There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
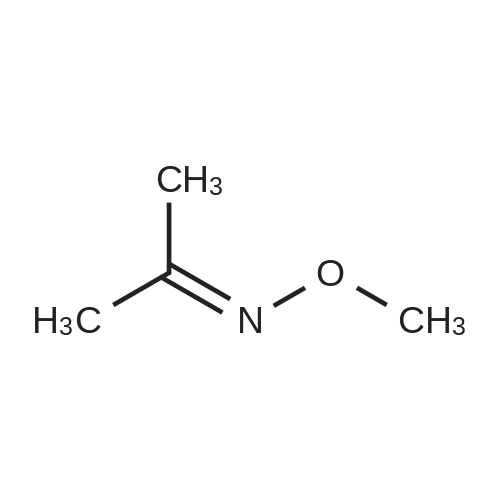
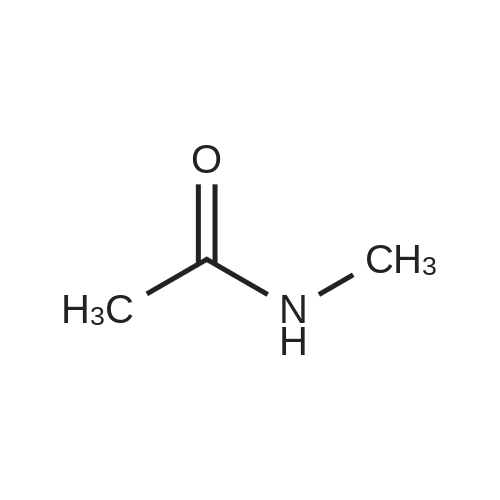
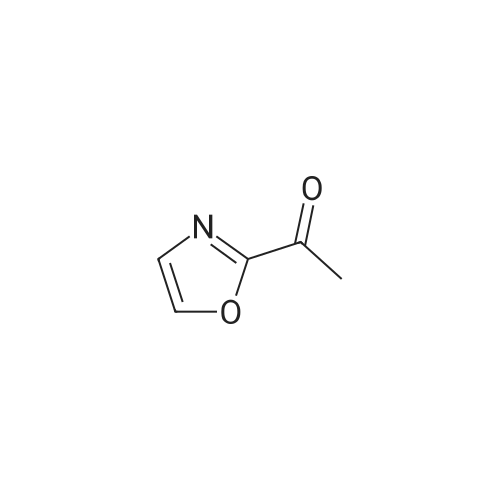
Structure of 78191-00-1 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
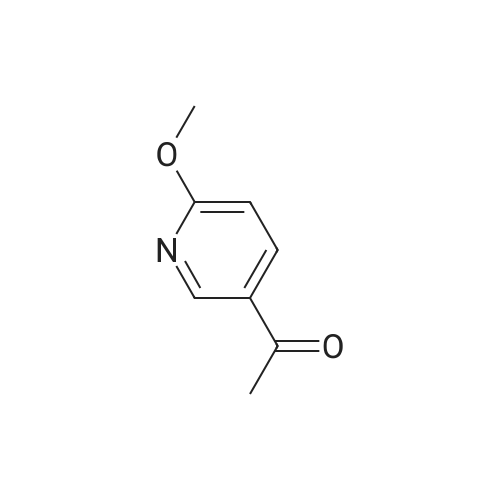
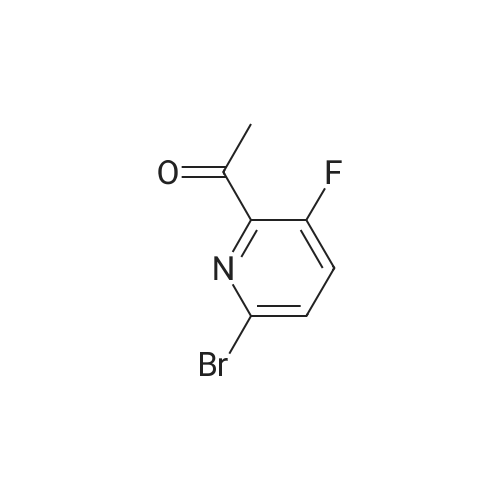
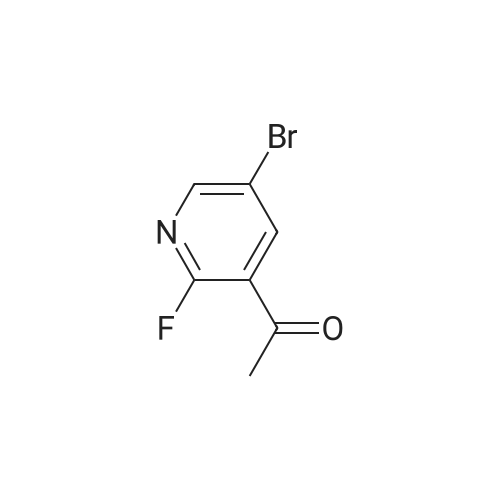
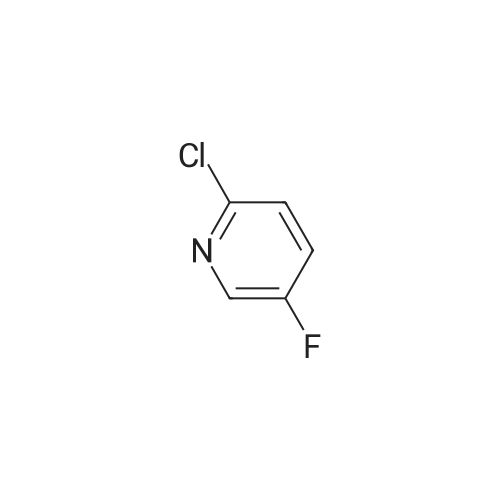
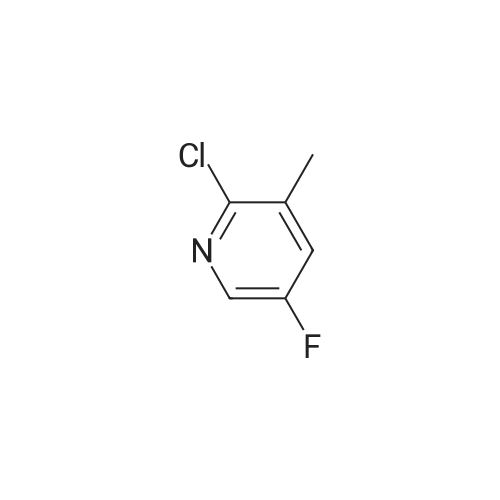
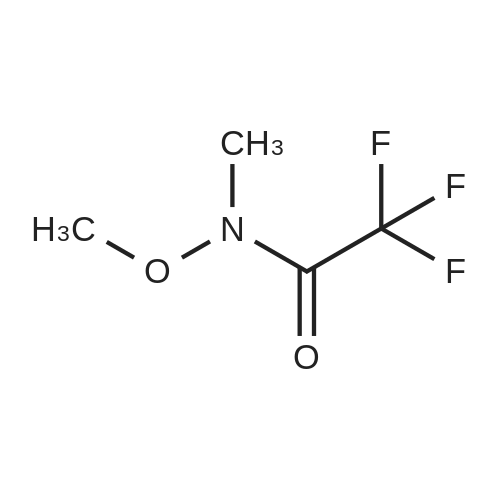
Stage #1: With TurboGrignard In tetrahydrofuran at 0℃; for 0.25 h; Inert atmosphere Stage #2: at 0 - 20℃; Inert atmosphere
A round bottomed flask was charged with 5-bromo-2-chloropyridine (5.30 g, 27.6 mmol) in THF under N2 and cooled at 0° C. A solution of 1 M iso-propylmagnesiumchloride-lithium chloride complex in THF (40 mL) was added drop wise over 15 min. After 70 min N-methoxy-N-methylacetamide (4.1 mL, 38 mmol) was added drop wise. After stirring for 5 min at 0° C. the cooling bath was removed. The mixture was left stirring overnight and was then quenched by the addition of 100 mL saturated aqueous NH4Cl solution. The mixture was extracted with 3×100 mL EtOAc. The combined organic layers were washed with water followed by brine and dried over MgSO4. Evaporation of the volatiles at 80° C., 10 mbar for 1 h gave the title compound (3.596 g, 84) sufficiently pure for the next step.
With boron trifluoride diethyl etherate In 1,4-dioxane at 20 - 100℃; for 1 h; Inert atmosphere
General procedure: To a solution of o-diaminobenzene (5.0 mmol, 0.5405 g) and Weinreb amide (N-methoxy-Nmethylbenzamide, 5.0 mmol, 0.82595 g) in dioxane (10 mL), boron trifluoride diethyl etherate (5.0 mmol) was added at room temperature. The reaction mixture was stirred for the specified time (Table 3) at 100 °C. TLC revealed the complete consumption of starting material. Subsequently hydrolysis was achieved by the addition of saturated NH4Cl solution (50 mL). The aqueous layer was extracted with ethyl acetate (3 X 25 mL), washed with water (2 X 25 mL), brine solution (2 X 25 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to get crude product. This was purified by column chromatography over silica gel using hexane/ethyl acetate mixture in 1:1 ratios as eluent.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 23, p. 6231 - 6235
4
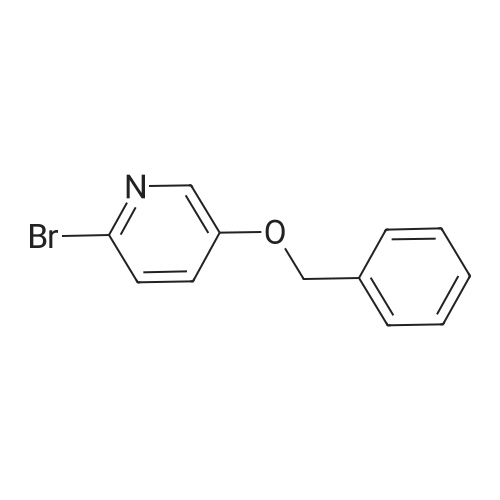
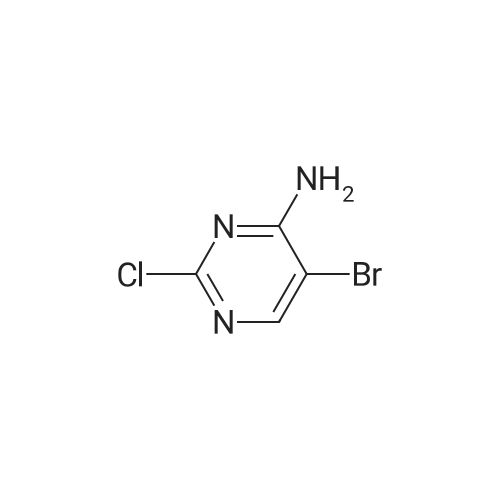
[ 78191-00-1 ]
[ 138343-75-6 ]
[ 23612-48-8 ]
Yield
Reaction Conditions
Operation in experiment
80%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -10℃; for 1 h; Inert atmosphere Stage #2: at 20℃; for 1 h; Inert atmosphere
tert-Butyl 3-methylpyridin-2-ylcarbamate (5.07 g, 23.6 mmol) was dissolved in 100 ml tetrahydrofuran under nitrogen and the mixture was cooled to -10 °C. n-Butyllithium (2.5 M in hexanes, 28 ml, 70 mmol) was added over 15 min with stirring, forming a dark red solution and the mixture was stirred for a further 1 h at -10 °C. N-Methoxy-N-methylacetamide (3.80 g, 35.7 mmol) was added and the mixture was stirred at room temperature for 1 h. The mixture was cooled to -10 °C, quenched with 13 ml concentrated hydrochloric acid and then stirred at 50 °C for 90 min. The biphasic mixture was allowed to cool and the organic phase was extracted twice with 2N hydrochloric acid. The combined aqueous was basified with 32percent sodium hydroxide solution and extracted three times with ethyl acetate. The organics were washed with brine, dried over anhydrous sodium sulphate, filtered and evaporated under reduced pressure. The residue was purified using the Isolera purification system (ethyl acetate-hexane gradient, 0:100 rising to 50:50) to give 95 (2.5 g, 18.9 mmol, 80percent yield) as a pale yellow solid. Purity 100percent. 1H NMR (400 MHz, CDCl3) δ ppm 2.55 (s, 3H, Me), 6.17 (s, 1H, H-3), 7.03 (dd, J = 4.9, 7.6 Hz, 1H, H-5), 7.82 (d, J = 7.4 Hz, 1H, H-4), 8.21 (d, J = 4.7 Hz, 1H, H-6), 11.96 (br. s., 1H, NH). UPLC/MS (3 min) retention time 0.68 min. LRMS: m/z 133 (M+1).
Reference:
[1] European Journal of Medicinal Chemistry, 2016, vol. 113, p. 102 - 133
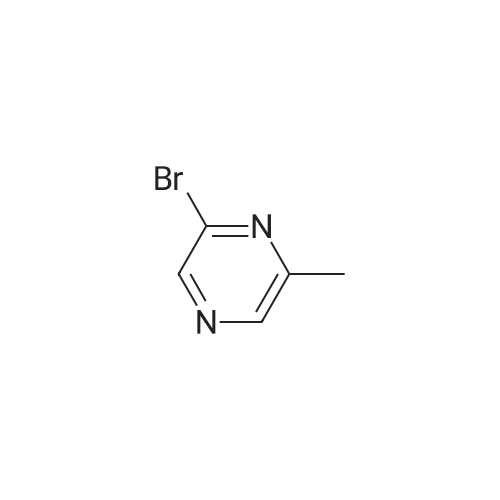
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5 h; Stage #2: at -78 - 0℃;
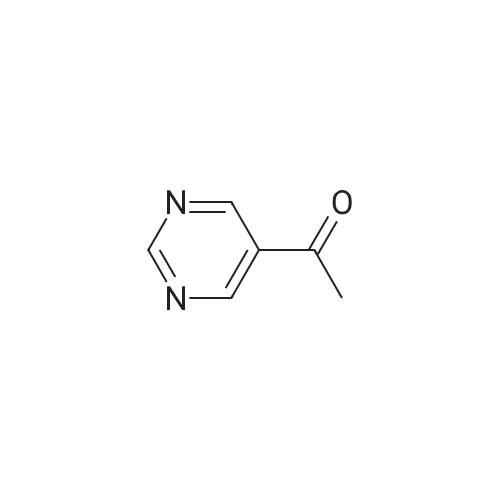
Step A: 5-Acetylpyrimidine 5-Bromopyrimidine (3.18 g, 20 mmol) was dissolved in 50 mL of tetrahydrofuran, While cooled to -78 °C, 15 mL of 1.6 M n-butolithium in hexane solution was added dropwise with stirring, After the solution was stirred for 30 minutes, a solution of N-methoxyl-N-methylacetamide (2.58 g, 25 mmol) in tetrahydrofuran solution (10 mL) was added slowly. The mixture was stirred at -78 °C for 1 hour and then allowed to be warmed slowly. When the temperature of the mixture was at 0 °C, aqueous ammonium chloride solution was added. The solution obtained was extracted with ethyl acetate for 3 times, The pooled extracts were washed with brine, dried over magnesium sulfate, concentrated under reduced pressure, and purified by silica gel column chromatography using 5percent methanol / methylene chloride as eluent to obtain 1 g of the title compound (45percent yield). MS(M+1)=123.05.
175 mg
Stage #1: With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h; Stage #2: at -78℃; for 1 h;
The5-bromo-pyrimidine (9.4 mmol) is dissolved in tetrahydrofuran steams again 30ml in. At -78 ° C lower, will n-BuLi (11.3 mmol) in drops to the reactionsystem, and stirring 30 minutes. N-methoxy-N-methyl acetamide (11.8 mmol) oftetrahydrofuran solution (15 ml) at -78 ° C in the system dropping, andstirring 1 hour. Under room temperature, add saturated ammonium chlorideaqueous solution, ethyl acetate extraction 3 times. Concentrated extract,column chromatography (ethyl acetate: petroleum ether = 1 the [...] 10)separated to obtain title compound 175 mg per litre. 1 H-NMR (CDCl 3)δ 9.37 (1H, s), 9.24 (2H, s), 2.66 (3H, s).
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1.5 h; Stage #2: at -78 - 0℃; for 4 h;
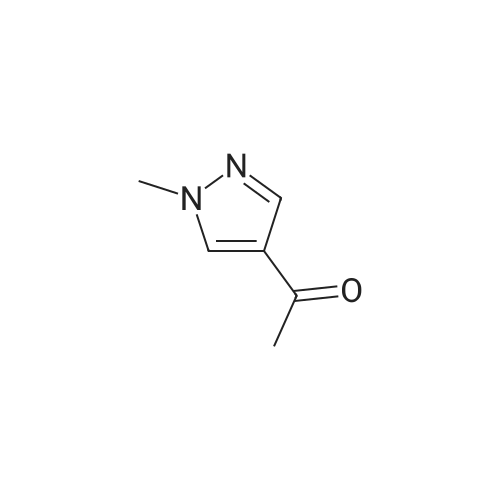
A. Synthesis of 1-(1-methyl-1H-pyrazol-4-yl)ethanone 4-Bromo-1-methyl-1H-pyrazole (41.3 mL, 400 mmol), was dissolved in tetrahydrofuran (750 mL) and cooled to -78° C. N-Butyllithium (2.5 M solution in hexanes, 160 mL, 400 mmol) was added drop-wise over 30 minutes, and the resulting mixture was stirred for 1 hour at -78° C. After drop-wise addition of a solution of N-methoxy-N-methylacetamide (40.9 mL, 400 mmol) in tetrahydrofuran (100 mL) to the -78° C. reaction mixture, the cooling bath was allowed to warm to 0° C. over 4 hours. The reaction was then quenched with saturated aqueous sodium chloride solution (50 mL), and volatiles were removed in vacuo. The residue was diluted with ethyl acetate (1000 mL), treated with magnesium sulfate, and stirred for 30 minutes before being filtered and concentrated in vacuo. Purification was carried out via silica gel chromatography (material was loaded in a minimum amount of dichloromethane; Gradient: 5percent to 100percent ethyl acetate in heptane) to provide a pale yellow oil that solidified on standing. Yield: 28.5 g, 230 mmol, 57percent. 1H NMR (500 MHz, CDCl3) δ 2.37 (s, 3H), 3.90 (s, 3H), 7.83 (s, 1H), 7.84 (s, 1H).
Reference:
[1] Patent: US2012/214791, 2012, A1, . Location in patent: Page/Page column 21
[2] Journal of Medicinal Chemistry, 2018, vol. 61, # 3, p. 1001 - 1018
8
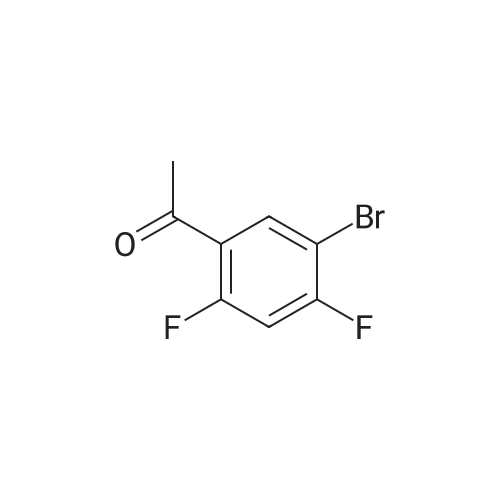
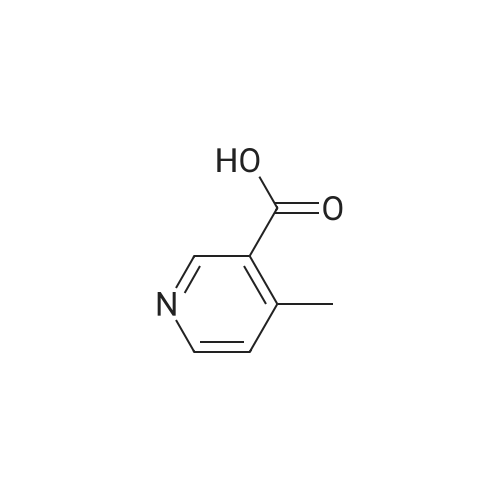
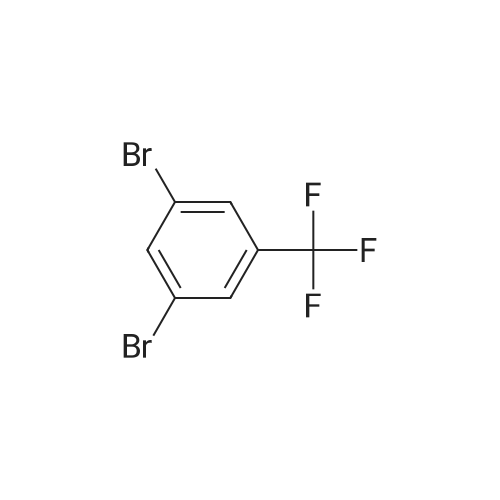
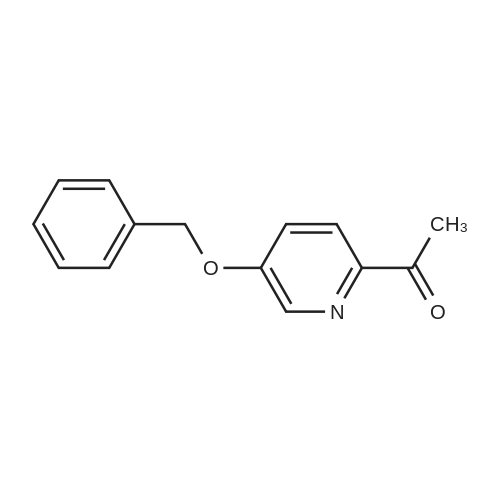
[ 39806-90-1 ]
[ 78191-00-1 ]
[ 37687-18-6 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 17, p. 3644 - 3649
Stage #1: With n-butyllithium In tetrahydrofuran at -60℃; for 0.5 h; Inert atmosphere Stage #2: at -30℃; for 3 h; Inert atmosphere
Into a 500-mL round-bottom flask purged and maintained with an inert atmosphere of N2 was placed a solution of 1-bromo-3- (propan-2-yl)benzene (5.0 g, 25.11 mmol, 1.00 equiv) in THF (250 mL). This was followed by the addition of n-BuLi (20 mL, 2.00 equiv) dropwise with stirring at -60 °C. The mixture was stirred at -60°C for 30 mm. To this was added N-methoxy-N-methylacetamide (3.9 g, 37.82 mmol, 1.50 equiv) dropwise with stirring at -30 °C. The resulting solution was stirred for 3 h at -30 °C in a liquid N2 bath. The reaction was then quenched by the addition of 20 mL of water. The resulting solution was diluted with 200 mL of EtOAc, washed with 2x100 mL of brine, dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was applied onto a silica gel column with EtOAc/petroleum ether (1:10). This resulted in 2.7 g (66percent) of the title product as colorless oil.
N-Methoxy-N-methyl-acetamide (105). Pyridine (10 mL, 124 mmol) was slowly added to a well stirred slurry of O,N-dimethylhydroxylamine hydrochloride (6 g, 62 mmol) and acetyl chloride (4.15 mL, 59 mmol) in CH2Cl2(85 mL) under argon at O°C. The mixture was warmed to room temperature, stirred for 3 hours and then partitioned between brine (55 mL) and diethyl ether (85 mL). Extraction with additional diethyl ether, drying of the combined organic layers over anhydrous Na2SO4, concentration, followed by distillation under reduce pressure gave Weinreb amide 105(4.54 g, 72 percent) as a colourless liquid. bp 42-44°C / 20 mmHg; IR (neat) 3497, 2971, 2941, 2824, 1663 cm-1; 1H NMR (CDCl3, 300 MHz) δ 3.62 (s, 3H), 3.11 (s, 3H), 2.05 (s, 3H); 13C NMR (CDCl3, 75.4 MHz) δ 171.8 (C), 61.0 (CH3), 31.9 (CH3), 19.6 (CH3). Spectral properties identical to samples already described in literature.
65%
With triethylamine In dichloromethane at 0 - 20℃; for 0.5 h; Cooling with ice
N,O-dimethyl-hydroxylamine HCl (100 g, 1025 mmol) was suspended under nitrogen in DCM (1000 mL) and cooled in ice. Triethylamine (300 mL, 2152 mmol) was added slowly, then acetyl chloride (76.5 mL, 1076 mmol) was added slowly, the temperature reached 20° despite ice cooling and slow addition. Stirring without cooling was continued for 30 min. Extraction: 1.x.DCM, 1.x.1N HCl, 2.x. saturated NaCl solution. Distillation at 42° C./20 mbar gave 69 g (65percent) of a colorless oil.
65%
With triethylamine In dichloromethane at 0 - 20℃; for 0.5 h;
N, O-dimethyl-hydroxylamine HC1 (100 g, 1025 mmol) was suspended under nitrogen in DCM (1000 mL) and cooled in ice. Triethylamine (300 mL, 2152 mmol) was added slowly, then acetyl chloride (76.5 mL, 1076 mmol) was added slowly, the temperature reached 20° despite ice cooling and slow addition. Stirring without cooling was continued for 30 min. Extraction: 1 x DCM, 1 x 1 N HC1, 2 x saturated NaCl solution. Distillation at 42 °C/20 mbar gave 69 g (65 percent) of a colorless oil.
61%
With triethylamine In dichloromethane at 0 - 20℃; for 4 h;
Compound 25-1 (0442) To a suspension of N,O-dimethylhydroamine hydrochloride (100 g, 1026 mmol) in DCM (1000 mL) was added triethylamine (300 mL, 2052 mmol) at 0° C. Acetyl chloride was added dropwise to the suspension for 2 h at 0° C. When the addition was complete, the mixture was allowed to warm to rt and stirred for 2 h. The mixture was washed with brine (1 L), 1 N HCl (500 mL), brine (200 mL) respectively and dried with magnesium sulfate, filtered and concentrated to afford brown oil, which was purified by distillation to afford 25-1 as a colourless liquid (65 g, 61percent).
56%
Stage #1: With triethylamine In dichloromethane for 0.333333 h; Inert atmosphere Stage #2: at 20℃; for 1 h;
To a stirred solution of N, O-dimethylhydroxylamine hydrochloride 3 (25 g, 256.4 mmol) in CH2C12 (400 mL) under inert atmosphere was added TEA (51.8 g, 512.8 mmol), and the reaction mixture was stirred for 20 mi Acetyl chloride (22.5 g, 282.0 mmol) was added to the reaction mixture at RT, and the reaction mixture stirred for 1 h. The reaction progress was monitored by TLC; after reaction completion, the reaction mixture quenched with saturated NaHCO3 solution (250 mL) and extracted with CH2C12 (2 x 250 mL). The combined organic extracts were washed with 1 N HC1 solution (100 mL), water (100 mL), brine solution (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to obtain the crude material. The crude material was distilled at 50°C to get 4(11.5 g, 56percent) as colorless liquid. 1HNMR (500 MHz, CDC13): ö 3.68 (s, 3H), 3.17 (s, 3H), 2.11 (s, 3H); LC-MS: 97.3percent; (M+H) Found=104.4; (column: X bridge C-18, 50 x 3.0 mm, 3.5 tm); RT 0.63 mm. 5 mM NH4OAc: ACN; 0.8 mL/min).
Reference:
[1] Organic Letters, 2012, vol. 14, # 9, p. 2250 - 2253
[2] European Journal of Organic Chemistry, 2017, vol. 2017, # 2, p. 278 - 286
[3] Patent: EP1300403, 2003, A1,
[4] Tetrahedron Letters, 1983, vol. 24, # 18, p. 1851 - 1854
[5] Tetrahedron Letters, 2007, vol. 48, # 3, p. 377 - 380
[6] Patent: US2005/197337, 2005, A1, . Location in patent: Page/Page column 10
[7] Patent: WO2005/94828, 2005, A1, . Location in patent: Page/Page column 19
[8] Patent: US9138427, 2015, B2, . Location in patent: Page/Page column 301
[9] Patent: WO2015/48301, 2015, A1, . Location in patent: Paragraph 00439
[10] Angewandte Chemie - International Edition, 2017, vol. 56, # 7, p. 1914 - 1918[11] Angew. Chem., 2017, vol. 129, p. 1941 - 1945,5
[12] Journal of Materials Chemistry C, 2014, vol. 2, # 24, p. 4879 - 4892
[13] Bulletin de la Societe Chimique de France, 1996, vol. 133, # 10, p. 1011 - 1021
[14] Synthesis, 2003, # 15, p. 2415 - 2426
[15] Collection of Czechoslovak Chemical Communications, 2004, vol. 69, # 7, p. 1472 - 1478
[16] Tetrahedron Letters, 2007, vol. 48, # 17, p. 3069 - 3072
[17] Patent: US2003/166628, 2003, A1,
[18] Synlett, 2008, # 19, p. 3036 - 3040
[19] Journal of the American Chemical Society, 2010, vol. 132, # 26, p. 8862 - 8863
[20] Patent: US2007/14733, 2007, A1, . Location in patent: Sheet 14
[21] European Journal of Organic Chemistry, 2011, # 1, p. 150 - 165
13
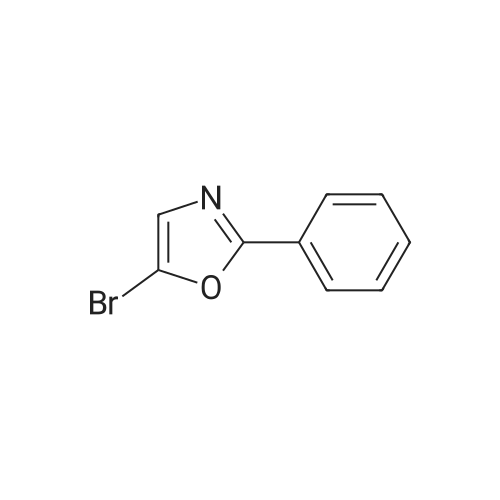
[ 457948-95-7 ]
[ 3536-96-7 ]
[ 860642-35-9 ]
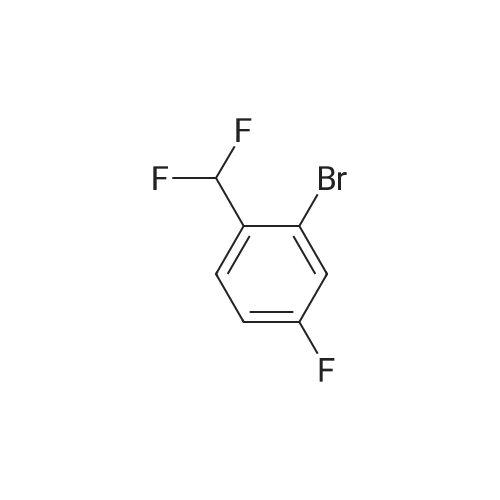
[ 119114-45-3 ]
[ 78191-00-1 ]
Yield
Reaction Conditions
Operation in experiment
100%
Stage #1: at -30℃; for 2 h; Stage #2: With acetic anhydride In tetrahydrofuran; toluene at -30℃; for 0.5 h; Stage #3: With ammonium chloride In tetrahydrofuran; water; toluene at -30 - 10℃;
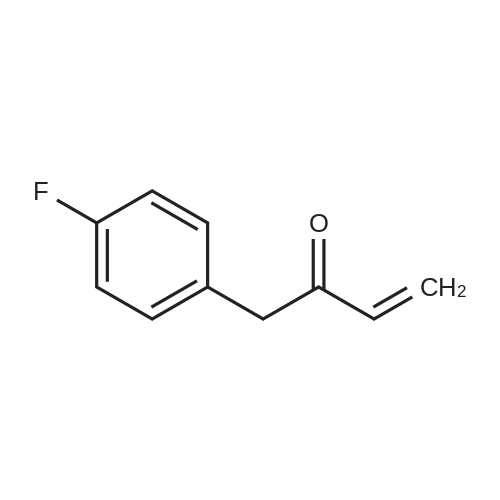
Step 2: 1-(4-Fluorophenyl)but-3-en-2-one (3) Summary: This reaction is very sensitive to the quality of the Grignard reagent and the quench method. Major side products have been identified (A, B, C), and are shown above. The product is unstable when concentrated to an oil, and has moderate stability in solution. The final toluene solution should be kept cold and used in the next step without delay. Procedure: FW: Amt. Moles Equiv. Weinreb amide (2) 197.2 157.2 g 0.800 mol 1.0 eq. Vinyl magnesium 1.6 M 700 mL 1.12 mol 1.4 eq. chloride in THFAc2O 102 151 mL 1.60 mol2.0 eq. NH4Cl (density 1.082 g/mL) (2.5 wt percent aq. solution) - 1.29 L - - Toluene - 1.54 L - - (density 0.865 g/mL) A 3 L round bottom flask equipped with an addition funnel was charged with the Weinreb amide 2 as a 61percent wt solution in toluene (262 g tot mass; 157.2 g 2, 105 g toluene). This solution was diluted to a 0.5 M solution of amide 2 in toluene by addition of 1.32 L of toluene (KF of solution150 ppm). This solution was cooled to -30° C., and vinyl magnesium chloride was slowly added. During the addition of vinyl magnesium chloride the batch temperature is maintained at -30° C. Typical addition time is around 60 minutes. After the vinyl Grignard addition was complete the reaction was allowed to age at -30° C. for 60 minutes. The reaction was checked by HPLC after this 60 minute age. Acetic anhydride (151 mL) was then slowly added to the reaction. Batch temperature is maintained at -30° C. during this addition to avoid impurities. Typical time is 30 minutes. Assay of the reaction at the end of this addition typically shows approximately 0.5percent LCAP of impurity B when compared to product. In a separate 5 L 3-neck round bottom flask a 2.5 wt percent solution of NH4Cl in water (1.29 L) was cooled to 10° C. The batch at -30° C. was cannulated to this vigorously stirred ammonium chloride solution. The final temperature of the batch is typically around 12-13° C. When the batch had reached ambient temperature the aqueous and organic layers were cut. The organic layer was then washed with water (1.3 L). The organic layer was dried with MgSO4 powder (100-200 g) until the KF of this solution reached at or below 1000 ppm. The solids were filtered away and washed with dry MeCN (4.x.50 mL) to provide a solution of the product in THF/MeCN/Toluene (2.0 L, KF 970 ppm, 1.80 kg, 7.29 wt percent, 131 g of 3, 100percent yield) which was used directly in the next step. The impurity profile shows 1.5 LCAP of impurity B and 9.1 LCAP of impurity C.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 2, p. 679 - 683
[2] Patent: WO2011/56725, 2011, A1, . Location in patent: Page/Page column 46-47
28
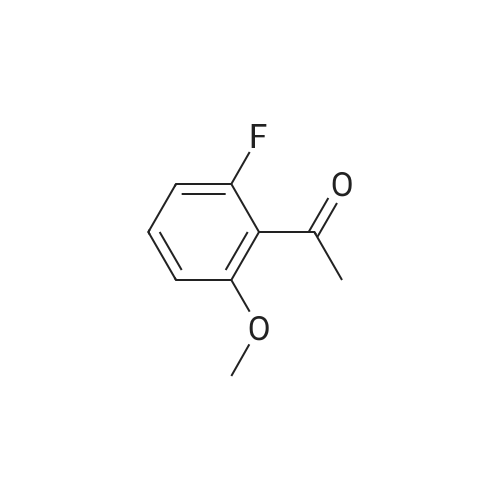
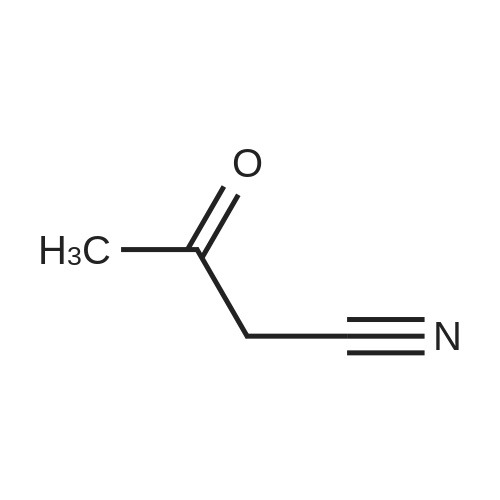
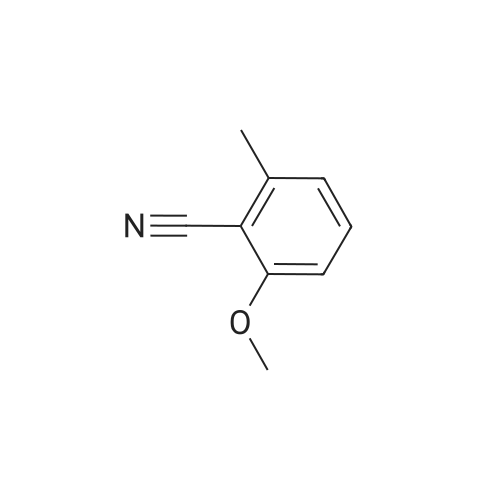
[ 372-47-4 ]
[ 78191-00-1 ]
[ 87674-20-2 ]
[ 87674-21-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 2, p. 679 - 683
[2] Patent: WO2011/56725, 2011, A1, . Location in patent: Page/Page column 46-47
29
[ 372-48-5 ]
[ 78191-00-1 ]
[ 116834-96-9 ]
Yield
Reaction Conditions
Operation in experiment
69%
Stage #1: With n-butyllithium; diisopropylamine In tetrahydrofuran; hexane at -78℃; for 1 h; Inert atmosphere Stage #2: With hydrazine hydrate In tetrahydrofuran; hexane at -78 - 60℃; for 1 h; Inert atmosphere
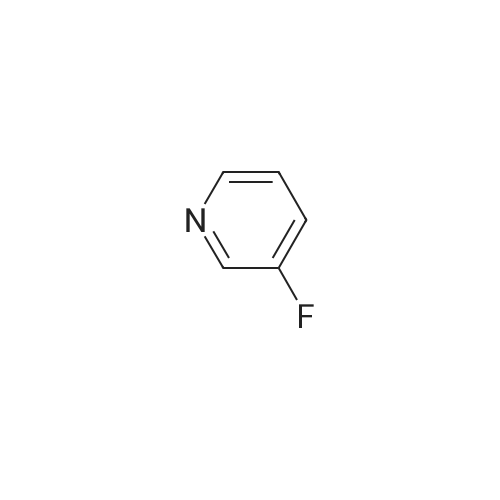
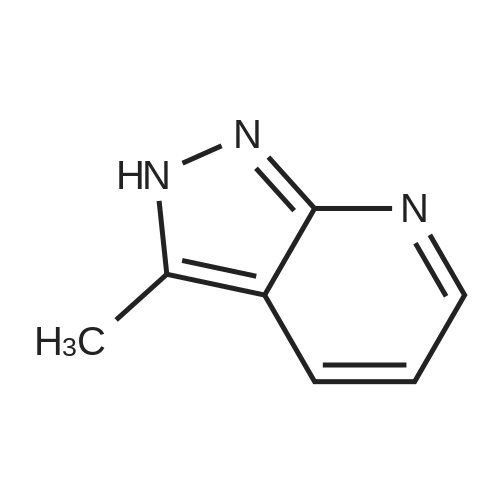
Example 2(1)3-Methyl-1H-pyrazolo[3,4-b]pyridine (2a)Normal-butyllithium (a 2.6 M solution in hexane, 87.0 mL) was dropwise added to a solution of N,N-diisopropylamine (11.2 mL) in THF (100 mL) at -78° C. under a nitrogen atmosphere, followed by increasing the temperature to 0° C. Then, the solution was cooled to -78° C., and a solution of 2-fluoropyridine (6.2 g) in THF (100 mL) was dropwise added thereto at -78° C., followed by stirring for 1 hr. Subsequently, a solution of N-methoxy-N-methylacetamide (8.46 mL) in THF (50 mL) was dropwise added to the reaction solution at -78° C., and then hydrazine monohydrate (31.9 mL) was added thereto, followed by stirring at 60° C. for 1 hr. After distillation of the solvent, water was added to the residue, followed by extraction with ethyl acetate. The organic layer was washed with saturated saline. The organic layer after the washing was dried over anhydrous sodium sulfate, and then the solvent was distilled away to obtain compound (2a) (5.90 g, 69percent) as a white solid.1H-NMR (DMSO-d6) δ 8.08 (1H, dd, J=4.27, 1.71 Hz), 7.79 (1H, dd, J=7.81, 1.71 Hz), 6.60 (1H, dd, J=7.81, 4.27 Hz), 2.44 (3H, s); LRMS (ESI) m/z 134 [M+H]+.
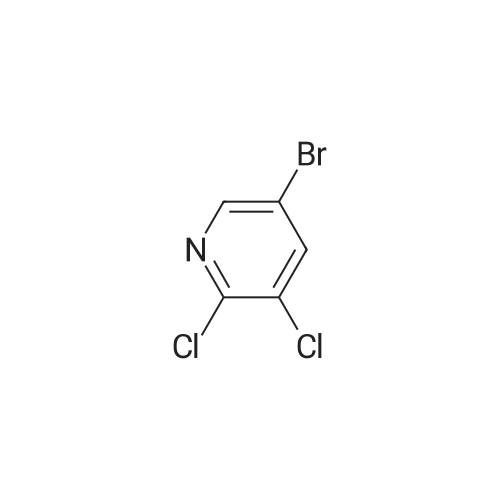
Stage #1: With TurboGrignard In tetrahydrofuran at 0℃; for 0.166667 h; Stage #2: at 0℃; for 2 h;
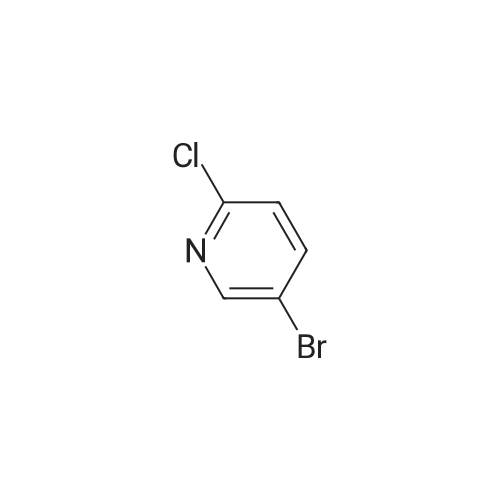
To a solution of 5-bromo-2,3-dichloropyridine (29) (702 mg, 3.09 mmol) in THF (7 ml) was dropwise added isopropylmagnesium chloride lithium chloride complex (3.09 ml, 4.02 mmol) at 0° C. for 10 min. Then N-methoxy-N-methylacetamide (658 μl, 6.19 mmol) in THF (2 ml) was dropwise added to the reaction mixture at the same temperature and stirred at for 2 hrs. Then the reaction mixture was quenched with aqueous solution and extracted with ethyl acetate. The resulting organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The resulting product was chromatographed on silica gel eluting with a gradient of hexane/ethyl acetate (5-15percent) to afford 232 mg of the desired product (30) in 40percent yield as a white solid. 1H-NMR (DMSO-d6) δ: 8.91 (1H, s), 8.55 (1H, s), 2.65 (3H, s).
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 2, p. 679 - 683
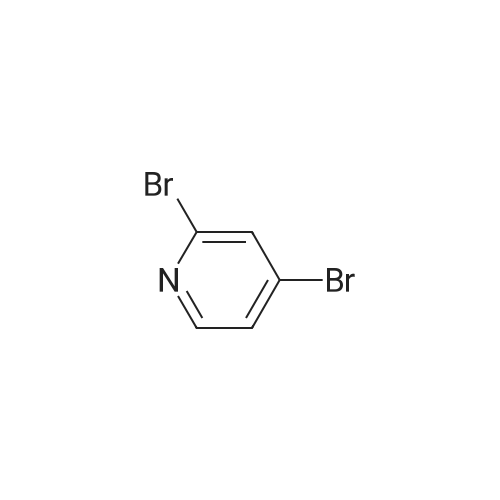
32
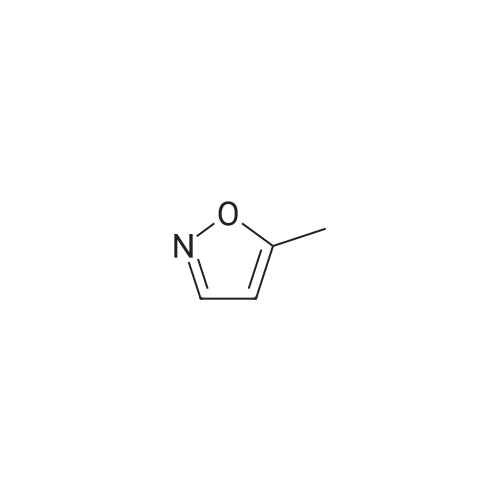
[ 13472-85-0 ]
[ 78191-00-1 ]
[ 213193-32-9 ]
Yield
Reaction Conditions
Operation in experiment
62%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -70℃; for 0.333333 h; Stage #2: at -70 - 20℃;
[0559] To a solution of III-1 (30 g, 0.162 mol, 1 eq.) in 300 mL of anhydrous THF was added dropwise a solution of n-BuLi (2.5M in hexane, 77.5 mL, 0.19 mol, 1.2 eq.) at −70° C. After completion of addition, the mixture was stirred at −70° C. for 20 min, followed by addition of a solution of N-methoxy-N-methylacetamide (33 g, 0.322 mol, 2 eq.) in 100 mL of anhydrous THF by drop wise, the solution was allowed to warm to rt and stirred for 2 hrs. The reaction was quenched with saturated aq. NH4Cl (100 mL), extracted with EtOAc (300 mL×3), the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with petroleum ether/EtOAc (100:1) to yield III-2 (14.8 g, 62percent yield) as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.81 (d, J=2.0 Hz, 1H), 8.16 (dd, J=2.4, 8.4 Hz, 1H), 6.90 (d, J=8.8 Hz, 1H), 3.93 (s, 3H), 2.55 (s, 3H). MS (ESI) m/z [M+H]+ 151.6.
62%
Stage #1: With n-butyllithium In tetrahydrofuran; hexane at -70℃; for 0.333333 h; Stage #2: at 20℃; for 2 h;
To a solution of Ill-i (30 g, 0.162 mol, 1 eq.) in 300 mL of anhydrous THF was added dropwise a solution of n-BuLi (2.5M in hexane, 77.5 mL, 0.19 mol, 1.2 eq.) at -70°C. After completion of addition, the mixture was stuffed at -70°C for 20 mm, followed by addition of a solution of N-methoxy-N-methylacetamide (33 g, 0.322 mol, 2 eq.) in 100 mL of anhydrous THF by drop wise, the solution was allowed to warm to rt and stirred for 2 hrs. The reaction was quenched with saturated aq. NH4C1 (100 mL), extracted with EtOAc (300 mLx3), the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with petroleum ether/EtOAc (100:1) to yield 111-2 (14.8 g, 62percent yield) as a white solid. ‘H NMR (DMSO-d6, 400 MHz) (58.81 (d, J= 2.0 Hz, 1H), 8.16 (dd, J= 2.4, 8.4 Hz, 1H), 6.90 (d, J = 8.8 Hz, 1H), 3.93 (s, 3H), 2.55 (s, 3H). MS (ES) m/z [M+H] 15 1.6.
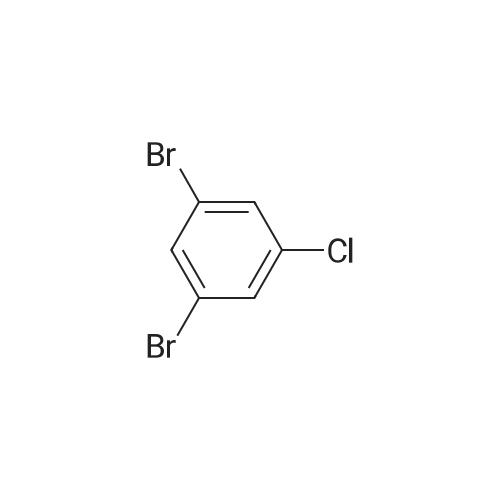
Stage #1: With n-butyllithium In di-isopropyl ether at -78℃; for 0.5 h; Inert atmosphere Stage #2: at -78 - 30℃; for 0.5 h;
1,3-Dibromo-5-fluoro-benzene (20 g, 78.77 mmol, 1 eq) was dissolved in i-Pr2O (200 mL) in a dried flask under nitrogen. The reaction mixture was cooled to -78 °C and stirred under nitrogen atmosphere. n-BuLi (2.5 M, 31.5 mL, 1 eq) was added drop wise to the above solution and the reaction mixture was stirred at -78 °C for 30 min. After complete addition of n-BuLi, N-methoxy-N-methyl-acetamide (9.75 g, 94.5 mmol, 10.05 mL, 1.2 eq) dropped to the above reaction mixture, while keeping the reaction mixture below -78 °C. After addition, the reaction mixture was warmed slowly to 30 °C for 30 min. The reaction mixture was poured into water (150 mL) and the reaction mixture was stirred for 15 min. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (150 mL), combined organic phase, dried over anhydrous Na2SO4, filtered and evaporated in vacuum to give residue (16 g crude). The residue was purified by flash silica gel chromatography (ISCO®; 120 g CombiFlash® Silica Flash Column, Eluent of 0~10percent Ethyl acetate/Petroleum ether gradient 85 mL/min). Compound was obtained as off-white solid (11.3 g, yield 66percent).1H NMR (400 MHz, CDCl3) δ ppm 7.91 - 7.84 (m, 1H), 7.63 - 7.54 (m, 1H), 7.45 (td, J=2.0, 7.8 Hz, 1H), 2.63 - 2.55 (m, 3H).
With n-butyllithium In diethyl ether; hexane at -78℃; for 0.5 h;
To a solution of 1,5-dibromo-2, 4-difluorobenzene (Nucleosides, Nucleotides nucleic Acid, 201 (1 and2), 11-40 (2001) ) (8.8 g, 32.4 mmol) in diethylether (60 ml), 1.6 M n-BuLi in hexane (24.3 ml, 1.2 eq) was added at-78 C under N2 atmosphere. After stirring the reaction mixture at-78 C for 30 min, N-methyl-N- (methyloxy) acetamide (5.0 g, 1.5 eq) was dropped into to quench the reaction. The reaction mixture was stirred at the same temperature for further 30 min. After added acetic acid ( (5.2 ml), water (78 ml), the reaction mixture was extracted with diethylether. The obtained organic phase was washed by 0.2 N HCI aqueous, water, saturated NaHC03 aqueous and saturated NaCI aqueous, and dried over MgS04. After removing the solvent under reduced pressure, the residue was purified by Silica gel chromatography (n-Hexane/EtOAc = 49/1). Desired compound was obtained as pale yellow oil (4.94 g, 65percent).
Stage #1: With n-butyllithium In diethyl ether; hexane at -78℃; for 2 h; Inert atmosphere Stage #2: at -78℃;
To a solution of 2-bromo-5-fluoropyridine (16.78 g, 95.3 mmol) in diethyl ether (348 ml) wasslowly added butyllithium (1.6 M in hexane) (65.6 ml, 105 mmol) at -78° C under argonatmosphere. The resulting yellow reaction mixture was stirred at -78° C for two hours and nmethoxy-n-methylacetamide (10.8 g, 11.2 ml, 105 mmol) was added. The reaction mixture was stirred at -78° C for another hour before quenching with lOml of a 4.OM aqueous solution of hydrochloric acid and the reaction was let to warm up to room temperature. The pH of themixture was ajusted to 7 by addition of 2M aqueous solution of hydrochloric acid. The reaction mixture was washed with 50m1 of brine and the organic phase was collected. The aqueous layer was back-extracted twice with ethyl acetate. Combined organic phases were dried over sodium sulfate and concentrated in vacuo. The crude material was purified by flash chromatography (silica gel, 20g, gradient 0percent to 7percent of ethyl acetate in heptane) to give the title compound (10.76g, 5 1.8percent) as light yellow oil. MS mle: 220.0 ([M+H]).
Stage #1: With n-butyllithium; N-ethyl-N,N-diisopropylamine In tetrahydrofuran at -78 - -65℃; for 1 h; Inert atmosphere Stage #2: at -78℃; for 1 h; Inert atmosphere
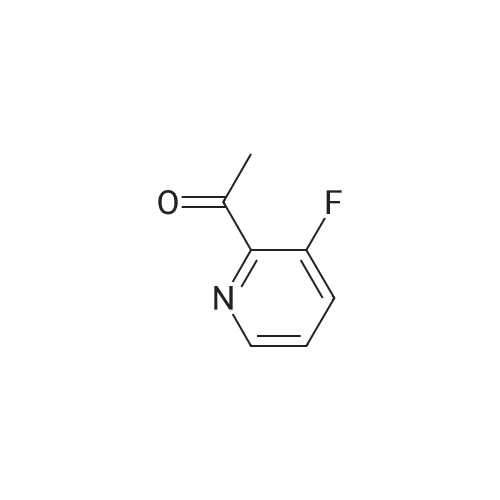
Preparative Example 7 Step 1: l-(5-bromo-2-fluoropyridin-3-yl)ethanone To a solution of diisopropylamine (46.3 g, 458.4 mmol) in tetrahydrofuran (lOOOmL) was added butyllithium (176 mL, 440 mmol, 2.5 M) at -78 °C under nitrogen. After addition, the reaction mixture was stirred for 30 min at -78 °C. 5-bromo-2-fluoropyridine (86.7 g, 442.3 mmol) was added (keeping the temperature under -65 °C). After addition, the mixture was stirred for 1 h. N-methoxy- N-methylacetamide (50 g, 485.4 mmol) was added and stirred at -78 °C for 1 h. The reaction mixture was quenched with water (lOOOmL), extracted with ethyl acetate (3 x 500 mL), washed with brine, dried over sodium sulfate and concentrated to dryness in vacuo. The resulting residue was purified by column chromatography (silica gel, 100-200 mesh, 0.5percent ethyl acetate in petroleum ether) affording 1- (5-bromo-2-fluoropyridin-3-yl)ethanone (43 g, 44.6 percent): H NMR (400 MHz, Chloroform-d): δ 8.46- 8.42 (m, 2H), 2.70 (s, 3H).
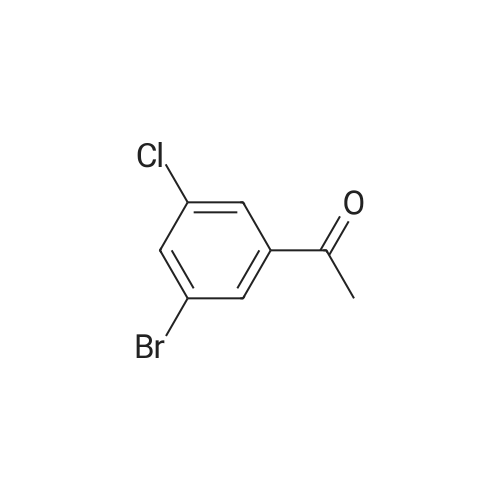
Stage #1: With n-butyllithium In diethyl ether at -78℃; for 0.5 h; Stage #2: at 20 - 24℃; for 0.166667 h;
Example 26: Preparation of 1-(3-bromo-5-chlorophenyl)ethan-1-one (C148) (0740) (0741) 656 1,3-Dibromo-5-chlorobenzene (5.0 g, 18.5 mmol) was dissolved in 167 diethyl ether (61.6 mL) and cooled to −78° C. Because the compound came out of solution, the mixture was removed from the cooling bath. As soon as stirring was again visible from temperature warming, 168 n-butyllithium (8.14 mL, 20.34 mmol) was added dropwise, and the solution was re-immersed in the cold bath. The solution took on a bright yellow color, and the mixture was stirred for 30 minutes. At this point a slight yellow precipitate was visible. 657 N-Methoxy-N-methylacetamide (2.359 mL, 22.19 mmol) was added dropwise, and the reaction mixture was stirred for 10 minutes, then warmed slowly to room temperature. The reaction mixture was quenched with 1 N hydrochloric acid and was extracted with diethyl ether. The combined organic extracts were washed with brine, dried over sodium sulfate and concentrated. The resulting oil was purified on silica running a 0-15percent gradient of 110 acetone in hexanes. The 658 title compound was isolated as a white solid (3.7 g, 86percent): mp 33-36° C.; 1H NMR (300 MHz, CDCl3) δ 7.97-7.95 (m, 1H), 7.85 (dd, J=1.5 Hz, 1H), 7.71 (t, J=1.8 Hz, 1H), 2.59 (s, 3H); IR (thin film) 1687 cm−1; ESIMS m/z 233 ([M+H]+).
N-Methoxy-N-methyl-acetamide (105). Pyridine (10 mL, 124 mmol) was slowly added to a well stirred slurry of O,N-dimethylhydroxylamine hydrochloride (6 g, 62 mmol) and acetyl chloride (4.15 mL, 59 mmol) in CH2Cl2(85 mL) under argon at O°C. The mixture was warmed to room temperature, stirred for 3 hours and then partitioned between brine (55 mL) and diethyl ether (85 mL). Extraction with additional diethyl ether, drying of the combined organic layers over anhydrous Na2SO4, concentration, followed by distillation under reduce pressure gave Weinreb amide 105(4.54 g, 72 percent) as a colourless liquid. bp 42-44°C / 20 mmHg; IR (neat) 3497, 2971, 2941, 2824, 1663 cm-1; 1H NMR (CDCl3, 300 MHz) delta 3.62 (s, 3H), 3.11 (s, 3H), 2.05 (s, 3H); 13C NMR (CDCl3, 75.4 MHz) delta 171.8 (C), 61.0 (CH3), 31.9 (CH3), 19.6 (CH3). Spectral properties identical to samples already described in literature.
65%
With triethylamine; In dichloromethane; at 0 - 20℃; for 0.5h;Cooling with ice;
N,O-dimethyl-hydroxylamine HCl (100 g, 1025 mmol) was suspended under nitrogen in DCM (1000 mL) and cooled in ice. Triethylamine (300 mL, 2152 mmol) was added slowly, then acetyl chloride (76.5 mL, 1076 mmol) was added slowly, the temperature reached 20° despite ice cooling and slow addition. Stirring without cooling was continued for 30 min. Extraction: 1.x.DCM, 1.x.1N HCl, 2.x. saturated NaCl solution. Distillation at 42° C./20 mbar gave 69 g (65percent) of a colorless oil.
65%
With triethylamine; In dichloromethane; at 0 - 20℃; for 0.5h;
N, O-dimethyl-hydroxylamine HC1 (100 g, 1025 mmol) was suspended under nitrogen in DCM (1000 mL) and cooled in ice. Triethylamine (300 mL, 2152 mmol) was added slowly, then acetyl chloride (76.5 mL, 1076 mmol) was added slowly, the temperature reached 20° despite ice cooling and slow addition. Stirring without cooling was continued for 30 min. Extraction: 1 x DCM, 1 x 1 N HC1, 2 x saturated NaCl solution. Distillation at 42 °C/20 mbar gave 69 g (65 percent) of a colorless oil.
61%
With triethylamine; In dichloromethane; at 0 - 20℃; for 4h;
Compound 25-1 (0442) To a suspension of N,O-dimethylhydroamine hydrochloride (100 g, 1026 mmol) in DCM (1000 mL) was added triethylamine (300 mL, 2052 mmol) at 0° C. Acetyl chloride was added dropwise to the suspension for 2 h at 0° C. When the addition was complete, the mixture was allowed to warm to rt and stirred for 2 h. The mixture was washed with brine (1 L), 1 N HCl (500 mL), brine (200 mL) respectively and dried with magnesium sulfate, filtered and concentrated to afford brown oil, which was purified by distillation to afford 25-1 as a colourless liquid (65 g, 61percent).
56%
To a stirred solution of N, O-dimethylhydroxylamine hydrochloride 3 (25 g, 256.4 mmol) in CH2C12 (400 mL) under inert atmosphere was added TEA (51.8 g, 512.8 mmol), and the reaction mixture was stirred for 20 mi Acetyl chloride (22.5 g, 282.0 mmol) was added to the reaction mixture at RT, and the reaction mixture stirred for 1 h. The reaction progress was monitored by TLC; after reaction completion, the reaction mixture quenched with saturated NaHCO3 solution (250 mL) and extracted with CH2C12 (2 x 250 mL). The combined organic extracts were washed with 1 N HC1 solution (100 mL), water (100 mL), brine solution (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to obtain the crude material. The crude material was distilled at 50°C to get 4(11.5 g, 56percent) as colorless liquid. 1HNMR (500 MHz, CDC13): oe 3.68 (s, 3H), 3.17 (s, 3H), 2.11 (s, 3H); LC-MS: 97.3percent; (M+H) Found=104.4; (column: X bridge C-18, 50 x 3.0 mm, 3.5 tm); RT 0.63 mm. 5 mM NH4OAc: ACN; 0.8 mL/min).
With potassium carbonate; In dichloromethane; water;
Example 15 Potassium carbonate (90 g) was added in portions at 0° C. over 10 minutes to a stirred solution of N,O-dimethylhydroxylamine hydrochloride (36.25 g) in water (325 ml), then dichloromethane (300 ml) was added and the mixture was stirred until the internal temperature reached 5° C. Acetyl chloride (25 ml) was added dropwise over 20 minutes, then the mixture was stirred at ambient temperature for 2 hours. The organic phase was separated, washed with water (250 ml) and saturated aqueous sodium chloride solution (250 ml), then it was dried (Na2SO4) and the solvents were removed in vacuo to give N-methoxy-N-methylacetamide (18.26 g) as a colourless oil which was used without further purification.
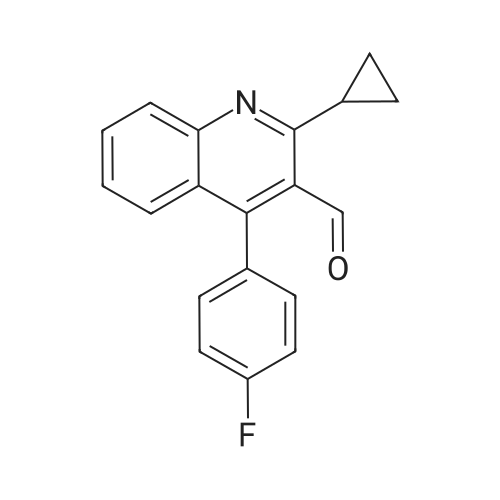
With n-butyllithium; diisopropylamine; In tetrahydrofuran;
REFERENTIAL EXAMPLE 11 STR40 Butyllithium (1.64M hexane solution, 7.54 ml, 12.4 mmol) was added to a THF (40 ml) solution of diisopropylamine (1.25 g, 12.4 mmol) at -78 C., and the mixture was stirred for 15 minutes. Thereto was added a THF (20 ml) solution of N-methoxy-N-methylacetamide (1.27 g, 12.3 mmol) at -78 C., and the resulting mixture was stirred at -78 C. for 15 minutes. To this mixture was added a THF (40 ml) solution of <strong>[121660-37-5]2-cyclopropyl-4-(4-fluorophenyl)-3-formylquinoline</strong> (3.00 g, 10.3 mmol). The reaction mixture was stirred at -78 C. to room temperature over a period of 3 hours before quenching with water and extraction with diethyl ether. The ethereal organic layer was washed with saturated sodium chloride aq solution, dried over magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography (hexane:ethyl acetate=2:1) to give N-methoxy-N-methyl-3 -{2-cyclopropyl-4-(4-fluorophenyl)quinoline-3-yl}-3-hydroxypropanamide (3.70 g, 91% yield). Rf=0.30 (hexane:ethyl acetate=2:1) IR (CHCl3): 3450, 3000, 1640, 1515, 1490, 1420, 1230, 1070, 780 cm-1. 1 H NMR (CDCl3): delta=1.02-1.16 (m, 3H), 1.74 1.79 (m, 1H), 2.66 (d, J=17.2 Hz, 1H), 3.17 (s, 3H), 3.16-3.24 (m, 1H), 3.52 (dd, J=17.2, 11.3 Hz, 1H), 3.62 (s, 3H), 4.14 (d, J=2.4 Hz, 1H), 5.35 (dt, J=11.3, 2.4 Hz, 1H), 7.12-7.35 (m, 6H), 7.58 (dd, J=6.8, 1.4 Hz, 1H), 7.92 (dq, J=8.4, 0.6 Hz, 1H). MS: m/z (rel. intensity) 394 (M+, 11), 363, (M+ -OMe, 46), 334 (58), 292 (100), 274 (38), 263 (37).
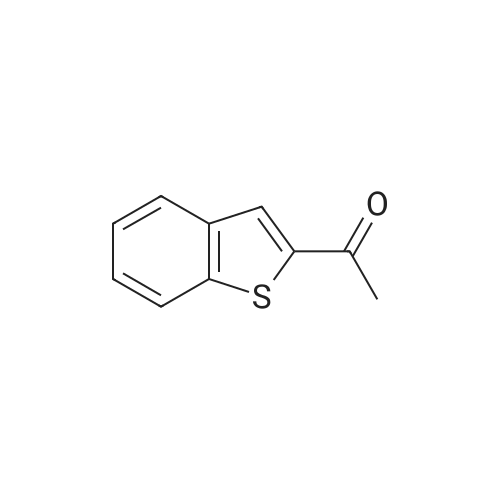
With n-butyllithium; In tetrahydrofuran; ethanol; pentane;
a. 2-Acetyl benzo[b]thiophene. Method a. Using the method described in Scheme 1, benzo[b]thiophene (10 g, 75 mmole) was dissolved in THF (50 mL) and cooled to -78° C. Butyl lithium (28 mL, 2.7 M in hexanes) was added. The mixture was stirred for 15 minutes and N,O-dimethyl acetohydroxamic acid was added. Following an additional 30 minutes of stirring, the reaction was quenched at -78° C. with ethanol and 2N HC1 solution and extracted into ether. The solvent was removed in vacuo and the residue chromatographed on silica gel eluding with 20percent ether in pentane to yield 6.9 g of the desired product as a white solid.
a) To a solution of 1-octyne in tetrahydrofuran (THF, 180 mL) previously cooled to -78 C. is added under nitrogen n-buyllithium (2.5 M solution in hexane, 33.2 mL, 83 mmol). The colorless mixture is stirred 1 h and is then treated with N-methoxy-methylacetamide in THF (20 mL). The reaction mixture is stirred at room temperature for 4 h. After that time 3M HCl solution (100 mL) is added. Tert-butyl-methyl ether (TBME) is added and the reaction mixture is extracted with TBME (3×100 mL). The combined organic layers are washed with saturated NaHCO3, dried over magnesium sulphate and the volatiles are removed on rotary evaporator. The residue is then purified by flash column chromatography (eluent: ethyl acetate/hexane 1:10) to obtain product 121 (colorless oil, 6.2 g, yield: 64%). (0445) NMR: 1H (400.1 MHz, CDCl3), delta=2.35 (2H, t, J=7.2 Hz), 2.31 (3H, s), 1.57 (2H, quint., J=7.2 Hz), 1.43-1.35 (2H, m), 1.33-1.25 (4H, m), 0.89 (3H, t, J=7.0 Hz); 13C (100.1 MHz, CDCl3), delta=184.9, 94.2, 81.4, 32.7, 31.2, 28.5, 27.6, 22.4, 18.9, 14.0.
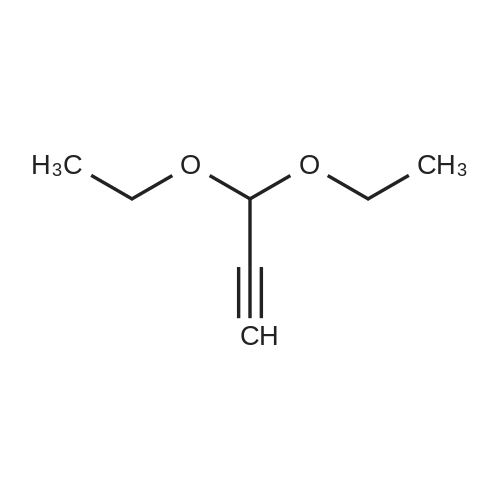
Propargylaldehyde diethylacetal (40 g, 312 mmol) was dissolved in THF (200 mL) under argon and cooled to -70° C. Then a 1.6 M solution of n-butyllithium in hexane (234 mL, 374 mmol) was added and stirring continued for 30 min at -30° C. Then N-methoxy-N-methylacetamide (38.6 g, 374 mmol) in THF (10 mL) was added. After 30 min at -30° C., the reaction was quenched by addition of saturated NH4Cl solution (20 mL). The product was extracted with AcOEt (2.x.200 mL), saturated solution of NH4Cl (2.x.200 mL), dried and concentrated. Chromatography on silica gel with a heptane/ethyl acetate gradient 100:0 to 95:5 gave 38.5 g (72percent) of a colorless oil. GC/MS: m/z=232(M).
72%
Propargylaldehyde diethylacetal (40 g, 312 mmol) was dissolved in THF (200 mL) under argon and cooled to-70 °C. Then a 1.6 M solution of n-butyllithium in hexane (234 mL, 374 mmol) was added and stirring continued for 30 min at-30 °C. Then N-methoxy-N- methylacetamide (38. 6 g, 374 mmol) in THF (10 mL) was added. After 30 min at-30 °C, the reaction was quenched by addition of saturated NH4C1 solution (20 mL). The product was extracted with AcOEt (2 x 200 mL), saturated solution of NH4Cl (2 x 200 mL), dried and concentrated. Chromatography on silica gel with a heptane/ethyl acetate gradient 100: 0 to 95: 5 gave 38.5 g (72 percent) of a colorless oil. GC/MS: m/z = 232 (M).
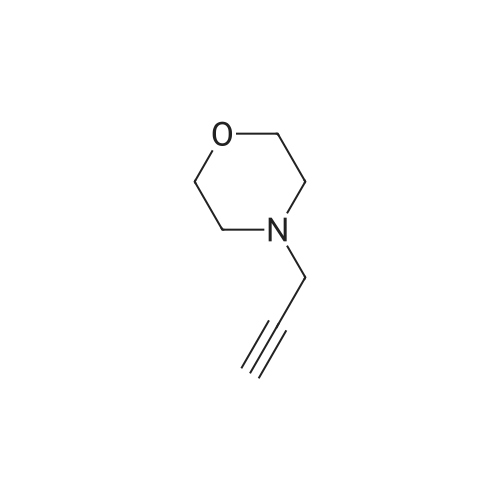
To a cooled ( -40 °C, CH3CN/C02) solution of 4-(2-propyn-1-yl)morpholine (2.2g, 17.58 mmol) in THF (5 mL) was added dropwise via. syringe under N2 a solution of2M isopropylmagnesium chloride in THF (10 mL, 20.00 mmol). The reaction was stirredfor 1 hr then a solution ofN-methoxy-N-methylacetamide (2.2 mL, 20.69 mmol) in THF( 5 mL) was added in one portion. The reaction was stirred for 2 hr (allowed to slowlywarm toRT), quenched with aq. NH4Cl, extracted with EtOAc, washed with brine, dried(Na2S04), filtered and evaoprated to dryness under vacuum. The remaining was purified by silica gel chromatography (Analogix, SF25-60 g, 0 to 80percent EtOAc in hexanes ). The pure fractions were combined and evaporated to dryness to give the product 5-(4-morpholinyl)-3-pentyn-2-one (2.09 g, 12.50 mmol, 71.1percent yield) as a yellow oil. 1H NMR(400MHz, DMSO-d6) 8 3.62-3.57 (m, 4 H), 3.56 (s, 2 H), 2.49-2.43 (m, 4 H), 2.34 (s, 315H). MS(ES)+ m/e 168.0 [M+H]+
71.1%
To a cooled (?40° C., CH3CN/CO2) solution of 4-(2-propyn-1-yl)morpholine (2.2 g, 17.58 mmol) in THF (5 mL) was added dropwise via. syringe under N2 a solution of 2 M isopropylmagnesium chloride in THF (10 mL, 20.00 mmol). The reaction was stirred for 1 hr then a solution of N-methoxy-N-methylacetamide (2.2 mL, 20.69 mmol) in THF (5 mL) was added in one portion. The reaction was stirred for 2 hr (allowed to slowly warm to RT), quenched with aq. NH4Cl, extracted with EtOAc, washed with brine, dried (Na2SO4), filtered and evaporated to dryness under vacuum. The remaining was purified by silica gel chromatography (Analogix, SF25-60g, 0 to 80percent EtOAc in hexanes). The pure fractions were combined and evaporated to dryness to give the product 5-(4-morpholinyl)-3-pentyn-2-one (2.09 g, 12.50 mmol, 71.1percent yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) delta 3.62-3.57 (m, 4H), 3.56 (s, 2H), 2.49-2.43 (m, 4H), 2.34 (s, 3H). MS(ES)+ m/e 168.0 [M+H]+.
53%
4-Prop-2-ynyl-morpholine (22 g, 176 mmol) was dissolved under nitrogen in THF (40 mL) and cooled to -40° C. Then a 2 M solution of isopropyl magnesium chloride in THF (97 mL, 193 mmol) was added while keeping the temperature below-20° C. Stirring at -40° C. to -30° C. was continued for 30 min. In a separate flask, N-methoxy-N-methylacetamide (20 g, 193 mmol) was dissolved under nitrogen in THP (40 mL) and cooled to -10° C. in ice/MeOH. The Grignard solution prepared above was transferred to the Weinreb amide solution at -10° C. via teflon tubing under slightly positive nitrogen pressure in vessel 1. There was no exotherm. Stirring at -10° C. to 0° C. was continued for 2 h. Ther resulting white suspension was poured on a 1:1-mixture of ice and saturated NH4Cl solution (400 mL). Extraction: 2.x.AcOEt, 1.x. saturated NaCl solution. One obtained a yellow oil (26.1 g, 89percent). Chromatography on silica gel in heptane/ethyl acetate 1:2 gave 19.4 g (66percent) of a brown oil which was distilled in the Kugelrohr at 130° C./0.2 mbar. One obtained 15.8 g (53percent) of a yellow oil.
53%
4-Prop-2-ynyl-morpholine (22 g, 176 mmol) was dissolved under nitrogen in THF (40 mL) and cooled to-40 °C. Then a 2 M solution of isopropyl magnesium chloride in THF (97 mL, 193 mmol) was added while keeping the temperature below-20 °C. Stirring at - 40 °C to-30 °C was continued for 30 min. In a separate flask, N-methoxy-N- methylacetamide (20 g, 193 mmol) was dissolved under nitrogen in THF (40 mL) and cooled to-10 °C in ice/MeOH. The Grignard solution prepared above was transferred to the Weinreb amide solution at-10 °C via teflon tubing under slightly positive nitrogen pressure in vessel 1. There was no exotherm. Stirring at-10 °C to 0 °C was continued for 2 h. Ther resulting white suspension was poured on a 1 : 1-mixture of ice and saturated NH4Cl solution (400 mL). Extraction: 2 x AcOEt, 1 x saturated NaCl solution. One obtained a yellow oil (26. 1 g, 89 percent). Chromatography on silica gel in heptane/ethyl acetate 1: 2 gave 19.4 g (66 percent) of a brown oil which was distilled in the Kugelrohr at 130 °C/0. 2 mbar. One obtained 15.8 g (53 percent) of a yellow oil.
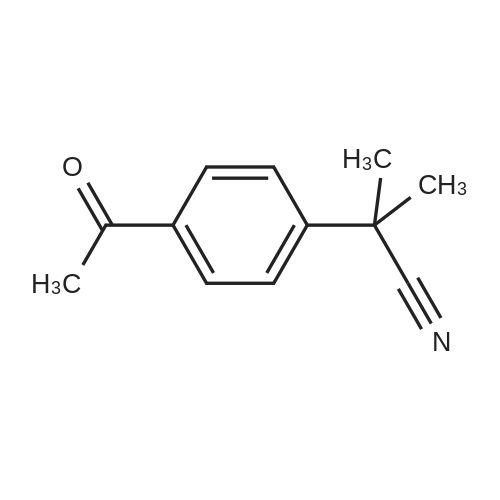
Step b) intermediate 21Synthesis of 2-(4-acetylphenyl)-2-methylpropanenitrile.2-(4-bromophenyl)-2-mefhylpropanenitrile (Ig, 4.46 mmol) is dissolved in anhydrous THF (75ml) the solution is cooled down to -1000C with a diethyl ether-liquid nitrogen bath, n- butyl lithium 2M in c-hexane (4.0 ml, 8.0 mmol) is added and this reaction mixture stirred for 10 min., then N-methoxy-N-mefhyl acetamide (1.6g, 15.6 mmol) is added and the reaction is then left to slowly warm-up to room temperature. After work-up (washing with acidic brine) and concentration the crude mixture is purified on silica gel using a 0 to 50 percent ethyl acetate in hexane gradient, to give the desired compound (660 mg, 78percent) as a clear oil.IH NMR (400 MHz, CHLOROFORM-D) delta ppm 1.76 (s, 6 H) 2.62 (s, 3 H) 7.59 (d, /=8.79 Hz, 2 H) 7.99 (d, /=8.79 Hz, 2 H)
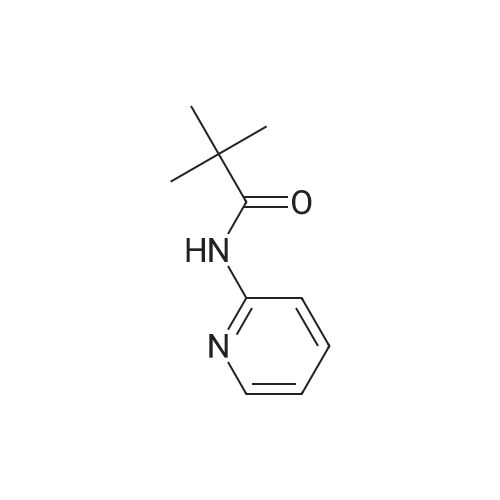
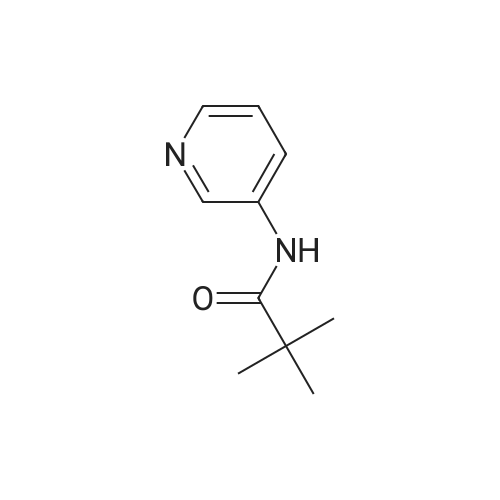
N-(3-Acetylpyridin-2-yl)pivalamide: To a solution of N-(pyridin-2-yl)pivalamide (2.0 g, 11.22 mmoles) in THF at -78 C was added 9.4 mL (2.1 eq) of n-BuLi. The resulting mixture was stirred at 0 C for 3 hours. The reaction mixture was then cooled to -78 C after which N- methoxy-N-methylacetamide (1.2 g, 1.1 eq) was added as a solution in THF. The resulting mixture was stirred at room temperature for 2 hours. It was then quenched into ice-H20. The resulting mixture was extracted with CH2C12 (3X 20 mL) and the combined organic extracts were washed with H20 (2x 20 mL), brine (lx 20 mL), then dried over MgSC^. Concentration and purification by MPLC gave N-(3-acetylpyridin-2-yl)pivalamide (1.62 g, 65%).Spectroscopic data: 1H NMR (300 MHz, CDC13) delta ppm 1.36 (s, 9 H) 2.65 (s, 3 H) 7.09 (dd, J=7.76, 4.83 Hz, 1 H) 8.17 (dd, J=7.76, 1.90 Hz, 1 H) 8.64 (dd, J=4.69, 2.05 Hz, 1 H) 11.49 (br. s., 1 H).
50%
To a solution of 2-PivNH pyridine (2.82 g, 16 mmol) in THF (40 mL) was added n-BuLi dropwise at -78 0C. After addition, the mixture was warmed to 0 0C (ice bath) and stirred for 2 h. The mixture was cooled to -78 0C, and a solution of 7V-methoxy-N- 5 methylacetamide (2.0 g, 19 mmol) in THF (10 mL) was added. The mixture was stirred for 10 min and warmed to rt overnight. The reaction was quenched by the addition of NH4Cl (30 mL), extracted with Et2O (30 mL), washed NaHCO3, brine, and dried over Na2SO4. The crude product was purified by flash chromatography (50%?100% EtOAc in hexanes) to afford a white solid (1.75 g, 50%). mp: 67-68 0C. IR (neat, cm"1): 3268, 10 2967, 1710, 1694, 1663, 1580, 1504, 1450, 1263, 1153. 1H NMR (300 MHz, CDCl3) delta 11.53 (IH, s), 8.66 (IH, dd, J=4.7, 1.8 Hz), 8.19 (IH, dd, J=7.9, 2.0 Hz), 7.10 (IH, dd, J = 7.9, 4.8 Hz), 2.67 (3H, s), 1.37 (9H, s). 13C NMR (100 MHz, CDCl3) delta 201.1, 176.9, 153.7, 152.2, 140.0, 118.1, 40.7, 28.1, 27.5. HRMS (ESI) calc'd for Ci2Hi6N2O2 ([M]+) 220.1211. Found: 220.1211
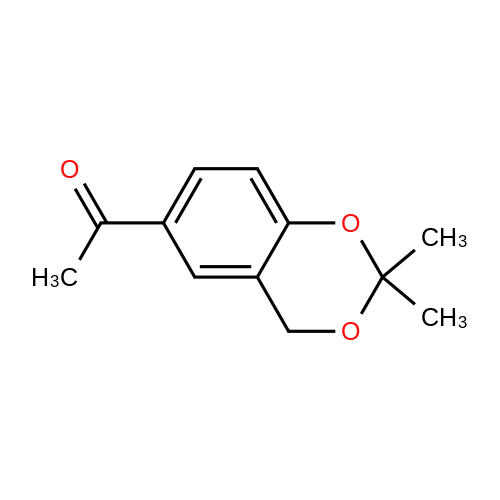
Step 5 1-(2,2-Dimethyl-4H-benzo[d][1,3]dioxin-6-yl)ethanone: At about -78° C. and under an atmosphere of nitrogen, n-butyllithium in tetrahydrofuran (14.8 mL; 2.5M) was added to a solution of 6-bromo-2,2-dimethyl-4H-benzo[d][1,3]dioxine (8.2 g; 33.07 mmol; 1.00 equiv) in tetrahydrofuran (200 mL). The mixture was stirred at -40 to -60° C. for about 2 hours. At about -78° C., N-methoxy-N-methylacetamide (5.2 g; 49.5 mmol; 1.50 equiv) was added, the mixture was stirred at about -78° C. for about 2 hours, and then quenched by adding a solution of saturated ammonium chloride (50 mL). Following standard extractive workup with ethyl acetate, the crude residue was purified by silica gel column chromotagraphy with ethyl acetate/petroleum ether (1/10) to give the title product as a white solid (4.1 g; 60percent yield). 1H NMR (300 MHz, CDCl3) delta: 7.82 (dd, J=2.1, 2.1 Hz, 1H), 7.68 (s, 1H), 6.88 (d, J=8.7 Hz, 1H), 4.91 (s, 2H), 2.57 (s, 3H), 1.59 (s, 6H).
53%
Example 2 Preparation of 1-(2,2-dimethyl-4H-1,3-benzodioxin-6-yl) ethanone (IV) Compound III (1.0eqt) was dissolved in THF (10V) at 25-30° C. and was cooled to -70 to -75° C. n-Butyl lithium (1.5 eqt) was added slowly the above reaction mass at the set temperature and stirred over a period of 60 min. N-methoxy-N-methylacetamide (1.5eqt) in THF (1V) was added to the above reaction mass at -70 to -75° C. The reaction mixture was further allowed for completion. After the completion of reaction, the temperature was raised to -30 to -20° C. and treated with 1N HCl. The reaction mass was further raised to 20-25° C. and ethyl acetate was added. The organic fractions were collected and treated with water. The combined organic fractions were dried over sodium sulfate and concentrated under vacuum to obtain the compound. This was then co distilled with heptane (5V). The crude compound was slurried with diisopropylether-heptane (1:3) at 0-10° C. The resultant was filtered and washed with heptane and suck dried under vacuum. The resultant compound was dried under vacuum over a period of 4 hr to obtain the desired compound (IV). Yield: 53percent; purity by HPLC: 99.97percent
With n-butyllithium; In tetrahydrofuran; hexane; at -78℃; for 2.5h;
(b) 6-Acetyl-2,2-dimethyl-4H-benzo[1,3]dioxine To the product of step (a) (110 g, 0.46 mol) in 1.0 L of THF at -78° C. was added 236 ML (0.51 mol) of 2.14 M n-butyllithium in hexanes via a dropping funnel.After 30 minutes, N-methyl-N-methoxy acetamide (71 g, 0.69 mol, available from TCI) was added.After 2 hours, the reaction mixture was quenched with water, diluted with 2.0 L of 1.0 M aqueous phosphate buffer (PH=7.0) and extracted once with diethyl ether.The diethyl ether phase was washed once with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to give a light orange oil.The oil was dissolved in a minimum volume of ethyl acetate, diluted with hexanes, and to give the title compound as a crystalline solid.
To 110 g (0.46 mol) of compound xx in 1.0 L of THF at -78° C. was added 236 mL (0.51 mol) of 2.14 M n-BuLi in hexanes via a dropping funnel. After 30 minutes, 71 g (0.69 mol) of N-Methyl-N-methoxyacetamide (available from TCI) was added. After 2 hours, the reaction was quenched with water, diluted with 2.0 L of 1.0 M aqueous phosphate buffer (pH=7.0), and extracted once with diethyl ether. The diethyl ether phase was washed once with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a light orange oil. The oil was dissolved in a minimum volume of ethyl acetate, diluted with hexanes, and the product crystallized to give compound yy as a white solid. 1H NMR (300 MHz, CDCl3) delta 7.79 (m, 1H), 7.65 (m, 1H), 6.85 (d, 1H), 4.88 (s, 2H), 2.54 (s, 3H), 1.56 (s, 6H).
To the product of step (a) (110 g, 0.46 mol) in 1.0 L OF THF AT-78 °C was added 236 mL (0.51 mol) of 2.14 M n-butyllithium in hexanes via a dropping funnel. After 30 minutes, N-methyl-N-methoxy acetamide (71 g, 0.69 mol, available from TCI) was added. After 2 hours, the reaction mixture was quenched with water, diluted with 2.0 L of 1.0 M aqueous phosphate buffer (pH = 7. 0) and extracted once with diethyl ether. The diethyl ether phase was washed once with brine, dried over NA2S04, filtered and concentrated under reduced pressure to give a light orange oil. The oil was dissolved in a --122-- minimum volume of ethyl acetate, diluted with hexanes, and to give the title compound as a crystalline solid.
56 g
3L three bottles, Compound 1-9 (140 g, 0.576 mol) was dissolved in tetrahydrofuran (1.3 L), cooled to -78 ° C, A n-hexane solution of n-butyllithium (270 mL, 2.5 M, 0.675 mol) was slowly added dropwise under nitrogen atmosphere, Plus after the reaction at _78 ° C for 1 hour, N-methoxy N-methylacetamide (88.8 g, 0.862 mol 1) In tetrahydrofuran (200 mL), and reacted at 78 ° C for 4 hours. Slowly add 500 mL of water to quench the reaction, The temperature does not exceed 10 ° C, ethyl acetate (500mL) extraction, The organic phase was washed with saturated brine (500 mL) Sodium sulfate, filter, Concentrated to give an oil, Adding n-hexane (800 mL) Stir for 2 hours, White solid precipitation, Filtration gave 30 g of a white solid, Mother liquor concentration, N-hexane (500 mL) was added, Stirring at -50 ° C for 2 hours, White solid precipitation, Filtered to give 26 g of a white solid, A total of 56 g of a white solid was obtained.
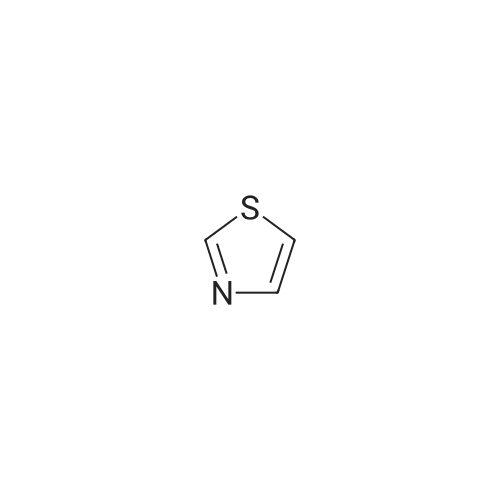
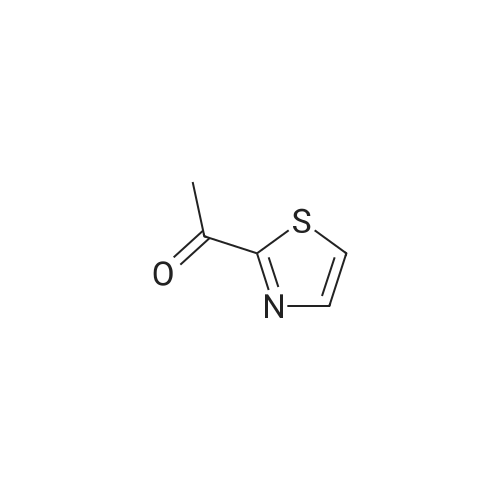
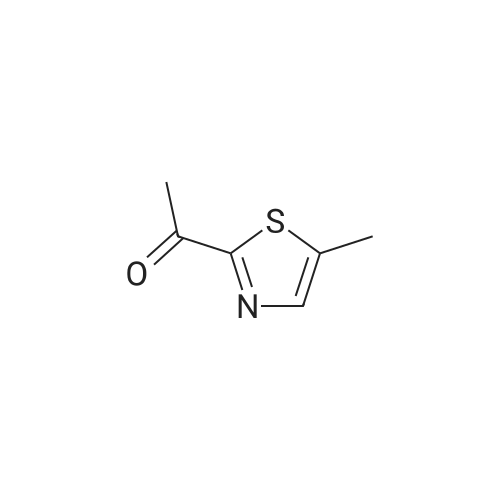
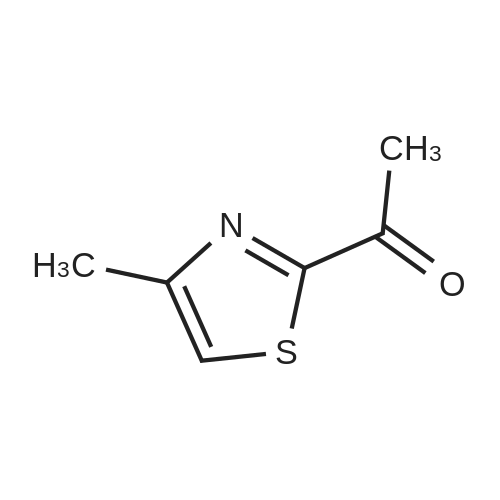
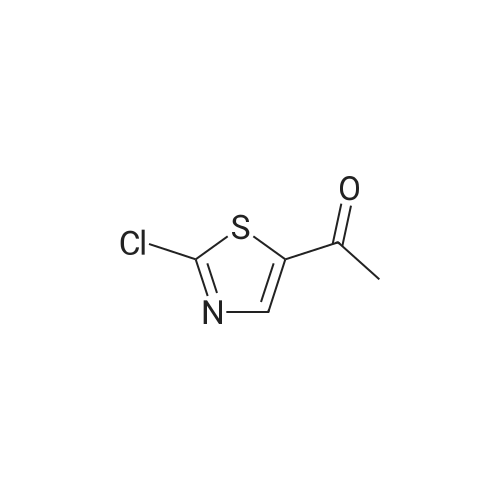
Production Example 4: 1-(5-Methyl-1,3-thiazol-2-yl)-1-ethanone Commercially available 5-methyl-1,3-thiazole (218 mg) was dissolved in tetrahydrofuran (5 ml) to prepare a solution which was then cooled to -78°C. A hexane solution (1.56 M) (1.4 ml) of n-butyllithium was slowly added to the cooled solution over a period of 10 min, and the mixture was stirred at -78°C for 2 hr. <strong>[78191-00-1]N-Methoxy-N-methylacetamide</strong> (206 mg) was dissolved in tetrahydrofuran (2 ml) to prepare a solution which was then slowly added thereto over a period of 10 min, followed by stirring at -78°C for 2 hr. The cooling bath was removed, an aqueous saturated ammonium chloride solution (5 ml) was added, and the mixture was stirred for 30 min. Further, water (5 ml) was added thereto, and the reaction solution was extracted with ethyl acetate. The organic layer was washed with saturated brine and was dried over sodium sulfate. The solvent was removed, and the residue was purified by chromatography on silica gel using hexane/ethyl acetate for development to give the title compound (267 mg, 86percent). 1H-NMR (CDCl3): delta 2.56 (d, J = 1.0 Hz, 3H), 2.67 (s, 3H), 7.65 (d, J = 1.0 Hz, 1H)
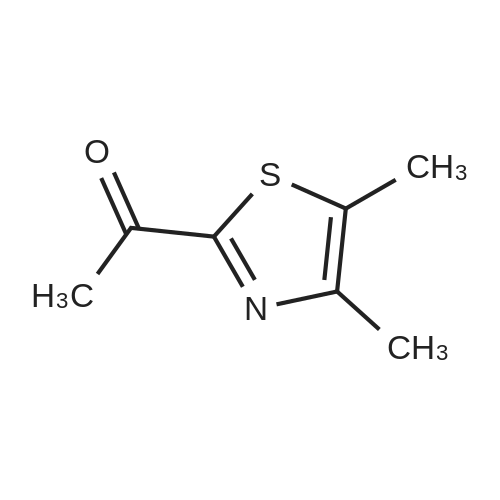
Production Example 5: 1-(4,5-Dimethyl-1,3-thiazol-2-yl)-1-ethanone Commercially available 4,5-dimethyl-1,3-thiazole (453 mg) was dissolved in tetrahydrofuran (10 ml) to prepare a solution which was then cooled to -78°C. A hexane solution (1.56 M) (2.8 ml) of n-butyllithium was slowly added over a period of 10 min, and the mixture was stirred at -78°C for 2 hr. <strong>[78191-00-1]N-Methoxy-N-methylacetamide</strong> (454 mg) was dissolved in tetrahydrofuran (2 ml) to prepare a solution which was then slowly added over a period of 10 min, and the mixture was stirred at -78°C for 2 hr. The cooling bath was removed, an aqueous saturated ammonium chloride solution (10 ml) was added, and the mixture was stirred for 30 min. Further, water (10 ml) was added, and the reaction solution was extracted with ethyl acetate. The organic layer was washed with saturated brine and was dried over sodium sulfate. The solvent was removed, and the residue was purified by chromatography on silica gel using hexane/ethyl acetate for development to give the title compound (609 mg, 98percent). 1H-NMR (CDCl3): delta 2.40 (d, J = 0.7 Hz, 3H), 2.44 (s, 3H), 2.64 (d, J = 0.7 Hz, 3H)
With n-butyllithium; In diethyl ether; hexane; at -78℃; for 0.5h;
To a solution of 1,5-dibromo-2, 4-difluorobenzene (Nucleosides, Nucleotides nucleic Acid, 201 (1 and2), 11-40 (2001) ) (8.8 g, 32.4 mmol) in diethylether (60 ml), 1.6 M n-BuLi in hexane (24.3 ml, 1.2 eq) was added at-78 C under N2 atmosphere. After stirring the reaction mixture at-78 C for 30 min, N-methyl-N- (methyloxy) acetamide (5.0 g, 1.5 eq) was dropped into to quench the reaction. The reaction mixture was stirred at the same temperature for further 30 min. After added acetic acid ( (5.2 ml), water (78 ml), the reaction mixture was extracted with diethylether. The obtained organic phase was washed by 0.2 N HCI aqueous, water, saturated NaHC03 aqueous and saturated NaCI aqueous, and dried over MgS04. After removing the solvent under reduced pressure, the residue was purified by Silica gel chromatography (n-Hexane/EtOAc = 49/1). Desired compound was obtained as pale yellow oil (4.94 g, 65percent).
n-BuLi (13.4 mL of a 1.6 M solution, 1.1 equiv) was added slowly to a solution of the tert-butyl-(3,5-dibromo-phenoxy)-dimethyl-silane (70, 7.16 g, 19.5 mmol) in Et2O (60 mL) cooled to -78° C. under a blanket of N2. The solution was stirred for 25 min, and N-methoxy-N-methyl-acetamide was added via syringe. The solution was warmed slowly to RT, added to saturated ammonium chloride, and extracted with ether. The combined organic layers were washed with brine and dried (MgSO4), filtered and concentrated in vacuo. The crude product material was dissolved in THF (50 mL), and a solution of Bu4NF (approximately 1.2 equiv) was added. The solution was stirred for 2 h and partitioned between EtOAc and brine. The organic layer was washed with water, brine, and dried (MgSO4) to afford 3.35 g (80percent) of 72a.
A suspension of 6-bromo-3-isopropyl[1 ,2,4]triazolo[4,3-a]pyridine hydrochloride (1.00 g, 3.62 mmoi) in THF (18.0 mL) was charged with a positive stream of nitrogen and cooled to 0 0C. The resulting suspension was then treated with commercially available solution of isopropylmagnesium chloride in diethyl ether (2.0 M THF solution, 3.5 mL, 7.0 mmol). The internal temperature of the reaction was not allowed to exceed 0 0C. The resulting dark solution was allowed to stir for 1 hour and then the reaction was treated with N-methoxy-N-methyl acetamide. After 4 hours, the reaction was quenched with 100 mL of saturated ammonium chloride solution and was extracted with ethyl acetate (3 X 250 mL). The resulting organic extract was Na2SO4 dried, filtered, and concentrated in vacuo to a residue that was directly subjected to normal phase silica chromatography (60 percent ethyl acetate, 30 percent hexanes, 10 percent MeOH) to furnish a gum (743 mg, 85 percent). 1H NMR (300 MHz, O4-MeOH) delta 9.02 (s, 1 H), 7.87 (dd, J= 9.7, 1.5 Hz, 1 H), 7.68 (dd, J= 9.6, 1.1 Hz, 1 H), 3.72 (septet, J = 6.8 Hz, 1 H), 2.68 (s, 3H), 1.51 (d, J= 6.8 Hz1 6H); LC/MS C-18 column, tr = 0.48 minutes (5 to 95percent acetonitrile/water over 5 minutes at 1 ml/min with detection 254 nm, at 50 0C). ES-MS m/z 204 (M+H). ES-HRMS m/z 204.1158 (M+H calcd for C11H14N3O requires 204.1131).
85%
A suspension of 6-bromo-3-isopropyl[1,2,4]triazolo[4,3-a]pyridine hydrochloride(1.00 g, 3.62 mmol) in THF (18.0 ml_) was charged with a positive stream of nitrogen and cooledto 0 °C. The resulting suspension was then treated with commercially available solution ofisopropylmagnesium chloride in diethyl ether (2.0 M THF solution, 3.5 ml, 7.0 mmol). Theinternal temperature of the reaction was not allowed to exceed 0 °C. The resulting dark solutionwas allowed to stir for 1 hour and then the reaction was treated with N-methoxy-N-methylacetamide. After 4 hours, the reaction was quenched with 100 ml_ of saturated ammoniumchloride solution and was extracted with ethyl acetate (3 X 250 ml). The resulting organic extractwas Na2SO4 dried, filtered, and concentrated in vacuo to a residue that was directly subjected tonormal phase silica chromatography (60 percent ethyl acetate, 30 percent hexanes, 10 percent MeOH) to furnisha gum (743 mg, 85 percent). 1H NMR (300 MHz, c/4-MeOH) 5 9.02 (s, 1H), 7.87 (dd, J= 9.7, 1.5 Hz,1H), 7.68 (dd, J= 9.6, 1.1 Hz, 1H), 3.72 (septet, J= 6.8 Hz, 1H), 2.68 (s, 3H), 1.51 (d, J= 6.8 Hz,6H); LC/MS C-18 column, tr = 0.48 minutes (5 to 95percent acetonitrile/water over 5 minutes at 1ml/min with detection 254 nm, at 50 °C). ES-MS m/z 204 (M+H). ES-HRMS m/z 204.1158 (M+Hcalcd for CnH14N3O requires 204.1131).
(b) 4-Cyclopropy 1-4-(2,2-dimethylhydrazono)butan-2-one; To a solution of diisopropylamine (14.8 inL, 105 mmol) in THF (80 mL) cooled to 0°C was added slowly BuLi (1.60 M in pentane, 65.4 mL, 105 mmol). The reaction mixture was stirred for 20 minutes and then cooled to -78°C. This solution was added drop-wise to a solution of 2-( 1-cyclopropylethylidene)-l, 1-dimethylhydrazine (12.0 g, 95.1 mmol) in THF (200 mL) cooled to -78°C. The reaction became heterogenous, was diluted with THF (100 mL) and stirred for 15 minutes. N-methoxy-N-methylacetamide (9.81 g, 95.1 mmol) was added drop- wise. The reaction was stirred for 15 minutes at -78°C, warmed to 0°C for 15 minutes and then quenched with saturated NH4Cl (100 mL). The combined organic layers from extractions with EtOAc (3 x 200 mL) were washed with brine, (200 mL), dried (Mg5psi4), passed through silica gel and concentrated to furnish4-cyclopropyl-4-(2,2-dimethylhydrazono)butan-2-one as an oil. Yield: 16.0 g (100 percent). M5 m/z: 169 (M+l).
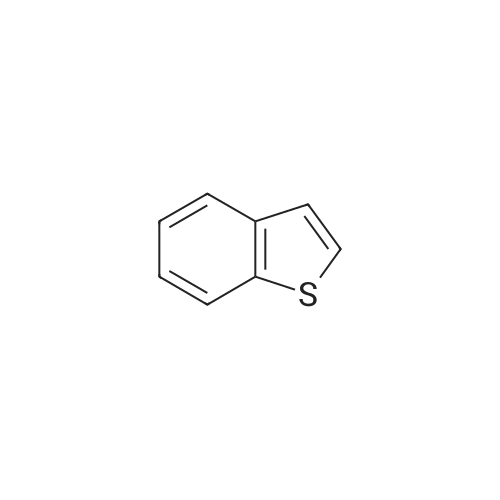
n-Butyllithium (1.6 M solution in hexanes; 74.6 ml) was added dropwise under nitrogen at 0° C. to a stirred solution of benzo[b]thiophene (25.0 g) in ether (350 ml), then the mixture cooled to -70° C. and a solution of N-methoxy-N-methylacetamide (19.21 g) in ether (100 ml) was added dropwise. The mixture was stirred at ambient temperature for 18 hours, then it was poured into saturated aqueous ammonium chloride solution (400 ml). The organic phase was separated, washed with water (300 ml) and saturated aqueous sodium chloride solution (300 ml), dried (Na2SO4) and the solvents were removed in vacuo. The residue was triturated with petroleum ether (b.p. 60-80° C.) (50 ml) and the resulting solid was collected by filtration and dried in vacuo to give 1-(benzo[b]thiophen-2-yl)ethan-1-one (18.7 g) as a brown solid, m.p. 78-81° C.
n-BuLi (1.6 M; 16.4 mL, 26.2 mmol) was added dropwise to a cooled (-70 °C) solution of 4 (7.0 g, 11.9 mmol) in anhydrous THF (150 mL) , and the solution was stirred for 20 min at -70 °C. A solution of N-methoxy-N-methylacetamide (2.45 g, 23.8 mmol) in anhydrous THF (10 mL) was added, and the reaction mixture was stirred for 20 min at -70 °C. After dilution with water, the resulting mixture was extracted with AcOEt (.x.2). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/THF = 6:1-3:1) to give 5 (3.53 g, 54percent) as a colorless powder. The analytical sample was obtained by recrystallization from i-Pr2O (colorless powder). 1H NMR (CDCl3) delta: 0.74 (3H, d, J = 6.8 Hz), 0.97 (3H, d, J = 6.8 Hz), 2.48-2.61 (1H, m), 2.72 (3H, s), 3.75 (1H, br s), 6.83 (1H, d, J = 1.4 Hz), 7.10-7.17 (6H, m), 7.30-7.37 (10H, m), 7.65 (1H, dd, J = 1.6, 8.8 Hz), 7.84 (1H, d, J = 8.8 Hz), 7.87 (1H, d, J = 8.8 Hz), 8.01 (1H, dd, J = 1.6, 8.8 Hz), 8.07 (1H, s), 8.42 (1H, s). IR (KBr): 3519, 2963, 1671, 1275, 1231, 1140, 747, 700 cm-1. Anal. Calcd for C38H34N2O2: C, 82.88; H, 6.22; N, 5.09. Found: C, 82.96; H, 6.17; N, 5.37.
With n-butyllithium; In tetrahydrofuran; hexane; water;
(i) Production of 1-{6-[1-Hydroxy-2-methyl-1-(1-trityl-1H-imidazol-4-yl)propyl]naphthalen-2-yl}-1-ethanone 1-(6-Bromonaphthalen-2-yl)-2-methyl-1-(1-trityl-1H-imidazol-4-yl)-1-propanol (7.0 g) was dissolved in THF (150 ml). The solution was cooled to -70° C. To the mixture was slowly added a solution of n-butyl lithium in hexane (1.6 M; 16.4 ml), and the mixture was stirred at -70° C. for 20 min. To the mixture was added dropwise a solution of <strong>[78191-00-1]N-methyl-N-methoxyacetamide</strong> (2.45 g) in THF (10 ml), and the mixture was stirred at -70° C. for 20 min. To the mixture was added water, and the mixture was extracted with ethyl acetate, washed with saturated sodium chloride solution and dried. The solvent was distilled off and the residue was purified column chromatography (eluent, hexane:THF=6:1-->3:1) to give the titled compound (3.53 g) as a colorless powder. 1H-NMR (CDCl3) delta: 0.74 (3H, d, J=6.8 Hz), 0.97 (3H, d, J=6.8 Hz), 2.48-2.61 (1H, m), 2.72 (3H, s), 3.75 (1H, br s), 6.83 (1H, d, J=1.4 Hz), 7.10-7.17 (6H, m), 7.30-7.37 (10H, m), 7.65 (1H, dd, J=1.6, 8.8 Hz), 7.84 (1H, d, J=8.8 Hz), 7.87 (1H, d, J=8.8 Hz), 8.01 (1H, dd, J=1.6, 8.8 Hz), 8.07 (1H, s), 8.42 (1H, s). IR (KBr): 3519, 2963, 1671, 1480, 1445, 1360, 1275, 1265, 1231, 1190, 1157, 1140 cm-1.
With hydrogenchloride; n-butyllithium; In tetrahydrofuran; hexane;
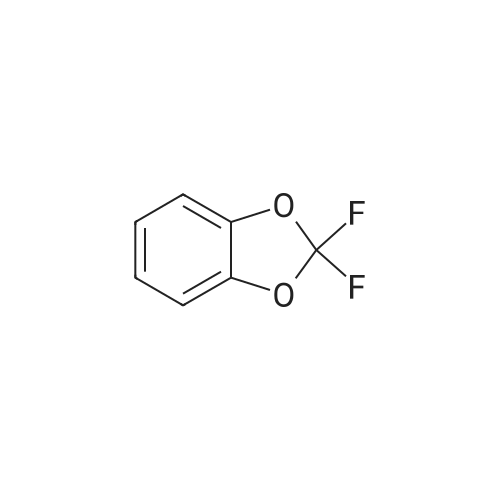
EXAMPLE 3 Preparation of 4-acetyl-<strong>[1583-59-1]2,2-difluoro-1,3-benzodioxole</strong> 6.2 g (55 mmol) of potassium tert.-butoxide dissolved in 40 ml of tetrahydrofuran (THF) are added dropwise at -30 C. under argon, over a period of 20 minutes, to a solution of 8.0 g (50 mmol) of <strong>[1583-59-1]2,2-difluoro-1,3-benzodioxole</strong> in 10 ml of THF in a 250 ml 3-necked flask. The mixture is then cooled to -90 C. (methanol/liquid nitrogen), and 35 ml (55 mmol) of n-butyllithium (1.58M in hexane) are added thereto over a period of 30 minutes. The deep red solution of 2,2-difluoro-1,3-benzodioxol-4-yl-potassium is maintained at -78 C. for 20 minutes. Then, 5.2 g (50 mmol) of N-methyl-N-methoxy-acetamide in 20 ml of THF are added thereto over a period of 15 minutes at -78 C. When the dropwise addition is complete, the beige reaction mixture is allowed to warm up to -10 C. and is then hydrolyzed with 40 ml of 10% hydrochloric acid. The hydrolyzed mixture is extracted three times with 70 ml of diethyl ether each time. The organic solutions are washed three times with 30 ml of 1N HCl each time, dried over Na2SO4 and concentrated in a vacuum rotary evaporator. Upon distillation of the residue, 7.1 g (79%) of the product distil over in the form of a colourless oil at boiling point (b.p.) 114-116 C./30 mbar. 1 H NMR (CDCl3; 300 MHz): 7.65 (d x d; J 7.9; 1.5; 1H); 7.27 (d x d; J 7.9; 1.5; 1H); 7.18 (t; J 7.9; 1H); 2.68 (s; 3H).
4-Methy-2-acetylimidazole-1-dimethylaminosulfonamide (3): To a -78° C. solution of the imidazole 2 (1.99 g, 10.54 mmol) in tetrahydrofuran (70 ml) was added slowly a solution of n-BuLi in hexane (2.5M, 11.60 ml). After 40 minutes, N-methoxy-N-methylacetamide (1.30 g, 12.65 mmol) was added dropwise to the cooled solution. The reaction was allowed to warm to room temperature and maintained for 2 hours. At completion, the reaction was quenched by addition of saturated aqueous NH4Cl (20 ml), then diluted with water (20 ml). The layers were separated, and the organic layer was washed with ethyl acetate (2.x.30 ml). The combined organics were washed with brine (20 ml), then dried over MgSO4 and concentrated. The crude product was purified by flash chromatography over silica (60-80percent ethyl acetate:hexane eluent) to provide 3 as an oil (1.85 g, 76percent yield): M+1=232.1.
EXAMPLE 36; 3-chloro-5-[2-chloro-5-(1H-pyrazolo[3,4-b]pyridin-3-ylmethoxy)phenoxy]benzonitrile (36-5); Step 1: 1-(2-fluoropyridine-3-yl)ethanone (36-1); A solution of 3.13 mL (30.90 mmol) of freshly distilled diisopropylamine in 10 mL of anhydrous THF under nitrogen cooled to -78 C. was treated dropwise with 19.31 mL (30.90 mmol) of a 1.6 M solution of n-BuLi in hexanes. The resulting solution was stirred at -78 C. for approximately 20 minutes, and was briefly (5-10 minutes) warmed to 40 C., then recooled to -78 C. At 30 minutes post addition, 3.00 g (30.90 mmol) of 2-fluoropyridine was added dropwise to the reaction. The resulting solution was stirred at -78 C. for 30 minutes. The reaction was treated dropwise with a solution of 3.16 mL (30.90 mmol) of the Weinreb amide in 30 mL of THF. The resulting solution was stirred 18 hours, allowing the bath to slowly evaporate and the reaction temperature to rise to room temperature. The reaction was treated with 5 mL of 1N HCl, and was concentrated to remove most of the THF. The residue was extracted twice with EtOAc, and the combined extracts were washed with 1N HCl, saturated aqueous NaHCO3 solution, and brine, and were dried over anhydrous MgSO4. Filtration and concentration of the filtrate provided a crude orange oil, which was purified by flash chromatography over silica gel with 3:1 Hexanes/EtOAc to provide 1.10 g of the title product as an orange oil. 1H NMR (CDCl3): 2.72(s,3H), 7.33(m,1H), 8.34(m,1H), 8.41(m,1H).
INTERMEDIATE 1 1 -(tert-Butyl)-3 -(bromomethyl)- 1 H-pyrazolo [3 ,4-b]pyridine- 1 -carboxylateIntermediate I was prepared in accordance with the procedure described in Henke et al, J. Med. Chem. 1997, 40: p. 2706. More particularly:Step 1 : l-(2-fluoropyridine-3-yl)ethanone; A solution of 3.13 mL (30.90 mmol) of freshly distilled diisopropylamine in 10 mL of anhydrous THF under nitrogen cooled to -780C was treated dropwise with 19.31 mL (30.90 mmol) of a 1.6 M solution of n-BuLi in hexanes. The resulting solution was stirred at - 780C for approximately 20 minutes, and was briefly (5-10 minutes) warmed to -4O0C, then recooled to -780C. At 30 minutes post addition, 3.00g (30.90 mmol) of 2-fluoropyridine was added dropwise to the reaction. The resulting solution was stirred at -780C for 30 minutes. The reaction was treated dropwise with a solution of 3.16 mL (30.90 mmol) of the Weinreb amide <n="27"/>(i.e., N-methoxy-n-methylacetamide) in 30 mL of THF. The resulting solution was stirred 18 hours, allowing the bath to slowly evaporate and the reaction temperature to rise to room temperature. The reaction was treated with 5 mL of IN HCl, and was concentrated to remove most of the THF. The residue was extracted twice with EtOAc, and the combined extracts were washed with IN HCl, saturated aqueous NaHCtheta3 solution, and brine, and were dried over anhydrous MgSO-J. Filtration and concentration of the filtrate provided a crude orange oil, which was purified by flash chromatography over silica gel with 3:1 Hexanes/EtOAc to provide 1.10 g of the title product as an orange oil. lH NMR (CDCI3): delta 2.72(s,3H), 7.33(m,lH), 8.34(m,lH),8.41(m,lH).
To a stirred solution of (2R,6S)-4-[6-(tert-Butyl-diphenyl-silanyloxymethyl)-2,3-difluoro- phenyl]-2,6-dimethyl-morpholine (Intermediate 7, 20.0 g, 30.0 mmol) in THF (150 mL) was added sec-butyllithium (1.4 M in cyclohexane, 66.4 mL, 93 mmol) at -78 0C under a nitrogen atmosphere. After stirring for 1 hour at this temperature, N-methoxy-N-methyl-acetamide (3.4 mL, 45 mmol) was added. After stirring for 30 minutes at -78 0C, the solution was allowed reach room temperature and stirring was continued for 12 hours. The reaction mixture treated with saturated aqueous NH4Cl solution and the aqueous layer extracted by EtOAc (2 X 100 mL). Organic phases were combined and dried over anhydrous sodium sulfate. The solution was filtered and the filtrate concentrated. The residue thus obtained was purified over a silica gel flash column using a gradient of ethyl acetate in petroleum ether to give the title compound as a yellow solid. Yield: 13.2 g (84percent). MS(ES)MH+: 538.6 for C3IH37F2NO3Si 1U NMR (400 MHz , CDCl3) delta: 1.1 (s, 15H), 2.8 (m, 4H), 3.4 (m, 2H), 4.6 (s, 2H), 7.3 (t, 4H), 7.4 (t, 2H), 7.6 (d, 4H), 7.8 (s, IH), 10.2 (s, IH).
82%
To a stirred solution of (2R,6S)-4-[6-([tert-Butyl(diphenyl)silyl]oxy}methyl)-2,3- difluorophenyl]-2,6-dimethylmorpholine (Intermediate 3, 15.0 g, 30.0 mmol) in THF (150 mL) was added sec-butyllithium (1.4 M in cyclohexane, 66.4 mL, 93 mmol) at -78 °C under a nitrogen atmosphere. After stirring for 1 h, N-methoxy-N-methyl-acetamide (3.4 mL, 45 mmol) was added and, after stirring for 1A h at -78 °C, the solution was allowed reach room temperature with stirring continuing for 12 hours. The reaction mixture treated with saturated aqueous NH4Cl solution and the aqueous layer extracted by EtOAc (2 X 100 mL). The organic phases were combined, dried (Na2SO4) and concentrated. The residue was purified over a silica gel flash column using a gradient of ethyl acetate in pet. ether to give the title compound as yellow solid.Yield: 13.2 g (82percent)MS (ES) MH+: 538.6 for C31H37F2NO3Si1H NMR (400 MHz, CDCl3) delta: 1.1 (s, 15H), 2.8 (m, 4H), 3.4 (m, 2H), 4.6 (s, 2H), 7.3 (t, 4H), 7.4 (t, 2H), 7.6 (d, 4H), 7.8 (s, 1H), 10.2 (s, 1H).
To a stirred solution of <strong>[3581-89-3]5-methylthiazole</strong> 75 (9 g, 90.90 mmol) in anhydrous THF (200 mL) under inert atmosphere was added n-butyl lithium (40 mL, 99.99 mmol) dropwise for 30 min at -78 oC. To this was added N-methoxy-N-methylacetamide (11.24 mL, 109.1 mmol) dropwise for 20 min at -78 oC, followed by warming to 0 oC and stirring for 16 min. The reaction was monitored by TLC. After completion of the reaction, the reaction mixture was quenched with saturated ammonium chloride solution and extracted using EtOAc. The combined organic extracts were dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to obtain the crude. The crude was purified through silica gel column chromatography using 3% EtOAc/ hexanes to afford compound 76 (12 g, 94%) as pale yellow liquid. TLC: 10% EtOAc/ hexanes (Rf: 0.5); 1H NMR (DMSO-d6, 400 MHz): delta 7.83 (s, 1H), 2.58 (s, 3H), 2.54 (s, 3H).
51%
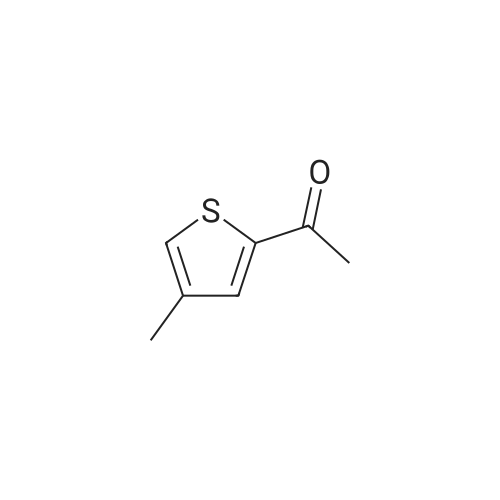
<strong>[3581-89-3]5-methyl-thiazole</strong> (2.00g, 20.2mmol) was dissolved in tetrahydrofuran (50 mL) , at -78C, under nitrogen gas protection was slowly added dropwise n-butyl lithium (2.5M in tetrahydrofuran, 9.68mL, 24.2mmol). The reaction solution was stirred for 0.5 hours at -78C, and dissolved in tetrahydrofuran was slowly added dropwise (ImL) of N- methoxy -N- methyl-acetamide (2.50g, 24.2mmol). The reaction was warmed to 0C stirred for 1.5 h. At 0C slow addition of water (10mL) to the reaction mixture, and extracted with ethyl acetate(30mLx3) . The combined organic phase was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure, the obtained product was purified by preparative high performance liquid chromatography (1: 1 petroleum ether / ethyl acetate, Rf = 0.7) to give the product 1-(5-methylthiazol-2-yl)ethanone (1.45 g of, as a yellow solid), yield: 51%.
43.2%
Intermediate 8: To a solution of <strong>[3581-89-3]5-methylthiazole</strong> (0.179 mL, 2.017 mmol) in ether (13.63 mL) was added nBuLi (1.387 mL, 2.219 mmol, 1.6 M in hexanes) dropwise at -78 C and the reaction mixture was stirred at this temperature for 15 mm. A solution of N-methoxy-N-methylacetamide (229 mg, 2.2 19 mmol) in THF (4.0 mL) was addeddropwise and the reaction mixture warmed to rt and stirred for 1 h. The reaction mixture was quenched with sat. NH4C1. The organic layer was separated and washed with water, brine, dried over anhydrous Na2SO4, filtered and concentrated. The crude product was purified by chromatography to yield Intermediate 8 (123 mg, 0.87 1 mmol, 43.2% yield) as a pale yellow solid. ?H NMR (500 MHz, CDC13) oe 7.66 (s, 1H), 2.68 (s, 3H), 2.57 (s,3H). LCMS Anal. Calc?d for C6H7NOS 141.0, found [M+H] 141.9.
To a stirred solution of n-BuLi (2M in pentane ; 22.4 ML9 0.045 mol) in 70 ML of dry ether AT-78°C was added dropwise 4-METHYLTHIAZOLE (3.7 g, 0.037 mol) in 30 ML of ether over a period of 30 minutes. The mixture was stirred for LH, then N-METHOXY-N- methylacetamide (4.37 mL, 0.041 mol) was added dropwise over 10 minutes. After Ih of stirring AT-78°C THE REACTION MIXTURE WAS WASHED WITH SAT D NAHCO3 AND EXTRACTED WITH ether. The organic layer was dried with NA2S04 and concentrated in vacuo to give 4. 38 g (85percent) of 2a as an oil that was used directly for the next STEP. 1H NMR (CDC13) 6 7.2 (s, 1H), 2.75 (s, 3H), 2.5 (s, 3H).
39%
With n-butyllithium; In hexane; at -78℃; for 2.5h;
Step 1-Synthesis of 1-(4-methyl-1,3-thiazol-2-yl)ethan-1-oneTo a solution of n-butyllithium (2.5M in hexane, 14.4 mL) maintained under nitrogen at -78° C. was added a solution of 4-methyl-1,3-thiazole (3 g, 30.26 mmol) in ether (20 mL).The reaction mixture was stirred at -78° C. for 20 min. <strong>[78191-00-1]N-Methoxy-N-methylacetamide</strong> (3.4 g, 32.97 mmol) was then added dropwise to the reaction mixture over 10 min.The resulting solution was stirred at -78° C. for 2 hr and then quenched by the addition of aqueous sodium bicarbonate (20 mL).The resulting solution was extracted with ether (3*50 mL).The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo.The residue was purified on a silica gel column with ethyl acetate/petroleum ether (1/5) to give 2.1 g (39percent) of 1-(4-methyl-1,3-thiazol-2-yl)ethan-1-one as a yellow oil: 1H NMR (300 MHz, CDCl3) delta 7.25 (s, 1H), 2.71 (s, 3H), 2.51 (s, 3H).
A 2.5 M solution of butyllithium in hexanes (4.67 mmol) was added dropwise to a -78 C. solution of compound 210 (0.90 g, 4.4 mmol) in THF (9 mL) and then stirred for 90 minutes. N-Methoxy-N-methylacetamide (0.92 g, 8.9 mmol) was then added dropwise to this mixture and stirred for 90 min. Upon completion of reaction, the mixture was warmed to approximately 25 C., diluted with NaHCO3 (aqueous) and extracted with Et2O. The organics were combined, dried over anhydrous MgSO4, and concentrated in vacuo, leaving 735 mg of a dark yellow oil, which was used without further purification.
Preparation of 1-(2-Chlorothiazol-5-yl)ethanone A solution of 2-chlorothiazole (2.5 g, 21 mmol) in THF (10 mL) was added dropwise to a chilled (-75° C.) 2.5 M hexanes solution of n-BuLi (9.2 mL, 23 mmol) in THF (70 mL). After the addition was complete the reaction mixture was stirred for 1r. Then a solution of N-methoxy-N-methylacetamide (2.4 g, 23 mmol) in THF (5 mL) was added. The reaction was allowed to stir overnight while warming to 25° C. The reaction was diluted with H2O and thrice extracted with ethyl ether. The organic portions were combined, washed with brine and then dried (Na2SO4). Filtration and removal of solvent gave a residue that was purified by flash chromatography on silica eluding with 5 percent Et2O in pentane, affording product as a white solid, 2.5 g, 15.5 mmol, 73 percent yield.
73%
solution of 2-chlorothiazole (2.5 g, 21 mmol) in THF (10 mL) was added dropwise to a chilled (-75°C) 2.5 M hexanes solution of n-BuLi (9.2 mL,23 mmol) in THF (70 mL). After the addition was complete the reaction mixture was stirred for lhr. Then a solution of N-methoxy-N-methylacetamide (2.4 g, 23 mmol) in THF (5 mL) was added. The reaction was allowed to stir overnight while warming to 25°C. The reaction was diluted with H2O and thrice extracted with ethyl ether. The organic portions were combined, washed with brine and then dried (Na2SO4). Filtration and removal of solvent gave a residue that was purified by flash chromatography on silica eluting with 5 percent Et2O in pentane, affording product as a white solid, 2.5 g, 15.5 mmol, 73 percent yield.
A solution of 2-chlorothiazole (5.0 g, 42 mmol) in THF (10 mL) was added dropwise to a -78° C. solution of n-BuLi (2.5 M in hexanes; 18.4 mL, 46 mmol) in THF (140 mL). The solution was stirred for 1 hour, and then N-methoxy-N-methylacetamide (4.7 g, 46 mmol) was added. The mixture was stirred for another hour, and then it was warmed to approximately 25° C. The reaction was quenched by the addition of a saturated aqueous ammonium chloride solution and extracted with ether (3*75 mL). The organics were combined, washed with brine, filtered and then concentrated. The residue was purified by flash chromatography (20 percent ether/pentane on silica gel), yielding 5.9 grams of yellow semi-solid.
To a STIRRED solution of n-BuLi (2M in pentane ; 15.24 mL, 0.0305 mol) in 70 mL of dry ether at-78°C was added dropwise 29 (6.3 g, 0.0277 mol) in 30 mL of ether over a period of 30 minutes. The mixture was stirred for lh, then N-methoxy-N-methylacetamide (3.83 mL, 0.036 mol) was added dropwise over 10 minutes. After lh of stirring at-78°C the reaction mixture was washed with sat'd NAHCO3 and extracted with ether. The organic layer was dried with NA2S04 and concentrated in vacuo to give 6.66 g (89percent) of 16 as an oil that was used directly for the next step.
To a solution of 2-1 (2.45 g, 6.3 mmol) in TEtaF(20 mL) was slowly added 2.0M /-PrMgCl in Et2O (3.2 mL) at -78 0C. After stirring at -78 0C for 1 h, the reaction mixture was added N-methoxy-N-methylacetamide (779 mg, 7.6 mmol). Subsequently, the mixture was slowly warmed up to it and diluted with EtOAc (100 mL). The mixture was washed with water (20 mL x 3) and dried with anhydrous Na2SO4. The solvent was removed and the residue was purified by silica gel column chromatography (Petroleum ether/EtOAc = 10/1 (v/v)) to give 2-2 (1.25 g, 65percent yield). 1H NMR (CDCl3, 500 MHz): delta 7.56 (d, IH, J= 10.0 Hz), 7.16 (d, IH, J= 10.5 Hz), 3.95 (s, 3H), 3.88 (s, 3H), 2.61 (s, 3H) ppm; LC-MS (ESI): m/z 307.0 [M+H]+.
65%
Referring to scheme 5-3, to a solution of compound 24 (2.45 g, 6.3 mmol) in THF (20 mL) was slowly added 2.0M z-PrMgCl in Et20 (3.2 mL) at -78°C. After stirring at -78°C for 1 h, the reaction mixture was added N-methoxy-N-methylacetamide (779 mg, 7.6 mmol). Subsequently, the mixture was slowly warmed up to rt and diluted with EtOAc (100 mL). The mixture was washed with 3/40 (20 mL x 3) and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel column chromatography (Petroleum ether/EtOAc = 10/1 (v/v)) to give compound 25 (1.25 g, 65percent yield). NMR (CDCI3, 500 MHz): delta 7.56 (d, 1H, J= 10.0 Hz), 7.16 (d, 1H, J= 10.5 Hz), 3.95 (s, 3H), 3.88 (s, 3H), 2.61 (s, 3H) ppm; LC-MS (ESI): m/z 307.0 (M+H)+.
65%
Example 2. Synthesis of 2-10, dimethyl (2S. 2,S -l.l '-((2S.2,S -2.2,-(5.5,-(2.3-bis(2- methoxyethoxy)biphenyl-4,4'-diyl)bis(lH-imidazole-5,2-diyl))bis(pyrrolidine-2,l- diyl))bis(3-methyl-l-oxobutane-2J-diyl)dicarbamate as shown Scheme 2[00196] Step 1. Preparation of 2-2. To a solution of 2-1 (2.45 g, 6.3 mmol) in THF(20 mL) was slowly added 2.0M -PrMgCl in Et20 (3.2 mL) at -78 °C. After stirring at -78 °C for 1 h, the reaction mixture was added N-methoxy-N-methylacetamide (779 mg, 7.6 mmol). Subsequently, the mixture was slowly warmed up to rt and diluted with EtOAc (100 mL). The mixture was washed with water (20 mL x 3) and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel column chromatography (Petroleum ether/EtOAc = 10/1 (v/v)) to give 2-2 (1.25 g, 65percent yield). 1H NMR (500 MHz , CDCls): delta 7.56 (d, 1H, J= 10.0 Hz), 7.16 (d, 1H, J= 10.5 Hz), 3.95 (s, 3H), 3.88 (s, 3H), 2.61 (s, 3H) ppm; LC-MS (ESI): m/z 307.0 [M+H]+.
65%
Step a. Referring to scheme 5-3, to a solution of compound 24 (2.45 g, 6.3 mmol) in THF (20 mL) was slowly added 2.0M /-PrMgCl in Et2O (3.2 mL) at -78°C. After stirring at -78°C for 1 h, the reaction mixture was added N-methoxy-N-methylacetamide (779 mg, 7.6 mmol). Subsequently, the mixture was slowly warmed up to rt and diluted with EtOAc (100 mL). The mixture was washed with H2O (20 mL x 3) and dried with anhydrous Na2SO4. The solvent was removed and the residue was purified by silica gel column chromatography (Petroleum ether/EtOAc = 10/1 (v/v)) to give compound 25 (1.25 g, 65percent yield). 1H NMR (CDCl3, 500 MHz): delta 7.56 (d, 1H, J= 10.0 Hz), 7.16 (d, 1H, J= 10.5 Hz), 3.95 (s, 3H), 3.88 (s, 3H), 2.61 (s, 3H) ppm; LC-MS (ESI): m/z 307.0 (M+H)+.
a) synthesis of l-(3-Fluoro-pyridin-2-yl)-ethanone (2a)[0205] A solution of BuLi (39.5 ml, 98.7 mmol, 2.5 M in hexane) in 100 ml THF was added to a solution of 3-fluoropyridine (1) (8g, 82.3 mmol) in 20 ml THF of at - 78 C. The reaction mixture was stirred at -78 C for 30 min. Then, N-methyl-N- methoxyacetamide 10.9 ml (106.9 mmol) was added at - 78 C. The reaction mixture was slowly warmed up to room temperature and stirred for 1 hour. The reaction was quenched with ice-cold saturated aqueous ammonium chloride solution. The reaction mixture was diluted with ethyl acetate and washed with water. The organic layer was dried with magnesium sulfate and concentrated under reduced pressure. The residue was purified via column chromatography (Ethyl acetate: n-Hexane = 1 : 6) and a 1 : 1 mixture of (2a) and (2b) was obtained 4.78 g (yield: 42%).
(S)-Methyl 6-(methoxy(methyl)amino)-4,6-dioxo-3-(tritylamino)hexanoate (13).; A round bottom flask equipped with a stirbar was charged with HN(TMS)2 (1.8 mL, 8.67 mmol, 3.81 equiv) and THF (4 mL) and was brought to 0 °C (ice/water bath). w-BuLi (3.53 mL of a 2.24 M solution in hexanes, 3.48 equiv) was next added and the mixture was permitted to stir for 1 h. The solution was cooled to -78 °C (acetone/dry ice). In a separate flask, N- methoxy-N-methylacetamide (0.74 mL, 8.00 mmol, 3.52 equiv) in THF (6 mL) was cooled to - 78 °C and LHMDS solution (from above) was added via cannula. The resulting anion solution was stirred for 1 h at -78 °C. In a separate flask, Tr-LAsp(OMe)-Im (12, 1.0 g, 2.28 mmol, 1.0 equiv) in THF (8 mL, 0.28 M) was cooled to -78 °C and the anion solution was added via cannula to the solution of the amino acid imidazolide. The reaction mixture was warmed to -41 °C (CH3CN/dry ice) and stirred for 45 min. The reaction was quenched with saturated aqueous NH4CI and extracted with EtOAc. The combined organics were washed with brine and dried over Na2S04. The reaction was filtered and concentrated in vacuo. Purification by silica gel flash column chromatography (20percent EtOAc/hexanes) afforded 13 (0.78 g, 72percent) as a sticky27orange foam and a mixture of keto-enol forms. \\p \\D = 9.4 (c = 4.7, CHC13); Reported as a mixture of keto:enol (8:2) forms: 1H NMR (500 MHz, CDC13) delta 13.51 (s, 1H); 7.46 - 7.53 (m, 6H); 7.14 - 7.28 (m, 9H); 5.20 (s, 1H); 3.79 (q, J = 5.5 Hz, 1H), 3.64 (s, 3H); 3.63 (s, 3H); 3.60 (s, 1H); 3.56 (s, 3H); 3.55 (s, 3H); 3.39 (d, J = 8.7 Hz, 1H); 3.26 (d, J = 16.2 Hz, 1H); 3.15 (s, 3H); 3.14 (s, 3H); 2.51 (dd, J = 4.2, 16.5 Hz, 1H); 2.47 (dd, J = 4.3, 15.4 Hz, 1H); 2.22 (dd, J = 6.3, 16.4 Hz, 1H); 2.02 (dd, J = 7.9, 15.3 Hz, 1H); 13C NMR (125 MHz, CDC13) delta 205.71, 176.86, 172.04, 171.82, 168.07, 146.56, 146.441, 146.18, 128.97, 128.19, 128.07, 126.86, 126.64, 87.05, 71.43, 61.48, 59.55, 54.33, 51.94, 51.64, 45.13, 38.86, 37.80, 32.11; IR (thin film): 3057, 2950, 1732, 1659, 1595, 1489, 1447, 1438, 1358, 1266, 1204, 1113, 902, 775, 736, 708 cm"1; HRMS [M+K] for C28H30N2O5K, calcd., 513.17863, found, 513.17760 (Error = 2.0105 ppm).
72%
(S)-Methyl 6-(methoxy(methyl)amino)-4,6-dioxo-3-(tritylamino)hexanoate (13).A round bottom flask equipped with a stirbar was charged with HN(TMS)2 (1.8 mL, 8.67 mmol, 3.81 equiv) and THF (4 mL) and was brought to 0° C. (ice/water bath).n-BuLi (3.53 mL of a 2.24 M solution in hexanes, 3.48 equiv) was next added and the mixture was permitted to stir for 1 h.The solution was cooled to -78° C. (acetone/dry ice).In a separate flask, N-methoxy-N-methylacetamide (0.74 mL, 8.00 mmol, 3.52 equiv) in THF (6 mL) was cooled to -78° C. and LHMDS solution (from above) was added via cannula.The resulting anion solution was stirred for 1 h at -78° C. In a separate flask, Tr-LAsp(OMe)-Im (12, 1.0 g, 2.28 mmol, 1.0 equiv) in THF (8 mL, 0.28 M) was cooled to -78° C. and the anion solution was added via cannula to the solution of the amino acid imidazolide.The reaction mixture was warmed to -41° C. (CH3CN/dry ice) and stirred for 45 min.The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc.The combined organics were washed with brine and dried over Na2SO4.The reaction was filtered and concentrated in vacuo.Purification by silica gel flash column chromatography (20percent EtOAc/hexanes) afforded 13 (0.78 g, 72percent) as a sticky orange foam and a mixture of keto-enol forms. [alpha]D27=9.4 (c=4.7, CHCl3); Reported as a mixture of keto:enol (8:2) forms: 1H NMR (500 MHz, CDCl3) delta 13.51 (s, 1H); 7.46-7.53 (m, 6H); 7.14-7.28 (m, 9H); 5.20 (s, 1H); 3.79 (q, J=5.5 Hz, 1H), 3.64 (s, 3H); 3.63 (s, 3H); 3.60 (s, 1H); 3.56 (s, 3H); 3.55 (s, 3H); 3.39 (d, J=8.7 Hz, 1H); 3.26 (d, J=16.2 Hz, 1H); 3.15 (s, 3H); 3.14 (s, 3H); 2.51 (dd, J=4.2, 16.5 Hz, 1H); 2.47 (dd, J=4.3, 15.4 Hz, 1H); 2.22 (dd, J=6.3, 16.4 Hz, 1H); 2.02 (dd, J=7.9, 15.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) delta 205.71, 176.86, 172.04, 171.82, 168.07, 146.56, 146.441, 146.18, 128.97, 128.19, 128.07, 126.86, 126.64, 87.05, 71.43, 61.48, 59.55, 54.33, 51.94, 51.64, 45.13, 38.86, 37.80, 32.11; IR (thin film): 3057, 2950, 1732, 1659, 1595, 1489, 1447, 1438, 1358, 1266, 1204, 1113, 902, 775, 736, 708 cm-1; HRMS [M+K] for C28H30N2O5K, calcd., 513.17863, found, 513.17760 (Error=2.0105 ppm).
n-Butyllithium (2.5 M in hexane, 2.31 rnL, 5.8 mmol) was added to a solution of 1,1-dimethylethyl 7-bromo-2,3-dihydro-l,4-benzoxazepine-4(5H)-carboxylate (1.56 g, 4.76 mmol ) in tetrahydrofuran (20 mL) and the resulting mixture was stirred at -78 0C for one hour. A solution of Lambda/-methoxy-iV-methylacetamide (0.97g, 9.4 mmol) in tetrahydrofuran (5 mL) was added to the reaction mixture dropwise then warmed to room temperature and stirred for an additional hour. Water (50 mL) was added and the resulting mixture was extracted with ethyl acetate (3x 40 mL). The combined organic extract was washed with water, then brine solution (80 mL each), dried over sodium sulfate, filtered, concentrated to give 1,1-dimethylethyl 7-acetyl-2,3-dihydro-l,4-benzoxazepine-4(5H)- carboxylate (1.34g, 97percent yield ); MS (EI) for Ci6Eta2iNO4: 292 (MH+).
Toluene-4-sulfonic acid 6-bromo-pyridin-3-yl ester (XVI) (980 mg, 2.99 mmol) was dissolved in tetrahydrofuran (THF) (3 mL) and the resulting solution was cooled to -78 °C. 2.0 M lithium diisopropylamide (LDA) (2.24 mL, 4.48 mmol) dissolved in tetrahydrofuran was slowly added dropwise to the solution and the solution was stirred at -78 °C for 3 hrs. And then, N-methoxy-N-methyl acetamide (609.77 muL, 5.97 mmol) was slowly added dropwise to the solution and the solution was stirred for 2 hrs. Saturated sodium hydrogen carbonate (NaHCO3) solution was added dropwise to the solution and the solution was diluted with ethylacetate (EA). Then, the organic solvent was dried over anhydrous magnesium sulfate (MgSO4), filtered, and evaporated under reduced pressure to concentrate. The resulting residue was isolated and purified by silica gel column chromatography (n-Hex/EA = 4/1) to give the title compound (570 mg, 52 percent). 1H NMR (400 MHz, CDCl3) delta 7.97 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.65 (s, 1H), 7.37 (d, J = 8.0 Hz, 2H), 2.58 (s, 3H), 2.49 (s, 3H).
44%
A 1-L 3-necked round bottom flask equipped with a nitrogen inlet, rubber septum, and an internal temperature probe was charged with a solution of 109 (10.9 g, 33.2 mmol) in tetrahydrofuran (130 mL). The resulting solution was cooled to -78 °C under a nitrogen atmosphere and lithium di-isopropyl amide solution (1 M in tetrahydrofuran/hexanes, 50 mL, 1.5 eq.) was slowly added via a cannula at such a rate that the internal temperature did not exceed -74 °C. After the resultingslurry was stirred at -78 °C for 2 h, N-methoxy-N-methyl-acetamide (6.0 g, 58.1 mmol) was added dropwise maintaining the internal temperature below -75 °C. The mixture was stirred at -78 °C for 1.5 h and then poured into an ice-cold saturated aq NaHCO3 solution (120 mL). The mixture was diluted with water (50 mL) and extracted with ethyl acetate (3x). The combined organic layers were dried (Na2SO4) and concentrated to leave cmde product as ared-brown oil (12.6 g). The oil was dissolved in 50 mL of dichloromethane and the solution was mixed with 70 g of silica gel. The solvent was allowed to evaporate and the residue was dry loaded on top of a 500 mL fritted funnel containing 65 g of flash silica gel wetted with 300 mL of dichloromethane. Washing the column with dichloromethane (500 mL) followed by concentration gave 110 (5.36 g, 44percent) as a colorless oil. ?H NMR (400 MHz, chloroform-d) oe 7.97 (s, 1H), 7.73 - 7.67 (m, 2H), 7.65 (s, 1H), 7.41 - 7.33 (m, 2H), 2.58 (s, 3H), 2.49 (s,3H).
28.9%
Example 73 2-amino-6'-(3-chlorophenyl)-l,2',2'-trimethyl-2',3'-dihydrospiro[imidazole-4,4'- pyrano[2,3-c]pyridin]-5(lH)-oneStep A: 2.5M n-BuLi in hexanes (59.7 mL, 149 mmol) was added by syringe to a -78°C solution of diisopropylamine (21.8 mL, 155 mmol) in ether (200 mL). The reaction mixture was stirred for 35 minutes, and then a solution of 6-bromopyridin-3-yl 4-methylbenzene-sulfonate (9.80 g, 29.9 mmol; prepared as described in Serban, Georgeta, Hitoshi Abe, Yasuo Takeuchi, and Takashi Harayama. "A New Approach to the Benzopyridoxepine Core by Metal Mediated Intramolecular Biaryl EtherFormation." Heterocycles. Vol. 75, Issue 12 (2008): 2949-2958) in ether (450 mL) was added via canula over 50 minutes. The reaction mixture was stirred at -78°C for 2 hours, and then a -78°C solution of N- methoxy-N-methylacetamide (12.7 mL, 1 19 mmol) in ether (20 mL) was added by canula. The reaction mixture was stirred another 1 hour, after which saturated NH4CI (200 mL) was added, and the reaction mixture was removed from the cold bath and allowed to warm to ambient temperature. The mixture was diluted with water and extracted with ether (2 X). The combined extracts were dried (NaaSC^), filtered, and concentrated. The crude was purified on silica gel (5-40percent EtOAc in hexanes gradient) to give recovered 6-bromopyridin-3-yl 4-methylbenzenesulfonate (4.4 g, 44.9percent yield) as a solid and 4-acet l-6- bromopyridin-3-yl 4-methylbenzene-sulfonate (3.20 g, 28.9percent yield) as an oil.
4-(2-Oxo-propyl)-nicotinic acid To <strong>[3222-50-2]4-methyl-nicotinic acid</strong> (1.37 g, 10 mmol) in THF (75 mL) at -78 C., lithium diisopropyl amide (0.020 mol, prepared from a 2M solution of n-butyl lithium in hexane, 0.02 mol and diisopropyl amine, 0.02 mol) was added dropwise, and the solution was stirred at -78 C. for 0.5 hour, the mixture was warmed up to -20 C. for 0.5 hours, and then cooled to -78 C., a solution of N-methoxy-N-methyl acetamie (1.03 g, 10 mmol) in THF (50 mL) was added at -78 C. After the addition was complete, the mixture was stirred at -78 C. for 0.5 hours, then allowed to warm to room temperature and stirred for another 2 hours, and then water (10 mL) was added. The reaction mixture was concentrated in vacuo and cooled to form a solid, which was used for the next step directly.
To <strong>[3222-50-2]4-methyl-nicotinic acid</strong> (1.37 g, 10 mmol) in THF (75 mL) at -78 C., lithium diisopropyl amide (0.020 mol, prepared from a 2M solution of n-butyl lithium in hexane, 0.02 mol and diisopropyl amine, 0.02 mol) was added dropwise, and the solution was stirred at -78 C. for 0.5 hour, the mixture was warmed up to -20 C. for 0.5 hours, and then cooled to -78 C., a solution of N-methoxy-N-methyl acetamie (1.03 g, 10 mmol) in THF (50 mL) was added at -78 C. After the addition was complete, the mixture was stirred at -78 C. for 0.5 hours, then allowed to warm to room temperature and stirred for another 2 hours, and then water (10 mL) was added. The reaction mixture was concentrated in vacuo and cooled to form a solid, which was used for the next step directly.
Step A: 5-Acetylpyrimidine 5-Bromopyrimidine (3.18 g, 20 mmol) was dissolved in 50 mL of tetrahydrofuran, While cooled to -78 C, 15 mL of 1.6 M n-butolithium in hexane solution was added dropwise with stirring, After the solution was stirred for 30 minutes, a solution of N-methoxyl-N-methylacetamide (2.58 g, 25 mmol) in tetrahydrofuran solution (10 mL) was added slowly. The mixture was stirred at -78 C for 1 hour and then allowed to be warmed slowly. When the temperature of the mixture was at 0 C, aqueous ammonium chloride solution was added. The solution obtained was extracted with ethyl acetate for 3 times, The pooled extracts were washed with brine, dried over magnesium sulfate, concentrated under reduced pressure, and purified by silica gel column chromatography using 5% methanol / methylene chloride as eluent to obtain 1 g of the title compound (45% yield). MS(M+1)=123.05.
175 mg
The5-bromo-pyrimidine (9.4 mmol) is dissolved in tetrahydrofuran steams again 30ml in. At -78 C lower, will n-BuLi (11.3 mmol) in drops to the reactionsystem, and stirring 30 minutes. N-methoxy-N-methyl acetamide (11.8 mmol) oftetrahydrofuran solution (15 ml) at -78 C in the system dropping, andstirring 1 hour. Under room temperature, add saturated ammonium chlorideaqueous solution, ethyl acetate extraction 3 times. Concentrated extract,column chromatography (ethyl acetate: petroleum ether = 1 the [...] 10)separated to obtain title compound 175 mg per litre. 1 H-NMR (CDCl 3)delta 9.37 (1H, s), 9.24 (2H, s), 2.66 (3H, s).
Example 2(1)3-Methyl-1H-pyrazolo[3,4-b]pyridine (2a)Normal-butyllithium (a 2.6 M solution in hexane, 87.0 mL) was dropwise added to a solution of N,N-diisopropylamine (11.2 mL) in THF (100 mL) at -78 C. under a nitrogen atmosphere, followed by increasing the temperature to 0 C. Then, the solution was cooled to -78 C., and a solution of 2-fluoropyridine (6.2 g) in THF (100 mL) was dropwise added thereto at -78 C., followed by stirring for 1 hr. Subsequently, a solution of N-methoxy-N-methylacetamide (8.46 mL) in THF (50 mL) was dropwise added to the reaction solution at -78 C., and then hydrazine monohydrate (31.9 mL) was added thereto, followed by stirring at 60 C. for 1 hr. After distillation of the solvent, water was added to the residue, followed by extraction with ethyl acetate. The organic layer was washed with saturated saline. The organic layer after the washing was dried over anhydrous sodium sulfate, and then the solvent was distilled away to obtain compound (2a) (5.90 g, 69%) as a white solid.1H-NMR (DMSO-d6) delta 8.08 (1H, dd, J=4.27, 1.71 Hz), 7.79 (1H, dd, J=7.81, 1.71 Hz), 6.60 (1H, dd, J=7.81, 4.27 Hz), 2.44 (3H, s); LRMS (ESI) m/z 134 [M+H]+.
A. Synthesis of 1-(1-methyl-1H-pyrazol-4-yl)ethanone 4-Bromo-1-methyl-1H-pyrazole (41.3 mL, 400 mmol), was dissolved in tetrahydrofuran (750 mL) and cooled to -78 C. N-Butyllithium (2.5 M solution in hexanes, 160 mL, 400 mmol) was added drop-wise over 30 minutes, and the resulting mixture was stirred for 1 hour at -78 C. After drop-wise addition of a solution of N-methoxy-N-methylacetamide (40.9 mL, 400 mmol) in tetrahydrofuran (100 mL) to the -78 C. reaction mixture, the cooling bath was allowed to warm to 0 C. over 4 hours. The reaction was then quenched with saturated aqueous sodium chloride solution (50 mL), and volatiles were removed in vacuo. The residue was diluted with ethyl acetate (1000 mL), treated with magnesium sulfate, and stirred for 30 minutes before being filtered and concentrated in vacuo. Purification was carried out via silica gel chromatography (material was loaded in a minimum amount of dichloromethane; Gradient: 5% to 100% ethyl acetate in heptane) to provide a pale yellow oil that solidified on standing. Yield: 28.5 g, 230 mmol, 57%. 1H NMR (500 MHz, CDCl3) delta 2.37 (s, 3H), 3.90 (s, 3H), 7.83 (s, 1H), 7.84 (s, 1H).
An oven-dried 100 mL three-necked round-bottomed flask under a nitrogen atmosphere was charged with compound enyne S15 (0.5 g, 1.4 mmol) and THF (20 mL). The solution was cooled to -78° C. in a dry ice/acetone bath for 15 min, then LDA (0.7 mL of 2.0 M heptane/THF/ethylbenzene solution, 1.4 mmol) was added via syringe. The mixture was stirred at -78° C. for 1 h, then N-methoxy-N-methylacetamide (0.16 mL, 1.54 mmol) was added. The solution was warmed to rt and was stirred for 4 h. The consumption of the starting material was monitored by TLC (AcOEt/n-hexane 1:9). The reaction was quenched by adding sat'd aq ammonium chloride solution (40 mL). The layers were separated and the aqueous phase was extracted with ether (3*30 mL). The combined organic layers were dried over Na2SO4, gravity filtered, and concentrated under reduced pressure. The reaction residue was purified by silica gel flash chromatography, eluting with AcOEt/n-hexane 2:8, to provide 0.47 g of the title compound as a colorless oil in 86percent yield. Data for 5m (EB-079) 1H NMR (400 MHz, CDCl3) 7.53 (d, J=7.2 Hz, 1H), 7.39 (t, J=7.7 Hz, 1H), 7.36-7.14 (m, 2H), 6.98 (d, J=15.6 Hz, 1H), 6.11 (dt, J=15.6, 7.6 Hz, 1H), 4.33 (q, J=7.1 Hz, 4H), 3.11 (s, 2H), 3.07 (d, J=7.6 Hz, 2H), 2.40 (s, 3H), 1.36 (t, J=7.1 Hz, 6H) ppm 13C NMR (100 MHz, CDCl3) 183.9, 169.2 (2C), 134.9, 132.7, 131.3, 129.6, 128.7, 126.9, 126.9, 125.9, 87.7, 83.6, 62.1 (2C), 56.8, 36.3, 32.9, 23.4, 14.1 (2C) ppm IR (thin film) 2982, 2936, 2213, 1734, 1679, 1203 cm-1 HRMS (TOF MS ES+) [M+H]+ calcd for C21H24O5Cl, 391.1312; found, 391.1299
(1) 4-(tert-Butyldiphenylsilyloxymethyl)-5-methyl-1H-imidazole, and 5-(tert-butyldiphenylsilyloxymethyl)-4-methyl-1H-imidazole (5-Methyl-1H-imidazol-4-yl)methanol hydrochloride, and (4-methyl-1H-imidazol-5-yl)methanol hydrochloride (5.00 g) were suspended in dichloromethane (100 mL), followed by addition of triethylamine (14.1 mL) and TBDPSCl (13.1 mL) under stirring, and the stirring was continued for 3 days. The reaction solution was diluted with water, and extracted with chloroform. The combined extract was washed with saturated brine, dried over anhydrous sodium sulfate, and subsequently the solvent was distilled off under reduced pressure. The resulting residue was purified by flash chromatography (methanol/chloroform=0%?5%) using silica gel column (product name: cartridge 40M, manufactured by Biotage, Ltd.) to afford the mixture (7.77 g) of the title compound.1H-NMR (CDCl3) delta: 1.05 (9H, s), 2.03 (3H, s), 4.68 (2H, s), 7.37-7.47 (7H, m), 7.66-7.68 (4H, m).(2) 4-(tert-Butyldiphenylsilyloxymethyl)-5-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazole, and 5-(tert-butyldiphenylsilyloxymethyl)-4-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazole The mixture (35.4 g) obtained in (1) was dissolved in acetonitrile (500 mL), followed by addition of TEA (37.8 mL) and then 2-trimethylsilylethoxymethyl chloride (hereinafter, also referred to as SEMCl) (21.4 mL) while stirring under ice cooling, followed by stirring at 70 C. for 12 hours. The reaction mixture was allowed to cool to room temperature, and the solvent was distilled off under reduced pressure. The residue was diluted with ethyl acetate, subsequently washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by flash chromatography (ethyl acetate/hexane=15%?50%) using silica gel column (product name: cartridge 65i, manufactured by Biotage, Ltd.) to afford 4-(tert-butyldiphenylsilyloxymethyl)-5-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazole (17.9 g) as a low polar component, and 5-(tert-butyldiphenylsilyloxymethyl)-4-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazole (9.42 g) as a high polar component.Low polar component: 4-(tert-butyldiphenylsilyloxymethyl)-5-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazole1H-NMR (CDCl3) delta: -0.02 (9H, s), 0.89 (2H, t, J=8.2 Hz), 1.04 (9H, s), 2.10 (3H, s), 3.45 (2H, t, J=8.2 Hz), 4.65 (2H, s), 5.14 (2H, s), 7.34-7.45 (3H, m), 7.75-7.70 (4H, m).MS (ESI) m/z: 481 [M+H]+.High polar component: 5-(tert-butyldiphenylsilyloxymethyl)-4-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazole1H-NMR (CDCl3) delta: -0.03 (9H, s), 0.89 (2H, t, J=8.0 Hz), 1.01 (9H, s), 1.92 (3H, s), 3.46 (2H, t, J=8.3 Hz), 4.68 (2H, s), 5.38 (2H, s), 7.37-7.47 (3H, m), 7.66-7.68 (4H, m).MS (ESI) m/z: 481 [M+H]+.(3) 1-[5-(tert-Butyldiphenylsilyloxymethyl)-4-methyl-1-(2-trimethylsilylethoxymethyl)-1H-imidazol-2-yl]ethanone The mixture (2.24 g) obtained in (2) was dissolved in THF (30 mL), followed by cooling to -78 C. while stirring under an argon atmosphere. To the present solution was added dropwise n-butyl lithium (2.69 M hexane solution, 2.82 mL) slowly using a syringe, followed by stirring at the same temperature for 30 minutes. Then, N-methoxy-N-methylacetamide (0.95 mL) was added using a syringe, and stirring was continued for 17 hours while the temperature was raised to room temperature. A saturated aqueous ammonium chloride solution was added to the reaction solution to terminate the reaction, followed by extraction with ethyl acetate. The combined extract was washed with saturated brine, dried over anhydrous sodium sulfate, and subsequently the solvent was distilled off under reduced pressure. The resulting residue was purified by flash chromatography (ethyl acetate/hexane=0%?15%) using silica gel column (product name: SNAP Cartridge KP-Sil 100 g, manufactured by Biotage, Ltd.) to afford the title compound (1.85 g).1H-NMR (CDCl3) delta: -0.05 (9H, s), 0.86 (2H, t, J=8.3 Hz), 1.02 (9H, s), 1.97 (3H, s), 2.65 (3H, s), 3.50 (2H, t, J=8.0 Hz), 4.73 (2H, s), 5.93 (2H, s), 7.38-7.47 (6H, m), 7.65-7.66 (4H, m).
A stirred mixture of l,3-dibromo-5-tert-butyl-benzene (11 g, 37.67 mmol, 1 eq) in anhydrous i-PnO (160 mL) was cooled to -78 C under nitrogen atmosphere was treated drop wise with n-BuLi (2.5 M, 15.1 mL, 1 eq). The resulting mixture was stirred at -78 C for 30 min. After complete addition of n-BuLi, N-methoxy-N-methyl-acetamide (4.66 g, 45.20 mmol, 4.81 mL, 1.2 eq) was added dropwise, while keeping the mixture below -78 C. After addition, the mixture was warmed slowly to 10 C for 30 min. The reaction mixture was quenched with saturated NH4CI (200 mL) then seperated and extracted with ethyl acetate (200 mL*2). The combined organic layers were washed with H2O (200 mL*2) and brine (200 mL), dried over Na2S04, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash silica gel chromatography (ISCO; 120 g CombiFlash Silica Flash Column, Eluent of 0-10% Ethyl acetate/Petroleum ether gradient 85 mL/min). The title was obtained as a light yellow solid (2 batches, 7.3 g, average yield: 64%). NMR (400MHz, CDCL) d = 7.94 - 7.90 (m, 1H), 7.88 (t, J=L7 Hz, 1H), 7.71 (t, J=L8 Hz, 1H), 2.60 (s, 3H), 1.37 - 1.31 (m, 9H).
a) Synthesis of l-(2-Chloro-5-fluoro-pyridin-4-yl)-ethanone (2)[0224] A solution of BuLi (7.3 ml, 2.5M in Hexane, 1.2 eq.) in THF (20 ml) was added to a solution of 2-Chloro-5-fluoro-pyridine (1.5ml, 15.2 mmol) in THF (5ml) at -78 C. The reaction mixture was stirred at -78 C for 30 min. N-Methyl-N- methoxyacetamide (2.04 g, 1.3 eq.) was added at -78 C. The resulting mixture was slowly warmed to room temperature and stirred for lh. The reaction was quenched with ice-cold saturated aqueous ammonium chloride solution. The organic layer was separated and the aqueous layer was extracted with EA (ethyl acetate). The combined organic layers were washed with NaHC03 and dried with MgS04. Removal of the solvent followed by the purification of the residue by flash column chromatography on silica gel (Hexand/EtOAc 10: 1) afforded l-(2-Chloro-5-fluoro-pyridin-4-yl)- ethanone as a liquid. (1.613 g, 61%). 1H NMR (300MHz, CDC13): delta 8.41 (d, J = 1.8 Hz, 1H), 7.68 (d, J = 4.8 Hz, 1H), 2.67 (d, J = 4.2 Hz, 3H)
A round bottomed flask was charged with 5-bromo-2-chloropyridine (5.30 g, 27.6 mmol) in THF under N2 and cooled at 0 C. A solution of 1 M iso-propylmagnesiumchloride-lithium chloride complex in THF (40 mL) was added drop wise over 15 min. After 70 min N-methoxy-N-methylacetamide (4.1 mL, 38 mmol) was added drop wise. After stirring for 5 min at 0 C. the cooling bath was removed. The mixture was left stirring overnight and was then quenched by the addition of 100 mL saturated aqueous NH4Cl solution. The mixture was extracted with 3×100 mL EtOAc. The combined organic layers were washed with water followed by brine and dried over MgSO4. Evaporation of the volatiles at 80 C., 10 mbar for 1 h gave the title compound (3.596 g, 84) sufficiently pure for the next step.
In THF (5 mL) was dissolved 2-bromo-5-(1-(tert-butyldimethylsilyloxy)-2,2,2-trifluoroethyl)thiazole (515 mg, 1.37 mmol) obtained in Step 2. n-Butyllithium (1.65 mol/L solution in n-hexane, 1.24 mL, 2.05 mmol) was added dropwise at ?78° C. and the mixture was stirred at ?78° C. for 30 minutes. A solution of N-methoxy-N-methylacetamide (1.38 mL, 13.7 mmol) in THF (1 mL) was added to the reaction mixture and the mixture was stirred at ?78° C. for 2 hours. A saturated aqueous ammonium chloride solution and water were added to the reaction mixture. Extraction with ethyl acetate and drying over anhydrous magnesium sulfate were performed. After filtration, the solvent in the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give 1-(5-(1-(tert-butyldimethylsilyloxy)-2,2,2-trifluoroethyl) thiazol-2-yl)ethanone (437 mg, 940).
94%
Compound BG2 (515 mg, 1.37 mmol) was dissolved in THF (5 mL). n-butyllithium (1. 65 mol/L solution in nhexane,1.24 mL, 2.05 mmol) was added dropwise at -78°C and the mixture was stirred at -78°C for 30 minutes. Asolution of N-methoxy-N-methylacetamide (1.38 mL, 13.7 mmol) in THF (1 mL) was added to the mixture and the mixturewas stirred at -78°C for 2 hours. A saturated aqueous ammonium chloride solution and water were added to the mixtureand extraction with ethyl acetate was performed. The organic layer was dried over anhydrous magnesium sulfate andfiltered. The solvent was evaporated from the filtrate under reduced pressure. The residue was purified by silica gelcolumn chromatography (hexane/ethyl acetate = 4/1) to give compound BG3 (437 mg, 94percent).
With boron trifluoride diethyl etherate; In 1,4-dioxane; at 20 - 100℃; for 1h;Inert atmosphere;
General procedure: To a solution of o-diaminobenzene (5.0 mmol, 0.5405 g) and Weinreb amide (N-methoxy-Nmethylbenzamide, 5.0 mmol, 0.82595 g) in dioxane (10 mL), boron trifluoride diethyl etherate (5.0 mmol) was added at room temperature. The reaction mixture was stirred for the specified time (Table 3) at 100 °C. TLC revealed the complete consumption of starting material. Subsequently hydrolysis was achieved by the addition of saturated NH4Cl solution (50 mL). The aqueous layer was extracted with ethyl acetate (3 X 25 mL), washed with water (2 X 25 mL), brine solution (2 X 25 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to get crude product. This was purified by column chromatography over silica gel using hexane/ethyl acetate mixture in 1:1 ratios as eluent.
With boron trifluoride diethyl etherate; In 1,4-dioxane; at 20 - 100℃; for 1h;Inert atmosphere;
General procedure: To a solution of o-diaminobenzene (5.0 mmol, 0.5405 g) and Weinreb amide (N-methoxy-Nmethylbenzamide, 5.0 mmol, 0.82595 g) in dioxane (10 mL), boron trifluoride diethyl etherate (5.0 mmol) was added at room temperature. The reaction mixture was stirred for the specified time (Table 3) at 100 C. TLC revealed the complete consumption of starting material. Subsequently hydrolysis was achieved by the addition of saturated NH4Cl solution (50 mL). The aqueous layer was extracted with ethyl acetate (3 X 25 mL), washed with water (2 X 25 mL), brine solution (2 X 25 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to get crude product. This was purified by column chromatography over silica gel using hexane/ethyl acetate mixture in 1:1 ratios as eluent.
Into a 500-mL round-bottom flask purged and maintained with an inert atmosphere of N2 was placed a solution of 1-bromo-3- (propan-2-yl)benzene (5.0 g, 25.11 mmol, 1.00 equiv) in THF (250 mL). This was followed by the addition of n-BuLi (20 mL, 2.00 equiv) dropwise with stirring at -60 C. The mixture was stirred at -60C for 30 mm. To this was added N-methoxy-N-methylacetamide (3.9 g, 37.82 mmol, 1.50 equiv) dropwise with stirring at -30 C. The resulting solution was stirred for 3 h at -30 C in a liquid N2 bath. The reaction was then quenched by the addition of 20 mL of water. The resulting solution was diluted with 200 mL of EtOAc, washed with 2x100 mL of brine, dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was applied onto a silica gel column with EtOAc/petroleum ether (1:10). This resulted in 2.7 g (66%) of the title product as colorless oil.
To a solution of 1 -bromo-3-isopropylbenzene (10 g, 0.05 moi) in THF was added dropwise with BuLi (47 ml,, 0.075 niol) at -60 'C. After stirred 30 minutes, N-methoxy-N-methyiacetamide (6.22 g, 0.06 mol) was added. The mixture was stirred at -30C for 3 hours. Then the mixture was quenched with H20, the mixture was partitioned between EtOAc and water. The layer was separated and washed with water, brine, dried over Na2SC>4, and concentrated. The residue was purified by column chromatography to obtained the titled compound. ? NMR (CHLQRQFORM-d): delta 7.85 (s, i l l ). 7.79 (dt, j = 7.6, 1 .4 Hz, 1H), 7.33 - 7.53 (m, 2H), 3.00 (dt, J - 13.8, 6.9 Hz, 1H), 1.30 (d, J - 6.7 Hz, 6H)
To a solution of l-bromo-3-isopropylbenzene (10 g, 0.05 niol) in THF was added dropwise with BuLi (47 mL, 0.075 mol) at -60C. After stirred 30 minutes, -methoxy- -methylacetamide (6,22 g, 0.06 mol) was added. The mixture was stirred at -30C for 3 hours. Then the mixture was quenched with H20, the mixture was partitioned between EtOAc and water. The layer was separated and washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by column chromatography to obtained the titled compound. 'H NMR (CHLOR OFQRM-d) : delta 7.85 (s, 1H), 7.79 (dt, J = 7.6, 1.4 Hz, 1H), 7.33 - 7.53 (m, 2H), 3.00 (dt, J - 13.8, 6.9 Hz, 1H), 1.30 (d, J - 6.7 Hz, 6H) LC-MS : m/z 163(M+H)+
To a solution of Compound 59A (3.96 g, 20 mmol) in dry THF (20 mL) at - 78 C was dropped a solution of n-BuLi in THF (2.5 M, 8 mL, 18 mmol). After stirred at - 78 C for 1 hour, to the mixture was dropped N-methoxy-N-methylacetamide (1.87 g, 18 mmol). The mixture was stirred at -78 C for 0.5 hour, quenched with water (10 mL), and extracted with ethyl acetate (20 mL x 3). The combined organic layers was washed with brine (40 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified with flash column chromatography on silica gel (ethyl acetate in petroleum ether, from 0% to 8% v/v) to yield Compound 59B: LC-MS (ESI) m/z: 163 [M+H]+;1H-NMR (CDCl3, 400 MHz): delta (ppm) 1.27 (d, J =7.2.4 Hz, 6H), 2.60 (s, 3H), 2.93-3.00 (m, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.44 (t, J = 7.2Hz, 1H), 7.76 (d, J = 7.2 Hz, 1H), 7.83 (s, 1H).
To <strong>[58556-75-5]2,7-dibromonaphthalene</strong> (1 g, 3.50 mmol) in anhydrous tetrahydrofuran (18 mL), at - 78C and under an atmosphere of nitrogen, was added a solution of n-butyl lithium (2.5 M in hexanes, 1.5 mL, 3.67 mmol) dropwise. The reaction was stirred at -78C for 20 min after which /V-methoxy-/V-methylacetamide (409 mu, 3.85 mmol) was added. After 15 min, the reaction was warmed to RT and stirred for 30 min. The reaction was quenched with 2 M hydrochloric acid and extracted twice with dichloromethane. The combined organic layers were dried through a hydrophobic frit and concentrated in vacuo. The product was purified by silica flash chromatography (/'so-hexanes/ethyl acetate, 7/1 ) to afford the title compound (650 mg, 75%) as a colorless solid
75%
To <strong>[58556-75-5]2,7-dibromonaphthalene</strong> (1 g, 3.50 mmol) in anhydrous tetrahydrofuran (18 mL), at -78 C. and under an atmosphere of nitrogen, was added a solution of n-butyllithium (2.5 M in hexanes, 1.5 mL, 3.67 mmol) dropwise. The reaction was stirred at -78 C. for 20 min after which N-methoxy-N-methylacetamide (409 muL, 3.85 mmol) was added. After 15 min, the reaction was warmed to RT and stirred for 30 min. The reaction was quenched with 2 M hydrochloric acid and extracted twice with dichloromethane. The combined organic layers were dried through a hydrophobic frit and concentrated in vacuo. The product was purified by silica flash chromatography (iso-hexanes/ethyl acetate, 7/1) to afford the title compound (650 mg, 75%) as a colorless solid.
Compound 1 (30 g, 100 mmol) was dissolved in anhydrous i-Pr20 (300.0 mL) in a dried flask under nitrogen. The reaction mixture was cooled to -78 C and stirred under nitrogen atmosphere. A 2.5 M solution of n-BuLi in hexanes (40 mL, 100 mmol) was added dropwise to the above solution and the reaction mixture was stirred at -78 C for 30 min after complete addition of n-BuLi. N-methoxy-N- methylacetamide (12 g, 120.0 mmol) was added to above reaction mixture, while keeping the reaction mixture below -78 C. After addition of N-methoxy-N- methylacetamide was completed, the reaction mixture was warmed slowly to room temperature for 30 min. The reaction mixture was poured into 40 mL of aqueous NH4CI and the reaction mixture was stirred for 15 min. The organic phase was separated, dried over anhydrous MgSC , filtered and evaporated in vacuo to give compound 2 (25 g, crude)as a pale yellow viscous liquid.
Example 26: Preparation of 1-(3-bromo-5-chlorophenyl)ethan-1-one (C148) (0740) (0741) 656 1,3-Dibromo-5-chlorobenzene (5.0 g, 18.5 mmol) was dissolved in 167 diethyl ether (61.6 mL) and cooled to ?78° C. Because the compound came out of solution, the mixture was removed from the cooling bath. As soon as stirring was again visible from temperature warming, 168 n-butyllithium (8.14 mL, 20.34 mmol) was added dropwise, and the solution was re-immersed in the cold bath. The solution took on a bright yellow color, and the mixture was stirred for 30 minutes. At this point a slight yellow precipitate was visible. 657 <strong>[78191-00-1]N-Methoxy-N-methylacetamide</strong> (2.359 mL, 22.19 mmol) was added dropwise, and the reaction mixture was stirred for 10 minutes, then warmed slowly to room temperature. The reaction mixture was quenched with 1 N hydrochloric acid and was extracted with diethyl ether. The combined organic extracts were washed with brine, dried over sodium sulfate and concentrated. The resulting oil was purified on silica running a 0-15percent gradient of 110 acetone in hexanes. The 658 title compound was isolated as a white solid (3.7 g, 86percent): mp 33-36° C.; 1H NMR (300 MHz, CDCl3) delta 7.97-7.95 (m, 1H), 7.85 (dd, J=1.5 Hz, 1H), 7.71 (t, J=1.8 Hz, 1H), 2.59 (s, 3H); IR (thin film) 1687 cm?1; ESIMS m/z 233 ([M+H]+).
Compound 1 (30 g, 100 mmol) was dissolved in anhydrous i-Pr2O (300.0 mL) in a dried flask under nitrogen. The reaction mixture was cooled to -78 °C and stirred under nitrogen atmosphere. A 2.5 M solution of n-BuLi in hexanes (40 mL, 100 mmol) was added dropwise to the above solution and the reaction mixture was stirred at -78 °C for 30 min after complete addition of n-BuLi. N-methoxy-N-methylacetamide (12 g, 120.0 mmol) was added to the above reaction mixture, while keeping the reaction mixture below -78 °C. After addition of N-methoxy-N-methylacetamide was completed, the reaction mixture was warmed slowly to room temperature for 30 min. The reaction mixture was poured into 40 mL of aqueous NH4Cl and the reaction mixture was stirred for 15 min. The organic phase was separated, dried over anhydrous MgSO4, filtered and evaporated in vacuo to give the compound 2 (25 g, crude) as a pale yellow viscous liquid.
Step 1 Synthesis of compound 33 [0219] Compound 32 (60.3 g, 255 mmol) was dissolved in toluene (1800 mL) under nitrogen atmosphere, and the solution was cooled to -66 C. To the solution was added dropwise 2.67 mol/L of n-butyllithium in hexane (100 mL, 267 mmol), and the mixture was stirred for 40 minutes. To the mixture was added dropwise N-methoxy-N-methylacetamide (79 g, 764 mmol), and the mixture was stirred at the same temperature for 40 minutes. The mixture was allowed to warm to 0 C by removing the cooling bath, and then cooled to -55 C. To the mixture was added a saturated ammonium chloride solution (300 mL), which was allowed to warm to room temperature. To the mixture was added water (200 mL) and extracted. The organic layer was washed with water (500 mL) and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the resulting residue was purified by silica gel column chromatography eluting with hexane-ethyl acetate to afford compound 33 (34.2 g, Yield 67 %). 1 H-NMR (CDCl3) delta: 2.72 (3H, s), 7.64 (1H, dd, J = 5.1, 1.8 Hz), 8.20 (1H, dd, J = 1.8, 0.6 Hz), 8.50 (1H, dd, J = 5.1, 0.6 Hz)
With n-butyllithium; In tetrahydrofuran; toluene; at -60℃; for 1h;Inert atmosphere;
1-(5-Benzyloxy-pyridin-2-yl)-ethanone A mixture of <strong>[630120-99-9]5-benzyloxy-2-bromo-pyridine</strong> (1.0 g), N-methoxy-N-methyl-acetamide (780 mg) and THF (20 mL) under argon at -60 C. was added n-BuLi (2.6 M in toluene, 2.9 mL). After 1 hour the cooling bath was removed. After reaching room temperature the mixture was quenched by addition of saturated NH4Cl-solution. The mixture was distributed between EA and brine. The organic phase was dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography (silica gel, heptane to EA/heptane 3:7) to provide the subtitle compound. MS ESI+: m/z=228 [M+H]+.
With n-butyllithium; In tetrahydrofuran; toluene; at -60℃; for 1h;Inert atmosphere;
A mixture of <strong>[630120-99-9]5-benzyloxy-2-bromo-pyridine</strong> (1 .0 g), A/-methoxy-/V-methyl-acetamide (780 mg) and THF (20 ml_) under argon at -60C was added n-BuLi (2.6 M in toluene, 2.9 ml_). After 1 hour the cooling bath was removed. After reaching room temperature the mixture was quenched by addition of saturated NH CI-solution. The mixture was distributed between EA and brine. The organic phase was dried (Na2SO4), filtered and concentrated. The residue was purified by chromatography (silica gel, heptane to EA/heptane 3:7) to provide the subtitle compound. MS ESI+: m/z = 228 [M+H]+.
[0559] To a solution of III-1 (30 g, 0.162 mol, 1 eq.) in 300 mL of anhydrous THF was added dropwise a solution of n-BuLi (2.5M in hexane, 77.5 mL, 0.19 mol, 1.2 eq.) at -70 C. After completion of addition, the mixture was stirred at -70 C. for 20 min, followed by addition of a solution of N-methoxy-N-methylacetamide (33 g, 0.322 mol, 2 eq.) in 100 mL of anhydrous THF by drop wise, the solution was allowed to warm to rt and stirred for 2 hrs. The reaction was quenched with saturated aq. NH4Cl (100 mL), extracted with EtOAc (300 mL×3), the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with petroleum ether/EtOAc (100:1) to yield III-2 (14.8 g, 62% yield) as a white solid. 1H NMR (DMSO-d6, 400 MHz) delta 8.81 (d, J=2.0 Hz, 1H), 8.16 (dd, J=2.4, 8.4 Hz, 1H), 6.90 (d, J=8.8 Hz, 1H), 3.93 (s, 3H), 2.55 (s, 3H). MS (ESI) m/z [M+H]+ 151.6.
62%
To a solution of Ill-i (30 g, 0.162 mol, 1 eq.) in 300 mL of anhydrous THF was added dropwise a solution of n-BuLi (2.5M in hexane, 77.5 mL, 0.19 mol, 1.2 eq.) at -70C. After completion of addition, the mixture was stuffed at -70C for 20 mm, followed by addition of a solution of N-methoxy-N-methylacetamide (33 g, 0.322 mol, 2 eq.) in 100 mL of anhydrous THF by drop wise, the solution was allowed to warm to rt and stirred for 2 hrs. The reaction was quenched with saturated aq. NH4C1 (100 mL), extracted with EtOAc (300 mLx3), the organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with petroleum ether/EtOAc (100:1) to yield 111-2 (14.8 g, 62% yield) as a white solid. ?H NMR (DMSO-d6, 400 MHz) (58.81 (d, J= 2.0 Hz, 1H), 8.16 (dd, J= 2.4, 8.4 Hz, 1H), 6.90 (d, J = 8.8 Hz, 1H), 3.93 (s, 3H), 2.55 (s, 3H). MS (ES) m/z [M+H] 15 1.6.
Preparation 62 l-(5-bromo-3-chlorothiophen-2-yl)ethanone To a solution of diisopropylamine (6.62 ml, 46.4 mmol) in THF (57.2 ml) at -78 °C was added a solution of BuLi (18.57 ml, 46.4 mmol) dropwise. When the addition was complete, the reaction mixture was allowed to stir at -78 °C for an additional 30 min. A solution of 2-bromo-3-chlorothiophene (Preparation 61, 9.17 g, 46.4 mmol) in THF (1 1.43 ml) was then added rapidly in one aliquot. The resulting mixture was stirred at -78 °C for 30 min. A solution of N-methoxy-N-methylacetamide (1.915 g, 18.57 mmol) in THF (5.72 ml) was then added dropwise to the reaction mixture, which was then allowed to stir for an additional 30 min at -78 °C. The reaction mixture was then quenched by the slow addition of IN HC1 (120 mL). After coming to rt, the mixture was extracted with ether (3 x 50 mL). The combined extracts were washed with water (50 mL), dried over MgS04, filtered and concentrated in vacuo. Purification by flash chromatography (silica, EtOAc/Hexanes) gave l-(5-bromo-3-chlorothiophen-2-yl)ethanone (100percent yield); XH NMR (500MHz, CHLOROFORM-d) delta 7.03 (s, 1H), 3.86 (t, J=6.2 Hz, 1H), 3.50 (t, J=6.3 Hz, 1H), 2.65 (s, 3H).
1-(6-chloro-2-fluoro-pyridin-3-yl)-ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: A solution of LDA is prepared by adding dropwise a 2.5 M butyllithium solution in hexane (1.1 to 1.2 eq.) to a solution of DIPA (1.07 to 1.2 eq.) in THF under nitrogen at a temperature comprised between -78 C. and -5 C. The reaction mixture is stirred 15 mm to 30 mm at the same temperature. Then Intermediate Gen-6-a (ito 1.2 eq.) in THF is added dropwise between -78 C. and -60 C., and the reaction is stirred under nitrogen at -78 C. for 1 h to 1.33 h. Then Intermediate Gen-7 (1.1 to 1.2 eq.) is added dropwise or portionwise by monitoring the temperature. The mixture is stirred 1.5 h to 3 h between -78C. and -70C. and quenched with a saturated aqueous NH4C1 solution. EtOAc is added, then the organic layer is separated, dried over Na2SO4, filtered and evaporated to dryness. The residue is purified by chromatography of silica gel to give Intermediate Gen-8.
1-(1-Methyl-1H-pyrazol-4-yl)ethan-1-ol. To a resealable vial was added 4-iodo-1-methyl-1H-pyrazole (772 mg, 3.71 mmol), THF (6 mL), and the solution was cooled to 0 C. To this solution was added isopropylmagnesium chloride lithium chloride complex solution (3.42 mL, 4.45 mmol) and the reaction stirred for 30 min (complete disappearance of iodo-pyrazole as detected via LCMS) before cooling to -78 C and addition of N-methoxy-N-methylacetamide (1.18 mL, 11.13 mmol). The solution was stirred at -78 C for 1 h before removing the cold bath and warming to ambient temperature. The solution was diluted with EtOAc and quenched via the addition of 1N HCl. The layers were separated and the aqueous was extracted with EtOAc. The combined organics layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude residue was taken up in MeOH and cooled to 0 C before addition of NaBH4 (281 mg, 7.42 mmol). The reaction was stirred while warming to ambient temperature and quenched when LCMS showed disappearance of starting material. This solution was then diluted with 1N HCl and EtOAc. The layers were separated and the aqueous was extracted with EtOAc (3X). The combined organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified via silica gel chromatography (MeOH:DCM) to afford 1-(1-methyl-1H-pyrazol-4-yl)ethanol
1-(1-cyclopropyl-1H-pyrazol-4-yl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
Intermediate 10: To a stirred solution of Intermediate 1OA (460 mg, 1.965 mmol) in THF (5 mL) at 0 C was added iPrMgCl (1.08 1 mL, 2.162 mmol) quickly. After 30 mm, additional 0.15 eq of iPrMgCl was added and after 30 mm, the mixture was cooled to -78 C. N-Methoxy-N-methylacetamide (304 mg, 2.95 mmol) was added quickly andthe reaction was warmed to RT for 3h. The reaction mixture was concentrated in vacuo and diluted with EtOAc. The organic layer was washed with H20. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified to give Intermediate 10 (250mg, 1.665 mmol, 85% yield) as a clear liquid. LCMS Anal. Calc?d for C8H,0N20 150.8, found [M+H] 151.0.
To a solution of 5-bromo-2,3-dichloropyridine (29) (702 mg, 3.09 mmol) in THF (7 ml) was dropwise added isopropylmagnesium chloride lithium chloride complex (3.09 ml, 4.02 mmol) at 0 C. for 10 min. Then N-methoxy-N-methylacetamide (658 mul, 6.19 mmol) in THF (2 ml) was dropwise added to the reaction mixture at the same temperature and stirred at for 2 hrs. Then the reaction mixture was quenched with aqueous solution and extracted with ethyl acetate. The resulting organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The resulting product was chromatographed on silica gel eluting with a gradient of hexane/ethyl acetate (5-15%) to afford 232 mg of the desired product (30) in 40% yield as a white solid. 1H-NMR (DMSO-d6) delta: 8.91 (1H, s), 8.55 (1H, s), 2.65 (3H, s).
Compound 25-3 (0444) 25-2 (9.5 g, 30 mmol) was dissolved in anhydrous THF (100 mL) and cooled to -78 C. To the above solution was added dropwise n-BuLi (8 mL, 21 mmol) for 30 min and stirred for 30 min. 25-1 was added dropwise at -78 C., stirred for 30 min and allowed to warm to rt before quenching with saturated ammonium chloride. The organic phase was separated and washed with brine, dried with anhydrous Na2SO4, filtered and concentrated to afford yellow oil, which was purified by column chromatography (elution: PE/EA=10/1) afford 25-3 as a yellow solid (2.3 g, 28%).
1-(2-(difluoromethyl)-5-fluorophenyl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
Step 2: 1-(2-(difluoromethyl)-5-fluorophenyl)ethanone 1-Bromo-2-(difluoromethyl)-5-fluorobenzene(8.36 g, 37.5 mmol) was dissolved in anhydrous ether (100 mL), cooled to -78 C, and then 2.4 M n-butyllithium (18.7 mL, 45 mmol) was added dropwise under a nitrogen atmosphere. The resultant was stirred for 1 hour. While the temperature was kept at -78C, N-methyl-N-methoxylacetamide (7.73 g, 75 mmol) was added, and the resultant was then stirred for 2 hours. After the resultant was warmed up to room temperature, it was washed with saturated brine and extracted with ethyl acetate. The extract was dried, concentrated, and purified by silica gel column chromatography to give 1-(2-(difluoromethyl)-5-fluorophenyl)ethanone (4.2 g, 60% yield). 1H-NMR (400 MHz, CDCl3): delta= 7.45-7.40 (m, 1H), 7.31-7.27 (m, 1H), 7.20-7.12 (m,1H), 6.78-6.50 (t, J= 56 Hz, 1H), 2.42 (s, 3H).
To a solution of 1.0 g (5.3 mmol) of 3-bromo-4-methylthiophen (Tokyo Chemical Industry Co., Ltd.) in diethyl ether (23 ml) was added dropwise 4.0 ml (6.4 mmol) of a 1.6M n-butyllithium hexane solution at -78C under an argon atmosphere, and the mixture was stirred at -78C for 15 minutes. Then, a solution of 0.70 ml (6.9 mmol) of N-methoxy-N-methyl acetamide in diethyl ether (1 ml) was added dropwise at -78C, and the mixture was stirred at -78C for 15 minutes and then at room temperature for 23 hours. After completion of the reaction, to the reaction mixture was added a saturated aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The resulting organic layer was washed with water, subsequently dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (elution solvent; hexane:ethyl acetate = 80:20 (V/V)), and the fractions containing the desired compound were concentrated under reduced pressure to obtain 552 mg (3.94 mmol, yield 75%) of the title compound as a pale yellow oil.
75%
(Reference Example 17) 1-(4-Methylthiophen-3-yl)ethanone In an argon atmosphere at -78C, 4.0 ml (6.4 mmol) of a 1.6 M hexane solution of n-butyllithium was added dropwise to a diethyl ether (23 ml) solution of 1.0 g (5.3 mmol) of 3-bromo-4-methylthiophene (Tokyo Chemical Industry Co., Ltd.). The mixture was stirred at the temperature for 15 minutes. Next, a diethyl ether (1 ml) solution of 0.70 ml (6.9 mmol) of N-methoxy-N-methylacetamide was added dropwise at -78C, and the mixture was stirred at the temperature for 15 minutes and at room temperature for 23 hours. After the completion of the reaction, a saturated aqueous ammonium chloride solution was added to the reaction mixture liquid, and the resultant mixture was extracted with ethyl acetate. The organic phase was washed with water, dried with anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel column chromatography (eluting solvent: hexane:ethyl acetate = 80:20 (V/V)), and the fraction containing the target compound was concentrated under reduced pressure to give the title compound weighing 552 mg (3.94 mmol, yield 75%) as a light yellow oil. Mass spectrum (CI, m/z): 141 [M+1]+. 1H-NMR spectrum (400MHz, CDCl3) delta: 7.99 (1H, d, J = 3.1 Hz), 6.91 (1H, dq, J = 3.1, 1.0 Hz), 2.53 (3H, s), 2.46 (3H, d, J = 1.0 Hz).
Preparative Example 7 Step 1: l-(5-bromo-2-fluoropyridin-3-yl)ethanone To a solution of diisopropylamine (46.3 g, 458.4 mmol) in tetrahydrofuran (lOOOmL) was added butyllithium (176 mL, 440 mmol, 2.5 M) at -78 C under nitrogen. After addition, the reaction mixture was stirred for 30 min at -78 C. 5-bromo-2-fluoropyridine (86.7 g, 442.3 mmol) was added (keeping the temperature under -65 C). After addition, the mixture was stirred for 1 h. N-methoxy- N-methylacetamide (50 g, 485.4 mmol) was added and stirred at -78 C for 1 h. The reaction mixture was quenched with water (lOOOmL), extracted with ethyl acetate (3 x 500 mL), washed with brine, dried over sodium sulfate and concentrated to dryness in vacuo. The resulting residue was purified by column chromatography (silica gel, 100-200 mesh, 0.5% ethyl acetate in petroleum ether) affording 1- (5-bromo-2-fluoropyridin-3-yl)ethanone (43 g, 44.6 %): H NMR (400 MHz, Chloroform-d): delta 8.46- 8.42 (m, 2H), 2.70 (s, 3H).
Step 1 To a solution of diisopropylamine (16.2 mL, 114 mmol) in 25 mL THF at -78 C was added dropwise n-butyllithium (2.5 M in hexanes, 45.5 mL) and the mixture stirred 1 h at -78 C following complete addition. The resulting solution was added dropwise over 50 min. to a stirred solution of F-1 in 75 mL THF at -78 C and the mixture stirred 1 h at -78 C. A solution of F-2 in 20 mL THF was then added and the mixture was allowed to warm slowly to rt, stirring 12 h. The reaction mixture was quenched with 1.0 N HCl and diluted with EtOAc. The organic layer was separated and washed sequentially with water and brine, dried (Na2S04), filtered and concentrated. The resulting residue was purified by flash chiOmatography (220 g Si02; 0 to 20 % EtOAc in hexanes) to provide F-3. MS for F-3: m/e = 218 and 220 (M+l).
1-(4-bromo-3-(hydroxymethyl)phenyl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a stirred solution of <strong>[946525-30-0]2-bromo-5-iodobenzyl alcohol</strong> (5.0 g, 16.0 mmol) (Compound (8-1)) in dry THF (40 mL) was added a 2 M solution of isopropylmagnesium chloride in THF (17.6 mL, 35.2 mmol) while maintaining the internal temperature below about ?10° C. A white suspension was formed after 5 min. The mixture was stirred for about 1 h at or below about ?10° C. and then was added N-methoxy-N-methylacetamide (3.73 mL, 35.2 mmol) dropwise over a period of about 3 min. The reaction mixture was allowed to warm to about 20° C. over 1 h. The reaction mixture was cooled to about 0° C., quenched with 3N HCl (25 mL), and diluted with tert-butylmethyl ether (50 mL). The resulting biphasic mixture was stirred and the layers separated. The organic layer was washed with 1M HCl (50 mL) followed by water (50 mL) and concentrated under reduced pressure to afford Compound (M-a) as a crude product mixture which was used in next step without further purification. 1H NMR (400 MHz, CDCl3) delta 8.07 (s, 1H), 7.72 (d, J=8.4 Hz, 1H), 7.63 (d, J=8.4 Hz, 1H), 4.79 (s, 2H), 2.59 (s, 3H)
tert-Butyl 3-methylpyridin-2-ylcarbamate (5.07 g, 23.6 mmol) was dissolved in 100 ml tetrahydrofuran under nitrogen and the mixture was cooled to -10 C. n-Butyllithium (2.5 M in hexanes, 28 ml, 70 mmol) was added over 15 min with stirring, forming a dark red solution and the mixture was stirred for a further 1 h at -10 C. N-Methoxy-N-methylacetamide (3.80 g, 35.7 mmol) was added and the mixture was stirred at room temperature for 1 h. The mixture was cooled to -10 C, quenched with 13 ml concentrated hydrochloric acid and then stirred at 50 C for 90 min. The biphasic mixture was allowed to cool and the organic phase was extracted twice with 2N hydrochloric acid. The combined aqueous was basified with 32% sodium hydroxide solution and extracted three times with ethyl acetate. The organics were washed with brine, dried over anhydrous sodium sulphate, filtered and evaporated under reduced pressure. The residue was purified using the Isolera purification system (ethyl acetate-hexane gradient, 0:100 rising to 50:50) to give 95 (2.5 g, 18.9 mmol, 80% yield) as a pale yellow solid. Purity 100%. 1H NMR (400 MHz, CDCl3) delta ppm 2.55 (s, 3H, Me), 6.17 (s, 1H, H-3), 7.03 (dd, J = 4.9, 7.6 Hz, 1H, H-5), 7.82 (d, J = 7.4 Hz, 1H, H-4), 8.21 (d, J = 4.7 Hz, 1H, H-6), 11.96 (br. s., 1H, NH). UPLC/MS (3 min) retention time 0.68 min. LRMS: m/z 133 (M+1).
1-(2-fluoro-4-(trifluoromethoxy)phenyl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
n-Butyl lithium (6.17 mL, 9.87 mmol) was added drop-wise at -78 C. to a solution of <strong>[168971-68-4]1-bromo-2-fluoro-4-(trifluoromethoxy)benzene</strong> (2.13 g, 8.22 mmol) in diethyl ether (16.5 mL). The reaction was stirred for 30 minutes before drop-wise addition of N-methoxy-N-methylacetamide (1.272 g, 12.34 mmol). The reaction was stirred for 5 minutes at -78 C. then warmed to room temperature and stirred for 30 minutes. The solution was quenched with saturated NH4Cl, extracted with EtOAc, dried with Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash silica gel chromatography, eluting with 10% EtOAc in heptanes provided 1-(2-fluoro-4-(trifluoromethoxy)phenyl)ethanone as a clear oil (1.118 g, 61%)
To a solution of 2-bromo-5-fluoropyridine (16.78 g, 95.3 mmol) in diethyl ether (348 ml) wasslowly added butyllithium (1.6 M in hexane) (65.6 ml, 105 mmol) at -78 C under argonatmosphere. The resulting yellow reaction mixture was stirred at -78 C for two hours and nmethoxy-n-methylacetamide (10.8 g, 11.2 ml, 105 mmol) was added. The reaction mixture was stirred at -78 C for another hour before quenching with lOml of a 4.OM aqueous solution of hydrochloric acid and the reaction was let to warm up to room temperature. The pH of themixture was ajusted to 7 by addition of 2M aqueous solution of hydrochloric acid. The reaction mixture was washed with 50m1 of brine and the organic phase was collected. The aqueous layer was back-extracted twice with ethyl acetate. Combined organic phases were dried over sodium sulfate and concentrated in vacuo. The crude material was purified by flash chromatography (silica gel, 20g, gradient 0% to 7% of ethyl acetate in heptane) to give the title compound (10.76g, 5 1.8%) as light yellow oil. MS mle: 220.0 ([M+H]).
A solution of <strong>[70951-85-8]4-bromo-1-tert-butyl-1H-pyrazole</strong> (973 mg, 4.79 mmol) in anhydrous THF (9.5 ml) was cooled to -78 C. under argon. A solution of n-BuLi in Hexanes (1.6 M, 3.2 mL, 5.12 mmol) was then added dropwise over 5 min and reaction mixture was stirred at -78 C. for 80 min. A solution of N-methoxy-N-methylacetamide (0.55 ml, 5.17 mmol) in THF (3 mL) was added dropwise and mixture was warmed to 0 C. After 4 hours, reaction mixture was quenched via addition of NH4Cl (aq) (10 mL) and mixture was extracted with ethyl acetate. Combined organic layers were washed with 50% brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Resulting residue was purified by silica gel column chromatography (0-50% ethyl acetate in hexanes) to yield 1-(1-tert-butyl-1H-pyrazol-4-yl)ethanone 5.14.
Bromoimidazo [l, 2_b] pyridazine (6.8 g, 34.3 mmol) was dissolved in re-distilled anhydrous tetrahydrofuranLiter). A solution of ethyl magnesium bromide in tetrahydrofuran (1M, 51.5 ml) was slowly added dropwise at 0 C under nitrogen.And stirred for 30 minutes, a solution of N-methoxy-N-methylacetamide (7.1 g, 68.7 mmol) in tetrahydrofuran (30Ml) was slowly added dropwise to the system. The temperature of the reaction system was returned to room temperature and stirred for 10 hours. Water (50 ml) was added to the reactionAnd extracted with ethyl acetate. The extract was dried over anhydrous sodium sulfate, concentrated and purified by column chromatography (petroleum ether: ethyl acetate = 10: 1To 1: 1) afforded the title compound 3 ? 4 g.
4-[6-(6-acetyl-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-ylamino)pyridine-3-yl]piperazine-1-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
A 10-necked flask was charged with 10 (58.45 g, 100 mmol) and tetrahydrofuran (300 mL), stirred and cooled to -10-0 C,Adding isopropyl chlorideMagnesium chlorideOf the lithium complexesTetrahydrofuran solution(2.0 M, 110 mL, 220 mmol)Stirring at low temperature for 20 to 30 minutesStir to room temperature3 to 4 hours.N-methyl-N-methoxy-2-chloroacetamide (22.69 g, 220 mmol) was added dropwise to 0 to 5 C after basic conversion,Plus after the rise to room temperature 20 ~ 30 reaction 4 to 6 hours. At the end of the reaction, dilute hydrochloric acid was added(2 mol / L, 150 mmol), extracted with ethyl acetate (150 mL) three times, combined with organic saturated brine(150 mL), dried over anhydrous sodium sulfate and concentrated to give compound 12 by column chromatography on a mixture of petroleum ether and ethyl acetate (42.72 g, 78%).
1-(4-amino-2-chloro-5-pyrimidinyl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With n-butyllithium; In tetrahydrofuran; hexane; at -78℃; for 1.33333h;
1 L reaction flask was charged with 20.8 g (0.1 mol) of Compound I and 208 mL of tetrahydrofuran,After stirring and dissolving,The system was cooled to -78 C. After the temperature was stabilized, 62.5 mL of n-butyllithium-n-hexane (15% concentration) was added dropwise. A solution of 12.9 g (0.125 mol) of N-methoxy-N-methylacetamide and 129 mL of tetrahydrofuran was added dropwise while maintaining the temperature for 10 min. After stirring for 1 h, the system was warmed to 0 C and 100 mL of saturated ammonium chloride solution was added. After stirring for 30 min, the mixture was extracted with 3X 300 mL of ethyl acetate. The organic layers were combined and the organic layer was washed with 3X 300 mL of saturated brine and dried over anhydrous magnesium sulfate Dried, filtered and concentrated to give 15.8 g of crude yellow solid, yield: 91.9%, used directly in the next step.
In the three bottle,3-Pivalamidopyridine (20.0 g, 112.2 mmol) was dissolved in anhydrous tetrahydrofuran (200 mL).In nitrogen protection, the temperature drops to -78 C,N-butyllithium (17.8 g, 280.5 mmol) was added dropwise and the solution was dropped.Slowly warming to 0C for 3 hours,Then cool down to -78 C,N-methyl-N-methoxyacetamide (17.4 g, 168.3 mmol) was added dropwise.After completion of the dropwise addition, the reaction was slowly warmed to room temperature for 10 h.After the reaction was completed, the reaction was quenched with 1N hydrochloric acid, extracted with ethyl acetate (100 mL×3), and the organic phase was collected and washed with saturated brine (100 mL×1).The solvent was evaporated under reduced pressure to give crude N-(4-ethanhydrin-3-yl)trimethylacetamide.The crude product was applied on a silica gel column (mobile phase: PE:EA=5:1) to obtain 11.2 g of a pure product with a yield of 45.4%.
1-(1-methyl-1H-indazol-6-yl)ethan-1-one To a solution of <strong>[590417-94-0]6-bromo-1-methyl-1H-indazole</strong> (2.5 g, 11.85 mmol, 1.00 equiv) in THF (50 mL) under N2 at-78° C. was added n-BuLi (10 mL, 2.00 equiv) dropwise with stirring. The solution was stirred for 60 min at this temperature, then N-methoxy-N-methylacetamide (3 g, 29.09 mmol, 2.50 equiv) was added, and the solution was stirred for an additional 60 min at this temperature. The resulting solution was stirred for 1 h at rt, cooled to 0° C., quenched by the addition of HCl (1m), and extracted with 2*100 mL of EtOAc. The combined organic layers were washed with 50 mL of brine, dried over Na2SO4, concentrated under vacuum, and purified with silica gel chromatography using EtOAc/petroleum ether (1:10) to afford 1.5 g (73percent) of the title compound as a yellow oil. LC-MS: (ES, m/z): 174. 1H NMR: (400 MHz, DMSO-d6) delta 8.36 (q, J=1.1 Hz, 1H), 8.16 (d, J=1.0 Hz, 1H), 7.86 (dd, J=8.5, 0.8 Hz, 1H), 7.69 (dd, J=8.5, 1.4 Hz, 1H), 4.16 (s, 3H), 2.70 (s, 3H).
1-(1-Methyl-1H-indol-6-yl)ethan-1-one To a solution of <strong>[125872-95-9]6-bromo-1-methyl-1H-indole</strong> (3.4 g, 16.19 mmol, 1.00 equiv) in THF (100 mL) was added BuLi (12 mL, 2.00 equiv, 2.5 M) dropwise with stirring at -78 C. The solution was stirred for 1 h, then N-methoxy-N-methylacetamide (4.1 g, 39.76 mmol, 2.50 equiv) was added dropwise with stirring at -78 C. The solution was stirred for 30 min, then warmed to rt and stirred an additional 2 h. The reaction mixture was cooled to 0 C., then quenched by the addition of 1 M HCl and extracted with 2×200 mL of EtOAc. The combined organic layers were washed with 100 mL of brine, dried over Na2SO4, concentrated under vacuum, and purified with silica gel chromatography using EtOAc/petroleum ether (1:5) to afford 1.9 g (68%) of the title compound as a yellow solid. LC-MS: (ES, m/z): 173. 1H NMR: (300 MHz, DMSO-d6) delta 8.12 (dt, J=1.5, 0.8 Hz, 1H), 7.70-7.59 (m, 2H), 7.57 (d, J=3.0 Hz, 1H), 6.51 (dd, J=3.0, 0.9 Hz, 1H), 3.88 (s, 3H), 2.63 (s, 3H).
1,3-Dibromo-5-fluoro-benzene (20 g, 78.77 mmol, 1 eq) was dissolved in i-Pr2O (200 mL) in a dried flask under nitrogen. The reaction mixture was cooled to -78 C and stirred under nitrogen atmosphere. n-BuLi (2.5 M, 31.5 mL, 1 eq) was added drop wise to the above solution and the reaction mixture was stirred at -78 C for 30 min. After complete addition of n-BuLi, N-methoxy-N-methyl-acetamide (9.75 g, 94.5 mmol, 10.05 mL, 1.2 eq) dropped to the above reaction mixture, while keeping the reaction mixture below -78 C. After addition, the reaction mixture was warmed slowly to 30 C for 30 min. The reaction mixture was poured into water (150 mL) and the reaction mixture was stirred for 15 min. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (150 mL), combined organic phase, dried over anhydrous Na2SO4, filtered and evaporated in vacuum to give residue (16 g crude). The residue was purified by flash silica gel chromatography (ISCO; 120 g CombiFlash Silica Flash Column, Eluent of 0~10% Ethyl acetate/Petroleum ether gradient 85 mL/min). Compound was obtained as off-white solid (11.3 g, yield 66%).1H NMR (400 MHz, CDCl3) delta ppm 7.91 - 7.84 (m, 1H), 7.63 - 7.54 (m, 1H), 7.45 (td, J=2.0, 7.8 Hz, 1H), 2.63 - 2.55 (m, 3H).
1-(6-chloro-3-fluoro-5-methylpyridin-2-yl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a solution of <strong>[38186-84-4]2-chloro-5-fluoro-3-methylpyridine</strong> (582 mg, 4.00 mmol) in THF (10 mL) was added n-butyllithium (2.5 M, 2.50 mL, 6.25 mmol) at -78 °C under a nitrogen atmosphere. After stirring for 1 hr, N-methoxy-N-methylacetamide (495 mg, 4.80 mmol) was added to the reaction and the resulting mixture stirred at -78 °C for 1 h. The reaction was quenched with water (0.5 mL) and the volatiles were removed under reduced pressure. The residue was purified by silica gel chromatography (5percent THF /petroleum ether) to give the title compound.
7-Bromo-4-fluorobenzofuran (0103-2) (0.95 g, 4.42 mmol, 1.0 eq.)Dissolved in anhydrous toluene (15 mL), cooled to -78 C with dry ice acetone bath.Then n-butyllithium (2.5 M, 2.47 mL, 6.19 mmol, 1.4 eq.) was slowly added dropwise.After the completion of the dropwise addition, the mixture was stirred at -78 C for 1.5 hours.N-methoxy-N-methylacetamide (1.17 mL, 11.05 mmol, 2.5 eq.)It was added dropwise and then slowly warmed to room temperature and stirred for 4 hours.The reaction was quenched by the addition of saturated ammonium chloride solution (50 mL).The aqueous layer was extracted with ethyl acetate (30 mL×3).The organic layers were combined and dried over anhydrous sodium sulfate.Concentration, the obtained crude product was purified by silica gel column chromatography ( petroleum ether: ethyl acetate = 30:1)Purification of the pale yellow solid product 1-(4-fluorobenzofuran-7-yl)ethan-1-one(0.28 g, yield: 36%).
tert-butyl (2-acetylnaphthalen-1-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
General procedure: Under argon, to a solution of N-Boc-1-naphthylamine (1) or N-Boc-1-bromo-2-naphthylamine (4) (2.1 mmol) in dry Et2O (20 mL), at 20 C, tBuLi solution in pentane or BuLi solution in hexanes (2.2 equiv.) was added dropwise. The reaction mixture was stirred at this temperature for 2 hours. After this time, 1 equiv. of electrophile at 78 C (for Ac2O) or at 20 C (for iPrCOCl or PivCl) was added dropwise and whole was further stirred at appropriate temperature for next 2 hours. Next, to the reaction mixture 20 mL of methanol was added. Evaporation of solvents and purifications of the crude residue by flash chromatography gave the desired product 2 or 5. In the case of the reaction of 1 with Ac2O at 78 C from post-reaction mixture also substrate and compound 3 were isolated. On the other hand from reaction of 4 with Ac2O and iPrCOCl compounds 6, 7, 8 and a small amount of substrate were isolated. tert-Butyl (2-acetylnaphthalen-1-yl)carbamate (2a). Light yellow solid, yield 45%, 270 mg, mp 150-152 C; Rf (Hex/AcOEt 5:1) = 0.22. FTIR (KBr, numax, cm-1) 3257, 3055, 3003, 2979, 2928, 1709, 1689. 1H NMR (CDCl3): delta 9.26 (1H, s, NH), 8.07 (1H, d, J 8.4 Hz, ArH), 7.82 (1H, d, J 8.4 Hz, ArH), 7.78 (1H, d, J 8.6 Hz, ArH), 7.70 (1H, d, J 8.7 Hz, ArH), 7.56 (2H, m, ArH), 2.71 (3H, s, Me), 1.52 (9H, s, BocMe). 13C NMR (CDCl3): delta 202.0, 154.5, 136.6, 136.1, 129.1, 128.6, 127.9, 126.7, 126.7, 126.4, 125.5, 125.1, 81.0, 29.6, 28.4. HRMS (ESI) m/z calcd for C17H20NO3: 286.1438; found [M+H]+: 286.1441.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +
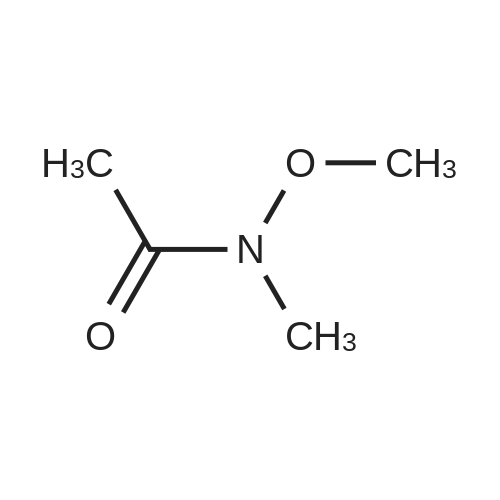

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping