* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 14, p. 2744 - 2748
4
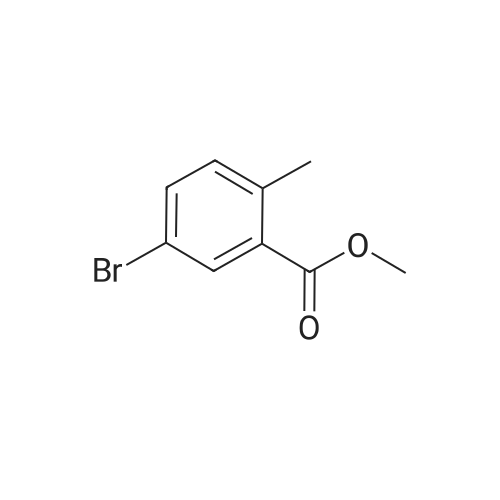
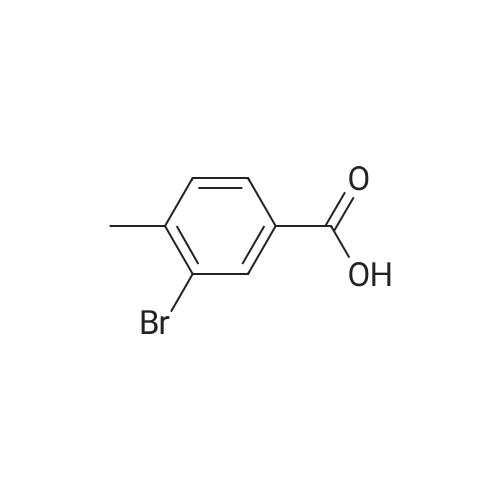
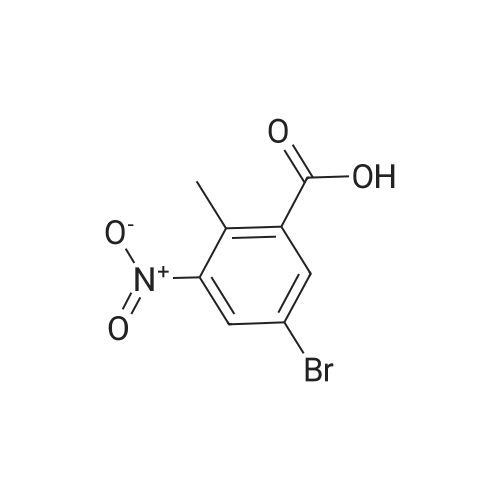
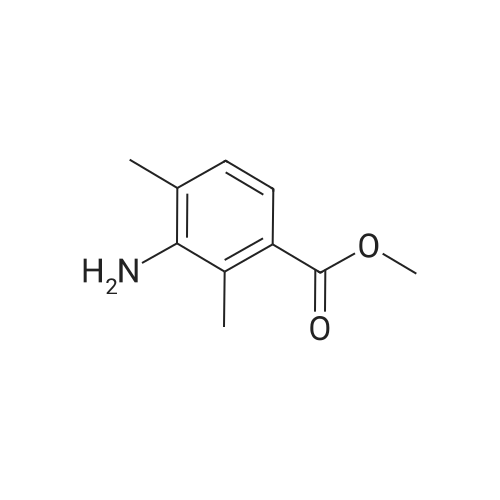
[ 76006-33-2 ]
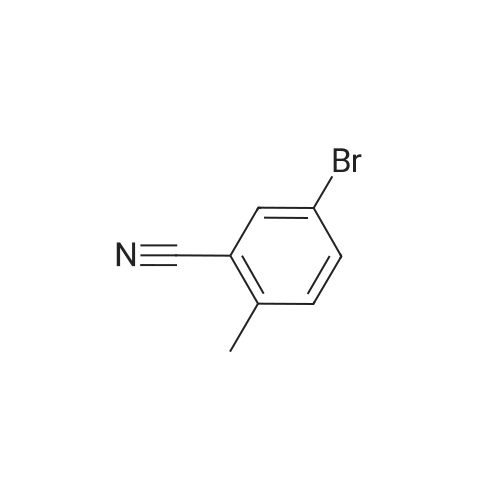
[ 79669-49-1 ]
[ 74-88-4 ]
[ 99548-54-6 ]
[ 79669-50-4 ]
Yield
Reaction Conditions
Operation in experiment
29%
With potassium carbonate In DMF (N,N-dimethyl-formamide) at 20℃; for 2 h;
A 60: 40 mixture of 5-bromo-2-methyl benzoic acid and 3-bromo-2-methyl benzoic acid (8.0 g, 0.037 mol) was dissolved in N,N'-dimethylformamide (130 mL). Methyl iodide (2.28 mL, 2.3 mol) and potassium carbonate (5.11 g, 0.037 mol) were added in sequence at room temperature. The mixture was stirred at room temperature for 2 hours at which point the reaction was determined to be complete by HPLC. The solvent was removed under high vacuum and the resulting residue was passed through a silica gel column using 5percent ethyl acetate in hexanes as the eluent. The mixture of isomers was obtained as an oil and then separated by preparative normal phase HPLC using 0.5percent isopropyl alcohol in hexanes as the eluent. The title compound was obtained as a white solid (1.38 g, 29percent). 1H NMR (300.132 MHz, CDC13) 8 8.04 (d, J= 2.2 Hz, 1H), 7.50 (dd, J= 8.2, 2.2 Hz, 1H), 7.12 (d, J= 8.2 Hz, 1H), 3.89 (s, 3H), 2.54 (s, 3H).
Stage #1: With diazomethyl-trimethyl-silane In hexane at 20℃; for 1.16667 h; Stage #2: With acetic acid In hexane for 0.75 h;
Place 5-bromo-2-methyl-benzoic acid (1.0 g, 4.7 mmol) in a 200 mL flask under an N2 atmosphere and add methanol via syringe. Add a 2M solution of diazomethyl- trimethyl-silane in hexane (3.5 mL, 23.0 mmol) drop wise over 10 minutes and stir for 1 hour at room temperature. Add glacial acetic acid (16 mL) and stir for 45 minutes. Dilute with ethyl acetate (100 mL) and wash with 1M aqueous sodium hydroxide solution (30 mL), saturated aqueous sodium bicarbonate solution (30 mL) and brine (30 mL). Dry the organic layer (Na2SO4), filter and concentrate in vacuo to obtain 1.01 g of 5-bromo-2- methyl-benzoic acid methyl ester (99 percent). Place 5-bromo-2-methyl-benzoic acid methyl ester (1.04 g, 4.5 mmol) in a 50 mL flask under a N2 atmosphere and add carbon tetrachloride (15 mL). Add N-bromo- succinamide (1.49 g, 8.3 mmol) and 2,2'-azobisisobutyronitrile (40 mg, 0.2 mmol) and fit flask with a condenser and reflux for 4 hours. Cool to room temperature and filter. Concentrate the filtrate and pre-adsorb the crude product onto silica gel. Chromatograph the residue on a Si02 column eluting with dichloromethane in hexane (0 to 50percent) to obtain 977 mg of 5-bromo-2-bromomethyl-benzoic acid methyl ester (70percent). Using 5-bromo-2-bromomethyl-benzoic acid methyl ester (0. 984 g, 3.20 mmol) and the procedure described in the I st paragraph for the alternative procedure for 5- (4,4, 5, 5-tetramethyl- [1, 3,2] dioxaborolan-2-yl) -2, 3-dihydro-isoindol-1-one, prepare 509 mg of the title compound (75 percent).
98%
Stage #1: at 0℃; for 3 h; Reflux; Inert atmosphere
Thionyl chloride (10 mL) was added to 5-bromo-2-methyl-benzoic acid (2.0 g, 9.3 mmol) at 0 °C and then DMF (one drop) was added. The mixture was heated at reflux for 3 h under nitrogen. The reaction mixture was evaporated and dry MeOH (5 mL) was added to the residue. The mixture was concentrated, and EtOAc was added. The mixture was washed with saturated aqueous NaHC03 solution and brine. The organic phase was dried (Na2S04), filtered, and evaporated to give 5-bromo-2-methyl-benzoic acid methyl ester (2.1 g, 98percent) as an off-white solid.
97%
at 20℃; for 5 h; Cooling with ice; Reflux
(1) Dissolve 300 g of 5-bromo-2-methylbenzoic acid in 2 L of methanol.Concentrated sulfuric acid 50 ml was added dropwise in an ice bath.Stir for 1 hour at room temperature and then heat to reflux for 4 hours. The solvent was concentrated and the residue was dissolved in 2 liters of dichloromethane.Wash three times with saturated sodium bicarbonate,It was dried and concentrated to give methyl 5-bromo-2-methylbenzoate (310 g, yield 97percent).
95.2%
at 0 - 20℃;
Example 2:; 5-Bromo-2-methyl benzoic acid methyl ester; The compound of example 1 (20.3 g, 0.0944 mol) was dissolved in 150 mL methanol and cooled to 0 °C. To this reaction mixture, thionyl chloride (28.07 g, 0.236 mol) was added slowly within 15-20 min. The reaction mixture was stirred at room temperature for 2- 3 h. After completion of the reaction, MeOH was removed under vacuum. The oily material obtained was dissolved in ethyl acetate and washed with sodium bicarbonate, water and brine and dried over anhydrous sodium sulfate. The organic layer was concentrated to obtain the title compound.Yield: 20.5 g (95.2 percent)Ref:-US2007/0213342 A1
95%
for 2 h; Reflux
A mixture of 5-bromo-2-methylbenzoic acid (1.00 g, 4.7 mmol) and thionyl chloride (1.40 g,11.6 mmol) in methanol (30 ml) was refluxed for 2 h. The solvent was removed underreduced pressure and the pH of the residue was adjusted to 7. The resultant was extractedwith ethyl acetate. The organic phase was dried over anhydrous Na2S04, filtered andconcentrated to give CAP-004-19-1 (1.02 g, 95 percent) as a pale yellow solid.
94%
With hydrogenchloride In 1,4-dioxane at 70℃;
(a) To a solution of 2.80 g (13 mmol) of 5-bromo-2-methylbenzoic acid in 11 mL of MeOH, 2.5 mL of HCl 4.0 M (10 mmol) in dioxane were added.The reaction was heated at 70 ºC and stirred at that temperature overnight.Then, the mixture was concentrated, cooled to 0ºC and neutralized with saturated NaHCO3. The resulting mixture was extracted with DCM and evaporated to obtain methyl 5-bromo-2-methylbenzoate as yellow oil (2.8 g, 94percent), which solidified into needles.
94%
With hydrogenchloride In 1,4-dioxane at 70℃;
a) To a solution of 2.80 g (13 mmol) of 5-bromo-2-methylbenzoic acid in 1 1 ml_ of MeOH, 2.5 ml_ of HCI 4.0 M (10 mmol) in dioxane were added. The reaction was heated at 70 5C and stirred at that temperature overnight.Then, the mixture was concentrated, cooled to 0 5C and neutralized with saturated NaHC03. The resulting mixture was extracted with DCM and evaporated to obtain methyl 5-bromo-2-methylbenzoate as yellow oil (2.8 g, 94percent), which solidified into needles.
94%
at 20 - 65℃; for 16 h; Inert atmosphere
Methyl 5-bromo-2-methylbenzoate To a solution of 5-bromo-2-methylbenzoic acid (15 g, 69.8 mmol) in MeOH (100 mL) was added H2S04(1 mL, 18.76 mmol) slowly under nitrogen at RT. The resulting reaction mixture was stirred at 65 °C for 16 h. Water (50 mL) was added to the reaction mixture and extracted with EtOAc (3 x 60 mL ). The combined organic layer was concentrated to afford the title compound methyl 5-bromo-2-methylbenzoate (15 g, 65.5 mmol, 94 percent yield). LC-MS MS m/z 231.1 (M+H)+, 1.98 (ret. time)
85%
With sulfuric acid In waterHeating / reflux
5-Bromo-2-methylbenzoateA solution of 5-bromo-2-methyl benzoic acid (120 g, 0.55 mole) in methanol (1.2 L)1 H2SO4 (50 mL, 0.93 mole) was heated at reflux overnight. The reaction mixture was cooled and the solvent was evaporated. The crude residue was dissolved in ethyl acetate (2 x 500 mL). The combined organic layers were washed with water (500 mL), brine (300 mL) and dried over anhydrous Na2SO4, filtered and the solvent was evaporated to give methyl 5- bromo-2-methylbenzoate as a creamy solid. (12Og, 85percent) 1H-NMR (CDCI3, 300 MHz): δ 8.1 (1H, s), 7.5 (1H, d), 7.1 (1H, d), 3.9 (3H, s), 2.6 (3H, s)
Reference:
[1] Patent: WO2005/73205, 2005, A1, . Location in patent: Page/Page column 27-28
[2] Patent: WO2014/76104, 2014, A1, . Location in patent: Page/Page column 62-63
[3] Patent: CN107474006, 2017, A, . Location in patent: Paragraph 0050-0052; 0056-0058; 0062-0064; 0068-0073
[4] Patent: WO2011/80718, 2011, A1, . Location in patent: Example 2
[5] Patent: WO2017/46318, 2017, A1, . Location in patent: Page/Page column 47
[6] Patent: EP2570125, 2013, A1, . Location in patent: Paragraph 0134
[7] Patent: WO2013/37960, 2013, A1, . Location in patent: Page/Page column 69; 70
[8] Patent: WO2015/92713, 2015, A1, . Location in patent: Page/Page column 350
[9] Patent: WO2008/20306, 2008, A2, . Location in patent: Page/Page column 38
[10] Justus Liebigs Annalen der Chemie, 1887, vol. 239, p. 84[11] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1888, vol. 106, p. 949
[12] Recueil des Travaux Chimiques des Pays-Bas, 1935, vol. 54, p. 73,74
[13] E-Journal of Chemistry, 2011, vol. 8, # 3, p. 1108 - 1113
[14] Inorganic Chemistry, 2014, vol. 53, # 6, p. 2932 - 2942
[15] Patent: WO2007/84451, 2007, A1, . Location in patent: Page/Page column 159
7
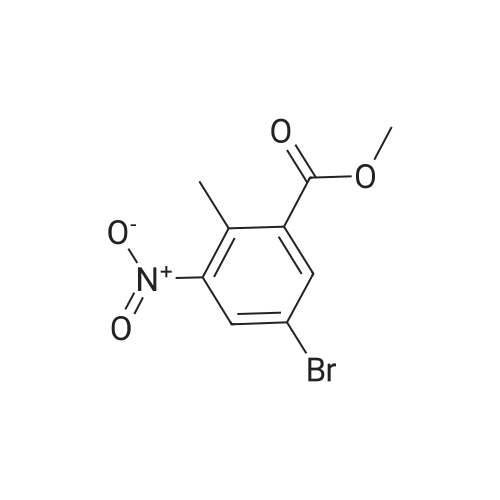
[ 79669-49-1 ]
[ 18107-18-1 ]
[ 79669-50-4 ]
Yield
Reaction Conditions
Operation in experiment
99%
With methanol In diethyl ether; toluene at 0 - 20℃; for 1 h;
5-Bromo-2-methylbenzoic acid (1 equiv) was dissolved in a 1:9 MeOH/toluene mixture (0.1 M). The reaction mixture was cooled to 0°C and a trimethylsilyldiazomethane (1.05 equiv) solution in diethylether (2M) was added slowly until a persistent yellow tinge was observed. The reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo. The resulting residue was sonicated in hexane, collected by vacuum filtration over a sintered funnel, dried and used without further purification. 5-Bromo-2-methyl~benzoic acid methyl ester: (99 percent yield, 100 percent purity) m/z (LC-MS, ESP): no ionisation R/T = 4.43 min
With potassium carbonate In DMF (N,N-dimethyl-formamide) at 20℃; for 2 h;
A 60: 40 mixture of 5-bromo-2-methyl benzoic acid and 3-bromo-2-methyl benzoic acid (8.0 g, 0.037 mol) was dissolved in N,N'-dimethylformamide (130 mL). Methyl iodide (2.28 mL, 2.3 mol) and potassium carbonate (5.11 g, 0.037 mol) were added in sequence at room temperature. The mixture was stirred at room temperature for 2 hours at which point the reaction was determined to be complete by HPLC. The solvent was removed under high vacuum and the resulting residue was passed through a silica gel column using 5percent ethyl acetate in hexanes as the eluent. The mixture of isomers was obtained as an oil and then separated by preparative normal phase HPLC using 0.5percent isopropyl alcohol in hexanes as the eluent. The title compound was obtained as a white solid (1.38 g, 29percent). 1H NMR (300.132 MHz, CDC13) 8 8.04 (d, J= 2.2 Hz, 1H), 7.50 (dd, J= 8.2, 2.2 Hz, 1H), 7.12 (d, J= 8.2 Hz, 1H), 3.89 (s, 3H), 2.54 (s, 3H).
Reference:
[1] Journal of Medicinal Chemistry, 2013, vol. 56, # 5, p. 1878 - 1893
13
[ 118-90-1 ]
[ 76006-33-2 ]
[ 79669-49-1 ]
Reference:
[1] Patent: WO2005/100351, 2005, A1, . Location in patent: Page/Page column 13; 17
[2] Patent: US2007/203116, 2007, A1, . Location in patent: Page/Page column 8; 10
[3] Journal of Organic Chemistry, 2008, vol. 73, # 5, p. 1732 - 1744
[4] Arkivoc, 2011, vol. 2011, # 6, p. 29 - 44
[5] Patent: WO2011/80718, 2011, A1, . Location in patent: Example 1
[6] Organic and Biomolecular Chemistry, 2017, vol. 15, # 48, p. 10172 - 10183
14
[ 118-90-1 ]
[ 79669-49-1 ]
Yield
Reaction Conditions
Operation in experiment
19%
Stage #1: at 20℃; for 2 h; Stage #2: With hydrogenchloride In methanol; water at 20℃;
2-Methylbenzoic acid (40.0 g, 290 mmol) was added to a suspension of Br2 (160 mL) and iron powder (3.20 g, 57.0 mol) under N2 atmosphere in an ice bath. The mixture was allowed to warm to room temperature and was stirred for 2 hours. The reaction mixture was poured into water and the reddish solid was collected by filtration. The solid was dried under vacuum at 50° C. The solid was dissolved in 400 mL of methanol before 640 mL of 0.1N aqueous HCl was added at room temperature. The mixture was stirred and a white solid was produced. This solid was recrystallized from ethanol to afford 5-bromo-2-methyl-benzoic acid (12.0 g, 19percent). 1H NMR (300M Hz, CDCl3) δ 8.17 (d, J=2.1, 1H), 7.56 (dd, J=8.1, 2.1, 1H), 7.15 (d, J=8.1, 1H), 2.59 (s, 3H).
Reference:
[1] Patent: US2009/143381, 2009, A1, . Location in patent: Page/Page column 55
[2] Chemische Berichte, 1883, vol. 16, p. 1959[3] Chemische Berichte, 1884, vol. 17, p. 164
[4] Justus Liebigs Annalen der Chemie, 1887, vol. 239, p. 84[5] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1888, vol. 106, p. 949
[6] Journal of the Indian Chemical Society, 1930, vol. 7, p. 503
[7] E-Journal of Chemistry, 2011, vol. 8, # 3, p. 1108 - 1113
[8] Inorganic Chemistry, 2014, vol. 53, # 6, p. 2932 - 2942
[9] Patent: CN103980263, 2016, B,
15
[ 118-90-1 ]
[ 76006-33-2 ]
[ 79669-49-1 ]
Reference:
[1] Patent: WO2005/100351, 2005, A1, . Location in patent: Page/Page column 13; 17
[2] Patent: US2007/203116, 2007, A1, . Location in patent: Page/Page column 8; 10
[3] Journal of Organic Chemistry, 2008, vol. 73, # 5, p. 1732 - 1744
[4] Arkivoc, 2011, vol. 2011, # 6, p. 29 - 44
[5] Patent: WO2011/80718, 2011, A1, . Location in patent: Example 1
[6] Organic and Biomolecular Chemistry, 2017, vol. 15, # 48, p. 10172 - 10183
16
[ 54811-38-0 ]
[ 79669-49-1 ]
Yield
Reaction Conditions
Operation in experiment
85%
Molecular sieve
2-methylbenzoic acid as the starting material,Under the action of metal reagent Fe powder and super acid catalyst trifluoromethanesulfonic acid,Adding a liquid bromine having a molar multiple of 1.05,And the solvent solution of bromine absolute molecular sieve without water pretreatment,Synthesis of intermediate 2 5-bromo-2-methyl benzoic acid, the yield of 85percent or more
Reference:
[1] Justus Liebigs Annalen der Chemie, 1941, vol. 546, p. 277,291
19
[ 87-41-2 ]
[ 79669-49-1 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1887, vol. 239, p. 84[2] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1888, vol. 106, p. 949
20
[ 939-83-3 ]
[ 79669-49-1 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1941, vol. 546, p. 277,291
21
[ 50670-64-9 ]
[ 79669-49-1 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1941, vol. 546, p. 277,291
22
[ 119-32-4 ]
[ 79669-49-1 ]
Reference:
[1] Chemische Berichte, 1895, vol. 28, p. 183
23
[ 118-90-1 ]
[ 7726-95-6 ]
[ 79669-49-1 ]
Reference:
[1] Chemische Berichte, 1895, vol. 28, p. 183
[2] Chemische Berichte, 1883, vol. 16, p. 1959[3] Chemische Berichte, 1884, vol. 17, p. 164
24
[ 79669-49-1 ]
[ 616-38-6 ]
[ 19725-82-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2008, vol. 51, # 12, p. 3507 - 3525
25
[ 79669-49-1 ]
[ 675109-26-9 ]
Reference:
[1] E-Journal of Chemistry, 2011, vol. 8, # 3, p. 1108 - 1113
[2] Patent: CN107474006, 2017, A,
[3] Patent: WO2007/84451, 2007, A1,
With sulfuric acid; nitric acid In acetone at -5 - 0℃; for 2.25 h; Cooling with acetone-ice
Step 1: Synthesis of 5-bromo-2-methyl-3-nitrobenzoic acid A solution of 5-bromo-2-methylbenzoic acid (5.0 g, 23 mmol) in concentrated H2SO4 (27 ml, 512 mmol) was cooled to 5° C. in an acetone/ice bath. A mixture of concentrated nitric acid (1.9 ml, 30 mmol) and concentrated H2SO4 (2.8 ml, 52 mmol) was added dropwise to the reaction mixture at -5 to 0° C. over 15 minutes. The yellow reaction mixture was stirred at -5 to 0° C. for 2 hours during which time a yellow precipitate formed. The reaction mixture was poured onto ice (150 g) and the precipitate was then collected by filtration. The precipitate was air dried to give the title compound (5.5 g, 52percent) as a pale yellow solid. LC-MS 57percent, 1.82 min (3.5 minute LC-MS method), no ionization; 1H NMR (500 MHz, DMSO-d6) δ ppm 8.29 (s, 1H) 8.13 (d, J=1.58 Hz, 1H) 2.43 (s, 3H).
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione; sulfuric acid;
To a mixture of 2-methylbenzoic acid 1 (15 g, 110.29 mmol)in conc. H2SO4 (60 ml), 1,3-dibromo-5, 5-dimethyl-2,4-imidazolidinedione (18.19 g, 60.66 mmol) was added andreaction mixture was stirred at room temperature for 5 h.After completion of reaction, reaction mixture was slowlypoured onto ice-cold water (400 ml). Solid was precipitatedout, filtered and dried under vacuum to afford compound 2.Yield: 88.00%. 1HNMR (DMSO-d6, 400 MHz): delta 13.16 (s,1H), 7.91 (s, 1H), 7.63 (d, J =8.0 Hz, 1H), 7.27 (d, J =8.4 Hz, 1H), 2.50 (s, 3H). LCMS: m/e 215/217 (M + 1).
19%
2-Methylbenzoic acid (40.0 g, 290 mmol) was added to a suspension of Br2 (160 mL) and iron powder (3.20 g, 57.0 mol) under N2 atmosphere in an ice bath. The mixture was allowed to warm to room temperature and was stirred for 2 hours. The reaction mixture was poured into water and the reddish solid was collected by filtration. The solid was dried under vacuum at 50 C. The solid was dissolved in 400 mL of methanol before 640 mL of 0.1N aqueous HCl was added at room temperature. The mixture was stirred and a white solid was produced. This solid was recrystallized from ethanol to afford 5-bromo-2-methyl-benzoic acid (12.0 g, 19%). 1H NMR (300M Hz, CDCl3) delta 8.17 (d, J=2.1, 1H), 7.56 (dd, J=8.1, 2.1, 1H), 7.15 (d, J=8.1, 1H), 2.59 (s, 3H).
Place <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (1.0 g, 4.7 mmol) in a 200 mL flask under an N2 atmosphere and add methanol via syringe. Add a 2M solution of diazomethyl- trimethyl-silane in hexane (3.5 mL, 23.0 mmol) drop wise over 10 minutes and stir for 1 hour at room temperature. Add glacial acetic acid (16 mL) and stir for 45 minutes. Dilute with ethyl acetate (100 mL) and wash with 1M aqueous sodium hydroxide solution (30 mL), saturated aqueous sodium bicarbonate solution (30 mL) and brine (30 mL). Dry the organic layer (Na2SO4), filter and concentrate in vacuo to obtain 1.01 g of 5-bromo-2- methyl-benzoic acid methyl ester (99 %). Place <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> methyl ester (1.04 g, 4.5 mmol) in a 50 mL flask under a N2 atmosphere and add carbon tetrachloride (15 mL). Add N-bromo- succinamide (1.49 g, 8.3 mmol) and 2,2'-azobisisobutyronitrile (40 mg, 0.2 mmol) and fit flask with a condenser and reflux for 4 hours. Cool to room temperature and filter. Concentrate the filtrate and pre-adsorb the crude product onto silica gel. Chromatograph the residue on a Si02 column eluting with dichloromethane in hexane (0 to 50%) to obtain 977 mg of 5-bromo-2-bromomethyl-benzoic acid methyl ester (70%). Using 5-bromo-2-bromomethyl-benzoic acid methyl ester (0. 984 g, 3.20 mmol) and the procedure described in the I st paragraph for the alternative procedure for 5- (4,4, 5, 5-tetramethyl- [1, 3,2] dioxaborolan-2-yl) -2, 3-dihydro-isoindol-1-one, prepare 509 mg of the title compound (75 %).
98%
Thionyl chloride (10 mL) was added to <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (2.0 g, 9.3 mmol) at 0 C and then DMF (one drop) was added. The mixture was heated at reflux for 3 h under nitrogen. The reaction mixture was evaporated and dry MeOH (5 mL) was added to the residue. The mixture was concentrated, and EtOAc was added. The mixture was washed with saturated aqueous NaHC03 solution and brine. The organic phase was dried (Na2S04), filtered, and evaporated to give <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> methyl ester (2.1 g, 98%) as an off-white solid.
97%
With sulfuric acid; at 20℃; for 5h;Cooling with ice; Reflux;
(1) Dissolve 300 g of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> in 2 L of methanol.Concentrated sulfuric acid 50 ml was added dropwise in an ice bath.Stir for 1 hour at room temperature and then heat to reflux for 4 hours. The solvent was concentrated and the residue was dissolved in 2 liters of dichloromethane.Wash three times with saturated sodium bicarbonate,It was dried and concentrated to give <strong>[79669-49-1]methyl 5-bromo-2-methylbenzoate</strong> (310 g, yield 97%).
95.2%
With thionyl chloride; at 0 - 20℃;
Example 2:; 5-Bromo-2-methyl benzoic acid methyl ester; The compound of example 1 (20.3 g, 0.0944 mol) was dissolved in 150 mL methanol and cooled to 0 C. To this reaction mixture, thionyl chloride (28.07 g, 0.236 mol) was added slowly within 15-20 min. The reaction mixture was stirred at room temperature for 2- 3 h. After completion of the reaction, MeOH was removed under vacuum. The oily material obtained was dissolved in ethyl acetate and washed with sodium bicarbonate, water and brine and dried over anhydrous sodium sulfate. The organic layer was concentrated to obtain the title compound.Yield: 20.5 g (95.2 %)Ref:-US2007/0213342 A1
95%
With thionyl chloride; for 2h;Reflux;
A mixture of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (1.00 g, 4.7 mmol) and thionyl chloride (1.40 g,11.6 mmol) in methanol (30 ml) was refluxed for 2 h. The solvent was removed underreduced pressure and the pH of the residue was adjusted to 7. The resultant was extractedwith ethyl acetate. The organic phase was dried over anhydrous Na2S04, filtered andconcentrated to give CAP-004-19-1 (1.02 g, 95 %) as a pale yellow solid.
94%
With hydrogenchloride; In 1,4-dioxane; at 70℃;
(a) To a solution of 2.80 g (13 mmol) of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> in 11 mL of MeOH, 2.5 mL of HCl 4.0 M (10 mmol) in dioxane were added.The reaction was heated at 70 ºC and stirred at that temperature overnight.Then, the mixture was concentrated, cooled to 0ºC and neutralized with saturated NaHCO3. The resulting mixture was extracted with DCM and evaporated to obtain <strong>[79669-49-1]methyl 5-bromo-2-methylbenzoate</strong> as yellow oil (2.8 g, 94%), which solidified into needles.
94%
With hydrogenchloride; In 1,4-dioxane; at 70℃;
a) To a solution of 2.80 g (13 mmol) of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> in 1 1 ml_ of MeOH, 2.5 ml_ of HCI 4.0 M (10 mmol) in dioxane were added. The reaction was heated at 70 5C and stirred at that temperature overnight.Then, the mixture was concentrated, cooled to 0 5C and neutralized with saturated NaHC03. The resulting mixture was extracted with DCM and evaporated to obtain <strong>[79669-49-1]methyl 5-bromo-2-methylbenzoate</strong> as yellow oil (2.8 g, 94%), which solidified into needles.
94%
With sulfuric acid; at 20 - 65℃; for 16h;Inert atmosphere;
Methyl 5-bromo-2-methylbenzoateTo a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (15 g, 69.8 mmol) in MeOH (100 mL) was added H2S04(1 mL, 18.76 mmol) slowly under nitrogen at RT. The resulting reaction mixture was stirred at 65 C for 16 h. Water (50 mL) was added to the reaction mixture and extracted with EtOAc (3 x 60 mL ). The combined organic layer was concentrated to afford the title compound <strong>[79669-49-1]methyl 5-bromo-2-methylbenzoate</strong> (15 g, 65.5 mmol, 94 % yield). LC-MS MS m/z 231.1 (M+H)+, 1.98 (ret. time)
94%
With sulfuric acid; at 65℃; for 16h;Inert atmosphere;
To a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (15 g, 69.8 mmol) in methanol (100 ml) was added H2SO4 (1 ml, 18.76 mmol) slowly under nitrogen at ambient temperature. The resulting reaction mixture was stirred at 65 C for 16 hrs. Water (50 mL) was added to the reaction mixture and extracted with ethyl acetate ( 3 x 60 ml ). The combined organic layer was concentrated to afford the title compound <strong>[79669-49-1]methyl 5-bromo-2-methylbenzoate</strong> (15 g, 65.5 mmol, 94 % yield). LC-MS MS m/z 231.1 (M+H)+, 1.98 (ret. time)
85%
With sulfuric acid; In water;Heating / reflux;
5-Bromo-2-methylbenzoateA solution of <strong>[79669-49-1]5-bromo-2-methyl benzoic acid</strong> (120 g, 0.55 mole) in methanol (1.2 L)1 H2SO4 (50 mL, 0.93 mole) was heated at reflux overnight. The reaction mixture was cooled and the solvent was evaporated. The crude residue was dissolved in ethyl acetate (2 x 500 mL). The combined organic layers were washed with water (500 mL), brine (300 mL) and dried over anhydrous Na2SO4, filtered and the solvent was evaporated to give methyl 5- bromo-2-methylbenzoate as a creamy solid. (12Og, 85%) 1H-NMR (CDCI3, 300 MHz): delta 8.1 (1H, s), 7.5 (1H, d), 7.1 (1H, d), 3.9 (3H, s), 2.6 (3H, s)
sulfuric acid; at 20℃; for 72h;
Example 1251Step 1 Compound 1251 A (2.0 g), MeOH (20 mL), and H2SO4 (cone, 2mL) were stirred at RT for three days. Hexane (50 mL) was added. The organic layer was washed by water, dried over Na2SO-J, concentrated by rotary evaporator. The product was purified by sgc (Hexane/EtOAc 10:1 ) to give compound 1251B (1.219 g)
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 3h;Inert atmosphere;
To a 50 mL flask, <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (4.3 g, 20 mmol) was added, then anhydrous dichloromethane (30 mL) was added after purging with N2. The reaction mixture was cooled to 0 C., then a catalytic amount of DMF (0.5 mL) was added, and then oxalyl chloride (5 mL, 58 mmol) was slowly added. The reaction mixture was warmed up to room temperature and stirred for 3 hours until the reaction solution became a clear solution, and then the stirring was stopped. Dichloromethane and excess oxalyl chloride were removed by rotary evaporation to give a pale yellow oil (4.8 g, 100.0%), which was used directly in the next step.
100%
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; at 0 - 20℃; for 6h;
To a solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> 4a (21.5 g, 100 mmol) in dichloromethane (200 mL)Oxalyl chloride (50.4 g, 397 mmol) was added slowly, then N,N-dimethylformamide (0.5 mL, 6.4 mmol) was added dropwise.The resulting mixture was stirred at room temperature for 6 hours. After the reaction was completed, it was concentrated under reduced pressure.The title compound 4b (23.3 g, pale yellow liquid) was obtained.Yield: 100%.
70%
With thionyl chloride; N,N-dimethyl-formamide; for 7h;Reflux;
5-Bromo-2-methyl benzoic acid (4.5 g, 7.0 mmol) was dissolved in 24 mL dried SOCl2 with a fewdrops DMF, the mixture was refluxed for 7 h. The solvent was evaporated under reduced pressureto give compound 1 as a transparent liquid. Dimethoxybenzene 4.5 mL (35.4 mmol) was addedto 30 mL dried CH2Cl2 and stirred at 0 C. Next, anhydrous AlCl3 (3.0 g, 22.7 mmol) was addedportion-wise. The obtained compound 1 was then added to the solution, which was allowed to warm to room temperature and stirred for 3 h and quenched with 30 mL distilled water. The organic phasewas separated, washed with 30 mL water and dried over anhydrous Na2SO4, and then concentratedvia rotary evaporation. The crude product was puried by silica gel chromatography with ethylacetate±petroleum ether (v/v, 1/8) as the eluent to afford compound 2. The product was recrystallizedfrom methanol to give a white powder in 63% total yield. m.p. 102.0-104.0 C; 1H-NMR (600 MHz,DMSO-d6) delta: 2.53 (s, 3H, Ar-2-CH3), 3.86 (s, 3H, Ar-5?-OCH3), 3.94 (s, 3H, Ar-4?-OCH3), 6.99 (s, 1H,Ar-6?-H), 7.05 (s, 1H, Ar-6-H), 7.20 (t, J = 10.4 Hz,1H, Ar-3?-H), 7.31(d, J = 11.4 Hz, 1H, Ar-3-H), 7.34 (d,J = 11.4 Hz, 1H, Ar-4-H), 7.41 (t, J = 10.4 Hz, 1H, Ar-2?-H); ESI-MS m/z (%): 334.88, 336.93 ([M + H]+,100, 98).
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; for 2h;
Example 16; 3H[(Cycloheptylmethyl)amino] carbonyl]-4'-methyl-[l,l '-biphenylJ-2-carboxylic acid; EPO <DP n="70"/>a) 5-Bromo-iV-(cycloheptylmethyl)-2-methyl-benzamide; To a stirred solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (US4282365) (1 g) in dichloromethane (20 mL) was added LambdayV-dimethylformamide (1 drop) followed by oxalyl chloride (1.6 mL). The reaction was stirred for two hours, the volatiles were removed under vacuum and dichloromethane (20 mL), cycloheptanemethanamine (649 mg) and triethylamine (1.29 mL) were added. The reaction was stirred for 30 minutes before the reaction was acidified with 2M hydrochloric acid. The aqueous phase was separated, the organic phase was washed once with brine, dried over magnesium sulphate, filtered and10 the solvent removed to afford the sub-title compound (1.48 g).MS: APCI(+ve) 324 (M+H1).1H NMR (300 MHz, ddelta-DMSO) delta 8.43 - 8.33 (IH, m), 7.50 (IH, dd), 7.45 - 7.40 (IH, m), 7.25 - 7.18 (IH, m), 3.10 - 3.02 (2H3 m), 2.30 - 2.24 (3H, m), 1.79 - 1.32 (HH, m), 1.31 -I5 1.11 (2H, m).
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 20 - 25℃;
STEP A:A 250 ml_ three-necked round bottom flask was charged with 5-bromo-2- methylbenzoic acid (22.5 g, 0.10 mol), CH2CI2 (100 ml_) and DMF (0.25 ml_) at ambient temperature (2O0C). Oxalyl chloride (12 ml_, 0.13 mol) was added such that the internal temperature was maintained below 250C. Vigorous gas evolution was observed. The reaction mixture was stirred overnight at ambient temperature, under argon, then the volatiles were removed under reduced pressure. The resulting residue (an acid chloride compound) was dissolved in DCM (50 ml_) and set aside under a nitrogen atmosphere
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 0.5h;
Oxalyl chloride (50 mL, 581.5 mmol) was added dropwise at room temperature to a solution of 14 (50 g, 232.5 mmol) in CH2C12 (750 mL) and DMF (1 mL, 11.6 mmol. The reaction mixture was stirred for 0.5 h at room temperature. The reaction mixture was concentrated under vacuum and the residue was used without further purification Total 65 g of the crude 15 was obtained.
With thionyl chloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 12h;
Step 1: Synthesis of(5-bromo-2-methylphenyl)(4-ethoxyphenyl)methanone:<strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (13 g) was suspended in dichloromethane (100 ml) and dimethylformamide (100 ml) then it was cooled to 0 C and SOCl2 (26.5 ml) was added slowly. The reaction mixture was slowly allowed to room temperature and stirred for about 12 hours. The solvent was removed under reduced pressure to give 5-bromo-2-methylbenzoyl chloride as oil. Ethoxybenzene (7 ml) was suspended in dichloromethane and cooled to -5 C then the mixture was added slowly to A1C13 (10.4 g) by portion wise and the above acid chloride in dichloromethane (- ) was added drop wise and stirred for about 1 hour at 0 C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ice- water. The organic layer was separated and aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous sodium sulphate and concentrated to give the crude compound. The crude product was purified via silica gel column chromatography with EtOAc and n- Hexane (1: 7) to afford the title compound as light yellow solid (15 g). Yield: 77.8 %; 1H NMR (CDCI3, 300 MHz): delta 7.76 (d, J = 8.7 Hz, 2H), 7.48 (dd, J = 2.1, 8.1 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.15 (d, / = 8.1 Hz, 1H), 6.92 (d, J = 8.7 Hz, 2H), 4.11 (q, J = 6.9 Hz, 2H), 2.22 (s, 3H), 1.45 (t, J = 6.9 Hz, 3H).
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 6h;
Synthesis of compound 43Ci8H12BrFOS M = 375.25 g.mof1 FN MR rCDCk 282.5MHz): - 111.3 (m, IF, Ar-F).Mass (GC-MS): 375.0 (97%); 376.0 (28%); 377.0 (100%); 416.0 (23%); 418.05-Bromo-2-methylbenzoic acid (725mg, 3.37mmol, leq) was suspended in dry dichloromethane (9.7mL). Oxalyl chloride (0.32mL, 3.74mmol, l . l eq) and N,N- dimethylformamide (0.013mL, 0.17mmol, 0.05eq) were then adde d at room temperature and the mixture was stirred for 6 hours. The solvent was then evaporated to give 5-bromo-2-methylbenzoyl chloride as yellow oil. This crude product was dissolved in dry dichloromethane (19.3mL), A1C13 (49.5mg, 3.71mmol, l . leq) and 42 (600mg, 3.37mmol, leq) were then added at 0C (internal temperature). The resultant mixture was stirred at this temperature for 30 minutes and then at room temperature overnight. The reaction mixture was poured into ice and water, the organic layer was separated and the aqueous layer was extracted with dichloromethane. The organic layers were gathered, dried over MgS04, filtered and concentrated. The rsidue was taken up with n- hexane to form a precipitate which was collected by filtration, washed with n-hexane and dried to afford compound 43 (69% yield) as yellowish crystals
With oxalyl dichloride; N,N-dimethyl-formamide; In chloroform; at 20℃;
To a stirred suspension of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (11) (39 g, 181 mmol) in chloroform (390 mL) was added oxalyl chloride (20.7 mL, 218 mmol) dropwise at room temperature, followed by N,N-dimethylformamide (1 drop). The resultant mixture was stirred at the same temperature overnight. The mixture was concentrated in vacuo, dissolved in dichloromethane (650 mL), and to the mixture was added 2-chlorothiophene (21.8 mL, 236 mmol). To the mixture was added portion-wise aluminum chloride (26.6 g, 200 mmol) at 0 C. The resultant mixture was allowed to warm to room temperature, and stirred at the same temperature overnight. The reaction mixture was poured into ice-water, and extracted with chloroform. The organic layer was separated, washed with saturated aqueous sodium hydrogen carbonate solution, and brine, and dried over sodium sulfate prior to filtration. The solvent was evaporated under reduced pressure, and the residue was dissolved in methanol on heating, and treated with activated carbon. The insoluble was filtered off, and the filtrate was evaporated under reduced pressure. The residue was recrystallized from methanol to give titled compound 12 (52 g, 81%) as pale yellow powder.
With dmap; oxalyl dichloride; In dichloromethane; at 20℃;
2-Methyl-4- bromobenzoic acid (1, 26.0 g, 121 mmol) and oxalyl chloride (13.2 mL, 152 mmol) were suspended in 520 mL of CH2CI2. A catalytic amount of DMAP (0.5 mL) was added dropwise and the reaction was stirred at room temperature until the reaction become homogenous. The volatiles were removed in vacuo. The crude material was dissolved in 200 mL of CH2CI2 and N,0- dimethylhydroxylamine hydrochloride (23.6 g, 242 mmol) was added. The reaction was cooled to 0 C and triethylamine (55 mL, 399 mmol) was slowly added. Upon completion of addition of triethylamine, the reaction was warmed to room temperature and stirred overnight. The reaction was quenched with 50% saturated aqueous NaHS04. The aqueous layer was extracted twice with CH2CI2. The combined organic layers were washed with brine, dried over Na2S04, filtered, and the solvent removed in vacuo. The resulting Weinreb amide (31.3g, 99% yield) was used without further purification in the next step.
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; for 2h;
To a solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (1.0 g, 4.7 mmol) in THF (5.0 ml), CH2C12 (5.0 ml) and DMF (50.0 mu, 646.6 muiotaetaomicronepsilonbeta) is added dropwise oxalyl chloride (2.6 ml, 5.1 mmoles) at 0 C. The reaction mixture is allowed to warm gradually to ambient temperature. After 2 hours, the solvent is removed under reduced pressure. The residue is diluted with CH2C12 (5 ml), and the mixture is cooled to 0 C. Methyl 3-amino-2,4-dimethylbenzoate (957.9 mg, 4.70 mmol) is added, followed by N,N-dimethylpyridin-4-amine (28.4 mg, 0.232 mmoles) and pyridine (1.1 ml, 14.0 mmoles). The cooling bath is removed, and the clear solution is allowed to warm to ambient temperature. After 2 hours, the solution is concentrated. The residue is diluted with EtOAc and washed sequentially with IN HC1, saturated solution of sodium bicarbonate, and brine. The organic layer is dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. The residue is triturated with 30% EtOAc in Hexanes and the solids are filtered to provide the title compound (680.0 mg; 38.9%). Mass spectrum (m/z): 376.0 (M+H)+.
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 0 - 30℃; for 2h;
5-Bromo-2-methyl-benzoic acid (50.0 g, 233 mmol) was dissolved in dichloromethane (400 mL) at room temperature, and N, N-dimethylformamide (1.0 mL) was added and the mixture was cooled to 0 , oxalyl chloride (30 mL, 344 mmol) was added dropwise. After the end of addition, the mixture was stirred at room temperature for 2 hours. Then the mixture was concentrated under reduced pressure to give the title compound as a yellow semi-solid (55 g, 100%) . The crude product was directly used for the next reaction.
With methanol; In diethyl ether; toluene; at 0 - 20℃; for 1h;
5-Bromo-2-methylbenzoic acid (1 equiv) was dissolved in a 1:9 MeOH/toluene mixture (0.1 M). The reaction mixture was cooled to 0C and a trimethylsilyldiazomethane (1.05 equiv) solution in diethylether (2M) was added slowly until a persistent yellow tinge was observed. The reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo. The resulting residue was sonicated in hexane, collected by vacuum filtration over a sintered funnel, dried and used without further purification. 5-Bromo-2-methyl~benzoic acid methyl ester: (99 % yield, 100 % purity) m/z (LC-MS, ESP): no ionisation R/T = 4.43 min
With borane-THF; In tetrahydrofuran; diethyl ether; at 50℃; for 3h;
[00201] A solution of borane-tetrahydrofuran in tetrahydrofuran (1.0 M, 123 mL, 123 mmol, 1.2 equiv) was added dropwise to a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (9, 22.0 g, 102 mmol, 1 equiv) in ether (120 mL) (CAUTION: gas evolution). After 1 h, the reaction flask was placed in an oil bath preheated to 50 C. After 2 h, the oil bath was removed, and the reaction flask was allowed to cool to 23 C. Methanol (20 mL) was added dropwise (CAUTION: gas evolution), and the product solution was concentrated to remove the bulk of solvent. The residue was purified by flash-column chromatography (10: 1 hexanes-ethyl acetate initially, grading to 5: 1 hexanes-ethyl acetate), furnishing (5-bromo-2- methylphenyl)methanol (10) as a white solid (19.5 g, 95%).
92%
In the inert gas N2 atmosphere, after dehydration and deoxidation treatment to the reaction bottle after adding 2 - methyl -5 - bromo - benzoic acid (107.1 mg, 0.5 mmol, gun for adding [...] borane (289 mul, 2 mmol), reacting at room temperature 12 hours, the reaction out of the glove box, in order to have three methoxybenzene (83.77 mg, 0.5 mmol) as the internal standard, CDCl3 for dissolving, stirring 10 minutes, sampling, with nuclear magnetic resonance. Calculated 1 H and the yield is 99%. The product of nuclear magnetic data: The residue is added to the sample in 1 g silica gel, in order to 3 ml methanol as a solvent, 50 C lower reaction 3 h, the borate further hydrolysis alcohol, after the reaction, extracted with ethyl acetate three times, the combined organic layer, dried with anhydrous sodium sulfate, the solvent is removed under reduced pressure, through the silica gel (100 - 200 mesh) column chromatography purification, ethyl acetate/hexane (1:5) mixture as the eluent, to obtain the pure primary alcohol, separation and the yield is 92%.
74.9%
With dimethylsulfide borane complex; In tetrahydrofuran; toluene; at 0℃; for 16h;Inert atmosphere;
(5-Bromo-2-methylphenyl)methanol To a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (70 g, 326 mmol) in tetrahydrofuran (THF) (700 mL) stirred under nitrogen at 0 C was added a toluene solution of borane-methyl sulfide complex (244 mL, 488 mmol) drop wise during 15 min. The reaction mixture was stirred for 16 h. The reaction was cooled to 0 C and quenched with methanol (500 mL) drop wise. The reaction mixture was stirred at ambient temperature for 3 h and then concentrated. The crude residue was diluted with ethyl acetate (1 L) and washed with 1 N HCI (500 mL), brine solution (500 mL) and dried over Na2S04, filtered and concentrated to give the title compound (49 g, 244 mmol, 74.9 % yield). 1 H NMR (400 MHz, DMSO) delta= 7.52 (d, J = 2.6 Hz, 1 H), 7.31 (dd, J = 8.0, 2.2 Hz, 1 H), 7.12 - 7.03 (m, 1 H), 5.22 (td, J = 5.5, 1 .8 Hz, 1 H), 4.48 (dd, J = 5.1 , 1 .8 Hz, 2H), 2.17 (s, 3H).
74.9%
With borane dimethyl sulfide complex; In tetrahydrofuran; toluene; at 0℃; for 16h;Inert atmosphere;
To a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (70 g, 326 mmol) in tetrahydrofuran (THF) (700 mL) stirred under nitrogen at 0 C was added a toluene solution of borane-methyl sulfide complex (244 mL, 488 mmol) drop wise during 15 min. The reaction mixture was stirred for 16 h. The reaction was cooled to 0 C and quenched with methanol (500 mL) drop wise. The reaction mixture was stirred at ambient temperature for 3 h and then concentrated. The crude residue was diluted with ethyl acetate (1 L) and washed with 1 N HCI (500 mL), brine solution (500 mL) and dried over Na2S04, filtered and concentrated to give the title compound (49 g, 244 mmol, 74.9 % yield). 1H NMR (400 MHz, DMSO) delta= 7.52 (d, J = 2.6 Hz, 1 H), 7.31 (dd, J = 8.0, 2.2 Hz, 1 H), 7.12 - 7.03 (m, 1 H), 5.22 (td, J = 5.5, 1 .8 Hz, 1 H), 4.48 (dd, J = 5.1 , 1 .8 Hz, 2H), 2.17 (s, 3H).
74.9%
With dimethylsulfide borane complex; In tetrahydrofuran; toluene; at 0℃; for 16h;Inert atmosphere;
To a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (70 g, 326 mmol) in tetrahydrofuran (THF) (700 mL) stirred under nitrogen at 0 C was added a toluene solution of borane-methyl sulfide complex (244 ml_, 488 mmol) drop wise during 15 min. The reaction mixture was stirred for 16 h. The reaction was cooled to 0 C and quenched with methanol (500 ml.) drop wise. The reaction mixture was stirred at ambient temperature for 3 h and then concentrated. The crude residue was diluted with ethyl acetate (1 L) and washed with 1 N HCI (500 ml_), brine solution (500 ml.) and dried over Na2S04, filtered and concentrated to give the title compound (49 g, 244 mmol, 74.9 % yield). N1HMR (400 MHz, DMSO) delta= 7.52 (d, J = 2.6 Hz, 1 H), 7.31 (dd, J = 8.0, 2.2 Hz, 1 H), 7.12 - 7.03 (m, 1 H), 5.22 (td, J = 5.5, 1 .8 Hz, 1 H), 4.48 (dd, J = 5.1 , 1 .8 Hz, 2H), 2.17 (s, 3H).
73.4%
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0℃; for 2h;
(5-bromo-2-methylphenyl)methanolTo a stirred solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (10.0 g, 46.5 mmol) in THF (50 ml_) was added LAH (46.5 ml_, 46.5 mmol, 1 M in THF) dropwise at 0 C. After 0.5 h the reaction turned a milky white color and then stirred for another 1.5 h. The reaction was then quenched with the dropwise addition of saturated Na2S04in water at 0 C. The reaction was diluted with Et20 (100 ml_) and water (100 ml_). The organic phase was separated from the aqueous phase, dried over Na2S04and concentrated in vacuo. The residue was purified via silica gel chromatography with hexane ethylacetate to yield (5- bromo-2-methylphenyl)methanol (7.3 g, 34.1 mmol, 73.4 % yield) as a white solid. LC-MS m/z 182.8 (M-OH)+, 0.78 min (ret. time).
5.6 g
With borane-THF; In diethyl ether; at 0℃; for 0.333333h;Reflux;
To a mixture of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (6.0 g, 28 mmol) in 32 mL diethyl ether at 0 C was added a 1M solution of borane-tetrahydrofuran complex (34 mL, 34 mmol) over 10 minutes. The reaction mixture was warmed to room temperature and then heated to reflux for 10 minutes. Methanol was added to react with the excess borane and then the mixture was extracted five times with saturated aqueous sodium bicarbonate solution. The organic phases was combined, dried over magnesium sulfate and concentrated under reduced pressure to provide the title compound (5.6 g). H NMR (CDC13) delta 7.53 (d, 1H), 7.31 (m, 1H), 7.03 (d, 1H), 4.66 (s, 2H), 2.27 (s, 3H).
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 20℃; for 12h;
To a solution <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> 8d(91 g, 425 mmol) in THF (500 mL) at 0 oC was added borane-dimethylsulfide complex (2M in THF, 400 mL, 800 mmol). The resulting mixture wasstirred with gradual warming to ambient temperature over 12h, re-cooled to 0 oC,and quenched with MeOH and then water. The mixture was extracted with EtOAc.The organic layer was washed with brine, dried over anhydrous MgSO4,filtered and evaporated in vacuo to yield a white solid
5.6 g
With borane-THF; In diethyl ether; at 0℃; for 0.333333h;Reflux;
Step A: Preparation of 5-bromo-2-methylbenzenemethanol To a mixture of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (6.0 g, 28 mmol) in 32 mL diethyl ether at 0 C was added a 1M solution of borane-tetrahydrofuran complex (34 mL, 34 mmol) over 10 minutes. The reaction mixture was warmed to room temperature and then heated to reflux for 10 minutes. Methanol was added to react with the excess borane and then the mixture was extracted five times with saturated aqueous sodium bicarbonate solution. The organic phases was combined, dried over magnesium sulfate and concentrated under reduced pressure to provide the title compound (5.6 g). H NMR (CDC13) delta 7.53 (d, 1H), 7.31 (m, 1H), 7.03 (d, 1H), 4.66 (s, 2H), 2.27 (s, 3H).
97 g
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 20℃; for 24h;
A stirred solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (100 g, 465 mmol) in tetrahydrofuran (1 .2 L) was cooled in an ice bath to 0C. 2N borane-methyl sulfide complex in THF (302 mL, 605 mmol) was added dropwise via additional funnel over 90 min. The reaction mixture was allowed to warm to room temperature and was stirred for 24 hr. Reaction mixture was cooled to 0 C, quenched with methanol (200 ml) and was stirred for 1 hr. The solvents were removed under reduced pressure and the resultant oil was partitioned between diethyl ether (1 L) and 1 N HCI (1 L). The layers were separated and the aqueous extracted with diethyl ether (2 x 500 mL). The combined organic extracts were washed with 1 N HCI (2 x 500ml), brine, dried over sodium sulfate, filtered and concentrated to afford a soft yellow solid (5-bromo-2-methylphenyl)methanol (97 g, 482 mmol, 104 % yield). NMR (CHCI3-d) delta: 7.54 (s, 1 H), 7.33 (d, J=8.0 Hz, 1 H), 7.05 (d, J=8.3 Hz, 1 H), 4.66 (s, 2H), 2.28 (s, 3H), 1 .99 (br. s., 1 H). LC-MS: m/z 183.0 (M-OH)+
With borane; In tetrahydrofuran; at 0 - 20℃;
Intermediate 9 (5-Bromo-2-methylphenyl)methanol. Borane (10.80 mL of a 1 M solution in THF, 10.80 mmol) was added to a cooled solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (116 mg, 0.54 mmol) in THF (15 mL), under nitrogen, at 0C (ice water bath) and the resulting mixture allowed to warm to rt overnight. The mixture was then treated with MeOH (10 mL) followed by aqueous HCl (2M, 20 mL) and the mixture stirred for about 15 minutes, concentrated under vacuum and then partitioned with EtOAc. The organic layer was washed with aqueous HCl (2M), water and brine, dried (MgS04), filtered and reduced to give the title compound as a colourless oil (90 mg). LC/MS: Rt 3.09 min, no molecular ion observed.
With potassium carbonate; In DMF (N,N-dimethyl-formamide); at 20℃; for 2h;
A 60: 40 mixture of 5-bromo-2-methyl benzoic acid and 3-bromo-2-methyl benzoic acid (8.0 g, 0.037 mol) was dissolved in N,N'-dimethylformamide (130 mL). Methyl iodide (2.28 mL, 2.3 mol) and potassium carbonate (5.11 g, 0.037 mol) were added in sequence at room temperature. The mixture was stirred at room temperature for 2 hours at which point the reaction was determined to be complete by HPLC. The solvent was removed under high vacuum and the resulting residue was passed through a silica gel column using 5% ethyl acetate in hexanes as the eluent. The mixture of isomers was obtained as an oil and then separated by preparative normal phase HPLC using 0.5% isopropyl alcohol in hexanes as the eluent. The title compound was obtained as a white solid (1.38 g, 29%). ¹H NMR (300.132 MHz, CDC13) 8 8.04 (d, J= 2.2 Hz, 1H), 7.50 (dd, J= 8.2, 2.2 Hz, 1H), 7.12 (d, J= 8.2 Hz, 1H), 3.89 (s, 3H), 2.54 (s, 3H).
Step B Methyl 5-Bromo-o-toluate Reflux a mixture of 54.30 gm. (0.252 mole) of 5-bromo-o-toluic acid, 400 ml. of methanol and 5 ml. of concentrated sulfuric acid for 18.5 hours. Remove the methanol under vacuum. Dissolve the residue in benzene, wash with water, aqueous sodium bicarbonate solution, water and dry over anhydrous magnesium sulfate. Remove the solvent under vacuum and distill the residual oil to obtain the title product together with the 3-bromo isomer as a colorless oil (yield 41.6 gm., b.p. 132-135 C./0.1 mm). Chill the oil in a refrigerator for 1 hour and separate the solids to obtain 12.53 gm. of white solid (m.p. 42-46 C., mainly the 5-bromo isomer).
In methanol;
Example 126 5-Bromo-2-[2-(2,3-difluoro-phenyl)-imidazo[4,5-d]pyridazin-5-ylmethyl]-benzoic acid methyl ester (Compound 312) A solution of 5-bromo-2-methyl-benzoic acid (1.0 g, 4.7 mmol) in MeOH (15 mL) was treated with TMS-diazomethane (2 M in hexanes, ca. 5 mL) until TLC showed consumption of starting material. The solvents were removed to give crude 5-bromo-2-methyl-benzoic acid methyl ester as a white solid used without further purification.
With diazomethyl-trimethyl-silane; In methanol; toluene; at 0℃;
2-Methyl-5-bromobenzoic acid (2.89 g, 13.44 mmol) was dissolved in toluene (44 ml) and methanol (22 ml). After cooling to 0C, trimethylsilyl diazomethane (10.8 ml, 21.6 mmol) was slowly added and the reaction wasstirred overnight. Upon completion, the volatiles were evaporated and the crude product was dried under high vacuum overnight. The resulting crude product was used without further purification. NMR oe (ppm)(CDC13): 8.03 (d, 1H), 7.50 (dd, 1H), 7.11 (d, 1H), 3.89 (s, 3H), 2.56 (s, 3H).
5-Bromo-2-methyl-benzoic acid methyl ester (2); The crude 5-bromo-2-methylbenzoic acid (1) (60.8 g; 283 mmol) was stirred in methanol (400 mL) and concentrated sulfuric acid (6 mL) was added. The mixture was heated at gentle boiling for 20 hours. The solvent was evaporated and the residue was taken up into toluene and water. The solution was shaken and the phases were separated. The organic layer was washed with dilute aqueous NaHCO3, water, and brine. The solution was dried with MgSO4, filtered, and evaporated to get an orange oil. Fractional distillation at reduced pressure yields a mixture of the title compound and 3-bromo-2-methylbenzoic acid methyl ester (47.9 g; 74%).
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 4h;
To a solution of 5-bromo-2-methyl-benzoic acid (9.9 g, 46 mmol) in DMF (100 mL) was added K2CO3 (7.6 g, 55 mmol) and CH3I (20 g, 140 mmol) slowly. After stirring at room temperature for 4 h, the solvent was removed under vacuum. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine and dried over Na2SO4. The solvent was removed under vacuum to afford 5-bromo-2-methylbenzoic acid methyl ester (8.6 g, 82%), which was used in next step without further purification. 1H NMR (300 MHz, CDCl3) delta 8.04 (d, J=2.1, 1H), 7.50 (dd, J=8.1, 2.1, 1H), 7.12 (d, J=8.1, 1H), 3.89 (s, 3H), 2.53 (s, 3H).
With sulfuric acid; magnesium sulfate; In dichloromethane; at 20℃; for 24.0h;
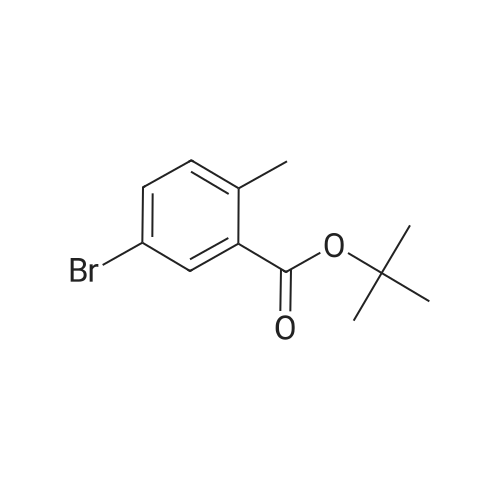
To a suspension of anhydrous magnesium sulfate (4.5 g, 37.28 mmol) in dichloromethane (37 ml.) is added concentrated H2SO4 (516 μl, 9.302 mmol) and the mixture is stirred vigorously for 15 min. 5-Bromo-2-methyl-benzoic acid (2 g, 9.302 mmol) is added, followed by t-butanol (4.39 ml_, 46.51 mmol). The flask is capped tightly and stirred at room temperature for 24 h. After adding saturated sodium bicarbonate, the resulting solution is extracted with ethyl acetate. The organic layer is washed with a saturated sodium chloride solution, dried over sodium sulfate, filtered and concentrated. The resulting residue is purified by silica gel flash chromatography (ethyl acetate- heptane, 0:1 to 3:7) to furnish 5-bromo-2-methyl-benzoic acid fert-butyl ester. 1H NMR (400 MHz, DMSO-cfe) δ ppm 1.59 (s, 9 H), 2.51 (s, 3 H), 7.08 (d, J=8.1 Hz, 1 H), 7.46 (dd, J=8.3, 2.3 Hz, 1 H), 7.92 (d, J=2.3 Hz, 1 H)
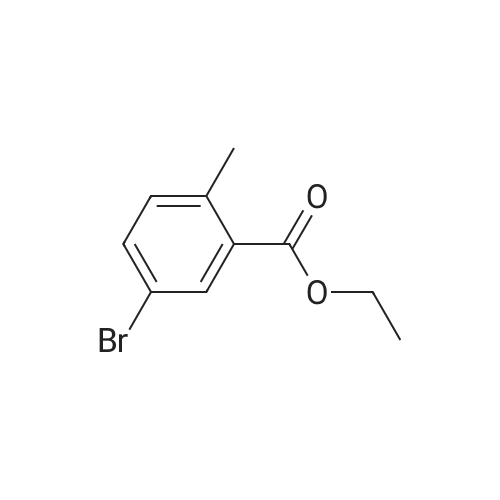
To a suspension of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (40 g. 0.19 mol) in EtOH (600 mL) was added 98% H2SO4 (3 mL) slowly. The reaction was heated at reflux overnight, then cooled to 0 C, and treated with aHC03 until no gas bubbled. The mixture was filtered through a pad of Celite and the filtrate was concentrated in vacuo. The residue was partitioned between EtOAc (300 mL) and H2O (200 mL). The separated aqueous phase was extracted with EtOAc (100 mL x 4). The combined organic phases were washed with saturated aqueous a2C03 (100 mL x 2) followed by brine (100 mL), dried over anhydrous a2S04, filtered and concentrated in vacuo to give the title compound as brown-red liquid (43.4 g, 95%). MS (ESI, pos. ion) m/z: 243.0 [M + H]+; NMR (400 MHz, CDC13) delta (ppm): 8.02 (d, J = 2.24 Hz, 1H), 7.48 (dd, J = 8.16 Hz, 2.20 Hz, 1H), 7.09 (d, J= 8.20 Hz, 1H), 4.31-4.38 (q, J= 7.16 Hz, 2H), 2.53 (s, 3H), 1.35-1.42 (t, J = 7.16 Hz, 3H).
95%
With sulfuric acid;Reflux;
5-Bromo-2-methylbenzoic acid (40 g, 0.19 mol)Suspended in ethanol (600 mL)To this was slowly added 98% sulfuric acid (3 mL).The reaction solution was refluxed overnight,After cooling to 0 C,Add sodium bicarbonate until no gas is produced.The mixture was filtered through celite and the filtrate was concentrated under reduced pressure.The residue was partitioned between ethyl acetate (300 mL) and water (200 mL)The separated aqueous phase was extracted with ethyl acetate (100 mL x 4).The combined organic phases were washed sequentially with saturated sodium carbonate solution (100 mL x 2), brine(100 ml), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.The title compound was obtained as a reddish brown solid (43.4 g, 95%).
69%
sulfuric acid; at 0 - 100℃; for 15h;Inert atmosphere;
Step 1 : Ethyl 5-bromo-2-methylbenzoate (3)To a mixture of acid 2 (50.0 g, 233 mmol) in EtOH (700 mL) was added c-H2S04 (50 mL) at 0 C. The mixture was warmed-up to room temperature and stirred at 100C for 15 h. The mixture was cooled to room temperature and evaporated in vacuo to remove EtOH. The residue was diluted with EtOAc and washed with H20, aq. saturated NaHC03 solution. The organic layer was dried over anhydrous MgS04, filtered, and concentrated in vacuo. The residue was purified by silica column chromatography to provide the intermediate 3 (39.0 g, 69%).1H NMR (400 MHz, CDC13) delta 8.02 (d, J = 1.6 Hz, 1H), 7.71 (dd, J = 8.0, 2.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 4.32 (q, J = 7.2 Hz, 2H), 2.55 (s, 3H), 1.40 (t, J = 7.2 Hz, 2H); MH+ 243.
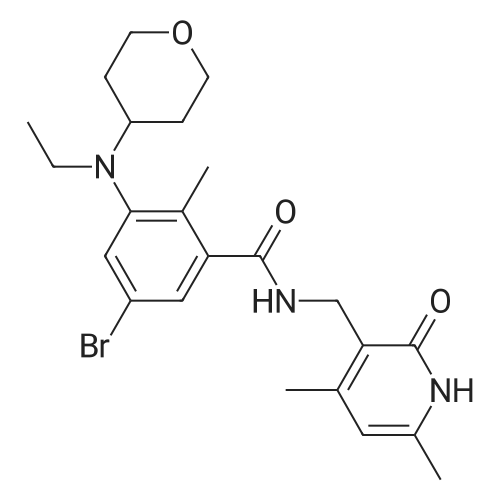
With ammonium hydroxide; N-ethyl-N,N-diisopropylamine; HATU; In N,N-dimethyl-formamide; at 20℃; for 16h;
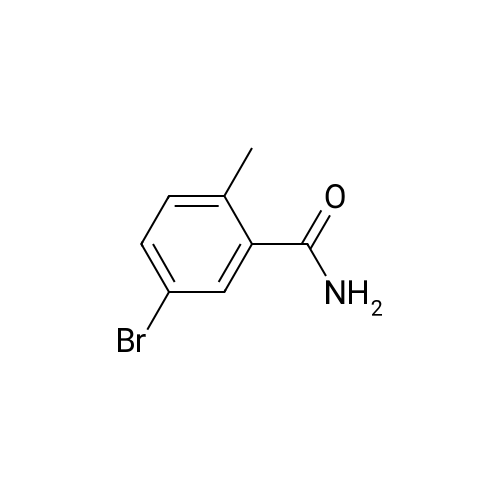
To a mixture of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (0.538 g, 2.50 mmol) and HATU(1.289 g, 3.390 mmo/) in DMF (8 mL) was added DIPEA (0.800 mL, 4.59 mmol). The mixture was stirred for 10 minutes before addition of NH OH (0.50 mL) and then left stirring for 16 hours at room temperature. The mixture was poured in to water and cooled at 0 C for 20 minutes before collecting the resulting precipitate via vacuum filtration to give the title compound (169) (0.292 g, 55%); 1H NMR (400 MHz, CDCI3) delta 7.58 (d, J = 2.1 Hz, 1 H), 7.45 (dd, J = 8.2, 2.1 Hz, 1 H), 7.12 (d, J = 8.2 Hz, 1 H), 5.68 (bs, 2H), 2.44 (s, 3H). LCMS Method C: rt 4.89 min; m/z 214, 216 (M+H]
55%
To a mixture of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (0,538 g, 2,50 mmo) and HATU (1.289 g, 3.390 mmoi) in DMF (8 mL) was added DPEA (0.800 mL, 4.59 mmoi). The mixture was stirred for 10 minutes before addition of NH4OH (0.50 mL) and then left stirring for 16 hours at room temperature. The mixture was poured in to water and cooled at 0 C for 20 minutes before collecting the resulting precipitate via vacuum filtration to give the title compound (169) (0.292 g, 55%); H N R (400 MHz, CDCI3) delta 7.58 (d, J = 2.1 Hz, 1 H), 7.45 (dd, J = 8.2, 2.1 Hz, 1 H), 7.12 (d, J = 8.2 Hz, 1 H), 5.68 (bs, 2H), 2.44 (s, 3H). LCMS Method C: rt 4.89 min; m/z 214, 216 [M+H]+.
To a stirred mixture 2 (20 g, 93.023 mmol) was added tocooled conc. H2SO4 (100 ml) at -10 C lot wise. After10 min nitrating mixture (prepared as mixing KNO3 (9.39 g,93.023 mmol) with conc. H2SO4 (100 ml) was added dropwise at -10 C. Resulting reaction mass was stirred at-10 C for 1 h. On completion, reaction mixture waspoured on ice-cold water; Solid was precipitated out, filteredand dried under vacuum to afford compound 3.Yield: 82.50%. 1HNMR (DMSO-d6, 400 MHz) delta 13.85(bs, 1H); 8.37 (s, 1H); 8.18 (s, 1H); 2.50 (s, 3H). LCMS:m/e 260/262 (M+ 1).
52%
With sulfuric acid; nitric acid; In acetone; at -5 - 0℃; for 2.25h;Cooling with acetone-ice;
Step 1: Synthesis of 5-bromo-2-methyl-3-nitrobenzoic acid A solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (5.0 g, 23 mmol) in concentrated H2SO4 (27 ml, 512 mmol) was cooled to 5 C. in an acetone/ice bath. A mixture of concentrated nitric acid (1.9 ml, 30 mmol) and concentrated H2SO4 (2.8 ml, 52 mmol) was added dropwise to the reaction mixture at -5 to 0 C. over 15 minutes. The yellow reaction mixture was stirred at -5 to 0 C. for 2 hours during which time a yellow precipitate formed. The reaction mixture was poured onto ice (150 g) and the precipitate was then collected by filtration. The precipitate was air dried to give the title compound (5.5 g, 52%) as a pale yellow solid. LC-MS 57%, 1.82 min (3.5 minute LC-MS method), no ionization; 1H NMR (500 MHz, DMSO-d6) delta ppm 8.29 (s, 1H) 8.13 (d, J=1.58 Hz, 1H) 2.43 (s, 3H).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; N,N-dimethyl-formamide; at 100℃; for 4h;Inert atmosphere;
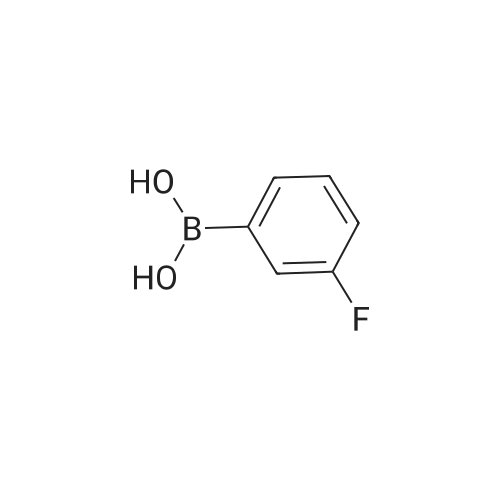
To a solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (1 g, 5.12 mmol, 1.0 eq) and (3- fluorophenyl)boronic acid (720 mg, 4.65 mmol, 1.1 eq) in a mixture of EtOH (5 mL), DMF (20 mL) and H20 (5 mL) were added Na2C03 (1.98g, 18.68 mmol, 4 eq) and Pd(PPh3)4 (270 mg, 0.23 mmol, 0.05 eq). The mixture was heated at 100C under N2 for 4h and the reaction quenched by addition of 1M HCl. The aqueous phase was extracted with EtOAc and the combined organic extracts washed with brine, dried (Na2SC>4), filtered and evaporated in vacuo. The residue was purified by chromatography (petroleum ethenEtOAc, 20: 1 to 1 : 1) to give the title compound as a white solid (780 mg, 68 %).LC-MS: m z 229.0 [M-H]" 1H NMR (400 MHz, DMSO-d6) ? 13.01 (br s, 1H), 8.08 (d, J= 1.9 Hz, 1H), 7.78 (dd, J = 7.9, 1.9 Hz, 1H), 7.56 - 7.47 (m, 3H), 7.41 (d, J= 8.0 Hz, 1H), 7.25 - 7.16 (m, 1H), 2.55 (s, 3H)
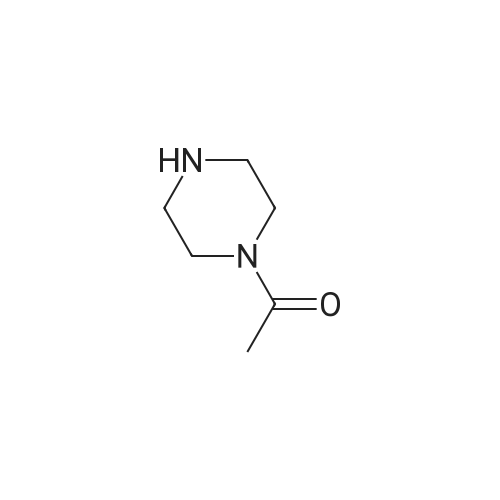
To a mixture of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (2.0 g, 9.30 mmol) in DCM (25 mL) was added DIPEA (3.25 mL, 18.60 mmol) and HBTU (4.23 g, 1 1.16 mmol) at rt. The reaction mixture was stirred at rt for 20 min. To the mixture was then added 1 -(piperazin-1 - yl)ethanone (1.31 1 g, 10.23 mmol) and the reaction mixture was stirred at rt for 1 hour. The reaction was quenched with a saturated aqueous solution of NaHC03 and extracted with DCM. The organic layer was washed twice with brine, dried by passing through a phase separating cartridge and evaporated. Purification by Flash chromatography using Biotage Isolera system (amine functionalized silica KP-NH, eluting with Cyclohexane/EtOAc 0 to 100%) gave the title compound (2.475 g, 82% yield) as a white foam. MS: 325.4 [M+1]+, Rt (2) = 0.94 min.
82%
To a mixture of <strong>[79669-49-1]5-bromo-2-methyl benzoic acid</strong> (2.0 g, 9.30 mmol) in DCM (25 ml_) was added DIPEA (3.25 ml_, 18.60 mmol) and HBTU (4.23 g, 11.16 mmol) at rt. The reaction mixture was stirred at rt for 20 min. To the mixture was then added 1-(piperazin-1-yl)ethanone (1.31 1 g, 10.23 mmol) and the reaction mixture was stirred at rt for 1 hour. The reaction was quenched with a saturated aqueous solution of NaHC03 and extracted with DCM. The organic layer was washed twice with brine, dried by passing through a phase separating cartridge and evaporated. Purification by Flash chromatography using Biotage Isolera system (amine functionalized silica KP-NH, eluting with Cyclohexane/EtOAc 0 to 100%) gave the title compound (2.475 g, 82% yield) as a white foam. MS: 325.4 [M+1]+, Rt (2) = 0.94 min.
82%
1-[4-(5-Bromo-2-methyl-benzoyl)-piperazin-1-yl]-ethanone To a mixture of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (2.0 g, 9.30 mmol) in DCM (25 mL) was added DIPEA (3.25 mL, 18.60 mmol) and HBTU (4.23 g, 11.16 mmol) at rt. The reaction mixture was stirred at rt for 20 min. To the mixture was then added 1-(piperazin-1-yl)ethanone (1.311 g, 10.23 mmol) and the reaction mixture was stirred at rt for 1 hour. The reaction was quenched with a saturated aqueous solution of NaHCO3 and extracted with DCM. The organic layer was washed twice with brine, dried by passing through a phase separating cartridge and evaporated. Purification by Flash chromatography using Biotage Isolera system (amine functionalized silica KP-NH, eluting with Cyclohexane/EtOAc 0 to 100%) gave the title compound (2.475 g, 82% yield) as a white foam. MS: 325.4 [M+1], Rt(2)=0.94 min.
Scheme 8. step A. To a solution of 5-bromo-2-methylbenzoic acid (2.0 g, 0.0093mol) in CH2Ch (20 mL) at 0 C are added methyl3-amino-2,4-dimethylbenzoate (1.49 g,0.0084 mol, see preparation 10) and N,N-diisopropylethylamine (4.79 g, 0.0372 mol). After stirring the reaction mixture for 10 minutes, 1-propanephosphonic acid cyclicanhydride (50% solution in ethyl acetate, 8.87 g, 0.028 mol) is added via syringe andstirred at 50C. After 16 hours, the solvent is removed under reduced pressure and theresidue is diluted with water and extracted twice with ethyl acetate. The organic layersare combined and dried over anhydrous Na2S04, filtered, and concentrated under reduced10 pressure. The resulting residue is purified by flash chromatography (silica gel) using 10%ethyl acetate in hexane to give the title compound as a white solid (2.80 g, 80%). Massspectrum (m/z): 376.1 (M+1).
Scheme 11, step A. To a solution of 5-bromo-2-methyl-benzoic acid (2.0 g,0.0093 mol) in CH2Ch (20 ml) at room temperature are added methyl3-amino-3,5-dimethylbenzoate (1.49 g, 0. 0083 mol, see preparation 12) and N,N-diisopropylethylamine (4.79 g, 0.037 mol). After stirring the reaction mixture for 10minutes, 1-propanephosphonic acid cyclic anhydride (50percent solution in ethyl acetate, 8.87g, 0.027 mol) is added via syringe and heated at 50°C. After 16 hours, the reactionmixture is diluted with CH2Ch, washed with water and brine. The organic layers arecombined and dried over anhydrous Na2S04, filtered, and concentrated under reduced5 pressure. The residue is purified by flash chromatography (silica gel) using 10percent EtOAcin hexanes to give the title compound as a white powder (2.8 g, 80 percent).
Scheme 9, step A. To a solution of 5-bromo-2-methylbenzoic acid (2.0 g, 0.008mol) in CH2Ch (20 mL) at 0 °C are added methyl4-amino-3,5-dimethylbenzoate (1.28 g,0.0072, see preparation 12) and N,N-diisopropylethylamine ( 4.12 g, 0.032 mol). Afterstirring the reaction mixture for 10 minutes, 1-propanephosphonic acid cyclic anhydride (50percent solution in ethyl acetate, 7.63 g, 0.024 mol) is added via syringe and stirred at 50°C.After 16 hours, the solvent is removed under reduced pressure and the residue is dilutedwith water and extracted twice with ethyl acetate. The organic layers are combined anddried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Theresulting residue is purified by flash chromatography (silica gel) using 12percent ethyl acetate in hexane to give the title compound as a white solid (2.9 g, 97 percent). Mass spectrum (m/z):376.0 (M+ 1).
Scheme 13, step A. To a solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (2.45 g,11.16 mmol) in THF (10 ml), CH2Ch (10 ml) and DMF (20.00 ~-tl, 258.65 ~-tmoles) isadded dropwise oxalyl chloride (1.16 ml, 13.39 mmoles) at 0 oc and the reaction mixture is slowly allowed to warm to ambient temperature. After 2 hours, the solvent is removedunder reduced pressure. To the residue is added CH2Ch ( 40 ml) and the reaction mixtureis cooled to 0 C, then ethyl4-amino-3,5-dimethyl-benzoate (2 g, 11.16 mmol) is addedfollowed by N,N-dimethylpyridin-4-amine (13.63 mg, 0.11 mmoles) and pyridine (2.71ml, 33.48 mmoles). The cooling bath is removed and the clear solution is allowed to warm to ambient temperature. After 2 hours, the solvent is removed and the reactionmixture is diluted with ethyl acetate and washed with IN HCI, a saturated solution ofsodium bicarbonate, and brine. The organic layers are combined, dried over anhydrousNa2S04, filtered, and concentrated under reduced pressure to give the title compound as a light yellow solid (4.0 g; 95.5%). Mass spectrum (m/z): 376.0 (M+ 1).
Scheme 12. step A. To a solution of <strong>[79669-49-1]5-bromo-2-methyl-benzoic acid</strong> (1.35 g, 6.15mmol) in THF (15 ml), CH2Ch (15 ml) and DMF (14.27 ~-tl, 184.57 ~-tmoles) is added dropwise oxalyl chloride (587.12 ~-tl, 6.77 mmoles) at 0 oc and reaction mixture is slowlyallowed to warm to ambient temperature. After 2 hours, the solvent is removed underreduced pressure. To the residue CH2Ch (30 ml) is added and reaction mixture is cooledto 0 C, then ethyl4-amino-3,5-dimethyl-benzoate (1.19 g, 6.15 mmol) is added followedby 4-pyridinamine, N,N-dimethyl- (37.58 mg, 307.61 ~-tmoles) and pyridine (1.49 ml,15 18.46 mmoles). The cooling bath is removed and the clear solution is allowed to warm toambient temperature. After 5 hours, the solvent is removed and the reaction mixture isdiluted with ethyl acetate and washed with IN HCl, saturated solution of sodiumbicarbonate, and brine. The organic layers are combined and dried over anhydrousNa2S04, filtered, and concentrated under reduced pressure. The resulting residue istriturated with hexanes and the resulting solids were collected by filtration to give the title compound as a white solid (1.80 g; 75.2%). Mass spectrum (m/z): 390.2 (M+ 1).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In water; acetonitrile; at 80℃; for 18h;Inert atmosphere;
To a suspension of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (4000 mg, 18.6 mmol) in AcN (40 mL) under Ar atmosphere, 3-chlorophenylboronic acid (4360 mg, 27.9 mmol), Pd(PPh3)4 (1075 mg, 0.93 mmol), and 2M Na2CO3 aqueous solution (28 mL, 55.8 mmol) were added. The reaction mixture was stirred at 80 C for 18 h. Organic solvents were evaporated under reduced pressure. The aqueous layer was washed twice with EtOAc and the pH of the solution was adjusted to 2 by adding 10 % HCl aqueous solution. The resulting mixture was filtered and the collected solids were washed with water and dried overnight in the oven to afford 3360 mg of the desired product (73.2 % yield). LC-MS (method 1): tR = 1.46 min; m/z= 245 (MH-).
73.2%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In water; acetonitrile; at 80℃; for 18h;Inert atmosphere;
To a suspension of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (4000 mg, 18.6 mmol) in AcN (40 mL) under Ar atmosphere, 3-chlorophenylboronic acid (4360 mg, 27.9 mmol), Pd(PPh3)4 (1075 mg, 0.93 mmol), and 2M Na2C03 aqueous solution (28 mL, 55.8 mmol) were added. The reaction mixture was stirred at 80 C for 18 h. Organic solvents were evaporated under reduced pressure. The aqueous layer was washed twice with EtOAc and the pH of the solution was adjusted to 2 by adding 10 % HCI aqueous solution. The resulting mixture was filtered and the collected solids were washed with water and dried overnight in the oven to afford 3360 mg of the desired product (73.2 % yield). LC-MS (method 1 ): tR = 1.46 min; m/z = 245 (MH").
With tri-tert-butyl phosphine; palladium diacetate; In tetrahydrofuran; 1-methyl-pyrrolidin-2-one; toluene; at 100℃; for 18h;Inert atmosphere;
To a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (600 mg, 2.79 mmol) in NMP (6 mL) under Ar atmosphere, 0.5 M (cyclohexylmethyl)zinc(ll) chloride solution in THF (16.7 mL, 8.37 mmol), Pd(AcO)2 (31 mg, 0.14 mmol), and 1 M PtBu3 solution in toluene (0.28 mL, 0.28 mmol) were added. The reaction mixture was stirred at 100 C for 18 h. Organic solvents were evaporated under reduced pressure. The aqueous layer was washed twice with EtOAc and water. The combined organic phases were dried over Na2S04 and concentrated to dryness. The crude residue was cromatographed on a silica gel flash system (Biotage SP1 ) using hexane/EtOAC mixtures of increasing polarity as eluent to afford 468 mg of the desired product (72.2 % yield). LC-MS (method 1 ): tR = 1.71 min; m/z = 231 (MH").
72.2%
With tri-tert-butyl phosphine; palladium diacetate; In tetrahydrofuran; 1-methyl-pyrrolidin-2-one; toluene; at 100℃; for 18h;Inert atmosphere;
To a solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (600 mg, 2.79 mmol) in NMP (6 mL) under Ar atmosphere, 0.5 M (cyclohexylmethyl)zinc(II) chloride solution in THF (16.7 mL, 8.37 mmol), Pd(AcO)2 (31 mg, 0.14 mmol), and 1 M PtBu3 solution in toluene (0.28 mL, 0.28 mmol) were added. The reaction mixture was stirred at 100 C for 18 h. Organic solvents were evaporated under reduced pressure. The aqueous layer was washed twice with EtOAc and water. The combined organic phases were dried over Na2SO4 and concentrated to dryness. The crude residue was cromatographed on a silica gel flash system (Biotage SP1) using hexane/EtOAC mixtures of increasing polarity as eluent to afford 468 mg of the desired product (72.2 % yield). LC-MS (method 1): tR = 1.71 min; m/z= 231 (MH-).
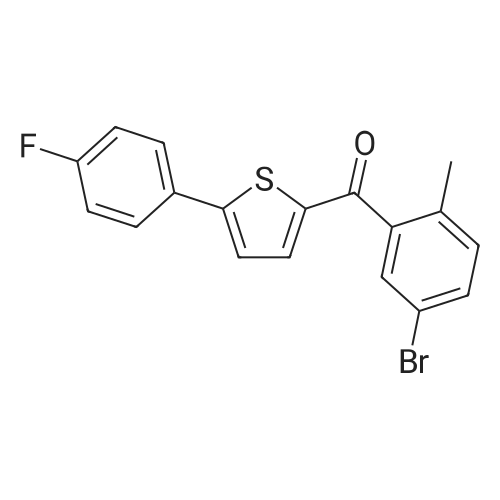
into the reaction flask add 30g of 5-bromo-2-methylbenzoic acid, 6mL DMF and 400mL Dichloromethane, stirring. Under the room temperature added the drops of 24.6g of thionyl chloride; after the drop is completed, the temperature is increased at 20-25 C; the reaction is monitored by TLC. After the reaction is completed, 25.2g of anhydrous aluminum chloride are added portion-wise into the reaction flask, stir for 30min, and cool down at 0-5 C, add the drops of 26.5g of a dichloromethane solution of <strong>[58861-48-6]2-(4-fluorophenyl)-thiophene</strong>; after dripping, warm up to 25-30 C and carry on reaction for 12 hours, TLC tracking reaction. After the reaction is over, the reaction solution is poured into 800mL ice water and quenched, stirring, static stratification. The aqueous phase is extracted with 300mL of dichloromethane; combine the organic phase, concentrated and then obtained Brown solid. The above solid is heated and dissolved with 8OOmL ethyl acetate, added the drops of 200mL of n-hexane, the crystals are stirred, filtered, and dried and then obtained 48.7g of a yellow solid product, yield is 93.2%.
80.4%
Oxalyl chloride (64.97 g) was slowly added to a solution of 5-bromo-2- methylbenzoic acid (100 g) in dichloromethane (1000 mL) and dimethylformamide (10 mL) at 25C to 30C under nitrogen atmosphere. The reaction mixture was stirred at the same temperature for about one hour. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a temperature of about 45 C under nitrogen atmosphere. The reaction mass obtained was dissolved in dichloromethane (1000 mL), and then cooled to 0C under nitrogen atmosphere. 2-(4-Fluorophenyl)thiophene (82.86 g; Example 1) and aluminum chloride (68 g) were added to the reaction mixture at 0C to 5C under nitrogen atmosphere. The reaction mixture was stirred for 60 minutes to 90 minutes at 10C to 15C under nitrogen atmosphere. After completion of the reaction, the reaction mixture was slowly added to water (1000 mL) at 0C to 10C. The reaction mixture was stirred at 25C to 30C for 10 to 15 minutes. The reaction mixture was allowed to settle for 15, minutes and then the layers were separated. Dichloromethane (200 mL) was added to the aqueous layer and the mixture was stirred for 10 minutes to 15 minutes. The mixture was allowed to settle for 15 minutes and then the layers were separated. The organic layers were combined, and then washed with an aqueous solution of sodium bicarbonate (50 g in 500 mL of de-ionized water). The organic layer was washed with an aqueous solution of sodium chloride (10 g in 200 mL of de-ionized water). The organic layer was concentrated under reduced pressure at a temperature of about 45C to obtain a residue. Hexanes (500 mL) were added to the residue, and then the reaction mixture was stirred for 15 minutes to 20 minutes at 25 C to 30C. The reaction mixture was cooled to 0C to 5C, and then stirred for 60 minutes to 120 minutes at the same temperature to obtain a solid. The solid was filtered under reduced pressure, then washed with hexanes (100 mL; pre-cooled), and then dried under reduced pressure at 40C to 45 C to obtain (5-bromo-2-methylphenyl)[5-(4-fluorophenyl)thiophen-2-yl]methanone. Yield: 80.4%
80%
In a 500-ml three-necked flask, 22 g (0.1 mol) of 2-methyl-5-bromobenzoic acid and 200 ml of dichloromethane were added, and 18.8 g (0.15 mol) of oxalyl chloride was slowly added dropwise with stirring, and the mixture was stirred at room temperature for 2 hours. The reaction was completely detected by TLC. Evaporation of excess oxalyl chloride under reduced pressure, cooling of the ice bath to -15C, addition of 200 ml of methylene chloride, 17.8 g (0.1 mol) of <strong>[58861-48-6]2-(4-fluorophenyl)thiophene</strong> prepared according to Example 3 and anhydrous trichlorin Aluminium 16g (0.12mol) was added and naturally warmed and controlled at a temperature of 25-35C for 4 hours. TLC detection reaction was completed, adding ice water 500ml to the reaction solution, stirring, standing to separate the organic layer, the aqueous layer was extracted with dichloromethane 100ml once, the organic phase was combined, the saturated saline solution was washed, and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain (5-bromo-2-methylphenyl)[<strong>[58861-48-6]2-(4-fluorophenyl)thiophene</strong>]methanone as a crude product, which was recrystallized from acetone to give 30.1 g of the product. The yield was 80%, and the purity was 99.3% by HPLC. .
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃;Inert atmosphere;
Into a 50-mE round-bottom flask, was placed a solution of<strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (1.5 g, 6.98 mmol, 1.00equiv) in dichloromethane (15 mE), 4-aminobenzonitrile(820 mg, 6.94 mmol, 1.00 equiv), EDCI (2 g, 10.43 mmol,1.50 equiv), 4-dimethylaminopyridine (1.28 g, 10.48 mmol,1.50 equiv). The resulting solution was stirred overnight atroom temperature. The resulting mixture was concentratedunder vacuum. The residue was dissolved in 100 mE of ethylacetate. The resulting mixture was washed with 3x30 mE ofhydrogen chloride (2.4 mol/E) and 2x30 mE of watet Theresulting mixture was washed with 2x30 mE of sodiumbicarbonate (aq.) and 2x30 mE of brine. The mixture wasdried over anhydrous sodium sulfate and concentrated undervacuum. This resulted in 1.93 g (88%) of 5-bromo-N-(4-cyanophenyl)-2-methylbenzamide as a light yellow solid.
With trifluorormethanesulfonic acid; bromine; iron;Molecular sieve;
2-methylbenzoic acid as the starting material,Under the action of metal reagent Fe powder and super acid catalyst trifluoromethanesulfonic acid,Adding a liquid bromine having a molar multiple of 1.05,And the solvent solution of bromine absolute molecular sieve without water pretreatment,Synthesis of intermediate 2 5-bromo-2-methyl benzoic acid, the yield of 85% or more
With [D]-sodium hydroxide; In water-d2; at 20 - 160℃; for 6.5h;
2-Methyl-5-bromobenzoic acid III-1 (5.0 g, 23.3 mmol) was weighed into a microwave pressure tube,NaOD (40%, 28.0 mmol),24 ml (24 g, 1.2 mol) of deuterated water was added,The mixture was stirred at room temperature for 30 minutes,The mixture was heated to 160 C and stirred for 4 hours.Remove the heavy water, then add deuterium water 24ml. The mixture was heated to 160 C and stirred for 2 hours. After cooling to 0 C, add DCl to pH = 3, and extract the reaction solution (3 × 30 ml) with CH2Cl2. After drying MgSO4, the filtrate was distilled off the solvent under reduced pressure to give an oily residue III-2 (4.6 g, yield 90%).
With 1-hydroxybenzotriazol-hydrate; triethylamine; dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;Inert atmosphere;
2-Methyl-5-bromobenzoic acid (2.15 g, 10 mmol) was added to a 50 ml single-necked flask,Dichloromethane (25 ml) was added sequentially,N, O-dimethylhydroxylamine hydrochloride (1.07 g, 11 mmol),Dicyclohexylcarbodiimide (2.26 g, 11 mmol),1-hydroxybenzotriazole monohydrate (1.48 g, 11 mmol).Triethylamine (1.67 ml, 12 mmol) was added under nitrogen.The reaction mixture was stirred overnight at room temperature. TLC showed that the starting material 2-methyl-5-bromobenzoic acid was still abundant. N, O-dimethylhydroxylamine hydrochloride (1.07 g, 11 mmol), dicyclohexylcarbodiimide (1.50 g, 7.3 mmol), triethylamine (1.0 mL, 7.2 mmol). Stirring at room temperature for 5 hours showed no significant residual TLC. The reaction solution was filtered under reduced pressure and the filter cake was washed twice with dichloromethane (5 ml x 2). The filtrate was successively added with 20 ml of dichloromethane and 20 ml of water, and the aqueous layer was partitioned and extracted with 10 ml of dichloromethane. The combined organic layers were washed with saturated brine, dried over anhydrous sodium sulfate and distilled under reduced pressureThe crude product. The crude product was purified by column chromatography on silica gel eluting with petroleum ether-ethyl acetate (20: 1, v / v) to give pale yellow viscous liquid II-3 (1.42 g, 5.48 mmol) in 54.8% yield.
2-amino-1-(4-(dibenzo[b,f][1,4]thiazepin-11-yl)piperazin-1-yl)-3-phenylpropan-1-one[ No CAS ]
[ 79669-49-1 ]
5-bromo-N-(1-(4-(dibenzo[b,f][1,4]thiazepin-11-yl)piperazin-1-yl)-1-oxo-3-phenylpropan-2-yl)-2-methylbenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
79%
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 12h;Inert atmosphere;
General procedure: General Procedure: To a 25 mL RBFwas charged Compound14 (0.200 gm,0.452 mmol), carboxylic acid15 (0.474 mmol), TBTU (0.189 gm,0.587 mmol), DMF (2.0 mL, 10 Vol) and DIPEA (0.24 mL, 1.356 mmol) at RT undernitrogen atmosphere. The reaction was stirred for 12 hr at RT then monitored byTLC, SM was absent. The reaction was diluted with water (20 mL) and extractedwith EtOAc (20 mL x 2) then dried with Na2SO4. Thesolution was distilled under vacuum at 40 C and dried to get crude product.The crude product was purified by Column chromatography to obtained product.
With potassium bromide; In dichloromethane; at 40℃;Inert atmosphere;
Intermediate 72: 6-Bromo-3H-isobenzofuran-l-one[0613] Iodosobenzene-l,l-diacetate (5.39g) was added in portions to a stirred, degassed suspension of 5-bromo-2-methylbenzoic acid (3.0g) and potassium bromide (1.66g) in dry DCM (70ml) under an atmosphere of nitrogen. The resultant mixture was stirred and heated at 40C overnight. After cooling to room temperature, the mixture was washed with NaHC03 (saturated aqueous solution), sodium sulphite (saturated aqueous solution), dried (Na2S04) and filtered. The filtrate was evaporated to dryness and the residue was purified by chromatography on silica, eluting with a mixture of ethyl acetate and cyclohexane with a gradient of 0-30% to give 6-bromo-3H-isobenzofuran-l-one (1.3g) as a white solid.[0614] NMR (CDC13) delta 8.1 (d, 1H), 7.8 (dd, 1H), 7.35 (d, 1H), 5.3 (s, 2H).
2-((5-Bromo-2-methylbenzyl)oxy)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
98%Spectr.
With lithium anilide; In tetrahydrofuran; at 20℃; for 1.25h;Inert atmosphere;
In an inert gas atmosphere,Add <strong>[79669-49-1]2-methyl-5-bromo-benzoic acid</strong> to the reaction vial after dehydration and deoxidation(107.1 mg, 0.5 mmol, using a pipette to add pinacol borane (289 muL, 2 mmol), and finally adding anilidine lithium (0.8 mol%) in tetrahydrofuran solution, reacting at room temperature for 75 minutes, the reaction solution The air was contacted and the solvent was removed to give the product boronic ester. The yield of 1H was calculated to be 98%.Add 1 g of silicon to the system for removing the solvent after the hydroboration reactionGlue, 3mL methanol, reaction at 50 C for 135 minutes, after the reaction is over,The organic layer was combined and dried over anhydrous sodium sulfate.The solvent was removed under reduced pressure and purified by silica gel (100-200 mesh) column chromatography.Using ethyl acetate/hexane (1:5 by volume) mixture as eluent,An alcohol compound is obtained. The nuclear magnetic yield was 91%.
99%Spectr.
With n-butyllithium; In tetrahydrofuran; at 20℃; for 0.75h;Inert atmosphere; Autoclave;
In an inert gas atmosphere,Add to the reaction bottle after dehydration and deoxidation<strong>[79669-49-1]2-methyl-5-bromo-benzoic acid</strong> (107.1 mg, 0.5 mmol,Add the pinacol borane (289 muL, 2 mmol) with a pipette.Finally, a solution of n-butyllithium (0.5 mol%) in tetrahydrofuran is added.After reacting for 45 minutes at room temperature, the reaction solution was exposed to air, and the solvent was removed to give the product boric acid ester, which was calculated to have a yield of 99%. Nuclear magnetic data of the product:1H NMR (400 MHz, CDCl3): delta 6.90 (d, 1H, ArCH), 7.19 (d, 1H, ArCH), 7.47 (s, 1H, ArCH), 4.77 (s, 2H, OCH2), 2.12 (s, 3H, CH3), 1.17 (s, 36H, CH3).To the system in which the solvent was removed after the hydroboration reaction, 1 g of silica gel and 3 mL of methanol were added.After the reaction was carried out at 50 C for 2 h, EtOAc / EtOAc (EtOAc) A mixture of hexane (1:5 by volume) was used as an eluent to give an alcohol compound. The nuclear magnetic yield was 95%.Nuclear magnetic data of the product: 1H NMR (400 MHz, CDCl3): delta 6.91 (d, 1H, ArCH), 7.22 (d, 1H, ArCH), 7.57 (s, 1H, ArCH), 4.44 (s, 2H, OCH2) , 2.15 (s, 3H, CH3), 2.25 (br s, 1H, OH).
83.77 mg
With lithium anilide; In tetrahydrofuran; at 20℃; for 1.25h;Inert atmosphere;
In an inert gas atmosphere, To the reaction flask after dehydration and deoxygenation, <strong>[79669-49-1]2-methyl-5-bromo-benzoic acid</strong> (107.1 mg, 0.5 mmol) was added to the pinacol borane (289 muL, 2 mmol) using a pipette.Finally, a solution of lithium anilide (0.8 mol%) in tetrahydrofuran is added.After reacting at room temperature for 75 minutes, the reaction solution was contacted with air to terminate the reaction, and the solvent was removed under reduced pressure to give the product boric acid ester, using tris-methoxybenzene (83.77 mg, 0.5 mmol) as an internal standard.Dissolve with CDCl3, stir for 10 minutes, sample,With nuclear magnetic. The 1H yield was calculated to be 99%.
99%Spectr.
at 20℃; for 12h;Inert atmosphere; Green chemistry;
Add <strong>[79669-49-1]2-methyl-5-bromo-benzoic acid</strong> (107.1 mg, 0.5 mmol) to the reaction flask after dehydration and deoxidation under an inert gas atmosphere of N2, and add pinacol borane (289 muL, using a pipette). 2 mmol), react at room temperature for 12 hours, remove the reaction from the glove box, use trimethyl benzene (83.77 mg, 0.5 mmol) as internal standard, dissolve with CDCl3, stir for 10 minutes, sample, and equilibrate with NMR. Calculated 1H The yield was 99%.
5-bromo-2-methyl-N-(6-methylpyridin-2-yl)benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57%
A solution of <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (5.0 g, 23.25 mmol) in thionyl chloride(15 mL) was heated at reflux for 3 hours. The completion of reaction was observedby TLC. The thionyl chloride was evaporated and the residue was charged to thereaction mixture contained 6-methylpyridin-2-amine (2.5 g, 23.14 mmol), triethylamine (5.8 g, 57.34 mmol) in dry ethyl acetate (50 mL) at 0C. The reactionmixture was stirred at RT (room temperature) for 12 hours. The completion ofreaction was observed by TLC. The reaction mixture was taken in water (200 mL),extracted with ethyl acetate (2 x 100 mL). The combined organic layer was washed with water (2 x 100 mL), evaporated. The crude was purified acetate in petroleum ether to pyridyl)benzamide intermediate 3 (4 g,brine, dried over sodium sulphate and by column chromatography using ethyl afford 5-bromo-2-methyl-N-(6-methyl-2- 57%) as an off white solid.
5-bromo-N-(3-hydroxypropyl)-N,2-dimethylbenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
5-Bromo-2-methylbenzoic acid (504 mg, 2.344 mmol), HATU (1053 mg, 2.77 mmol) and DIPEA (0.5 mL, 2.86 mmol) in DMF (4 mL) were stirred at room temp. for 30 min after which 3- (methylamino)propan-1-ol (0.275 mL, 2.83 mmol) was added. After 3 h the mixture was partitioned between ethyl acetate (20 mL) and water (15 mL). The separated aqueous layer was further extracted with ethyl acetate (15 mL). The combined organic layers were washed with 5% aqueous lithium chloride solution (2 x 10 mL), water (10 mL) and brine (10 mL). The organic layer was passed through a hydrophobic frit and the solvent removed in vacuo. The crude was purified by silica gel column chromatography eluting with 50 - 100% ethyl acetate in cyclohexane to give the title compound. LCMS (method F): rt = 0.80, [M+H]+ = 286.
(R)-5-bromo-N-(1-hydroxypropan-2-yl)-N,2-dimethylbenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Stage #1: 5-bromo-2-methylbenzoic acid With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate In tetrahydrofuran for 0.25h;
Stage #2: (2R)-2-(methylamino)propan-1-ol In tetrahydrofuran
5-Bromo-N-ethyl-N-(2-hydroxyethyl)-2-methoxybenzamide
General procedure: DIPEA (1.2 mL, 6.87 mmol) and HATU (980 mg, 2.58 mmol) were added to a stirred solution of 5-bromo-2-methoxybenzoic acid (500 mg, 2.164 mmol) in 2-MeTHF (10 mL). After 15 min 2- (ethylamino)ethan-1-ol (0.25 mL, 2.56 mmol) was added and the reaction left to stir overnight. The mixture was partitioned between ethyl acetate (50 mL) and saturated aqueous sodium bicarbonate (50 mL). The aqueous phase was further extracted with ethyl acetate (50 mL). The combined organic phase was washed with saturated aqueous sodium bicarbonate (50 mL) and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography eluting with 0 - 100% ethyl acetate: ethanol (3:1, containing 1% triethylamine) in cyclohexane to give the title compound. LCMS (method F): rt = 0.8, [M+H]+ = 302.
5-bromo-N-(1-hydroxypropan-2-yl)-N,2-dimethylbenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Stage #1: 5-bromo-2-methylbenzoic acid With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate In tetrahydrofuran for 0.25h;
Stage #2: 2-(methylamino)propan-1-ol In tetrahydrofuran
5-Bromo-N-ethyl-N-(2-hydroxyethyl)-2-methoxybenzamide
General procedure: DIPEA (1.2 mL, 6.87 mmol) and HATU (980 mg, 2.58 mmol) were added to a stirred solution of 5-bromo-2-methoxybenzoic acid (500 mg, 2.164 mmol) in 2-MeTHF (10 mL). After 15 min 2- (ethylamino)ethan-1-ol (0.25 mL, 2.56 mmol) was added and the reaction left to stir overnight. The mixture was partitioned between ethyl acetate (50 mL) and saturated aqueous sodium bicarbonate (50 mL). The aqueous phase was further extracted with ethyl acetate (50 mL). The combined organic phase was washed with saturated aqueous sodium bicarbonate (50 mL) and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography eluting with 0 - 100% ethyl acetate: ethanol (3:1, containing 1% triethylamine) in cyclohexane to give the title compound. LCMS (method F): rt = 0.8, [M+H]+ = 302.
<strong>[79669-49-1]2-methyl-4-bromobenzoic acid</strong> (1,100 g, 465 mmol) dissolved in 500 mL of dichloromethane and added oxalyl chloride (52 mL, 605 mmol) at room temperature Stir for 10 minutes. A catalytic amount of N,N-dimethylformamide (DMF, 3 mL) was added dropwise. Connect the exhaust gas absorption device, The reaction was stirred at room temperature until the reaction solution became homogeneous. Distilling off the volatile solutes under reduced pressure, The mixture was then dissolved in 1000 mL of dichloromethane. N,O-dimethylhydroxylamine hydrochloride (92 g, 930 mmol) was added. Cool the reaction solution to below 0 C, then triethylamine (215 mL, 1.533 mol) was slowly added with vigorous stirring. After the addition of triethylamine, The reaction was slowly warmed to room temperature and stirred overnight. The TCL plate was quenched with saturated ammonium chloride solution after completion of the reaction. The aqueous layer was extracted three times with dichloromethane, and the organic layers were combined. It was washed twice with saturated brine and dried over anhydrous sodium sulfate. It was filtered, and the solvent was evaporated under reduced pressure in vacuo. The resulting Weinreb amide (110 g, 97% yield) did not require purification for the next reaction.
tert-butyl (3R,4S)-3-amino-4-fluoropyrrolidine-1-carboxylate[ No CAS ]
tert-butyl (3R,4S)-3-(5-bromo-2-methylbenzamido)-4-fluoropyrrolidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92%
With (benzotriazo-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 2h;
A mixture of tert-butyl (3R,4S)-3-amino-4-fluoropyrrolidine-1- carboxylate (1 g, 4.90 mmol), <strong>[79669-49-1]5-bromo-2-methylbenzoic acid</strong> (1.053 g, 4.90 mmol), Huenig's base (2.57 mL, 14.69 mmol) and BOP (2.60 g, 5.88 mmol) in DMF (10 mL) was stirred at rt for 2 h. Water was added to the reaction mixture. The resulting precipitate was filtered and washed with water. It was dried to obtain tert-butyl (3R,4S)-3-(5-bromo-2- methylbenzamido)-4-fluoropyrrolidine-1-carboxylate (1.8 g, 4.49 mmol, 92 % yield) as a white solid. (1704) MS ESI m/z 424.8 (M+Na).
With oxalyl dichloride; In dichloromethane; at 0 - 20℃; for 1h;Inert atmosphere;
Compound BB-4A-1 (1.00 g, 4.65 mmol) and N,N-dimethylformamide (33.99 mg, 465.00 mumol) were dissolved in dichloromethane (10.00 mL) under nitrogen atmosphere, followed by dropwise addition of oxalyl chloride (1.48 g, 11.63 mmol) at 0 C. The reaction mixture was stirred for 1 hour at room temperature under nitrogen atmosphere. Methanol (1.49 g, 46.50 mmol) was added dropwise to the reaction solution. The reaction solution was concentrated under reduced pressure to remove solvent to give the crude compound BB-4A-2 (1.00 g, yield 93.88%).
Multi-step reaction with 3 steps
1.1: sulfuric acid / 16 h / 70 °C
2.1: 2,2'-azobis(isobutyronitrile); N-Bromosuccinimide / 8 h / 80 °C
2.2: 8 h / 0 - 25 °C
3.1: hydrogenchloride / ethanol / 1 h / 120 °C
Multi-step reaction with 4 steps
1: sulfuric acid / 16 h / 70 °C
2: 2,2'-azobis(isobutyronitrile); N-Bromosuccinimide / tetrachloromethane / 2 h / 85 °C
3: sodium hydride / N,N-dimethyl-formamide / 0.5 h / 0 - 25 °C / Inert atmosphere
4: hydrogenchloride / ethanol / 1 h / 120 °C
Multi-step reaction with 4 steps
1: sulfuric acid / 16 h / 70 °C
2: 2,2'-azobis(isobutyronitrile); N-Bromosuccinimide / tetrachloromethane / 8 h / 80 °C
3: sodium hydride / N,N-dimethyl-formamide / 0.5 h / 0 - 25 °C / Inert atmosphere
4: hydrogenchloride / ethanol / 1 h / 120 °C






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping