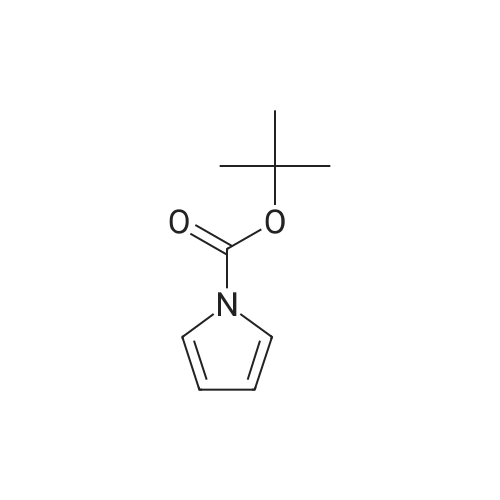
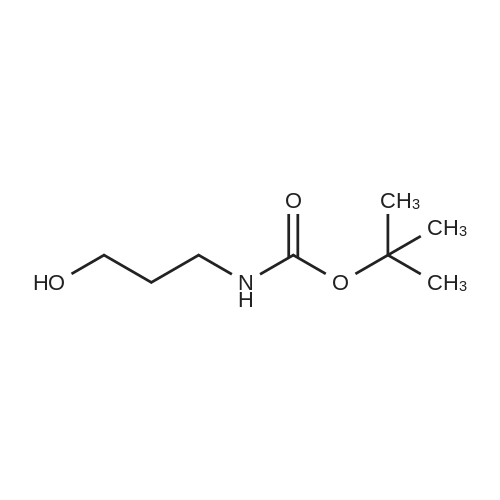
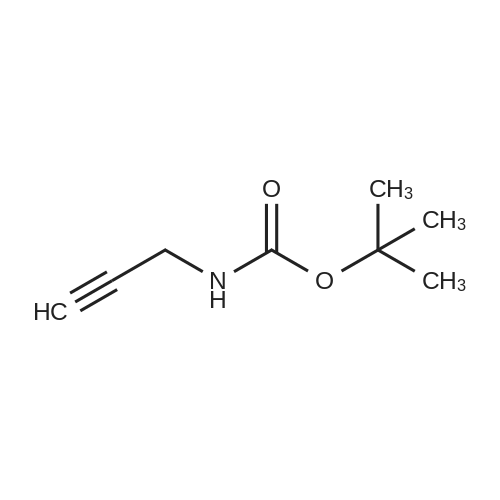
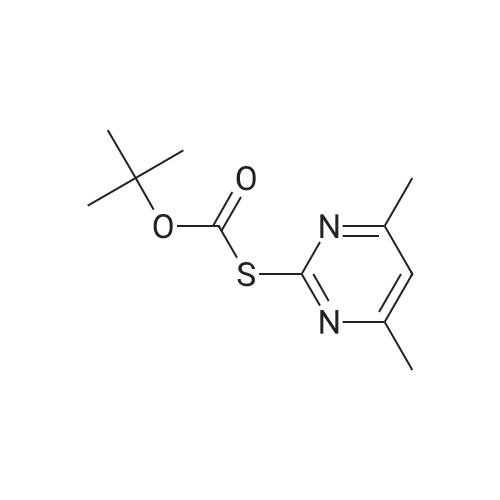
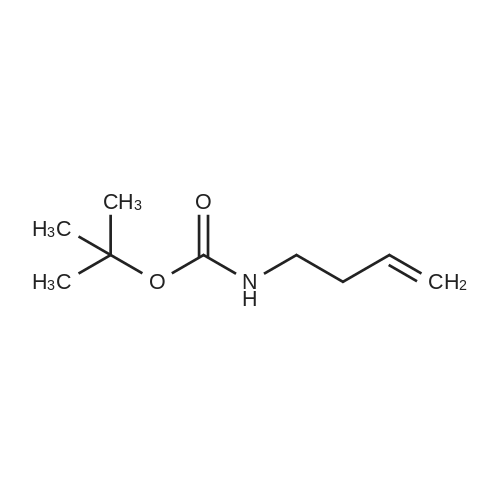
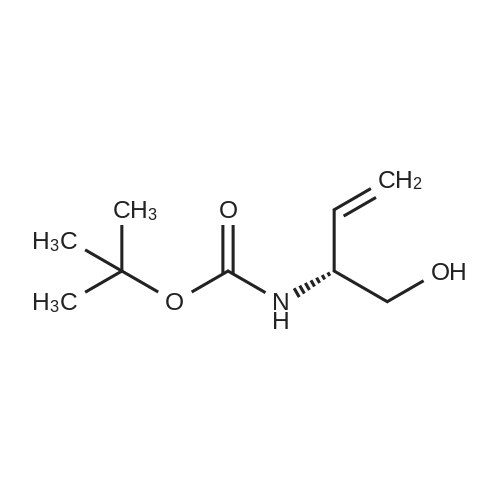
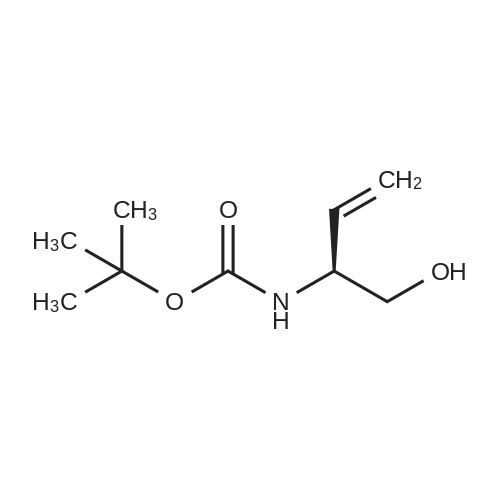
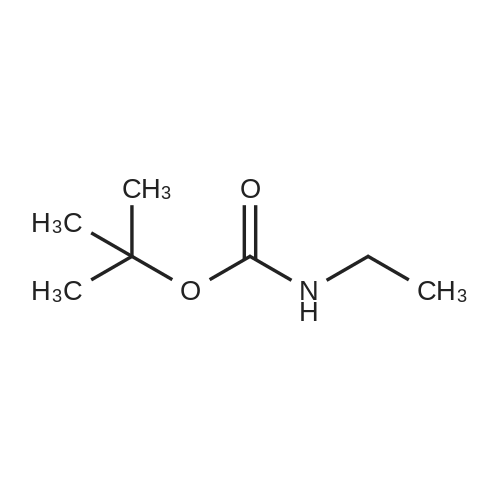
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
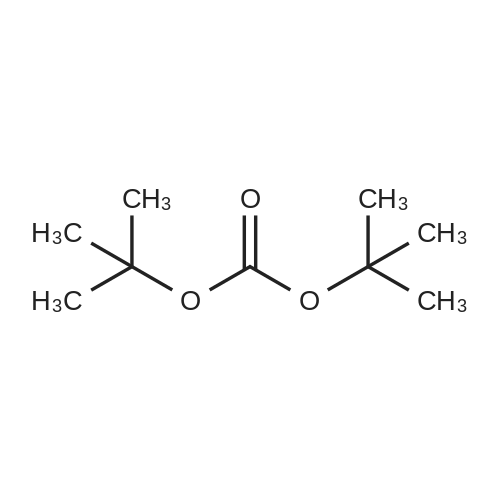
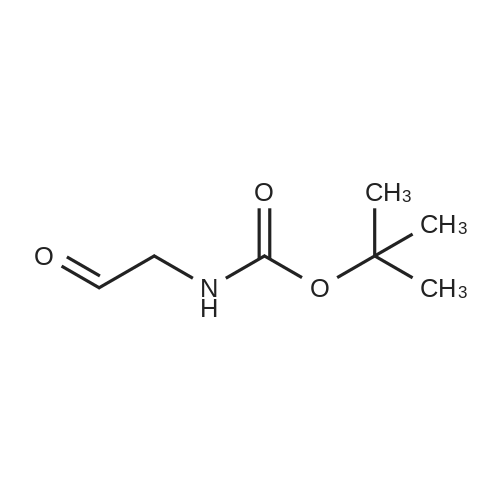
General procedure: The reactions were carried out in a 50 mL RB flask under reduced pressure for 10 min at 80°C unless reported differently. In a typical experiment, 5 mmol of amine was added to 5 mmol of BOC anhydride, and the reaction was allowed to proceed for 10 min. The desired product was obtained in a rotary evaporator under vacuum conditions.
91%
With triethylamine In dichloromethane at 0 - 20℃; for 5.5 h;
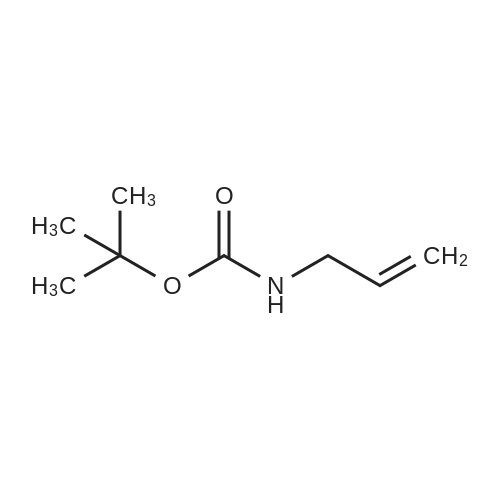
To a solution of compound 144 (8.80 g, 154.1 mmol) in CH2Cl2 (150 mL) was added TEA (32.2 mL, 231.2 mmol) and Boc2O (40.4 g, 185.3 mmol) at 0° C. The reaction mixture was continued to be stirred at 0° C. for 0.5 h, allowed to be warmed to room temperature and stirred for 5 h. Then the mixture was partitioned between CH2Cl2 (150 mL) and water (150 mL). The aqueous layer was separated and extracted with CH2Cl2 (2×150 mL). The combined organic extracts were washed with brine, dried over Na2SO4, concentrated to afford desired compound 145 (22.0 g, 91percent) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 5.90-5.77 (m, 1H), 5.17 (dq, J=17.1, 1.7 Hz, 1H), 5.10 (dq, J=10.4, 1.4 Hz, 1H), 4.64 (brs, 1H), 3.74 (t, J=5.2 Hz, 2H), 1.45 (s, 9H).
84.74%
at 0 - 20℃; for 4 h;
Example 235. Synthesis of 3-fluoro-2-(4-(6-hydroxy-4,5,6,7- tetrahydropyrazolo [1 ,5-a] pyrimidin-2-yl)-5-oxo-6,7-dihydro-5H-pyrrolo [3,4-b] pyridin-2- yl)benzonitrile 1-235 Synthesis of compound 235.2. To a solution of prop-2-en-l -amine (15.0g, 263mmol, 1.0 eq) in DCM (300mL) was added di-tert-butyl dicrabonate (57.3g, 263mmol, l.Oeq) at 0 °C. Reaction mixture was stirred at room temperature for 4 h. Upon completion of reaction; reaction mixture was washed with 5percent citric acid solution followed by brine. Organic layer was dried over Na2S04 and concentrated under reduced pressure to pressure to obtain pure 235.2 (35g, 84.74percent).
81%
With N-ethyl-N,N-diisopropylamine In tetrahydrofuran for 1 h; Inert atmosphere
General procedure: To a solution of allylamine (6; 3.0 mL, 39.8 mmol) in THF (20 mL) was added i-Pr2NEt (13 mL, 80.0 mmol) and Boc2O (9.0 mL, 39.9 mmol).After stirring 1 h, the reaction mixture was diluted with Et2O (50 mL),the organic layer was separated, and washed with sat. aq NH4Cl(3 × 20 mL), H2O (20 mL) and brine (20 mL), dried (Na2SO4), pouredover a silica gel plug, and concentrated under reduced pressure. Purification by flash column chromatography (hexanes–EtOAc, 90:10) delivered the corresponding Boc-allylamine (5.16 g, 81percent) as a clear oil.To a solution of this Boc-allylamine (2.60 g, 16.4 mmol) in p-xylene(16 mL) was added dioxinone 43(3.6 mL, 24.7 mmol). The reactionmixture was heated to reflux (150 °C) for 1.5 h, then allowed to coolto 23 °C, and concentrated under reduced pressure. Purification byflash column chromatography (hexanes–EtOAc, 80:20) delivered thecorresponding β-keto amide (2.35 g, 59percent) as a yellow oil. To a solutionof this β-keto amide (2.66 g, 11.0 mmol) in MeCN (55 mL) was addedp-ABSA (4.01 g, 16.5 mmol) and Et3N (6.2 mL, 44.1 mmol) at 0 °C. Thereaction was allowed to warm to 23 °C and stirred for 16 h. The mixture was concentrated and triturated with hexanes–EtOAc (1:1),poured over silica gel, and concentrated under reduced pressure. Purification by flash column chromatography (hexanes–EtOAc, 80:20) delivered 38bas a yellow oil; yield: 2.45 g (83percent)
80%
With triethylamine In dichloromethane at 0 - 20℃; for 12 h;
To a suspension of ally amine (2.0 g, 35.08 mmol) in dichloromethane was added Bocanhydride (8.4 g, 38.59 mmol), followed by triethylamine (5.3 g, 52.63 mmol) at 0 °C. The resulting reaction mixture was stirred at room temperature for 12 h. The reactionmixture was evaporated under reduced pressure to give the residue. Residue was purified by column chromatography on silica gel (hexanes/ethyl acetate =hexanes/ ethyl acetate =90/10) to give the titled compound (2.2 g, 80 percent) as a liquid.
80%
With 1H-imidazole In ethanol at 20℃;
Allylamine (0.75 mL, 10.02 mmol), abs. EtOH (50 mL), and B0C2O (2.40 g, 1 1.00 mmol) were stirred at room temperature under air until the effervescence subsided. Imidazole (0.81 g, 1 1.90 mmol) was then added and the reactioncontinued at room temperature under air for 1 h. The reaction was then concentrated in vacuo. The crude residue was purified via MPLC (silica, 10-20percent hexanes/EtOAc) to provide KSC-288- 055-1 (1.2637 g, 8.04 mmol, 80 percent yield). lR NMR (500 MHz, Chloroform-d) δ 5.82 (ddt, J = 15.8, 10.6, 5.4 Hz, 1H), 5.15 (dq, J = 17.2, 1.6 Hz, 1H), 5.08 (dq, J = 10.3, 1.4 Hz, 1H), 4.73 (s, 1H), 3.79-3.60 (m, 2H), 1.43 (s, 9H); 13C NMR (126 MHz, CDC13) δ 155.77, 134.91, 115.59, 79.25, 43.02, 28.36; HRMS (ESI-TOF) m/z: [M - C5H902 + 2H]+ Calcd for C3H8N 58.0651; Found 58.0661
68%
at 0 - 20℃; for 4 h;
Synthesis of tert-Butyl N-allylcarbamate (3) Tert-butyl N-allylcarbamate was synthesised according to the method of Rocheblave (30). 2.3 mL (30 mmol, 1 equiv) of freshly distilled allylamine 2 in 10 mL CH2Cl2 was cooled in an ice bath (0° C.). To this cold solution was added 6.54 g Boc2O (30 mmol, 1 equiv) in 20 mL CH2Cl2. The solution was brought to room temperature and stirred for 4 hours. The reaction mixture was then diluted with additional CH2Cl2 and washed with 5percent citric acid solution followed by brine. The organic layer was dried over Na2SO4 and concentrated in vacuo, yielding 3.29 g (68percent yield) of 3. 1H NMR (500 MHz, CDCl3): δ1.38 (s, 9H), 3.68 (brs, 2H), 4.53 (brs, 1H), 5.02-5.16 (m, 2H), 5.72-5.84 (m, 1H).
68%
at 20℃; for 4 h; Cooling with ice
Freshly-distilled allylamine (2.3 mL, 30 mmol, 1 eq) and 10 mL of anhydrous CH2Cl2 were combined and cooled in an ice bath. Di-tert-butyl dicarbonate (6.54 g, 30.0 mmol, 1.00 eq) in 20 mL anhydrous CH2Cl2 was added, and the mixture was stirred at room temperature for 4 h. The reaction mixture was diluted with additional CH2Cl2 and washed with 5percent citric acid solution, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to afford 3.3 g of tert-butyl allylcarbamate 9 with a yield of 68percent.1H NMR (400 MHz, CDCl3):δ 5.90-5.81 (m, 1H, NH), 5.24-4.96 (m, 2H, =CH2), 4.63 (s, 1H, =CH), 3.75 (s, 2H, CH2), 1.45 (s, 9H, CH3).
58%
at 0 - 20℃; for 1 h;
A solution of 5.88 ml (27.5 mmol, 1.1 eq.) of Boc2O in 50 ml of dry CH2Cl2 was addeddropwise to a solution of 1.88 ml (25.0 mmol, 1.0 eq.) of allyl amine in 60 ml of dry CH2Cl2at 0°C. The reaction mixture was stirred at r.t. for 1 h. After TLC showed completeconsumption of starting material, solvent was removed under reduced pressure. The crudeproduct was purified by flash column chromatography (cHex/EtOAc 12:1). The product wasobtained as a light yellow solid (yield: 2.26 g, 14.395 mmol, 58 percent).
Reference:
[1] Tetrahedron Letters, 2007, vol. 48, # 35, p. 6163 - 6166
[2] Angewandte Chemie - International Edition, 2014, vol. 53, # 41, p. 11075 - 11078
[3] Tetrahedron Letters, 1998, vol. 39, # 15, p. 2099 - 2102
[4] Journal of the Chemical Society. Perkin Transactions 1, 2001, # 16, p. 1916 - 1928
[5] Tetrahedron Letters, 2009, vol. 50, # 46, p. 6244 - 6246
[6] Journal of Organic Chemistry, 1998, vol. 63, # 26, p. 9904 - 9909
[7] Journal of Organic Chemistry, 2006, vol. 71, # 21, p. 8283 - 8286
[8] Journal of Medicinal Chemistry, 2002, vol. 45, # 15, p. 3321 - 3324
[9] Chemistry - A European Journal, 2016, vol. 22, # 35, p. 12274 - 12277
[10] Research on Chemical Intermediates, 2017, vol. 43, # 3, p. 1355 - 1363
[11] Tetrahedron, 2007, vol. 63, # 21, p. 4472 - 4490
[12] Tetrahedron Letters, 2009, vol. 50, # 23, p. 2716 - 2718
[13] Patent: US5534542, 1996, A,
[14] Journal of the American Chemical Society, 2006, vol. 128, # 50, p. 16323 - 16331
[15] Bulletin of the Chemical Society of Japan, 2009, vol. 82, # 12, p. 1520 - 1527
[16] ACS Combinatorial Science, 2013, vol. 15, # 7, p. 344 - 349
[17] Synthetic Communications, 1995, vol. 25, # 14, p. 2135 - 2143
[18] Journal of Organic Chemistry, 1988, vol. 53, # 15, p. 3457 - 3465
[19] Patent: US2014/171447, 2014, A1, . Location in patent: Paragraph 0427; 0428
[20] Polish Journal of Chemistry, 2006, vol. 80, # 6, p. 907 - 912
[21] Tetrahedron, 1990, vol. 46, # 6, p. 1975 - 1986
[22] Journal of Organic Chemistry, 2011, vol. 76, # 19, p. 7737 - 7749
[23] Patent: WO2017/40757, 2017, A1, . Location in patent: Paragraph 001058-001059
[24] Tetrahedron, 2009, vol. 65, # 24, p. 4751 - 4765
[25] Journal of Medicinal Chemistry, 1984, vol. 27, # 6, p. 711 - 712
[26] Synthesis (Germany), 2015, vol. 47, # 18, p. 2756 - 2766
[27] Patent: WO2014/111871, 2014, A1, . Location in patent: Page/Page column 29
[28] Patent: WO2014/201016, 2014, A2, . Location in patent: Paragraph 00169
[29] MedChemComm, 2015, vol. 6, # 5, p. 919 - 925
[30] Chemical Communications, 2017, vol. 53, # 26, p. 3713 - 3716
[31] Patent: US2008/312440, 2008, A1, . Location in patent: Page/Page column 9
[32] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 4, p. 949 - 954
[33] Synlett, 2018, vol. 29, # 6, p. 785 - 792
[34] Organic and Biomolecular Chemistry, 2005, vol. 3, # 12, p. 2333 - 2343
[35] Journal of Heterocyclic Chemistry, 1990, vol. 27, # 7, p. 2077 - 2079
[36] Liebigs Annalen der Chemie, 1989, p. 985 - 990
[37] Recueil des Travaux Chimiques des Pays-Bas-Journal of the Royal Netherlands, 1996, vol. 115, # 1, p. 13 - 19
[38] Tetrahedron Letters, 1999, vol. 40, # 22, p. 4173 - 4176
[39] Patent: US5220047, 1993, A,
[40] Patent: EP237082, 1991, B1,
[41] Journal of Organic Chemistry, 2008, vol. 73, # 22, p. 8987 - 8991
[42] Patent: EP1203767, 2002, A1, . Location in patent: Page 68
[43] Chemistry - A European Journal, 2012, vol. 18, # 18, p. 5589 - 5605
[44] Tetrahedron Letters, 2014, vol. 55, # 19, p. 3041 - 3044
[45] Tetrahedron, 2015, vol. 71, # 4, p. 554 - 559
[46] Patent: WO2016/149628, 2016, A1, . Location in patent: Paragraph 000140
[47] Heterocycles, 2017, vol. 95, # 2, p. 994 - 1029
[48] Organic Letters, 2017, vol. 19, # 1, p. 86 - 89
4
[ 92136-39-5 ]
[ 78888-18-3 ]
Yield
Reaction Conditions
Operation in experiment
63%
Stage #1: With n-butyllithium; copper(l) cyanide In tetrahydrofuran at -78℃; for 0.25 h; Stage #2: With tri-n-butyl-tin hydride In tetrahydrofuran at -78℃; for 0.333333 h; Stage #3: at -78℃; for 1 h;
A solution of copper cyanide (1.1Sg, 12.9 mmol) in THF (30 mL) at ?78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at ?78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at ?780C for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2percent ethyl acetate/heptane to provide the desired product (3.66 g, 63percent). 1H NMR (400 MHz, CDCl3) ? 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
With ammonium hydroxide; n-butyllithium; ammonium chloride; tri-n-butyl-tin hydride In tetrahydrofuran; n-heptane; dichloromethane; ethyl acetate
EXAMPLE 314A tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78° C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78° C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78° C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite.(R).). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2percent ethyl acetate/heptane to provide the desired product (3.66 g, 63percent). 1H NMR (400 MHz, CDCl3) δ 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
Stage #1: With n-butyllithium; copper(I) cyanide In tetrahydrofuran at -78℃; for 0.25 h; Stage #2: With tri-n-butyl-tin hydride In tetrahydrofuran for 0.333333 h;
tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78° C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78° C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78° C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite.(R).). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2percent ethyl acetate/heptane to provide the desired product (3.66 g, 63percent). 1H NMR (400 MHz, CDCl3) δ 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
1) Synthesis of N-t-butoxycarbonylallylamine (27) Allylamine [2.855 g (50 mmol.)] was dissolved in chloroform (100 m), to which was added t-butyl S-(4,6-dimethylpyrimidin-2-yl)thiocarbonate [12.017 g (50 mmol.)], and then the mixture was stirred at room temperature for 24 hours. 1N Hydrochloric acid was added to the reaction mixture, which was subjected to extraction with chloroform. The organic layer was dried over anhydrous potassium carbonate, and then concentrated under reduced pressure. The crude product thus obtained was purified by column chromatography (silica gel: 240 g; eluent: hexane.ethyl acetate = 3/1) to obtain the desired product (27) [6.720 g (85.5percent, colorless prisms, m.p. 34 to 34.8°C)].
With sodium hydrogencarbonate; In ethyl acetate; at 20℃; for 72h;Inert atmosphere;
General procedure: To a suspension of alkene (100 mol-%) and sodium bicarbonate (1000 mol-%) in ethyl acetate (0.5 M relative to starting alkene) was added <strong>[74213-24-4]dibromoformaldoxime</strong> (150 mol-%) under an atmosphere of argon. The reaction mixture was stirred at rt until the completion of the reaction (as monitored by LC-MS or TLC analysis), therefore the resulting slurry was filtered through Celite and washed with ethyl acetate. The solvent was evaporated and the residue purified by flash chromatography.
With sodium hydrogencarbonate; In ethyl acetate; at 20℃; for 4h;
Step A: (3-Bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamic acid tert-butyl ester Following the procedure described in Tetrahedron 46, 1990, 1975-1986, N-BOC-allylamine (1.8 g) was dissolved in ethyl acetate and treated with sodium hydrogenocarbonate (4.38 g) and <strong>[74213-24-4]dibromoformaldoxime</strong> (2.55 g). The reaction mixture was stirred at room temperature for 4 hours, then poured into water, extracted with ethyl acetate, the organic layer was dried over sodium sulfate and the solvent removed in vacuo. The title crude product was thus obtained as a colorless oil (3.16 g). LCMS (Method F) 1.48 min, M+H 179/181 (M-BOC).
In neat (no solvent); at 80℃; for 0.166667h;Green chemistry;
General procedure: The reactions were carried out in a 50 mL RB flask under reduced pressure for 10 min at 80C unless reported differently. In a typical experiment, 5 mmol of amine was added to 5 mmol of BOC anhydride, and the reaction was allowed to proceed for 10 min. The desired product was obtained in a rotary evaporator under vacuum conditions.
94.8%
With hydrogenchloride; triethylamine;
Step 1 3-t-Butoxycarbonylamino-1-propene. To a solution of 38.24 g of di-t-butyldicarbonate (175 mmol) and 17.7 g of triethylamine (175 mmol) in 100 mL of ether, was slowly added a solution of 10.0 g of allylamine (175 mmol) in 50 mL of ether. When the addition was complete and gas evolution had ceased, the solution was washed with three 100-mL portions of 0.5N hydrochloric acid and a 100-mL portion of distilled water, then dried over MgSO4. Removal of the solvent gave the product as a colorless liquid which crystallized slowly on standing to give large colorless crystals, producing 26.15 g of the product at a calculated yield of 94.8%. [Characterization: 1 H NMR (CDCl3): delta=1.48 (s, 9H), 3.76 (br, 2H), 4.60 (br, 1H), 5.08-5.22 (m, 2H), 5.75-5.9 (m, 1H); IR (thin film on KBr plate): 3347 (ms), 3080 (w), 2976 (ms), 2927 (m), 1696 (s), 1522 (ms), 1392 (w), 1368 (m), 1278 (m) 1250 (m), 1174 (m).
91%
With triethylamine; In dichloromethane; at 0 - 20℃; for 5.5h;
To a solution of compound 144 (8.80 g, 154.1 mmol) in CH2Cl2 (150 mL) was added TEA (32.2 mL, 231.2 mmol) and Boc2O (40.4 g, 185.3 mmol) at 0 C. The reaction mixture was continued to be stirred at 0 C. for 0.5 h, allowed to be warmed to room temperature and stirred for 5 h. Then the mixture was partitioned between CH2Cl2 (150 mL) and water (150 mL). The aqueous layer was separated and extracted with CH2Cl2 (2×150 mL). The combined organic extracts were washed with brine, dried over Na2SO4, concentrated to afford desired compound 145 (22.0 g, 91%) as a colorless oil. 1H NMR (400 MHz, CDCl3): delta 5.90-5.77 (m, 1H), 5.17 (dq, J=17.1, 1.7 Hz, 1H), 5.10 (dq, J=10.4, 1.4 Hz, 1H), 4.64 (brs, 1H), 3.74 (t, J=5.2 Hz, 2H), 1.45 (s, 9H).
84.74%
In dichloromethane; at 0 - 20℃; for 4h;
Example 235. Synthesis of 3-fluoro-2-(4-(6-hydroxy-4,5,6,7- tetrahydropyrazolo [1 ,5-a] pyrimidin-2-yl)-5-oxo-6,7-dihydro-5H-pyrrolo [3,4-b] pyridin-2- yl)benzonitrile 1-235 Synthesis of compound 235.2. To a solution of prop-2-en-l -amine (15.0g, 263mmol, 1.0 eq) in DCM (300mL) was added di-tert-butyl dicrabonate (57.3g, 263mmol, l.Oeq) at 0 C. Reaction mixture was stirred at room temperature for 4 h. Upon completion of reaction; reaction mixture was washed with 5% citric acid solution followed by brine. Organic layer was dried over Na2S04 and concentrated under reduced pressure to pressure to obtain pure 235.2 (35g, 84.74%).
81%
With N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; for 1h;Inert atmosphere;
General procedure: To a solution of allylamine (6; 3.0 mL, 39.8 mmol) in THF (20 mL) was added i-Pr2NEt (13 mL, 80.0 mmol) and Boc2O (9.0 mL, 39.9 mmol).After stirring 1 h, the reaction mixture was diluted with Et2O (50 mL),the organic layer was separated, and washed with sat. aq NH4Cl(3 × 20 mL), H2O (20 mL) and brine (20 mL), dried (Na2SO4), pouredover a silica gel plug, and concentrated under reduced pressure. Purification by flash column chromatography (hexanes-EtOAc, 90:10) delivered the corresponding Boc-allylamine (5.16 g, 81%) as a clear oil.To a solution of this Boc-allylamine (2.60 g, 16.4 mmol) in p-xylene(16 mL) was added dioxinone 43(3.6 mL, 24.7 mmol). The reactionmixture was heated to reflux (150 C) for 1.5 h, then allowed to coolto 23 C, and concentrated under reduced pressure. Purification byflash column chromatography (hexanes-EtOAc, 80:20) delivered thecorresponding beta-keto amide (2.35 g, 59%) as a yellow oil. To a solutionof this beta-keto amide (2.66 g, 11.0 mmol) in MeCN (55 mL) was addedp-ABSA (4.01 g, 16.5 mmol) and Et3N (6.2 mL, 44.1 mmol) at 0 C. Thereaction was allowed to warm to 23 C and stirred for 16 h. The mixture was concentrated and triturated with hexanes-EtOAc (1:1),poured over silica gel, and concentrated under reduced pressure. Purification by flash column chromatography (hexanes-EtOAc, 80:20) delivered 38bas a yellow oil; yield: 2.45 g (83%)
80%
With triethylamine; In dichloromethane; at 0 - 20℃; for 12h;
To a suspension of ally amine (2.0 g, 35.08 mmol) in dichloromethane was added Bocanhydride (8.4 g, 38.59 mmol), followed by triethylamine (5.3 g, 52.63 mmol) at 0 C. The resulting reaction mixture was stirred at room temperature for 12 h. The reactionmixture was evaporated under reduced pressure to give the residue. Residue was purified by column chromatography on silica gel (hexanes/ethyl acetate =hexanes/ ethyl acetate =90/10) to give the titled compound (2.2 g, 80 %) as a liquid.
80%
With 1H-imidazole; In ethanol; at 20℃;
Allylamine (0.75 mL, 10.02 mmol), abs. EtOH (50 mL), and B0C2O (2.40 g, 1 1.00 mmol) were stirred at room temperature under air until the effervescence subsided. Imidazole (0.81 g, 1 1.90 mmol) was then added and the reactioncontinued at room temperature under air for 1 h. The reaction was then concentrated in vacuo. The crude residue was purified via MPLC (silica, 10-20% hexanes/EtOAc) to provide KSC-288- 055-1 (1.2637 g, 8.04 mmol, 80 % yield). lR NMR (500 MHz, Chloroform-d) delta 5.82 (ddt, J = 15.8, 10.6, 5.4 Hz, 1H), 5.15 (dq, J = 17.2, 1.6 Hz, 1H), 5.08 (dq, J = 10.3, 1.4 Hz, 1H), 4.73 (s, 1H), 3.79-3.60 (m, 2H), 1.43 (s, 9H); 13C NMR (126 MHz, CDC13) delta 155.77, 134.91, 115.59, 79.25, 43.02, 28.36; HRMS (ESI-TOF) m/z: [M - C5H902 + 2H]+ Calcd for C3H8N 58.0651; Found 58.0661
68%
In dichloromethane; at 0 - 20℃; for 4h;
Synthesis of tert-Butyl N-allylcarbamate (3) Tert-butyl N-allylcarbamate was synthesised according to the method of Rocheblave (30). 2.3 mL (30 mmol, 1 equiv) of freshly distilled allylamine 2 in 10 mL CH2Cl2 was cooled in an ice bath (0 C.). To this cold solution was added 6.54 g Boc2O (30 mmol, 1 equiv) in 20 mL CH2Cl2. The solution was brought to room temperature and stirred for 4 hours. The reaction mixture was then diluted with additional CH2Cl2 and washed with 5% citric acid solution followed by brine. The organic layer was dried over Na2SO4 and concentrated in vacuo, yielding 3.29 g (68% yield) of 3. 1H NMR (500 MHz, CDCl3): delta1.38 (s, 9H), 3.68 (brs, 2H), 4.53 (brs, 1H), 5.02-5.16 (m, 2H), 5.72-5.84 (m, 1H).
68%
In dichloromethane; at 20℃; for 4h;Cooling with ice;
Freshly-distilled allylamine (2.3 mL, 30 mmol, 1 eq) and 10 mL of anhydrous CH2Cl2 were combined and cooled in an ice bath. Di-tert-butyl dicarbonate (6.54 g, 30.0 mmol, 1.00 eq) in 20 mL anhydrous CH2Cl2 was added, and the mixture was stirred at room temperature for 4 h. The reaction mixture was diluted with additional CH2Cl2 and washed with 5% citric acid solution, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to afford 3.3 g of tert-butyl allylcarbamate 9 with a yield of 68%. 1H NMR (400 MHz, CDCl3):delta 5.90-5.81 (m, 1H, NH), 5.24-4.96 (m, 2H, =CH2), 4.63 (s, 1H, =CH), 3.75 (s, 2H, CH2), 1.45 (s, 9H, CH3).
58%
In dichloromethane; at 0 - 20℃; for 1h;
A solution of 5.88 ml (27.5 mmol, 1.1 eq.) of Boc2O in 50 ml of dry CH2Cl2 was addeddropwise to a solution of 1.88 ml (25.0 mmol, 1.0 eq.) of allyl amine in 60 ml of dry CH2Cl2at 0C. The reaction mixture was stirred at r.t. for 1 h. After TLC showed completeconsumption of starting material, solvent was removed under reduced pressure. The crudeproduct was purified by flash column chromatography (cHex/EtOAc 12:1). The product wasobtained as a light yellow solid (yield: 2.26 g, 14.395 mmol, 58 %).
In tetrahydrofuran;
EXAMPLE 7 Synthesis of Tert-butyl N-allyl carbamate The procedure of Example 5 was repeated using di-tert-butyl dicarbonate (40.14 gms, 0.19 mole), allyl amine (10.83 gms, 0.18 mole), and tetrahydrofuran (20.11 gms). The maximum temperature during the addition was 37 degrees C. The mixture was vacuum stripped to produce a white solid which was purified by distillation at 44 degrees C. at 0.32 mm Hg. NMR analysis confirmed the structure.
In dichloromethane;
PREPARATION 3 (Step D _ Scheme II) N-tert-butoxycarbonyl allyl amine Di-tert-butyldicarbonate, 64 gm, was added portion-wise at room temperature to allyl amine, 22 ml, in 400 ml of CH2Cl2 with stirring. The reaction mixture was left overnight and then concentrated to give an oil which solidified on standing. IR(KBr): 3340, 2962, 1690, 1509 cm-1. 1H NMR (80 MHz, CDCl3): delta 1.45 (s, 9H, t-butyl), 3.6-3,9 (m, 2H, CH2N), 4.34-4.8 (broad s, 1H, NH), 5.0-5.3 (m, 2H, CH2=C), 5.6-6.1 (m, 1H, -CH=C).
In tetrahydrofuran; at 20℃;
A solution of aniline (0.911 mL, 10.0 mmol) and di-tert-butyl dicarbonate (2.34 mL, 10.2 mmol) in THF(5.0 mL) was stirred for 1 h at room temperature. The resulting mixture was concentrated to obtain N-tert-butoxycarbonylaniline(10) (2.03 g, quant.) as colorless crystals. The crude product was used without purification. (Step 2) A solution of 10 (545 mg, 2.82 mmol) and (Z)-1-bromo-4-methoxybut-2-ene (11) (422 mg,2.56 mmol) in DMF (8.5 mL) was treated with sodium hydride (60 wt.% in oil, 154 mg, 3.85 mmol) at 0 C. The resulting mixture was stirred for 1 h at 50 C and quenched with saturated aqueous ammonium chloride at 0 C.Extractive workup and purification of the residue by chromatography on silica gel (hexane/ethyl acetate = 5/1 asthe eluent) afforded (Z)-tert-butyl (4-methoxybut-2-en-1-yl)(phenyl)carbamate (12) (654 mg, 92% yield) as acolorless oil. (Step 3) A solution of diisopropylamine (484 L, 3.45 mmol) in THF (4.2 mL) was treated with a1.6 M n-butyllithium hexane solution (2.30 mL, 3.7 mmol) at 0 C under an argon atmosphere and the mixture was stirred for 30 min at the same temperature. A solution of 12 (649 mg, 2.34 mmol) in THF (7.5 mL) was added to the solution at 0 C and the mixture was stirred for 3 h at the same temperature. The resulting mixturewas quenched with saturated aqueous ammonium chloride and extracted with ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, and concentrated. The residue was purified bychromatography on silica gel (hexane/ethyl acetate = 20/1 as the eluent) to give 2f (516 mg, 90% yield)
In tetrahydrofuran; at 0 - 20℃; for 2h;
(Step 1) Di-tert-butyl dicarbonate (1.2 mL, 5.2 mmol) was added to a solution of allylamine (0.41 mL, 5.5mmol) in THF (10 mL) at 0 C. The solution was stirred for 2 h at room temperature and concentrated toobtain crude tert-butyl allylcarbamate (26) (730 mg, 89% yield) as a colorless oil.
With triethylamine; In diethyl ether; at 20℃;Inert atmosphere; Cooling;
[000140] Step 1. fert-Butyl Allylcarbamate. To a solution of allylamine (29.9 g, 0.525 mol) and triethylamine (53.0 g, 0.525 mol) in dry Et20 (300 ml), a solution of Boc-anhydride (114 g, 0.525 mol) in dry Et20 (150 ml) was added portionwise upon stirring and cooling in a cold-water-bath. The reaction mixture was stirred at room temperature overnight and concentrated under a reduced pressure. The oily residue was dried in a vacuum (1-2 mm Hg) to afford the title compound which was used further without an additional purification. The yield: 82.4 g (100%); the product contains traces of B0C2O and i-BuOH according to H NMR data. FontWeight="Bold" FontSize="10" H NMR (delta, DMSO-d6): 1.45 (s, 9H), 3.50-3.56 (m, 2H), 4.99-5.04 (m, 1H), 5.05-5.11 (m, 1H), 5.71-5.81 (m, 1H), 6.98 (br. s, 1H).
With triethylamine; In dichloromethane; at 20℃; for 16h;Cooling with ice;
Allylamine (4.8 mL, 64.0 mmol) and triethylamine (13.5 mL, 96.9 mmol) were dissolved in CH2Cl2 (180mL), and cooled on ice. A solution of di-t-butyl dicarbonate (14.0 g, 64.1 mmol) in CH2Cl2 (20 mL) was added, and stirred at room temperature for 16 h. The reaction mixture was diluted with CH2Cl2, and washed twice with 5% aqueous HCl solution, once with saturated aqueous NaHCO3 solution, dried over anhydrous magnesium sulfate, and evaporated to give crude t-butyl allylcarbamate (10.1 g, 100%). Crude t-butyl allylcarbamate (5.76 g, 36.6 mmol) and benzyl bromide (6.6 mL, 55.5 mmol) were dissolved in DMF (110 mL), and cooled on ice. NaH (60% in oil, 2.36 g, 59.1 mmol) was added, and stirred at room temperature for 2 h. After cooled on ice, the reaction was quenched by adding water.The mixture was diluted with Et2O, washed with 5% aqueous Na2S2O3 solution and saturated aqueous NaHCO3 solution, dried over anhydrous magnesium sulfate, and evaporated. Purification by columnchromatography (n-hexane : Et2O = 15 : 1 v /v), and distillation (500 Pa, 125 C) gave 42i (5.93g, 66%) as colorless liquid. 1HNMR (400 MHz, CDCl3) 1.42~1.52 (br, 9H), 3.65~3.95 (m, 2H), 4.30~4.50 (br, 2H), 5.00~5.25 (m,2H), 5.65~5.87 (br, 2H), 7.19~7.39 (m, 5H). 13CNMR (100 MHz, CDCl3) 28.39, 48.61, 48.80~49.70(br), 79.79, 116.00~117.20 (br), 127.09, 127.30, 127.76, 128.43, 133.69, 138.30, 155.80. FABMS (YokudelR) m/z: 248.1 ([M+H]+), 192.1 (100%). HR-MS calcd. for C15H22NO2 ([M+H]+) 248.1650,found. 248.1660.
Stage #1: N-tert-butoxycarbonylprop-2-en-1-amine With n-butyllithium In tetrahydrofuran at -78 - 0℃; for 0.25h; Inert atmosphere;
Stage #2: di-<i>tert</i>-butyl dicarbonate In tetrahydrofuran at -78 - 20℃; for 1h; Inert atmosphere;
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃;
Tert-butyl N-(2-oxiranylmethyl)carbamate was synthesized according to the method of Rocheblave (30). 1 g (6 mmol, 1 equiv) of N-t-BOC protected 3 was dissolved in 50 mL dry CH2Cl2. The solution was brought to 0 C. and kept cold upon addition or 2.8 g (12 mmol, 2 equiv) MCPBA. The solution was then brought to room temperature and stirred overnight.About half of the reaction mixture was taken and diluted with additional 80 mL of CH2Cl2. The solution was washed with 10% Na2SO3, followed by washing with saturated NaHCO3 3 times, and finally by washing with water. The organic layer was dried over Na2SO4 and concentrated in vacuo, yielding crude epoxide 4. By 1H NMR, approximately 85% yield was achieved.1H NMR (400 MHz, CDCl3): delta 1.44 (s, 9H), 2.59-2.78 (brm, 2H), 3.04-3.54 (m, 3H), 4.75 (brs, 1H).
85%
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃;
Tert-butyl allylcarbamate (1.0 g, 6.4 mmol, 1 eq) was dissolved in 50 mL anhydrous CH2Cl2, and the mixture was cooled to 0 C. 3-Chlorobenzenecarboperoxoic acid (2.8 g, MCPBA, commercially available as 77% pure) was added drop-wise. The mixture was brought to room temperature, stirred overnight, diluted with CH2Cl2, and subsequently washed with 10% Na2SO3, saturated NaHCO3, and H2O. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to afford 10 with a yield of 85%. 1H NMR (400 MHz, CDCl3): delta4.78 (s, 1H. NH), 3.54 (d, J = 14.7 Hz, 1H, O-CH), 3.27-3.15 (m, 1H, CH2), 3.10 (s, 1H, CH2), 2.81-2.77 (m, 1H, O-CH2), 2.60 (dd, J = 4.7, 2.7 Hz, 1H, O-CH2), 1.45 (s, 9H, CH3).
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 5 - 20℃;Inert atmosphere;
[000141] Step 2. tert-Butyl (Oxiran-2-ylmethyl)carbamate. To a solution of the compound i<?ri-Butyl Allylcarbamate (82.4 g, 0.525 mol) in CH2CI2 (1300 ml), commercial (75% purity) m-chloroperbenzoic acid (157 g, 0.683 mol) was added portionwise upon stirring and cooling in an ice-bath. The reaction mixture was stirred at -5-7 C for 1 h, then overnight at room temperature. The white precipitate was filtered, the mother liquid was concentrated under a reduced pressure. The residue was dissolved in Et20 (1000 ml); the resulted solution was washed with 10% aq. Na2S03 (500 ml), sat. aq. NaHC03 (4 x 500 ml), dried over MgS04, filtered, and concentrated under a reduced pressure. The resulted oil was dried in a vacuum (1-2 mm Hg) to afford the titled compound which was used further without an additional purification. The yield: 70.4 g (77.5%); the product contains -5-7% of m- chlorobenzoic acid according to lH NMR data. lH NMR (delta, DMSO-d6): 1.38 (s, 9H), 2.45 (dd, Jj = 5.1 Hz, J2 = 2.7 Hz, 1H), 2.65-2.70 (m, 1H), 2.91-2.97 (m, 1H), 2.99-3.15 (m, 1H), 6.96-7.03 (m, 1H).
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃; for 16h;
Synthesis of compound 235.3. To a solution of compound 235.2 (35.0 g, 222 mmol, 1.0 eq) in DCM (600mL) was added 3-chloroperbenzoic acid (72.85g, 423.5mmol, 1.9eq) at 0 C portionwise. Reaction mixture was stirred at room temperature for 16 h. Upon completion of reaction, mixture was washed with 10% sodium sulfite solution followed by washing with saturated NaHCC solution and brine. Organic layers were dried over Na2S04 and concentrated under reduced pressure to pressure to obtain crude 235.3 (37g, 95%). This was directly used for next step without any purification.
With sodium hypophosphite hydrate; triethyl borane; In tetrahydrofuran; methanol; at 20℃;
Compound 2 was prepared according to a modified literature procedure (See, e.g., Deprele and Montchamp, Journal of Organic Chemistry 2001, 66, 6745-6755). Briefly, to a solution of sodium hypophosphite hydrate (4.00 g, 45.465 mmol) and t-butyl N-allylcarbamate (2.859 g, 18.186 mmol) in methanol (MeOH) was added 1 M triethylborane (18.186 mmol) in THF at room temperature in an open vessel. The reaction was allowed to stir at room temperature for 2 hours. The reaction mixture was concentrated in vacuo, and the residue was partitioned between aqueous KHSO4 and EtOAc. The aqueous layer was extracted twice with EtOAc, and the combined organic layers were dried over Na2SO4 and concentrated to afford the crude phosphinic acid (3.268 g, 80.5%). The crude material was used for the next step without further purification: 1H NMR (CDCl3) delta 1.42 (s, 9H), 1.55-1.93 (m, 4H), 2.92-3.29 (m, 2H), 6.18 (s, 1H), 8.00 (s, 1H).
Tert-butyl N-allylcarbamate (14.71 g, 93.54 mmol) was dissolved inTHF (170 mL) and cooled to 0 C. Butyl lithium (2.5M in hexanes,58.62 mL, 146.54 mmol) was added dropwise over a period of 12 minwhile keeping the internal temperature below 15 C. After addition, thereaction was kept in the ice bath and allowed to return to 0 C.Compound 22 (30 g, 85.03 mmol) was added in a single portion and thereaction was then allowed to warm to RT with stirring over 19 h. Thereaction was diluted with EtOAc (500 mL) and Hexanes (100 mL).Washed with water and brine to give the crude product which waspurified via silica gel chromatography using a 3-15% MeOH in CH2Cl2gradient to give the titled compound (34.41 g, 85%). 1H NMR(400 MHz, CDCl3): delta 7.67-7.55 (m, 15H), 5.88-5.87 (m, 1H), 5.17-5.10(m, 2H), 4.01 (d, J=6.0 Hz, 1H), 3.97-3.95 (m, 2H), 3.82-3.76 (m,2H), 1.43 (s, 9H). LCMS (ESI+) (m/z) 474 [M+H]+.
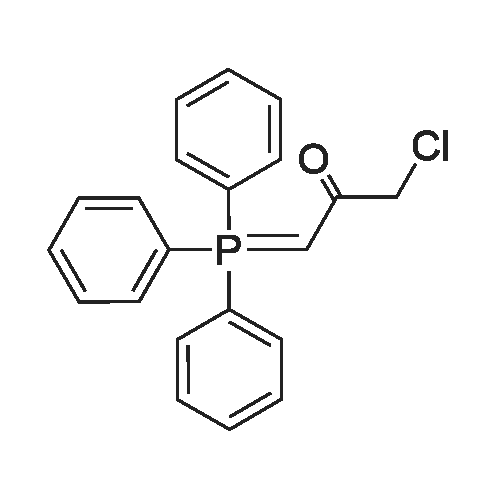
To an oven dried 250 mL round bottom flask under nitrogen was added tert-butyl allylcarbamate (6.81 g, 43.3 mmol) followed by tetrahydrofuran (80 ml). n-Butyl lithium (17.32 ml, 43.3 mmol) was added at room temperature, the mixture was stirred for 10 minutes and l-chloro-3- (triphenylphosphoranylidene)propan-2-one (13.89 g, 39.4 mmol) was added. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate, and the organic layer was dried with magnesium sulfate, filtered and concentrated to give the title compound.
[3-(2-formyl-3-hydroxy-4-methoxyphenyl)-prop-2-enyl]-carbamic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
With triethylamine;copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); for 0.5h;Heating / reflux;
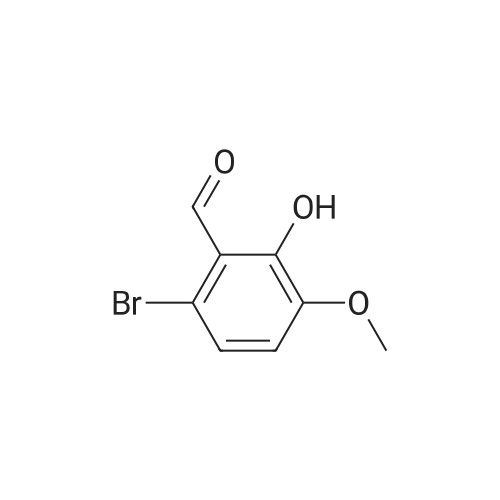
A solution of <strong>[20035-41-0]6-bromo-2-hydroxy-3-methoxybenzaldehyde</strong> (0.60 g, 2.6 mmoles) and prop-2-ynylcarbamic acid tert-butyl ester (0.44, 2.9 mmoles) in triethylamine (10 ml) was mixed with tetrakis(triphenylphosphine)palladium(0) (60 mg, 0.05 mmoles) and copper iodide (15 mg, 0.08 mmoles). The reaction mixture was refluxed for 30 minutes, cololed to ambient temperature, diluted (ethyl acetate 200 ml) and washed with 0.5N HCl (50 ml). The organic layer was dried (MgSO4), filtered and concentrated to yield a residue which was purified by flash chromatography (30% ethyl acetate/hexanes) to yield 0.36 g (46%) of the title compound.
A solution of copper cyanide (1.1Sg, 12.9 mmol) in THF (30 mL) at ?78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at ?78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at ?780C for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2% ethyl acetate/heptane to provide the desired product (3.66 g, 63%). 1H NMR (400 MHz, CDCl3) ? 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
With ammonium hydroxide; n-butyllithium; ammonium chloride; tri-n-butyl-tin hydride; In tetrahydrofuran; n-heptane; dichloromethane; ethyl acetate;
EXAMPLE 314A tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78 C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2% ethyl acetate/heptane to provide the desired product (3.66 g, 63%). 1H NMR (400 MHz, CDCl3) delta 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
63%
tert-butyl allylcarbamate A solution of copper cyanide (1.15 g, 12.9 mmol) in THF (30 mL) at -78 C. was treated slowly with n-butyllithium (16.9 mL, 27.1 mmol), stirred for 15 minutes at -78 C., treated with tributyltin hydride (7.88 g, 7.30 mL, 27.1 mmol) over a period of 5 minutes, stirred for 15 minutes, treated with tert-butyl 2-propynylcarbamate (2.00 g, 12.9 mmol) in tetrahydrofuran (7 mL), stirred at -78 C. for 1 hour, and treated with a 9:1 aqueous solution of ammonium chloride:ammonium hydroxide (250 mL) and dichloromethane (200 mL). The suspension was filtered through a short pad of diatomaceous earth (Celite). The organic phase of the filtrate was washed with brine and concentrated. The residue was purified on silica gel using 1-2% ethyl acetate/heptane to provide the desired product (3.66 g, 63%). 1H NMR (400 MHz, CDCl3) delta 6.08 (dt, B part of an AB system, J=19.3 Hz, 1.3 Hz, 1H); 5.93 (dt, A part of an AB system, J=19.3 Hz, 4.8 Hz, 1H), 4.59 (br s, 1H), 3.78 (br s, 2H), 1.45 (s, 9H), 1.32-1.26, (m, 12H), 0.90-0.85 (m, 15H).
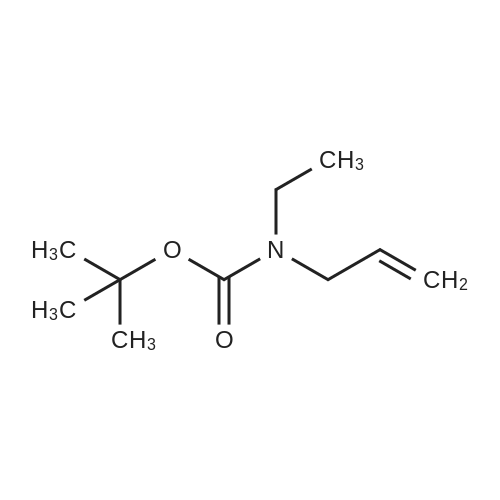
Sodium hydride (768 mg of 60% dispersion in mineral oil, 19.2 mmol) was washed twice with 15 mL portions of hexane and suspended in 15 mL anhydrous THF. Reaction mixture was cooled to 0C and tert-butyl allylcarbamate (1.0 g, 6.4 mmol) was added dropwise. Reaction mixture was warmed up to room temperature and stirred under argon for 30 min. Reaction mixture was cooled to 0C and freshly distilled ethyl iodide (1.02 mL, 12.8 mmol) was added dropwise. Reaction mixture was allowed to warm up to room temperature overnight, and it was then cooled to 0C and quenched with saturated ammonium chloride solution. The layers were separated and aqueous layer was extracted with Et2O. Organics were combined and washed with saturated NaCl solution, dried over MgSO4 and concentrated under reduced pressure. Crude reaction mixture was purified by flash column chromatography (hexanes:EtOAc 4:1) to give 1.0g (85%) of the title compound as a colorless oil. IR (neat) 3083, 3010, 2979, 2933, 2249, 1682 cm-1; 1H NMR (300 MHz, CDCl3): delta [ppm] 1.03-1.07 (t, 3H, J=7.1 Hz), 1.42 (s, 9H), 3.20 (br m, 2H), 3.77 (br m, 2H), 5.05-5.11 (m, 2H), 5.68-5.81 (m, 1H); 13C NMR (75 MHz, CDCl3): delta [ppm] 12.59, 27.55, 40.39, 48.19, 78.31, 115.10, 133.67, 154.46; m/z (relative intensity) 185(1), 129(25), 112(9), 84(10), 70(30), 57(100), 41(65), 29(25). Anal.Calcd. for C10H19NO2: C, 64.83; H, 10.34; O, 17.24. Found: C, 64.67; H, 10.60; O, 17.31.
N-allyl N-pent-4-enyl carbamic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
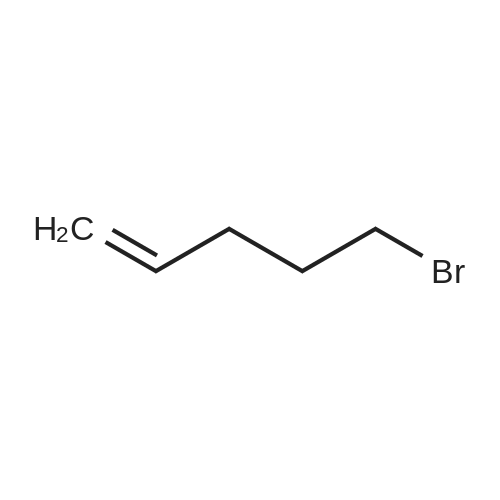
Step 1: To a suspension of sodium hydride (9.54 g, 238 mmol) in D F (50 mL), tert-butyl N- allylcarbamate (23.0 g, 146 mmol) in DMF (100 mL) was added in a drop wise fashion at 0 C. The mixture was stirred at room temperature for approximately for 30 minutes and cooled to 0 C whereupon 5-bromo-1-pentene (24.0 g, 175 mmol) in DMF (100 mL) was added in to it. After complete addition, the reaction mixture brought to room temperature and stirred for 1 h. The reaction mixture was partitioned between ethyl acetate and water. The organic layer washed with water, brine, dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was further purified by silica gel chromatography using 0-5% of ethyl acetatec/hexane to afford yellowish oil as product (Yield: 29.0 g, 88%). 1H NMR (300 MHz, DMSO-d6): delta 5.71-5.79 (m, 2H), 4.92-5.10 (m, 4H), 3.73 (d, J = 4.68 Hz, 2H), 3.08 (t, J = 7.10 Hz, 2H), 1 :92-1.99 (m, 2H), 1.52 (t, J = 7.14 Hz, 2H), 1.36 (s, 9H).
With NaH; In N,N-dimethyl-formamide;
a. Allyl-pent-4-enyl-carbamic acid tert-butyl ester To a suspension of NaH (3.05 g, 76.33 mmol of 60% NaH in oil; washed with hexanes) in DMF (30 mL) was added tert-butyl N-allylcarbamate (6.0 g, 38.2 mmol) in a dropwise fashion. The mixture was stirred at room temperature for approximately 10 minutes whereupon 5-bromo-1-pentene (6.78 mL, 57.24 mmol) was added in a dropwise fashion. The reaction was heated to 40 C. for approximately 2 hours whereupon the reaction was partitioned between ethyl acetate and water. The organic layer was washed with water (2*'s), brine, dried (MgSO4), filtered and concentrated to give 10 grams of the title compound as an oil: MS(EI) 226 (M+H+).
With NaH; In N,N-dimethyl-formamide;
a.) Allyl-pent-4-enyl-carbamic acid tert-butyl ester To a suspension of NaH (3.05 g, 76.33 mmol of 60% NaH in oil; washed with hexanes) in DMF (30 mL) was added tert-butyl N-allylcarbamate (6.0 g, 38.2 mmol) in a dropwise fashion. The mixture was stirred at room temperature for approximately 10 minutes whereupon 5-bromo-1-pentene (6.78 mL, 57.24 mmol) was added in a dropwise fashion. The reaction was heated to 40 C. for approximately 2 hours whereupon the reaction was partitioned between ethyl acetate and water. The organic layer was washed with water (2*'s), brine, dried (MgSO4), filtered and concentrated to give 10 grams of the title compound as an oil: MS(EI) 226 (M+H+).
P.11.1 1)
1) Synthesis of N-t-butoxycarbonylallylamine (27) Allylamine [2.855 g (50 mmol.)] was dissolved in chloroform (100 ml), to which was added t-butyl S-(4,6-dimethylpyrimidin-2-yl)thiocarbonate [12.017 g (50 mmol.)], and then the mixture was stirred at room temperature for 24 hours. 1N Hydrochloric acid was added to the reaction mixture, which was subjected to extraction with chloroform. The organic layer was dried over anhydrous potassium carbonate, and then concentrated under reduced pressure. The crude product thus obtained was purified by column chromatography (silica gel: 240 g; eluent: hexane.ethyl acetate = 3/1) to obtain the desired product (27) [6.720 g (85.5%, colorless prisms, m.p. 34 to 34.8°C)].
With hydrogenchloride In chloroform
P.11.1 (1)
(1) Synthesis of N-t-butoxycarbonylallylamine(27) Allylamine[2.855 g(50 mmol.)]was dissolved in chloroform (100 ml), to which was added t-butyl S-(4,6-dimethylpyrimidin-2-yl)thiocarbonate[12.017 g(50 mmol.)], and then the mixture was stirred at room temperature for 24 hours. 1N Hydrochloric acid was added to the reaction mixture, which was subjected to extraction with chloroform. The organic layer was dried over anhydrous potassium carbonate, and then concentrated under reduced pressure. The crude product thus obtained was purified by column chromatography(silica gel: 240 g;eluent:hexane.ethyl acetate=3/1) to obtain the desired product(27)[6.720 g(85.5%,colorless prisms, m.p.34 to 34.8° C.)]. TLC[Silica Gel;hexane/AcOEt(4/1)]:Rf=0.22. NMR(90 MHz,CDCl3) δ: 1.45(9H,s,CH3 x3), 3.72(2H,m,CH2), 4.79 (1H,br.NH), 5.15(2H,m,=CH2), 5.65 to 6.07[1H,m,CH=). IR(KBr)cm-1: 3320, 2960, 2900, 1670, 1510, 1360, 1245, 1150, 1040, 1015, 990, 950, 922, 860.
A mixture of N-tert-butoxycarbonyl prop-2-enylamine 2A(20 g, 127.39 mmoL) was dissolved in Nu, Nu-dimethylformamide (100 mL), sodium sulphate(7.6 g, 60 / , 191.08 mmoL) was treated with n-hexane and then added with N, N-dimethylformamide (400 mL) and added dropwise to the reaction solution at 0 C under nitrogen C for 30 minutes, and the reaction was continued at room temperature for 20 minutes.A mixture of proparagyl bromide (30.3 g, 245.78 mmoL) was added dropwise to the ice bath and the mixture was stirred at 0 C for 30 minutes. The reaction solution was slowly added to crushed ice to deal with excess sodium hydride, extracted with ethyl acetate (100 mL chi3), the organic phases were combined and washed with saturated brine (100 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure The The residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate (nu / nu) = 100: 1 to 50: 1) to give yellow liquid 2B (21 g, yield 85%).
85%
(20 g, 127.39 mmoL)Soluble in N,N-dimethylformamide (100mL),Sodium hydride (7.6g, 60%, 191.08mmoL)After treatment with n-hexane,Add N,N-dimethylformamide (400 mL),Under the protection of nitrogen,Add dropwise to the reaction solution at 0 C, and stir at 0 C for 30 minutes.The reaction was continued for 20 minutes at room temperature.Bromopropyne (30.3g, 245.78mmoL) was added dropwise under ice bath.Stir at 0 C for 30 minutes. The reaction solution was slowly added to crushed ice to treat excess sodium hydride.Extracted with ethyl acetate (100 mL × 3),The organic phases were combined and washed with saturated brine (100 mL×3).Dry over anhydrous sodium sulfate, filter,The filtrate was concentrated under reduced pressure.The residue was purified by silica gel column chromatography ( petroleum ether / ethyl acetate (v/v) = 100:1 - 50:1) to afford(21 g, yield 85%).
30mg
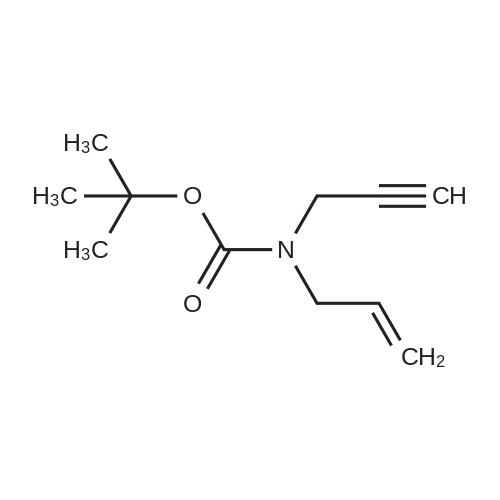
(Step 2) A solution of 26(730 mg, 4.64 mmol) in DMF (9.3 mL) was treated with sodium hydride (60 wt.% in oil, 0.22 g, 5.5 mmol) at0 C under an argon atmosphere. After stirring for 10 min at room temperature, 3-bromo-1-propyne (0.42mL, 5.6 mmol) was added to the mixture at 0 C. The mixture was stirred for 17 h at room temperature andquenched with saturated aqueous ammonium chloride at 0 C. Extractive workup and purification of theresidue by chromatography on silica gel (hexane/ethyl acetate = 10/1 as the eluent) afforded tert-butylallyl(prop-2-yn-1-yl)carbamate (27) (602 mg, 66% yield) as a yellow oil.
With diphenyl hydrogen phosphate; cyclopentadienylruthenium(II) trisacetonitrile hexafluorophosphate In acetone; toluene at 20℃; regioselective reaction;
(E)-tert-butyl (3-(2,6-dimethoxyphenyl)allyl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With palladium(II) trifluoroacetate; silver carbonate; In tetrahydrofuran; dimethyl sulfoxide; at 100℃; for 8h;Inert atmosphere;
General procedure: To an oven-dried pressure tube were sequentially added aryl carboxylic acid 1 (0.2 mmol), olefin 2 (0.24 mmol), Pd(TFA)2 (3.33 mg, 5.0 mol%), Ag2CO3 (55.2 mg, 0.2 mmol), THF (2.0 mL) and DMSO (0.10 mL) under nitrogen at room temperature. After degassing three times, the reaction mixture was heated at 100 C for 8 h, and then was cooled to room temperature. Water (20.0 mL)was added, and the mixture was extracted with ethyl acetate (3 5.0 mL). The combined organic layer was washed with brine, dried over anhydrious Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluant: hexane/ethyl acetate) to give the pure target product.
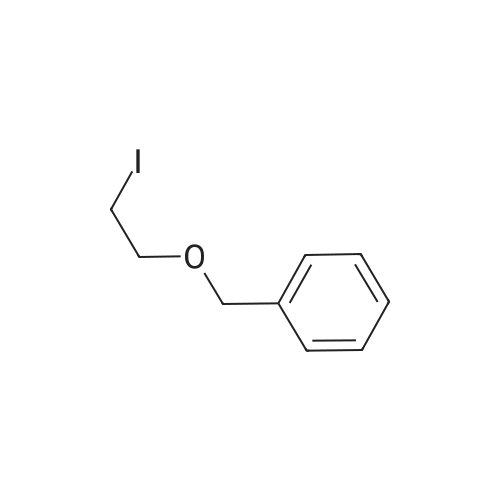
tert-butyl N-allyl-N-(2-benzyloxyethyl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
Stage #1: N-tert-butoxycarbonylprop-2-en-1-amine With sodium azide In N,N-dimethyl-formamide; mineral oil at 0℃; for 0.75h; Inert atmosphere;
Stage #2: benzyl iodoethyl ether In N,N-dimethyl-formamide; mineral oil at 23℃; for 16h; Inert atmosphere;
(E)-5-((tert-butoxycarbonyl)amino)-2-methylenepent-3-en-1-yl benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
54%
With tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidine][benzylidene]ruthenium(II) dichloride; ethene In dichloromethane at 0 - 20℃; for 24h; Inert atmosphere; Schlenk technique;
1.2.12.1 General metathesis procedure
General procedure: An inertized Schlenk flask was filled with 0.1 eq. of ruthenium catalyst and cooled to 0 °C. Itwas subsequently evacuated and flooded with ethylene. Then, 2.0 ml/mmol (regarding thealkyne) of dry CH2Cl2, 1.0 eq. of alkyne and 10.0 eq. of alkene were added and the reactionmixture was stirred at r.t. for 24 - 72 h. Residual catalyst was removed by stirring the reactionmixture over active charcoal.[13] The suspension was filtrated over silica and the solvent wasevaporated under reduced pressure. The crude product was purified via columnchromatography and the products were obtained as colourless oils.
ethyl 5-((tert-butoxycarbonyl)amino)-4-((ethoxycarbonothioyl)thio)pentanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With [4,4?-bis(1,1-dimethylethyl)-2,2?-bipyridine-N1,N1?]bis [3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate; In dimethyl sulfoxide; at 30℃; for 61h;Irradiation; Sealed tube; Inert atmosphere;
General procedure: To a 10 mL sealed tube was added ethyl 2-((ethoxycarbonothioyl)thio)acetate (1a,61.3 mg, 0.294 mmol), [Ir{dF(CF3)ppy}2(dtbbpy)](PF6) (8, 1.6 mg, 0.00143 mmol),and 1-octene (2a) (90 L, 0.573 mmol). Degassed anhydrous DMSO (290 L) wasadded and the reaction was sparged with Ar. The tube was sealed and surrounded byblue LEDs for 20 h. The reaction mixture was diluted with EtOAc and water. Theaqueous layer was separated and extracted twice with EtOAc. The combined organicextracts were washed with brine, dried over MgSO4, filtered and concentrated invacuo. The resulting crude material was purified by flash column chromatography(silica gel; hexane/EtOAc 200:1) to give 3aa (84.3 mg, 0.263 mmol) in 89% yield as acolorless oil.
58%
With tris[2-phenylpyridinato-C2,N]iridium(III); N,N-dimethyl-formamide; at 24℃;Inert atmosphere; UV-irradiation;
General procedure: In a 4.0mL glass vial equipped with a stirring bar, were added the corresponding xanthate (0.2mmol, 2.0 equiv.), the radical acceptor (0.1mmol, 1.0 equiv.) (alkene 2 or 5; or heterocycle 10-12), Ir(ppy)3 (0.002mmol, 0.02 equiv.) and DMF (0.4mL, [0.25M]). Next, the resulting solution was degassed by three consecutive freeze-pump-thaw cycles and placed under argon atmosphere. Finally, the reaction mixture was stirred and irradiated in a Blue LEDs reactor (12W) at 24C during 12-14h. After that, the mixture was concentrated in vacuo and the product was purified by flash column chromatography on silica gel.
ethyl 5-((tert-butoxycarbonyl)amino)-2,2-difluoro-4-iodopentanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
98%
With potassium carbonate; In acetone; at 25℃; for 16h;Irradiation;
To a 25 mL reaction tube, 82.9 mg (0.60 mmol) of K2CO3, 47.2 mg (0.30 mmol, 1 equivalent) of Compound A-1 were added, and after replacing with argon three times, 2 mL of acetone (Acetone) was added, and 66 muL (0.45 mmol) of the compound was injected. B-1. After stirring for 16 hours under the following three temperature environments, compound C-1 is obtained in the following yield (fluorescence spectrum yield, isolated yield in parentheses).
84%
With 2-hydroxybromobenzene; potassium acetate; In 1,2-dichloro-ethane; at 20℃; for 16h;Inert atmosphere; Irradiation;
To a 25 mL reaction tube, 58.9 mg (0.60 mmol) of KOAc, 47.2 mg (0.30 mmol, 1 equivalent) of A-11 was added, and the mixture was replaced with argon three times.2mL 1,2-dichloroethane (DCE),88 muL (0.60 mmol) of Compound B-1, 3.5 muL (0.03 mmol) of 2-bromophenol was injected, and after stirring for 16 hours under blue light irradiation,Compound C-11,The yield was 84%.
ethyl (E)-5-((tert-butoxycarbonyl)amino)-2,2-difluoropent-3-enoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With potassium acetate In N,N-dimethyl acetamide at 20℃; for 16h; Schlenk technique; Inert atmosphere; UV-irradiation;
General procedure for visible-light promoted atom transfer radicaladdition-elimination (ATRE) reaction for the synthesis of fluoroalkylatedalkenes using DMA as electron-donor
General procedure: To a 25 mL of Schlenk tube equipped with a Teflon septum were added KOAc (0.6 mmol, 2.0 equiv.) under Ar, followed by DMA(2 mL). Then, alkene (1) (0.3 mmol, 1.0 equiv.), and IRF (2) (0.45 mmol, 1.5 equiv.)were added subsequently. After stirring under purple light for 16 hours, the residuewas diluted with ethyl acetate, washed with H2O and brine, dried over Na2SO4,filtered and concentrated. The residue was purified with silica gel chromatography to provide pure product. 2. Data for compounds 3 Ethyl (E)-5-((tert-butoxycarbonyl)amino)-2,2-difluoropent-3-enoate (3a). This compound is known.1 The product (100 mg, 84% yield, Z:E =1:40) was purified withsilica gel chromatography (Petroleum ether/Ethyl acetate = 5:1) as colorless liquid. 1HNMR (400 MHz, CDCl3) δ 6.25 (d, J = 16.0 Hz, 1H), 5.85-5.73 (m, 1H), 4.80 (br,1H), 4.29 (q, J = 7.1 Hz, 2H), 3.84 (s, 2H), 1.42 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H). 19FNMR (376 MHz, CDCl3) δ -103.5 (s, 2F). 13C NMR (101 MHz, CDCl3) δ 163.7 (t, J= 34.6 Hz), 155.5, 136.1 (t, J = 8.7 Hz), 121.4 (t, J = 25.5 Hz), 112.1 (t, J = 249.2 Hz),79.8, 63.0, 41.0, 28.2, 13.8.
64%
With 2-hydroxybromobenzene; potassium carbonate In dimethyl sulfoxide at 20℃; for 16h; Schlenk technique; Inert atmosphere; Irradiation; regioselective reaction;
With bromine In ethanol at 0 - 20℃; for 16h; Inert atmosphere;
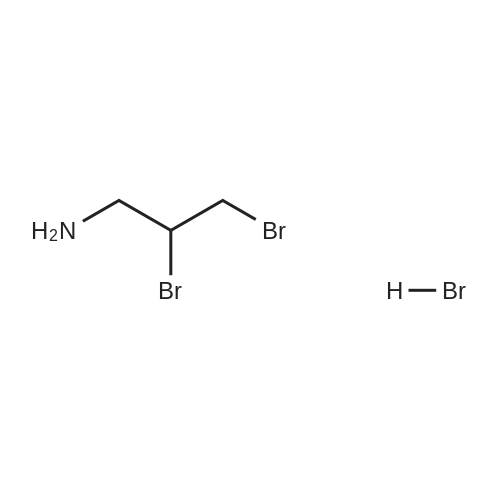
4B Preparation of 2,3-dibromopropan-1-amine hydrobromide (3)
[0518] To 1 (20.0 g, 1.0 equiv, 50 wt% in water) was added a solution of K2CO3 (29.55 g, 1.0 equiv) in water (100 mL) and a solution of Boc2O (93.32 g, 2.0 equiv) in EtOAc (100 mL) at 0 °C. The reaction mixture was stirred at 20 °C for 15 h. The organic layer was separated and solvent swapped to EtOH (100 mL) to provide a solution of 2 in EtOH. It was then added into a cold (0 °C) solution of Br2(71.74 g, 2.1 equiv) in EtOH (60 mL) at 0 °C. The reaction mixture was stirred at 20 °C for 16 h, filtered, washed with MTBE (60 mL), dried under vacuum at 40 °C for 15 h to provide 3 as a colorless solid (43.01 g, 66%):1H NMR (400 MHz, CD3OD) d 4.55 - 4.47 (m, 1H), 4.01 (dd, J = 10.9, 4.6 Hz, 1H), 3.86 (dd, J = 10.9, 8.7 Hz, 1H), 3.71 (dd, J = 13.9, 3.2 Hz, 1H), 3.39- 3.33 (m, 1H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping