated with no heating to remove tetrahydrofuran. The aqueous mixture was partitioned between ethyl acetate (300 mL), water (100 mL), and citric acid solution (50 ML, 10percent aqueous). The layers were separated, the organic layer washed with brine (50 mL), dried (magnesium sulfate), filtered and concentrated to an oil, which solidified on standing. The solid was slurried in hexanes and collected by filtration. 15.63 g, 98percent. MS (APCI-) 17 1/Z (percent): 268 (100). Calcd for C14H23NO4 : C, 62.43 ; H, 8.61 ; N, 5.20 Found: C, 62.33 ; H, 8.30 ; N, 5.15
97%
With sodium hydroxide In 1,4-dioxane; water at 20℃; for 18 h;
To a stirred solution of (2S, 3aS, 7AS)-OCTAHYDROINDOLE-2-CARBOXYLIC acid (30 g, 0.18 mol) in dioxane (180 ML) and 1 M NAOH (180 mL), was added di- TE7X-BUTYL DICARBONATE (48 G, 0. 22 mol). The reaction mixture was stirred for 18 hours at room temperature and then diluted with diethyl ether (500 mL) and water (500 mL). The resulting aqueous layer was removed and cooled to 0 °C. Methylene chloride (1 L) was added, and the mixture adjusted to pH 3 by the addition of 2 M HC1. The organic layer was removed and the aqueous phase extracted with methylene chloride (2 x 25Q ML). The combined organic layers were dried over sodium sulfate, filtered and concentrated in vacuo to afford A colorless oil. Trituration with ether and hexanes followed by removal of the excess solvents afforded the desired (2S, 3aS, 7AS)-OCTAHYDROINDOLE-1, 2- dicarboxylic acid 1-TERT-BUTYL ester (4 6. 3 g, 97percent), as a white solid : NNM (300 MHz, DMSO-d6) 8 1.00-1. 70 (m, 7H), 1.32, 1.38 (2 s, 9H), 1.76-1. 98 (m, 2H), 2.01-2. 13 (m, 1H), 2.18-2. 33 (m, 1H), 3.59-3. 69 (m, 1H), 3. 99-4. 10 (m, 1H); MS (CI) 170 [M + H-100 (Boc)] +, 270 [M + H] +.
94%
With triethylamine In dichloromethane
(a) To a solution of compound 5 in CH2Cl2 was added triethylamine and (Boc)2O. The reaction was monitored by TLC. After the starting materials were completely consumed, the mixture was diluted with CH2Cl2, washed sequentially with 1 N HCl and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to afford compound 6 as oil (94percent yield).
368 g
Stage #1: With sodium hydroxide In tetrahydrofuran; water at 0℃; for 0.25 h; Stage #2: at 0 - 20℃; for 12 h;
To a stirred solution of (2R,3aS,7aS)-octahydro-lH-indole-2-carboxylic acid (250 g, 1.0 equiv) (1) in THF (3 L) and water (1.5 L) at 0 °C was added dropwise a cooled aqueous solution of 2.5 M NaOH (1 L). The reaction mixture was stirred for 15 minutes at the same temperature. Di-tert-butyl dicarbonate (1.3 equiv) was added dropwise, maintaining the temperature at 0 °C. The resulting reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was washed with MTBE (3 times). The aqueous phase was acidified with IM aqueous citric acid and extracted with ethyl acetate (3 times). The combined organic layers were dried over sodium sulfate and concentrated to dryness to afford (2R,3a,S',7a)S)-l-(tert-butoxycarbonyl)octahydro-lH- indole-2-carboxylic acid (368 g) (2).
Reference:
[1] Patent: CN106083829, 2016, A, . Location in patent: Paragraph 0061; 0065; 0067; 0068
[2] Patent: WO2004/92132, 2004, A1, . Location in patent: Page 85
[3] European Journal of Organic Chemistry, 2008, # 5, p. 934 - 940
[4] Patent: WO2004/92132, 2004, A1, . Location in patent: Page 80-81
[5] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 23, p. 6684 - 6693
[6] Journal of Medicinal Chemistry, 1996, vol. 39, # 12, p. 2379 - 2391
[7] Patent: US2012/302538, 2012, A1, . Location in patent: Page/Page column 17
[8] Patent: WO2018/64585, 2018, A1, . Location in patent: Page/Page column 28; 29
With chloro-trimethyl-silane; 1,1,1,3,3,3-hexamethyl-disilazane; In water; ethyl acetate; acetonitrile;
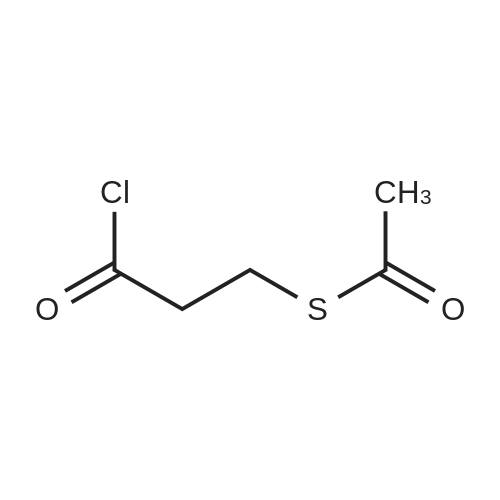
(b) (2alpha, 3abeta, 7abeta)-Octahydro-1-[3-(acetylthio)propanoyl]-1H-indole-2-carboxylic acid can also be prepared as follows. A mixture of (2alpha, 3abeta, 7abeta)-octahydro-1H-indole-2-carboxylic acid (3.0 g, 0.0177 mole), hexamethyldisilazane (3.0 g, 0.0186 mole) and one drop of chlorotrimethylsilane in 10 ml of acetonitrile is heated at reflux for three hours. The resulting solution is cooled in an ice bath and a solution of 2.9 g (0.0177 mole) of 3-(acetylthio)propanoyl chloride in 5 ml of acetonitrile is added dropwise. A volume of 15 ml of acetonitrile and volatiles is distilled off at atmospheric pressure. The solution is cooled and 0.35 ml of water is added and the mixture is heated at reflux for 5 minutes. The solution is cooled and filtered and then concentrated under reduced pressure to remove the remaining solvent to yield an oil which solidifies upon standing. The residue is dissolved in 60 ml of boiling ethyl acetate. The solution is filtered and cooled yielding 3.0 g of product; mp 131-133 C.
Stage 1E: (2S,3aS,7aS)-2-Carboxyoctahydroindole Place 25 kg of (S)-2-carboxyindoline, obtained previously, in 110 liters of methanol in a vessel. Keep stirred. Charge the rhodium (5% dry) catalyst into a mixer. Start up the stirring in a hydrogenator, charge the methanolic suspension of (S)-2-carboxyindoline by passing it through the mixer and rinse the assembly with water. Heat to 60 C. and pressurize with hydrogen (30 bars). Filter off the catalyst on a single-plate filter. Collect the hydroalcoholic liquors in a reactor and evaporate the methanol off under vacuum. After concentrating, charge approximately 300 kg of dioxane. Heat to boiling and add water until a solution is obtained. Allow to cool. Filter off and dry. 22.3 kg of crystals are obtained. Yield: 86.1%.
73.5%
With 8 % Pd/C; hydrogen; In methanol; at 55 - 65℃; under 37503.8 Torr;Autoclave;
In a 1 L autoclave,(S) -indoline-2-carboxylic acid (0.3 mol, 49.0 g) and methanol (600 mL)Then, 4.9 g of 8% wet palladium carbon wetted with methanol was added,Mechanical agitation. Through the nitrogen replacement air 3 times,And then hydrogen gas replacement nitrogen 3 times,Replace the kettle when the pressure is 0.5Mpa. after finishing,Continue to hydrogen to 5.0Mpa, set the heating temperature at 60 ,Maintaining the temperature of the reaction system at 55 to 65 C,TLC (developing solvent: chloroform: methanol: triethylamine = 6: 4: 1, ninhydrin)Tracking to the raw point of the reaction is complete. The hydrogen was vented and the hydrogen was replaced with nitrogen three times.Discharge, filter, take the filtrate under reduced pressure,A crude solid was obtained as a white solid. Finally, recrystallization from ethanol,A white solid was obtained in 37.3 g yield, 73.5% yield,Purity 98.6% (HPLC),
S-Indoline-2-carboxylic acid (50 gm) (0.31 mole), sodium hydroxide (12.27 gm) (0.31 mole), 5% rhodium on alumina (7.5 gm), were mixed with water (1 lit) in an autoclave and hydrogenated at a pressure of 12 bar at 50C. The catalyst was filtered off and the bed washed with water (200 ml). The reaction mass was cooled to 15-20C and acidified to pH 3.0 to 3.2 using conc. HCI and washed with (4 x 250 ml) ethyl acetate. The pH of the aqueous phase was adjusted to 6.5 with 10% sodium hydroxide solution. Water was then removed by distillation under vacuum and the traces were further removed by addition of toluene and continuing the distillation. The product was isolated by addition of acetonitrile (500 ml) and filtration. The crude product was recrystallized from methanol to give the title compound (40 gm).
With hydrogen;palladium 10% on activated carbon; In methanol; at 60℃; under 37503.8 Torr; for 40h;Product distribution / selectivity;
(S)-indoline-2-carboxylic acid (25 g) and 200 ml of methanol were hydrogenated in the presence of 2.5 g of 10 % Pd/C (dry basis) for 40 h at 60 C and at a pressure of 5 MPa. The catalyst was filtered off and the filtrate was concentrated by distilling off methanol. 50 ml of water were added to the residue and the mixture was heated to 70 C to obtain a clear solution. Then, at 70 C 140 ml of 1,2-dimethoxyethane were added dropwise to the clear solution. The mixture turned cloudy and was cooled spontaneously to room temperature. The precipitated solid was collected and dried to obtain 11 g of the title compound (optical rotation: -47.7). This solid was purified again to obtain 6 g of the title compound (optical rotation: -45.5). The same yield and optical rotation can be obtained by using 50 ml of water and 200 ml of THF at 65 C instead of water and 1,2-dimethoxyethane.
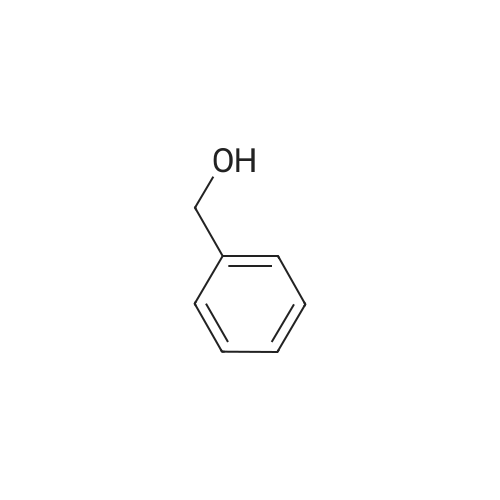
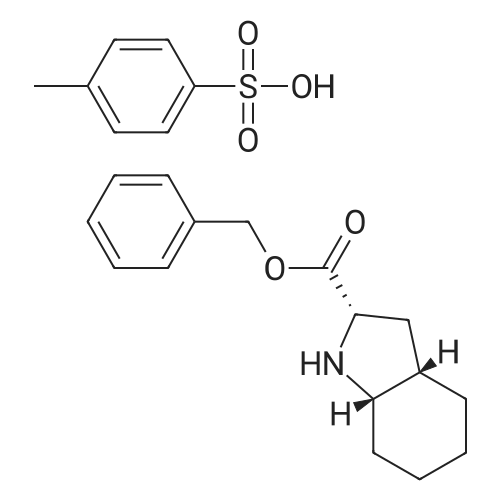
In a reactor, reflux 200 g of the compound of Example 1, 800 g of para-toluenesulphonic acid, 227 g of benzyl alcohol and 700 ml of toluene, the water formed being continuously removed with the aid of a separator. When no more water is separated off, cool, filter off the resulting precipitate under suction and dry. Benzyl (2S, 3aS, 7aS)-perhydroindole-2-carboxylate para-toluenesulphonate is obtained in that manner in a yield of 91% and an enantiomeric purity of 99%.
Compound 111 (20 g, 119 mmol) and K2C03 (23 g, 165 mmol) were dissolved in TIFF (60 mL) and H20 (60 mL) at room temperature. The reaction was stirred at room temperature for 5 minutes, the mixture was cooled to 0C and Cbz-CI (22 g, 131 mmol) was slowly added. The mixture was then stirred at room temperature for 2 hours. The mixture was acidified to Ph=3 and the product was extracted with dichloromethane (2 x 40 mL). The combined organic layer was concentrated, and the obtained residue dried in vacuo, resulting in compound 112 (34 g, 96 %)
69%
With sodium hydroxide; In tetrahydrofuran; water; at 20℃; for 4h;
Operation: NaOH (0.8 g, 20 mmol) was added to 50 ml of an aqueous solution at room temperature, cooled to room temperature,L-octahydroindole-2-carboxylic acid (1.69 g, 10 mmol) was added to the solution.A solution of CbzCl (2.05 g, 12 mmol) in 50 mL of THF was then slowly added dropwise and the reaction was maintained for 4 hours.Acidified to pH 3 with HC1 (2M) aqueous solution, extracted three times with ethyl acetate (3 x 100 ml)The organic layer was washed with saturated brine (50 ml), dried over anhydrous sodium sulfate, and dried. Using CHCl3: MeOH= 20:1 column to obtain 2.09g compound 17, yield 69%.
With N-ethyl-N,N-diisopropylamine; In water; N,N-dimethyl-formamide; at 0 - 20℃;
(2S,3aS,7aS)-Octahydro-indole-l,2-dicarboxylic acid 1-benzyl ester(2S,3aS,7aS)-Octahydro-indole-2-carboxylic acid (257.8 mg, 1.52 mmol) was dissolved in DMF (8 mL) and distilled water (8 mL). The solution was cooled to 00C, and DIPEA (400 muL, 2.30 mmol) was added followed by benzyl chloroformate (325 muL, 2.28 <n="101"/>mmol). The reaction was stirred at O0C and allowed to warm to ambient temperature overnight. The reaction was filtered and purified by reverse phase HPLC to give the desired product.
L-octahydroindole-2-carboxylic acid (1.69 g, 10 mmol) was added to the reaction flask,Add 30 mL of THF and 30 mL of distilled water to stir,A 7 mL aqueous solution of NaOH (0.68 g, 17 mmol) was added dropwise to the ice bath,Plus,The reaction was stirred for 10 min.(Boc) 2O (2.84 g, 13 mmol)Rose to room temperature for 12h.TLC detection reaction is completed, diluted with water, with methyl tert-butyl ether extraction impurity removal,The water phase with citric acid PH to acidic,Ethyl acetate was extracted three times, the organic phases were combined, washed with saturated brine, dried over Na2SO4,Filtration and concentration gave 3.21 g of compound 1 in 100% yield.
98%
With triethylamine; In tetrahydrofuran; water; at 20℃;
Triethylamine (8.25 mL, 59 mmol) was added to (S)-octahydroindole-2- carboxylic acid (10.0 g, 59 mmol) in a 1/1 volume/volume mixture of tetrahydrofuran/water (200 ML total), followed by addition of di-tert-butyl dicarbonate (14.0 g, 65 mmol). The reaction mixture was stirred overnight at room temperature, and then concentrated with no heating to remove tetrahydrofuran. The aqueous mixture was partitioned between ethyl acetate (300 mL), water (100 mL), and citric acid solution (50 ML, 10% aqueous). The layers were separated, the organic layer washed with brine (50 mL), dried (magnesium sulfate), filtered and concentrated to an oil, which solidified on standing. The solid was slurried in hexanes and collected by filtration. 15.63 g, 98%. MS (APCI-) 17 1/Z (%): 268 (100). Calcd for C14H23NO4 : C, 62.43 ; H, 8.61 ; N, 5.20 Found: C, 62.33 ; H, 8.30 ; N, 5.15
97%
With sodium hydroxide; In 1,4-dioxane; water; at 20℃; for 18h;
To a stirred solution of (2S, 3aS, 7AS)-OCTAHYDROINDOLE-2-CARBOXYLIC acid (30 g, 0.18 mol) in dioxane (180 ML) and 1 M NAOH (180 mL), was added di- TE7X-BUTYL DICARBONATE (48 G, 0. 22 mol). The reaction mixture was stirred for 18 hours at room temperature and then diluted with diethyl ether (500 mL) and water (500 mL). The resulting aqueous layer was removed and cooled to 0 C. Methylene chloride (1 L) was added, and the mixture adjusted to pH 3 by the addition of 2 M HC1. The organic layer was removed and the aqueous phase extracted with methylene chloride (2 x 25Q ML). The combined organic layers were dried over sodium sulfate, filtered and concentrated in vacuo to afford A colorless oil. Trituration with ether and hexanes followed by removal of the excess solvents afforded the desired (2S, 3aS, 7AS)-OCTAHYDROINDOLE-1, 2- dicarboxylic acid 1-TERT-BUTYL ester (4 6. 3 g, 97%), as a white solid : NNM (300 MHz, DMSO-d6) 8 1.00-1. 70 (m, 7H), 1.32, 1.38 (2 s, 9H), 1.76-1. 98 (m, 2H), 2.01-2. 13 (m, 1H), 2.18-2. 33 (m, 1H), 3.59-3. 69 (m, 1H), 3. 99-4. 10 (m, 1H); MS (CI) 170 [M + H-100 (Boc)] +, 270 [M + H] +.
94%
With triethylamine; In dichloromethane;
(a) To a solution of compound 5 in CH2Cl2 was added triethylamine and (Boc)2O. The reaction was monitored by TLC. After the starting materials were completely consumed, the mixture was diluted with CH2Cl2, washed sequentially with 1 N HCl and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to afford compound 6 as oil (94% yield).
51.52%
With triethylamine; In tetrahydrofuran; water; at 20℃; for 16h;
To a solution of compound 1 (50.00 g, 295.47 mmol) in THF (350 mL) and H20 (150.00 mL) was added TEA (89.70 g, 886.41 mmol, 122.88 mL), and then dropwise added (Boc)20 (77.38 g, 354.56 mmol, 81.45 mL) (a solution in 100 mL THF). The mixture was stirred at 20 C for 16 hr. TLC showed compound 1 was consumed and one new spot was detected. The resulting mixture was diluted with EtOAc (300 mL) and H20 (300 mL), and extracted with EtOAc (500 mL*3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give compound 2 (41.00 g, 5 1.52% yield) as a yellow oil. ?H NIVIR (400 MHz, CHLOROFORM-d) = 4.26 - 4.04 (m, 1H), 3.68 (br s, 1H), 3.03 (q, J=7.3 Hz, 2H), 2.31 -2.17 (m, 1H), 2.17- 1.89 (m, 3H), 1.68 - 1.53 (m, 2H), 1.51 - 1.32 (m, 11H), 1.27- 1.15 (m, 4H). TLC (Dichloromethane: Methanol=10:1)Rf= 0.1.
With sodium hydroxide; In tetrahydrofuran; water; at 0 - 20℃; for 12h;
To a stirred solution of (2R,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid (250 g, 1.0 equiv) (12) in THF (3 L) and water (1.5 L) at 0 C., was added dropwise a cooled aq solution of 2.5 M NaOH (1 L). The reaction mixture was stirred for 15 min at the same temperature. Then di-tert-butyl dicarbonate (1.3 equiv) was added dropwise, maintaining the temperature at 0 C. The resulting reaction mixture was stirred at rt for 12 h. The reaction mixture was washed with MTBE (3 times). The aq phase was acidified with 1 M aq citric acid and extracted with ethyl acetate (3 times). The combined organic layers were dried over sodium sulphate and concentrated to dryness to give (2R,3aS,7aS)-1-(tert-butoxycarbonyl)octahydro-1H-indole-2-carboxylic acid (368 g) (13).
368 g
To a stirred solution of (2R,3aS,7aS)-octahydro-lH-indole-2-carboxylic acid (250 g, 1.0 equiv) (1) in THF (3 L) and water (1.5 L) at 0 C was added dropwise a cooled aqueous solution of 2.5 M NaOH (1 L). The reaction mixture was stirred for 15 minutes at the same temperature. Di-tert-butyl dicarbonate (1.3 equiv) was added dropwise, maintaining the temperature at 0 C. The resulting reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was washed with MTBE (3 times). The aqueous phase was acidified with IM aqueous citric acid and extracted with ethyl acetate (3 times). The combined organic layers were dried over sodium sulfate and concentrated to dryness to afford (2R,3a,S',7a)S)-l-(tert-butoxycarbonyl)octahydro-lH- indole-2-carboxylic acid (368 g) (2).
Example 4 Preparation of (2S,3aS,7a5)-octahydroindole-2-carboxylic acid p-nitrobenzylic ester p-toluenesulfonate In a 1 L flask with mechanical stirring and equipped with a Dean-Stark apparatus were mixed 40 g of (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (0.236 mmole), 40 g of p-nitrobenzyl alcohol (~1.1 mol. eq.), 54 g of p-toluenesulfonic acid (1.2 mol. eq.) and 400 mL of toluene. The reaction was heated under reflux for 3 hours while water was removed by azeotropic distillation with a Dean-Stark apparatus. The reaction was cooled to room temperature then 400 ml of isopropanol were added and the solution was stirred at 0-5C for 1 hour. The solid was removed by filtration, rinsed with isopropanol and dried at 50C in vacuo for one night to give 89 g of ester (79%). NMR-1H: 8.28 (d, J = 8.6, 2H arom. APTS), 7.71 (d, J = 8, 2H arom. Bn), 7.68 (c, J = 8.6, 2H arom. APTS), 7.24 (d, J = 8, 2H arom. Bn), 5.44 (m, CH2 Bn), 4.59 (bt, J = 8.4, H), 3.73 (m, 1H), 2.53-2.45 (m, 2H), 2.37 (s, CH3 APTS), 2.28-2.17 (m, 1 H), 1.98-1.90 (m, 1 H), 1.76-1.36 (m, 7H).
With O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate; triethylamine; In DMF (N,N-dimethyl-formamide); acetonitrile; at 20℃; for 0.5h;
To 2,79 g of (S)-1- (N- (1- (ETHOXYCARBONYL)-3-PHENYLPROPYL)-L-ALANINE and 1,75 g of (2S, 3aR, 7AS)-OCTAHYDRO-1 H-INDOL-2-CARBOXYLIC acid in 100 mi of acetonitrile and 5 ml DMF first 2,9 ml of triethylamine and then with stirring 3,8 g O-(BENZOTRIAZOL-1- yl)-N, N, N', N'-tetramethyluronium HEXAFLUOROPHOSPHATE were and the stirring of the mixture was continued for 30 minutes at room temperature. Finally, 300 ml of saturated NACI solution was added and the mixture was extracted twice with 100 mi of ethyl acetate. After isolation of the product, which was done by the same way as in example 1,3, 96 g (92%) of trandolapril was obtained.
With sodium carbonate; In dichloromethane; for 16h;
To A stirred solution of (2S, 3aS, 7AS)-OCTAHYDROINDOLE-2-CARBOXYLIC acid (10.0 g, 59.1 mmol) and sodium carbonate (11.9 g, 112 mmol) in methylene chloride was added dropwise methyl chloroformate (5.0 ML, 65 mmol), at a rate sufficient to maintain a gentle reflux. The reaction mixture was stirred for 16 hours then diluted with methylene chloride (100 mL) and water (100 mL). The organic layer was removed and extracted with water (2 x 50 mL). The combined aqueous layers were then acidified to pH 3 using 3 M HCl and then extracted with methylene chloride (2 x 100 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated. The residue was then dissolved in thionyl chloride (25 mL), and the solution was heated to reflux for 45 minutes, cooled to room temperature and then co-evaporated with toluene (2 x 30 mL). The residue was dissolved in anhydrous methylene chloride and methanol (10 mL) was added. The mixture was stirred for 45 minutes and concentrated to dryness. The residue was dissolved in methylene chloride (50 mL) and washed successively with water (1 x 50 mL), saturated aqueous sodium bicarbonate solution (1 x 50 ML) and brine (1 x 50 mL), dried over sodium sulfate, filtered and concentrated. Purification by silica gel chromatography (eluant 80: 20 to 50: 50 hexanes/ethyl acetate) afforded the desired (2S 3aS, 7AS>OCTAHYDROINDOLE-1, 2- dicarboxylic acid dimethyl ester (9.1 g, 64%) as a light orange oil : 1H NMR (300 MHz, CDC13) 8 1.12-1. 74 (m, 8H), 1.93-2. 36 (m, 3H), 3.64, 3.70, 3.75 (3s, 6H), 3.71-3. 98 (m, 1H), 4.22-4. 34 (m, 1H) ; MS (ESI) FRALZ 242 [M+H] +.
To a stirred solution of (2S, 3aS, 7aS) -2, 3,3a, 4,5, 6,7, 7a-octahydroindole-2- carboxylic acid (4.0 g, 24 mmol) in water (50 mL), was added formalin (9.5 ML, 118 mmol). The mixture was stirred for 10 minutes and then added to a stirred solution of sodium borohydride (2.2 g, 59 mmol) in tetrahydrofuran (300 mL). After stirring at room temperature for 3 hours, the solvents were removed in vacuo. The residue was dissolved in methanol and then reduced in volume to approximately 15 mL. Silica gel chromatography (eluant 80 : 20 methylene chloride/methanol) afforded the desired product (4.15 g, 96%) as a gummy solid. The product was dissolved in ethyl acetate, and 2 M HCL/ETHER (13 ML) was added dropwise. After stirring and sonicating the mixture for 5 hours, the resulting precipitate was filtered to afford the desired (2S, 3aS, 7aS)-1-methyl- 2,3, 3a, 4,5, 6,7, 7a-octahydroindole-2-carboxylic acid hydrochloride (4.5 g, 90%) as a white solid: mp 177-180 C ; [OC] 25D-17. 7 (c 1.02, Methanol); 1H NMR (300 MHz, D20) 8 1.15-1. 98 (m, 9H), 2.39-2. 48 (M, 2H), 2. 82 (s, 3H), 3.42-3. 52 (m, 1H), 4.04 (dd, J = 10.0, 6.9 Hz, 1H) ; 13C NMR (75 MHZ, CD3OD) 6 23.0, 23. 3, 25.6, 27.6, 34.2, 38.0, 44.2, 71.0, 72.2, 168.0 ; MS (ESI) m/z 184 [M+H] +. Anal. Calcd. FOR CLOHL7NO2-HCL-0. L5 H2O : C, 54.00 ; H, 8. 29; N, 6.30 ; CL, 15.94. Found: C, 54.25 ; H, 8.37 ; N, 6.21 ; CL, 15.72.
With hydrogenchloride; In methanol; at 20℃; for 48h;
Anhydrous hydrogen chloride gas was bubbled into a mixture of (2S, 3aS, 7aS) -2, 3,3a, 4,5, 6,7, 7a-octahydroindole-2-carboxylic acid (17.14 g, 101 mmol) and methanol (500 ML) for about 10 minutes. The flask was stoppered and the mixture stirred for 2 days at room temperature. The solution was filtered and the solution concentrated to a solid that was used directly in the next step.
With thionyl chloride; In methanol; at 0 - 20℃; for 3.5h;
To a solution of 1 (5.10 g, 30.1 mmol, 1.00 eq.) in MeOH (30 mL) was slowly added thionyl chloride (4.4 mL, 60.2 mmol, 2.00 eq.) at 0 C over 30 mm. The mixture was stirred at room temperature for 3 h, then concentrated under reduced pressure. The residue was dissolved in DCM (50 mL) and added Et3N (12.5 mL, 90.3 mmol, 3.00 eq.) at 0 C. To the mixture, trityl chloride (8.40 g, 30.1 mmol, 1.00 eq.) was added at 0 C, and the mixture warmed to room temperature and continued stirring for 16 h. The mixture was filtered and washed with DCM (50 mL). Filtrate was washed with saturated NaHCO3 (50 mL*2), and back-extracted with DCM (30 mL*1). The combined organic layers were dried over Na2SO4, filtered, and concentrated to obtain compound 2 (13.73 g, crude, pale brown foam), which was used directly without further purification.
EXAMPLE 1; Step I: Preparation of silylated <strong>[80875-98-5](2S,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid</strong>; Trimethyl chlorosilane (10.86 g, 0.1M) was added to a mixture of octahydroindole-1H-2-carboxylic acid (15.2 g, 0.09M), anhydrous methylene chloride (300 ml) and triethylamine (10.1 g, 0.1M) and the mixture was stirred for 2 hours until silylated octahydro-1H-indole-2-carboxylic acid was formed.
With triethylamine; In dichloromethane; at 20℃; for 1.25h;Product distribution / selectivity;
Example 1; Preparation of perindopril erbumine; Stage I : Preparation of silylated (2S, 3aS, 7aS)-octahydroindole-2-carboxylic acid; To a suspension of 2.0 g (0.0077 mole) (2S, 3aS, 7aS)-octahydroindole-2- carboxylic acid in 24 ml of methylene chloride at room temperature are added 0.98 ml of trimethylchlorosilane (0.0077 mole) and 1.08 ml (0.0077 mole) of triethylamine under stirring. The mixture is stirred at room temperature for 75 minutes.
With 1H-imidazole; In dichloromethane; at 10 - 15℃; for 130h;
Step A Into a 4-neck round bottom flask, methylene chloride (50 ml) and (2S, 3aS, 7aS)-2-carboxyperhydroindole (5 g) were added and cooled to a temperature of about 10 C. Next, trimethyl silyl chloride (3.21 g) was added dropwise at a temperature below 10 C. While stirring for 10 minutes, imidazole (2 g) was added at a temperature below 10 C. The reaction mass was then stirred for about 2 hours at a temperature ranging from about 10 C. to about 15 C. to yield a trimethyl silyl ester of (2S, 3aS, 7aS)-2-carboxyperhydroindole.
With 1H-imidazole; In dichloromethane; at 10 - 15℃; for 2.16667h;
EXAMPLE 4; Preparation of Perindopril tert-butyl amine salt; Into a 4 necked RB flask, methylene chloride (50 ml) and (2S,3aS,7aS)-2-carboxyperhydroindole (5 gm) from Example 1 were added and cooled to about 10 C. Next, trimethyl silyl chloride (3.21 gm) was added dropwise while maintaining the temperature below 10 C. and stirred for 10 minutes. Imidazole (2 gm) was added maintaining the temperature below 10 C. The reaction mass was then stirred for about 2 hours at a temperature of about 10-15 C. to provide the trimethyl silyl ester of (2S,3aS,7aS)-2-carboxyperhyd Into another 4 necked RB flask, methylene chloride (100 ml) and N-1(S)-carboxyethylbutyl-(S)-alanyl chloride (8.85 gm) were added and cooled to about 10 C. Imidazole (8.04 gm) was added at below 10 C. and maintained for 1 hour at 10-15 C. The trimethyl silyl ester of (2S,3aS,7aS)-2-carboxyperhydroindole was then added dropwise slowly in 1 hour at 10 C. The reaction mixture was stirred for 2 hours at 10 C.
(2S,3aS,7aS)-octahydroindole-2-carboxylic acid-4-chlorophenylmethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2S,3aS,7aS-Octahydroindole-2-carboxylic acid (50 gm) (0.3 mole), 4-chlorobenzyl alcohol (46.37 gm) (0.33 mol), p-toluene sulfonic acid (67.52 gm) (0.35 mol), and toluene (400 ml) were heated to reflux temperature and water removed azeotropically. The contents were cooled and water (100ml) was added and stirred for 15 minutes. The lower aqueous layer was discarded and the toluene was distilled under vacuum at 60C to get an oil. Diisopropyl ether (200 ml) was added to the residue and stirred at room temp. The solids were filtered and the wet cake was added to dichloromethane (500 ml) and aqueous ammonia (60 ml) was added dropwise under stirring. The aqueous phase was separated and the organic phase washed with water till the washings were neutral. Dichloromethane was concentrated at 50C under vacuum to get oil (37.2 gm) which solidifies on standing.
(2S,3aS,7aS)-1-[(2R)-2-bromo-propionyl]-octahydro-1H-indole-2-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
EXAMPLE 2 (2S,3aS,7aS)-1-{(2S)-2-[(1S)-1-(Ethoxycarbonyl)butylamino]-propionyl}octahydro-1H-indole-2-carboxylic acid tert-butylamine salt Step A: (2S,3aS,7aS)-1-[(2R)-2-Bromopropionyl]octahydro-1H-indole-2-carboxylic acid Introduce into a reactor 200 g of <strong>[80875-98-5](2S,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid</strong>, 75 ml of water and 150 ml of toluene, and then bring the mixture to from 0 to 5 C. and add 250 ml of 5M sodium hydroxide solution, followed by a solution of 202 g of (2R)-2-bromopropionyl chloride in toluene, while maintaining the temperature below 10 C. and maintaining the pH of the mixture at 10 by adding 5M sodium hydroxide solution. After stirring for a further 1 hour at 10 C., add concentrated hydrochloric acid to adjust the pH of the mixture to 6. Separate off the toluene phase, and then add concentrated hydrochloric acid to the aqueous phase to adjust the pH to 2. The precipitate formed is then filtered off and dried to yield (2S,3aS,7aS)-1-[(2R)-2-bromopropionyl]octahydro-1H-indole-2-carboxylic acid.
With hydrogen; acetic acid;platinum on carbon; at 20℃; under 3750.38 Torr;
Introduce 200 g of the compound obtained in the previous Step, dissolved in acetic acid, and then 5 g of Pt/C 10% into a hydrogenator. Hydrogenate under a pressure of 5 bars at ambient temperature until the theoretical amount of hydrogen has been absorbed. Remove the catalyst by filtration and then cool to a temperature of from 0 to 5 C. and collect the resulting solid by filtration. Wash the filter cake and dry it to constant weight. (2S, 3aS, 7aS)-Perhydroindole-2-carboxylic acid is obtained in that manner in a yield of 87% and with an enantiomeric purity of 99%.
With 1H-imidazole; In dichloromethane; for 3h;Heating / reflux;
To a slurry of (2S, 3aS, 7aS)-2-carboxyperhydroindole (25 g) in methylene chloride (250 ml) hexamethyldisilazane (24 g) and imidazole (0.5 g) were added. The reaction mixture was refluxed for 3 hours and then cooled to O0C. Triethylamine (30 g) was then added to the reaction mixture and it was further cooled to -250C. To this reaction mixture wet N-[(S)-l-carbethoxybutyl]-(S)-alanine chloride hydrochloride obtained above was added at -250C to O0C over a period of 5-10 minutes. After addition, the reaction mixture was stirred at the same temperature for 1 hour. The reaction mixture was washed with water and concentrated to get crude perindopril.
The (2S,3aS,7aS)benzyl-perhydroindole-2-carboxylate (70 g) was added to methanol (350 ml) and charged with a solution of sodium hydroxide (13.4 g) in water (50 ml). The reaction mixture was refluxed for about 2 hours. After completion of reaction as determined by TLC, the pH of the reaction mixture was adjusted to a range of from about 6 to about 7 with dilute hydrochloric acid. The methanol was concentrated and charged with ethanol (700 ml). The reaction mixture was heated to a temperature of about 70 C. and the inorganics were filtered off. The ethanol was concentrated and the product was isolated with acetone (700 ml). The product was dried at a temperature of about 60 C. to yield (2S,3aS,7aS)-perhydroindole-2-carboxylic acid (42 g).
With triethylamine; In dichloromethane; at 20 - 25℃; for 3h;
Trimethyl chlorosilane (29.3 ml, 0.22M) and trietylamine (30.7 ml, 0.22 M) were added to a suspension of (2aS,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid (18,6 g 0.11M) in 400 ml of dichloromethane. The mixture was stirred for 3 h at 20 to 25C.
Ethyl (S)-indoline-2-carboxylate hydrochloride (30 g, 0.13 M), 300 ml of ethanol and 3 g of 7.5 % Pd/C were charged under nitrogen into a hydrogenator. The nitrogen atmosphere was 3 times replaced with fresh nitrogen and 5 times with hydrogen. The reaction mixture was hydrogenated at 60C and 50 bar under stirring until the hydrogen had been consumed. The catalyst was filtered off and 28 ml of sodium hydroxide (30 %) were added to the filtrate. The mixture was stirred at 30C for 5 h. After hydrolysis, the pH was adjusted to 4.5 using 37% aqueous HCl. Sodium chloride was removed by filtration and the filtrate was concentrated under reduced pressure. 250 ml of ethanol were added to the residue, the mixture was heated to reflux and again sodium chloride was filtered off. The filtrate was concentrated under reduced pressure. 160 ml of tetrahydrofurane and 30 ml of water were added to the dry residue and the mixture was heated to reflux to obtain a clear solution. This solution was cooled to 10C and aged at this temperature for 2 h. The precipitated crystals were filtered off, washed with 10 ml of tetrahydrofurane and dried at 70C for 4 h. 9 g (yield: 40.4 %) of (2S,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid as white powder were obtained. This product was employed in the next step without any additional treatment.; (S)-indoline-2-carboxylic acid ethyl ester hydrochloride (43 g), prepared according to Vincent et al., supra, and 320 ml of ethanol were hydrogenated in the presence of 4.5 g of 7.5 % Pd/C (dry basis) for 24 h at 60 to 65 C and at a pressure of 5 MPa. The catalyst was filtered off and 40 ml of an aqueous sodium hydroxide solution (30 %) were added to the filtrate. The mixture was hydrolyzed at 35C for 5 h. After hydrolysis, the pH was adjusted to 4.5 to 4.8 using 37% aqueous hydrochloric acid. Sodium chloride was removed by filtration and the filtrate was concentrated by distilling off ethanol and water. 160 ml of 1,2-dimethoxyethane and 40 ml of water were added to the dry residue and the mixture was heated to reflux to obtain a clear solution. This solution was cooled for crystallizing. The precipitated solid was collected, dried and purified again using a mixture of 1,2-dimethoxyethan and water as described above to obtain the title compound (assay by titration: 99 %; optical rotation: -44.7).
(2S,3aS,7aS)-(octahydro-indol-2-yl)-methanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With lithium aluminium tetrahydride; In tetrahydrofuran; diethyl ether;Inert atmosphere;
To a solution of (25,3a5,7a5)-octahydro-1 H-indole-2-carboxylic acid 5 (1 .69 g, 10.0 mmol) in ether (50 mL) was added a solution of LiAIH4 in THE (2M, 7.5 mL, 15 mmol) under argon, and the mixture was stirred overnight. After completion of the reaction, the mixture was quenched with Na25041 0H20, andthe solids were filtered off and washed with ethyl acetate. The filtrate was dried over anhydrous Na2504 and evaporated to give 1 .23 g (79%) of the crude compound 6 as a colorless oil. 1H NMR (500 MHz, CDCI3 ppm): O3.70 (1 H, dd, J 11.0, 3.5 Hz), 3.60 (1 H, dd, J 11.0, 6.0 Hz), 3.50-3.40 (1 H, m), 3.24 (1 H, q, J6.0 Hz), 2.13-2.08(1H, m), 1.94-1.86 (1H, m), 1.75-1.65 (1H, m), 1.65-1.40(6H, m), 1.35-1.23(2H, m); ESI MS for C9H11NO calculated 155.2, observed [M÷H]÷ 156.1.
Example 2 Perindopril erbumineTo a suspension of (2S, 3aS, 7aS) -octahydro-lH-indole-2- carboxylic acid (1.86 g; 0.011 M) in dichloromethane (40 mL) , there were added trimethylchlorosilane (2.93 mL; 0.22 M) and triethylamine (3.07 mL; 0.022 M) and it was continued with stirring for 3 hours at 20-25 0C. After 3 hours to the suspension cooled to -10 0C a cooled solution of previously prepared activated N- ( (S) -1-carbetoxybutyl) - (S) -alanine was added, which had been prepared to the following manner: A solution of N- ( (S) -1-carbethoxybutyl) - (S) -alanine (2.17 g; 0.01 M) and imidazole (2,73 g; 0.04 M) in dichloromethane (40 mL) was cooled to -10 0C and thionyl chloride (0.76 mL; 0.0115 M) was added thereto and it was continued with stirring for 2 hours at 20-25 0C. After two hours imidazole hydrochloride was filtered off and the filtrate was cooled to -10 0C.After the completed addition of activated N- ((S)-I- carbetoxybutyl) - (S) -alanine, it was continued with stirring for 3 hours at -10 0C and then for another 18 hours at 20-25 0C. After completed reaction water (25 mL) was added thereto, the pH was adjusted to 5-6 with a 20 % NaOH solution, the layers were separated and the aqueous layer was extracted two more times with dichloromethane (15 mL) . The volatile components of the combined organic layers were evaporated, the oily residue was suspended in acetonitrile (75 mL) , the undissolved part was filtered off and t-butylamine (1.11 mL; 0.01 M) was added to the filtrate. The suspension was heated to the boiling point of the solution, the clear solution was cooled to 20-25 0C and the precipitated crystals were filtered off. After drying at 40-45 0C, 2.69 g of perindopril erbumine were obtained.
Stage #1: (2S,3aS,7aS)-perhydroindole-2-carboxylic acid With chloro-trimethyl-silane; triethylamine In dichloromethane at 20 - 25℃; for 2h;
Stage #2: N-[1(S)-ethoxycarbonyl-1-butyl]-(S)-alanyl chloride hydrochloride In dichloromethane at -15 - 0℃; for 2h;
Stage #3: With sodium hydroxide In dichloromethane; water
1
Example 12-methylpropane-2-amine salt of perindopril (2S, 3aS, 7aS) -1- ( (2S) -2- ( ( (IR) -1-ethoxycarbonyl) butyl) amino-1- oxopropyl) octahydro-lH-indole-2-carboxylic acid) or perindopril erbumineTo (2S, 3aS, 7aS) -octahydroindole-2-carboxylic acid (7.44 g) in dichloromethane (80 mL) at 20-25 0C, trimethylchlorosilane (5.84 mL) and triethylamine (6.4 mL) were added and it was stirred at 20-25 0C for two hours. After two hours triethylamine (5.54 mL) was added, the suspension was cooled to -15 0C, a suspension of N- ( (S) -1-carbetoxybutyl) -L- alanylchloride hydrochloride (11 g) in dichloromethane (80 mL) , cooled to -15 0C, was poured thereto and the stirring was continued at a temperature from 0 0C to -5 0C for two hours. The reaction solution was heated to 0 0C, the precipitated triethylamine hydrochloride was filtered off and washed with dichloromethane (20 mL) , water (66 mL) in which NaOH (1.6 g) had been dissolved was added to the filtrate and the pH was adjusted to 4.2 with a 20 % NaOH solution. The organic phase was separated and the aqueous layer was washed two more times with dichloromethane (40 mL) . The combined dichloromethane layers were evaporated, the residue was dissolved in isopropyl acetate (250 mL) , the undissolved part was filtered off, and t-butylamine (4.4 mL) was added to the filtrate. The precipitated crystals were dissolved at the boiling point of the solution, the clear solution was cooled to 10-20 0C and it was continued with stirring for two hours. After two hours, the precipitated crystals were filtered off, washed with isopropyl acetate (30 mL) and dried at 35-40 0C in an air dryer. 15.3 g of perindopril erbumine in alpha form with a purity of more than 99 % were obtained; the content of single impurities was not above 0.1 %.
Method-II; A mixture of (S, S, S) -Octahydroindole-2-carboxylic acid (1.5 g, 0.0088 moles) in acetonitrile (15 ml) was stirred for 5 min. at 25-30C. To this was added hexamethyl disilazane (1. 8 g, 0. 011 moles), followed by a few drops of chlorotrimethylsilane. The reaction mixture was heated to reflux for 3-3. 5 hrs. When the evolution of ammonia gas evolved during the reaction ceased, the reaction mixture was cooled to 25-30C. Dichloromethane (20 ml) was added and the mixture was further cooled to 0-5 C. Triethylamine (1.4 ml) was added to the cooled reaction mixture and stirred for 5-10 min and the mixture further cooled to-10 to-20 C. To the cooled reaction mixture was added N- [l (S)-ethoxycarbonyl butyl]- (S)-alanine-2'- benzothiazolylthiol ester (3.5 g, 0.0106 moles, obtained from Examples 1 and 2), slowly over a period of 1 hr. After the addition was over the temperature was raised to 25-30 C and the mixture stirred at this temperature for 15-16 hrs. The reaction mixture was then concentrated under reduced pressure at 30-35 C to give an oily residue. The oil was dissolved in a mixture of water (15 ml) and diisopropyl ether (50 ml). The solution was cooled to 0-5 C and pH of the solution adjusted to 8. 3-8. 6 using 10% aqueous sodium hydroxide solution. The reaction mixture was stirred at this pH for 15-20 mins, filtered and the organic layer was separated. The aqueous layer was again cooled to 0-5 C and the pH adjusted to 2.2-2. 5 using 6N hydrochloric acid. The aqueous solution was extracted with diisopropyl ether (25 ml X 2). The layers were separated and the aqueous layer was cooled to 0-5 C and the pH adjusted to 3.5-3. 8 using 10% aqueous sodium hydroxide solution. Then it was extracted with dichloromethane (50 ml X 3). The dichloromethane layer was concentrated under reduced pressure to give 1.8 g (55 %) of Perindopril as a viscous oil.
20%
Step II : Preparatiofz of Perindopril Method-I; To a suspension of (S, S, S) -Octahydroindole-2-carboxylic acid (1.5 g, 0.0088 moles) in dichloromethane (15 ml) at 25-30C was added triethylamine (1.34 ml, 0.0096 moles) and the mixture stirred for 5-10 mns to get a clear solution. The solution was cooled to- 10 to-15'C and to the cooled solution was added Nul (S) -ethoxycarbonyl butyl]- (S)- alanine-2'-benzothiazolylthiol ester (3.5 g, 0.0106 moles, obtained from Examples 1 and 2), slowly over a period of 1 hr. After the addition was over the temperature was raised to of 25-30 C and the reaction, mixture stirred at this temperature for 15-16 hrs. The reaction mixture was then concentrated under reduced pressure at 30-35 C, to give an oily residue. The oil was dissolved in a mixture of water (15 ml) and diisopropyl ether (50 ml). The solution was cooled to 0-5 C and pH of the solution adjusted to 8.3-8. 6 using 10% aqueous sodium hydroxide solution. The reaction mixture was stirred at this pH for 15-20 mins, filtered and the organic layer was separated. The aqueous layer was again cooled to 0-5 C and the pH adjusted to 2.2-2. 5 using 6N hydrochloric acid. The aqueous solution was extracted with diisopropyl ether (25 ml X 2). The layers were separated and the aqueous layer was cooled to 0-5 C and the pH adjusted to 3.5-3. 8 using 10% aqueous sodium hydroxide solution. Then it was extracted with dichloromethane (50 ml X 3). The dichloromethane layer was concentrated under reduced pressure to give: 0.4 g (20 %) of Perindopril as a viscous oil.
N-((S)-1-carbethoxybutyl)-L-alanyl chloride hydrochloride[ No CAS ]
[ 80875-98-5 ]
[ 75-64-9 ]
[ 107133-36-8 ]
Yield
Reaction Conditions
Operation in experiment
86%
Example 3 Preparation of perindopril erbumin from isopropyl acetate ((2S,3aS,7aS)-l-((2S)-2-(((lS)-l- (ethoxycarbonyl)butyl)amino)-1-oxopropyl)octahydro-1H- indol-2-carboxylic acid 2-methylpropane-2-amine salt) ; Trimethylchlorosilane (2.92 ml) and triethyl amine (3.2 ml) were added to (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (3.72 g) in dichloromethane (40 ml) at 20 - 25C and it was stirred at 20 - 25C for two hours. After two hours triethyl amine (2.77 ml) was added, the suspension was cooled to-15C, a suspension of N-((S)-l-carbethoxybutyl)- L-alanyl chloride hydrochloride (5.5 g) in dichloromethane (80 ml), which had been cooled to-15C, was poured thereto and the stirring was continued at-15C for two hours. The reaction solution was heated to 0C, the precipitated triethylamine hydrochloride was filtered off and washed with dichloromethane (10 ml), water (33 ml), wherein NaOH (0.8 g) had been dissolved, was added to the filtrate, and the pH was adjusted to 4.2 with a 20% NaOH solution. The organic phase was separated and the aqueous layer was washed with dichloromethane (2 x 20 ml). The combined dichloromethane layers were evaporated, the residue was dissolved in isopropyl acetate (125 ml), the undissolved part was filtered off and t-butylamine (2.2 ml) was added to the filtrate. The precipitated crystals were dissolved at the boiling point of the solution, the clear solution was cooled to 10 - 20C and the stirring was continued for two hours. After two hours the precipitated crystals were filtered off, washed with isopropyl acetate (15 ml) and dried at 35 - 40C in an air drier. Perindopril erbumin (7.65 g; 86%) in an a form having a purity over 99% was obtained with individual impurities present in not more than 0.1% each.
80%
Example 2 Preparation of perindopril erbumin ((2S,3aS,7aS)-1-((2S)-2- (((lS)-l-(ethoxycarbonyl)butyl)amino)-l- oxopropyl) octahydro-1H-indol-2-carboxylic acid 2- methylpropane-2-amine salt) ; Trimethylchlorosilane (2.86 ml) and triethyl amine (3.08 ml) were added to (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (3.72 g) in dichloromethane (60 ml) at 20 - 25C and stirred at 20 - 25C for two hours. After two hours triethyl amine (2.8 ml) was added, the suspension was cooled to -15C, a suspension of N-((S)-l-carbethoxybutyl)- L-alanyl chloride hydrochloride (5.5 g) in dichloromethane (60 ml), which has been cooled to-15C, was poured thereto and the stirring was continued at-15C for two hours. The reaction solution was heated to 0C, brine (25 ml) with dissolved NaOH (0.8 g) was added, and the pH was adjusted to 4.2 with a 20% NaOH solution. The organic phase was separated and the aqueous layer was once more washed with dichloromethane (20 ml). The combined dichloromethane layers were evaporated, the residue was dissolved in ethyl acetate (100 ml), the undissolved part was filtered off and t-butylamine (2.2 ml) was added to the filtrate. The precipitated crystals were dissolved at the boiling point of the solution, the clear solution was cooled to 10 - 20C and the stirring was continued for two hours. After two hours the precipitated crystals were filtered off, washed ,with ethyl acetate (12 ml) and dried at 35 - 40C in an air drier. Perindopril. erbumin (7.1 g; 80%) in an a form having a purity over 99% was obtained with individual impurities present in not more than 0.1% each.
78%
Example Preparation of perindopril erbumin from N,N- dimethylformamide ((2S,3aS,7aS)-l-((2S)-2-(((lS)-l- (ethoxycarbonyl)butyl)amino)-l-oxopropyl)octahydro-lH- indol-2-carboxylic acid 2-methylpropane-2-amine salt) ; Trimethylchlorosilane (2.92 ml) and triethyl amine (3.2 ml) were added to (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (3.77 g) in dichloromethane (40 ml) at 20 - 25C and stirred at 20 - 25C for two hours. After two hours triethyl amine (2.77 ml) was added, the suspension was cooled to-15C, a suspension of N-((S)-l-carbethoxybutyl)- L-alanyl chloride hydrochloride (5.5 g) in dichloromethane (40 ml), which had been cooled to-15C, was poured thereto and the stirring was continued at-15C for two hours. The reaction solution was heated to 0C, the precipitated triethylamine hydrochloride was filtered off and washed with dichloromethane (10 ml), water (33 ml), wherein NaOH (0.8 g) had been dissolved, was added to the filtrate, and the pH was adjusted to 4.2 with a 20% NaOH solution. The organic phase was separated and the aqueous layer was washed with dichloromethane (2 x 20 ml). The combined dichloromethane layers were evaporated, the residue was dissolved in N, N-dimethylformamide (90 ml),. the undissolved part was filtered off and t-butylamine ,(2.2 ml) was added to the filtrate. The precipitated crystals were dissolved at 80C, the clear solution was cooled to 10 - 20C and the stirring was continued for two hours. After two hours the precipitated crystals were filtered off, washed with N,N- dimethylformamide (9 ml) and dried at 35 - 40C in an air drier. Perindopril erbumin (6.9 g; 78%) in an a form having a purity over 99% was obtained with individual impurities present in not more than 0.1% each.
EXAMPLE 2 Preparation of perindopril erbumine Into a 4-neck round bottom flask, methylene chloride (2000 ml) and N-1(S)-carboxyethylbutyl-(S)-alanyl chloride hydrochloride (177 g) were added and cooled to a temperature of about -5 C. To this mixture was added imidazole (161 g) and the temperature was maintained for about 1 hour at a temperature below about 0 C. Next, (2S, 3aS, 7aS)-2-carboxyperhydroindole (100 g) was added slowly in about 45 minutes. The reaction mass was then stirred for about 2 hours at a temperature of about -5 to 0 C. The temperature was raised to a temperature ranging from about 20 C. to about 25 C. and maintained for 2 hours. A mixture of acetic acid (106 g) in water (1 L) was added while the temperature was kept below about 5 C. and stirred for 30 minutes. The methylene chloride layer was separated and then washed with saturated brine solution (200 ml) and dried over sodium sulfate (10 g). The methylene chloride layer was charged in a round bottom flask and cooled to a temperature of about 10 C. Next, tert-butyl amine (65 ml) was charged in 30 minutes at a temperature below 10 C. and stirred further for 30 minutes at 35-40 C. The methylene chloride was then distilled off completely. A mixture of isopropyl alcohol (300 ml), acetone (600 ml) and acetonitrile (600 ml) was charged and added to the separated methylene chloride. The mixture was heated to a temperature ranging from about 65 C. to about 70 C. to get a clear solution. The reaction mass was cooled very slowly to a temperature of about 25 C. in 2 hours and then further cooled to a temperature ranging from about 5 C. to about 10 C. and filtered. The filtered material was then dried under vacuum at a temperature of about 40 C. (Weight: 124 gm, purity: >99.5% by HPLC). Specific optical rotation [alpha]n=-66 (C=1%, Methanol), IR (KBr) spectrum shows the following absorptions cm-1 3300, 2930, 1744, 1732 m 1644 m 1568. The 1H-NMR (CDCI3) shows the following signals at delta 4.28-4.12 (m, 1H), 4.18-4.09 (q,2H), 3.76 (m,2H) 3.53 (q, 1H), 3.1 (t, 1H), 2.32-2.14 (m, 2H), 2.01 (m, 1H), 1.75-1.62 (m, 4H), 1.32 (m, 2H), 1.30 (S, 9H), 1.28 (t, 3H), 0.88 (t, 3H). C.I. Mass shows m/z at 368 (base peak.) XRD matches with alpha-polymorph.
EXAMPLE 3 General procedure for production of perindopril (free acid) starting from (2S, 3aS, 7aS)-2-carboxy-perhydroindole and N-[1-(S)-ethoxy-carbonyl-butyl]-(S)-alanine by activation with N,N'-carbonyl-diimidazole N-[1-(S)-ethoxycarbonyl-butyl]-(S)-alanine (65 g, 1 eq.) and N,N'-carbonyldiimidazole (48 g, 1 eq.) are introduced and then acetonitrile (500 g) is added. The reaction mixture is then stirred at a temperature less than +10 C. for 3 hours. The reaction mixture is poured into (2S, 3aS, 7aS)-2-carboxyperhydroindole (50 g, 1 eq.); an amount of fresh acetonitrile (80 g) is used to rinse the apparatus. The reaction mixture is then stirred for 5 hours at a temperature less than +10 C. and is then clarified over a filter to obtain a clear solution.
EXAMPLE 6 L-arginine salt of perindopril-starting from (2S, 3aS, 7aS)-2-carboxy-perhydroindole and triphosgene [0055] N-[1-(S)-ethoxycarbonyl-butyl]-(S)-alanine (20 g, 1 eq.) and Na2HPO4.12 H2O (43 g, 1.3 eq.) are suspended in dichloromethane (212 g). The reaction mixture is heated to reflux and then a solution of triphosgene (9.55 g, 0.35 eq.) in dichloromethane (64 g) is poured in. After liquid/liquid washings of the organic phase with water, the dichloromethane is evaporated off to yield activated N-[1-(S)-ethoxycarbonyl-butyl]S)-alanine of formula (III) (22 g). The latter is then dissolved in acetonitrile (180 g). The solution is poured into (2S, 3aS, 7aS)-2-carboxyperhydroindole (15 g, 1 eq.) and the reaction mixture is stirred for about 5 hours in the presence of triethylamine (9.15 g, 1 eq.) at a temperature less than 10 C., and it is then clarified through a filter in order to obtain a clear solution. The L-arginine salt of perindopril is obtained by adding L-arginine (14.5 g, 0.90 eq.) suspended in DMSO (180 g) at a temperature of 50 C. After a contact time of about 5 hours, the mixture is seeded with 2% of the delta form and is then stirred overnight at 50 C. and filtered using a filtration cell. [0056] Perindopril L-arginine (43 g) is obtained in a yield of 89% relative to the (2S, 3aS, 7aS)-2-carboxyperhydroindole (seed subtracted). The HPLC quality of the isolated product is greater than 99%. [0057] The filtration rate of the mother liquors is about 2000 kg/h/m2.
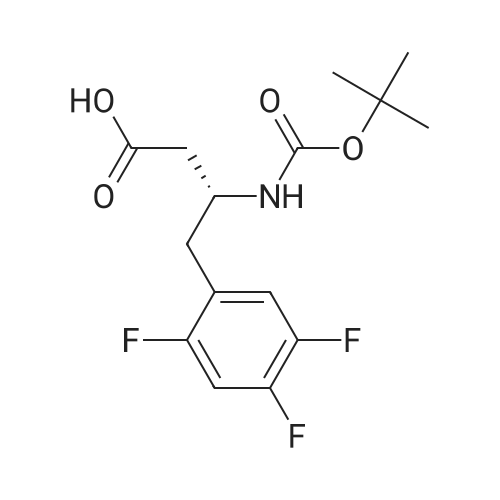
General procedure: (d) To a solution of (R)-3-(tert-butoxycarbonylamino)-4-(2,4,5-trifluorophenyl)butanoic acid 9 in CH2Cl2 was added HOBt, EDCI, DMAP, and triethylamine. After stirring for 30min 8a was added. The reaction mixture was stirred overnight. The reaction was monitored by TLC. After the starting materials were completely consumed, the mixture was diluted with CH2Cl2, washed sequentially with 1N HCl and brine. The organic layer was dried over Na2SO4, filtered, and evaporated. The residue was purified by a flash column chromatography (petroleum ether/ethyl acetate=4:1, v/v, as an eluent) to afford compound 10a as a white solid (72% yield).
5 mL of dichloromethane was added sequentially to a 1 L three-necked flask,(2S) -hydrohydroindole-2-carboxylic acid (0.2 mol, 33.8 g)And triethylamine (0.17 mol, 17.2 g)Mechanical stirring, nitrogen protection.Trimethylchlorosilane (0.21 mol, 22.8 g) was added,Stir at room temperature for 2 hours. Ice bath cooling to -5 ~ 5 , and then,A solution of N - [(S) -1-butyryl chloride] -S-pentamethyl ethyl ester in dichloromethane (0.21 mol, 63 g / 250 mL) was slowly added dropwise.After completion of the drop, the reaction was carried out under ice bath for 4 hours.HPLC monitoring to(2S) -hydro-indole-2-carboxylic acid content of less than 1%The reaction ends. filter,Take the filtrate with 10% sodium hydroxide solution to adjust the pH to 6,Layered.The aqueous phase was extracted twice with 100 mL of dichloromethane,Combined organic phase,Dried over anhydrous sodium sulfate,filter.The filtrate was concentrated under reduced pressure,To obtain a red oil,For perindopril.Add ethyl acetate to dissolve,Tert-butylamine (0.35 mol, 26.0 g) was slowly added dropwise,Precipitate a white solid. Stirred for 30 minutes, allowed to stand at room temperature for 3 hours,filter,Washed with ethyl acetate to give 95.0 g of crude perindopril tert-butylamine salt,Yield 78.8%, purity 98%, ee value 99%.
5-methoxy-1'-methyl-2'-(1-methyl-1H-indol-3-yl)-1'-nitro-1',2',4a',5',6',7',8',8a',9',9a'-decahydrospiro[indoline-3,3'-pyrrolo[1,2-a]indol]-2-one[ No CAS ]
5-fluoro-1'-methyl-2'-(1-methyl-1H-indol-3-yl)-1'-nitro-1',2',4a',5',6',7',8',8a',9',9a'-decahydrospiro[indoline-3,3'-pyrrolo[1,2-a]indol]-2-one[ No CAS ]
5-chloro-1'-methyl-2'-(1-methyl-1H-indol-3-yl)-1'-nitro-1',2',4a',5',6',7',8',8a',9',9a'-decahydrospiro[indoline-3,3'-pyrrolo[1,2-a]indol]-2-one[ No CAS ]
5-fluoro-2'-(1-methyl-2-phenyl-1H-indol-3-yl)-1'-nitro-1',2',4a',5',6',7',8',8a',9',9a'-decahydrospiro[indoline-3,3'-pyrrolo[1,2-a]indol]-2-one[ No CAS ]
To a test tube equipped with magnetic stirring, containing a mixture of CBD (79 mg, 0.251mmol, 3 eq.) and (2S,3aS,7aS)-octahydro-1 H-indole-2-carboxylic acid (14 mg, 0.084mmol), was added heptane (0.55 mL). The resulting mixture was stirred at roomtemperature overnight. The suspension was filtered through a sinter funnel (porosity n3) and washed with 3 x 0.08 mL of heptane. After drying under vacuum at room temperature, cocrystal Form VI (possibly contaminated with (2S,3aS,7aS)-octahyd ro-1 H-indole-2-carboxylic acid) of the present invention was obtained as a white solid (25.2 mg, 48%).
48%
In n-heptane; at 20℃;
To a test tube equipped with magnetic stirring, containing a mixture of CBD (79 mg, 0.251 mmol, 3 eq.) and (2S,3aS,7aS)-octahydro-1 H-indole-2-carboxylic acid (14 mg, 0.084 mmol), was added heptane (0.55 ml_). The resulting mixture was stirred at room temperature overnight. The suspension was filtered through a sinter funnel (porosity n3) and washed with 3 x 0.08 ml. of heptane. After drying under vacuum at room temperature, cocrystal Form VI (possibly contaminated with (2S,3aS,7aS)-octahydro-1 /-/-indole-2- carboxylic acid) of the present invention was obtained as a white solid (25.2 mg, 48%). The cocrystal Form VI thus obtained shows an X-ray powder diffractogram (XRPD) as in Fig. 12, a DSC as in Fig. 13. The cocrystal Form VI thus obtained also shows the 1H NMR disclosed below.
Stage #1: (2S,3aS,7aS)-perhydroindole-2-carboxylic acid With chloro-trimethyl-silane; triethylamine In 2-methyltetrahydrofuran at -5 - 5℃; Inert atmosphere;
Stage #2: N-[(S)-1-butyryl chloride]-S-pentanoic acid ethyl ester In 2-methyltetrahydrofuran at 0 - 5℃; Inert atmosphere;
Stage #3: <i>tert</i>-butylamine at 60 - 65℃;
5 Example 5
at room temperature,In a 2L four-necked flask, 600 mL of 2-methyltetrahydrofuran was added successively,67.6g of compound II and 35g of triethylamine were stirred and protected by nitrogen gas.Further, 50 g of trimethylchlorosilane was added, and stirred at room temperature for 2 hours.Cool down to -5-5°C in an ice bath, then,Slowly add N-[(S)-1-butyryl chloride]-S-pentanoic acid ethyl ester dropwise2-Methyltetrahydrofuran solution (126g/500mL). After the dropwise addition, react at 0-5°C for 4 hours. HPLC monitors that the compound II content is less than 1%, and the reaction ends. After filtering, the filtrate was concentrated to dryness, and 600 ml of dichloromethane was added to the residue, and the pH value was adjusted to 6 with 5% ammonia solution, and the layers were separated. The aqueous phase was extracted twice with 100 mL of dichloromethane, and the organic phases were combined, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to obtain a red oily substance, which was perindopril. Add 400ml of methyl isobutyl ketone to dissolve, slowly add 51.0g of tert-butylamine dropwise, and a white solid precipitates out. Raise the temperature to 60-65 and stir for 30 minutes,Slowly lower the temperature to 10-15° C. for crystallization for 3 hours, filter, and wash with methyl isobutyl ketone to obtain 209.8 g of crude perindopril tert-butylamine salt, with a yield of 87.1%, a purity of 99.6%, and an ee value of 99.7%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping