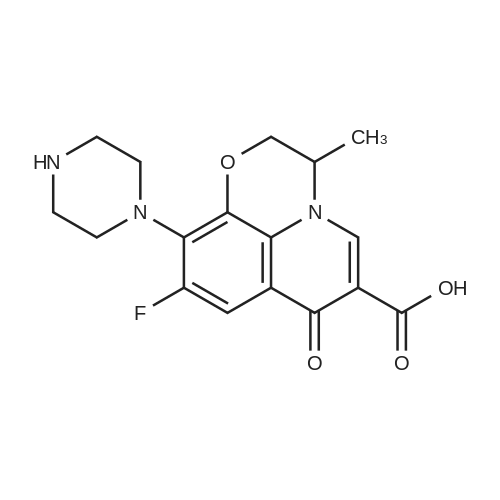
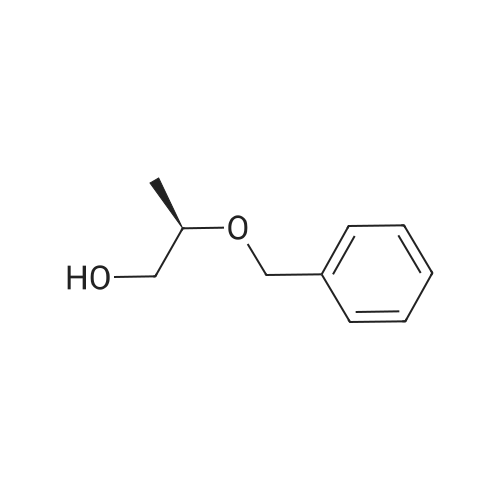
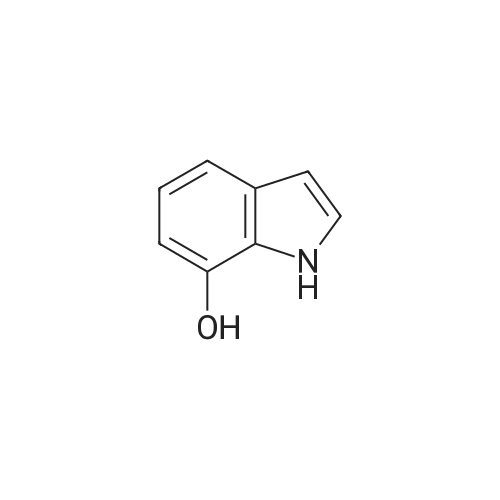
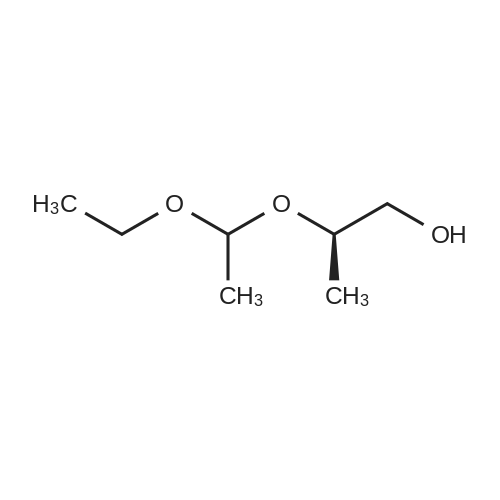
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
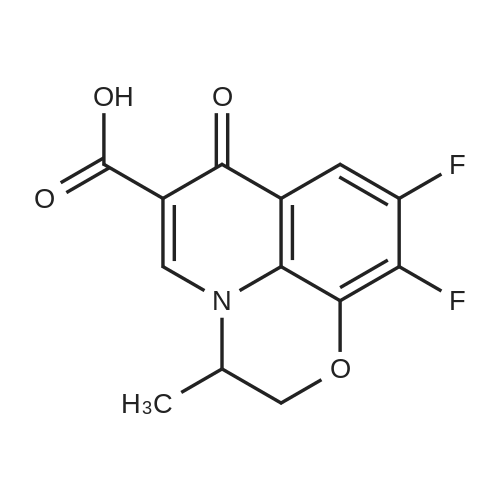
[1] Journal of the American Chemical Society, 2012, vol. 134, # 39, p. 16216 - 16227,12
2
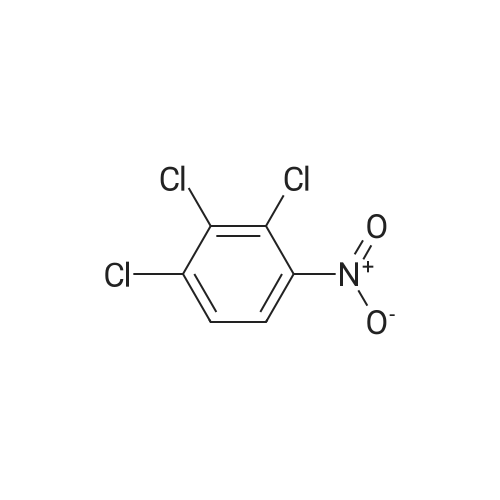
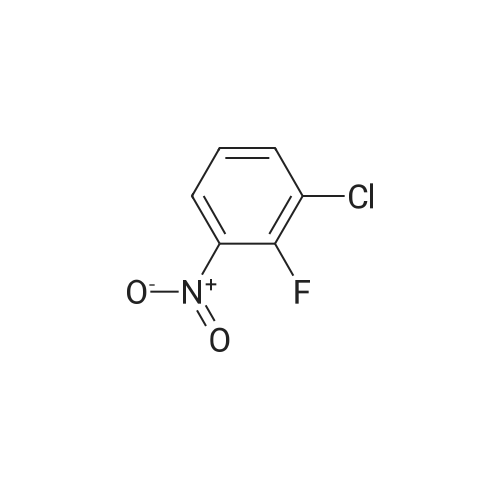
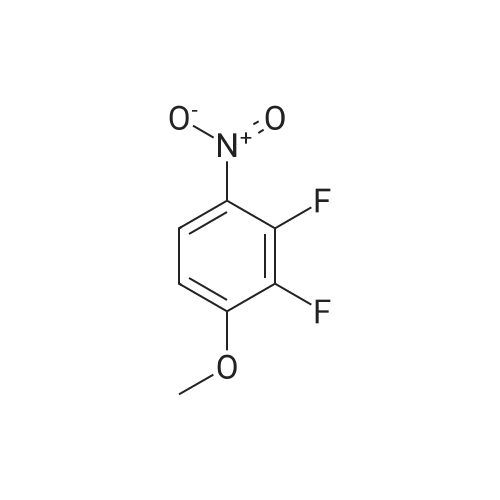
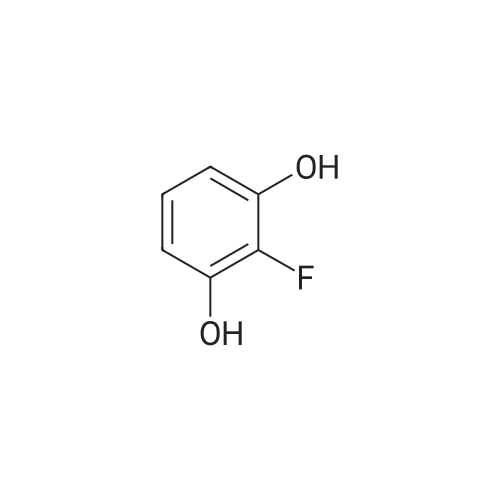
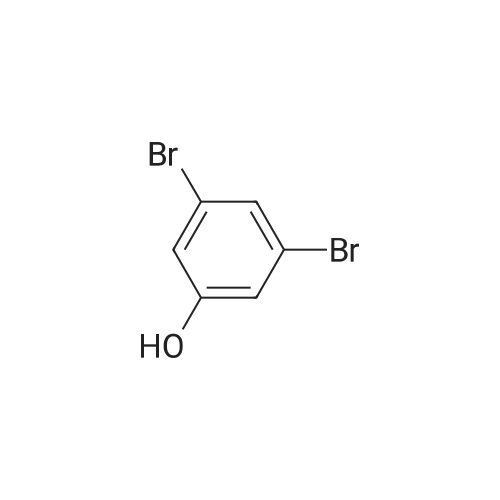
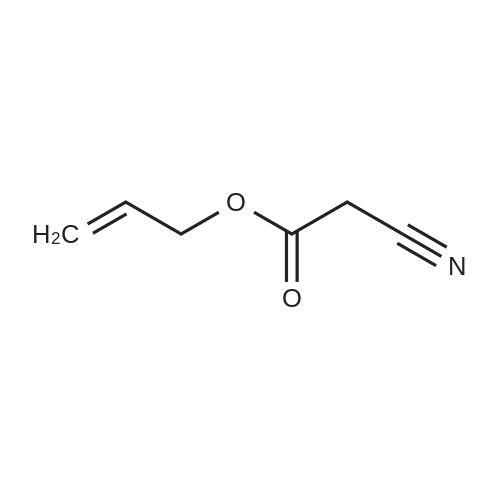
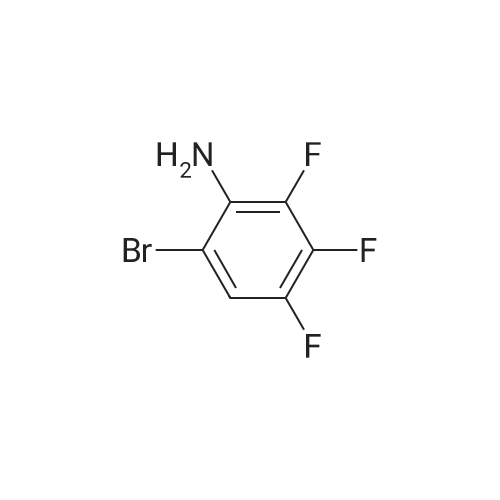
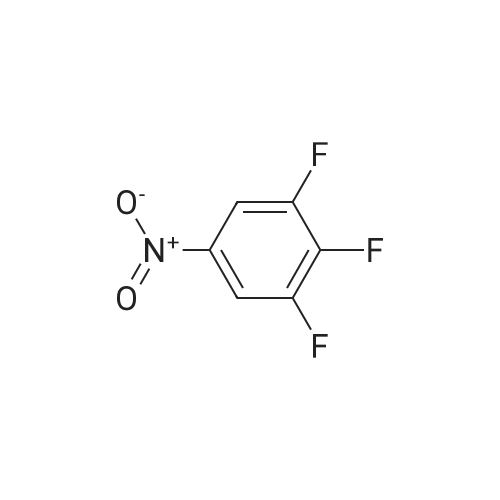
[ 771-69-7 ]
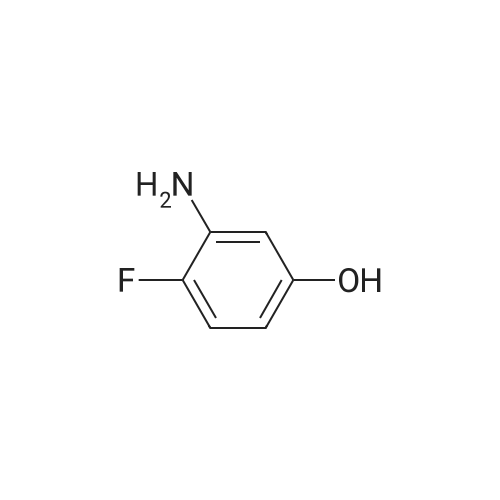
[ 6921-22-8 ]
Reference:
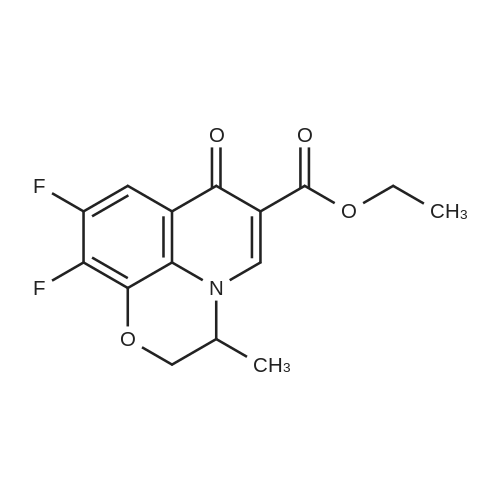
[1] Advanced Synthesis and Catalysis, 2012, vol. 354, # 8, p. 1529 - 1541
3
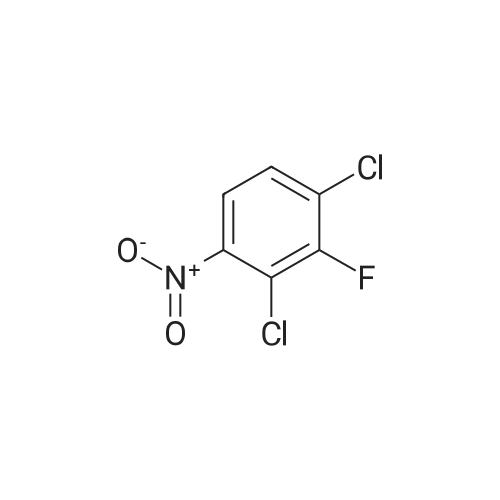
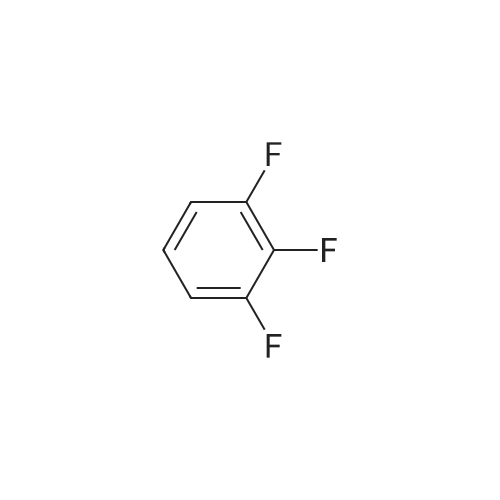
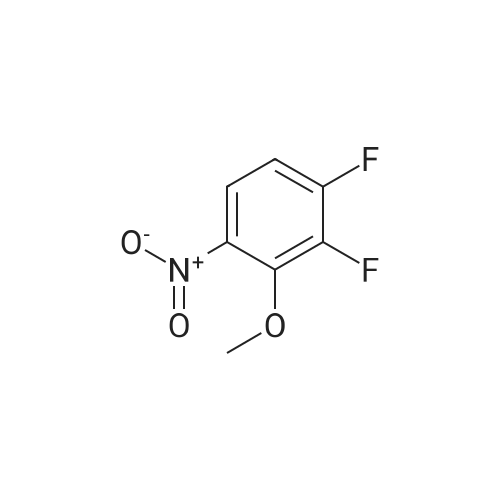
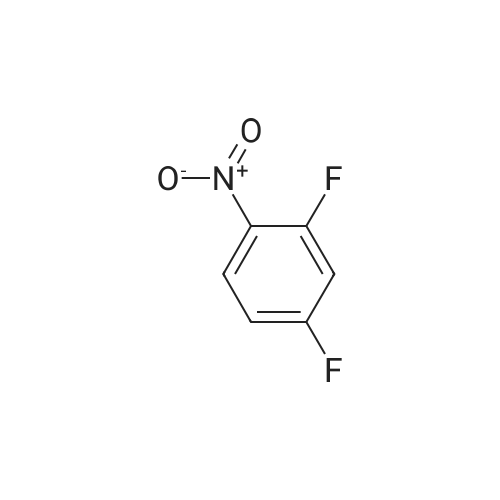
[ 771-69-7 ]
[ 369-34-6 ]
[ 6921-22-8 ]
[ 3862-73-5 ]
Reference:
[1] Angewandte Chemie - International Edition, 2013, vol. 52, # 11, p. 3203 - 3207[2] Angew. Chem., 2013, vol. 125, # 11, p. 3285 - 3289,5
4
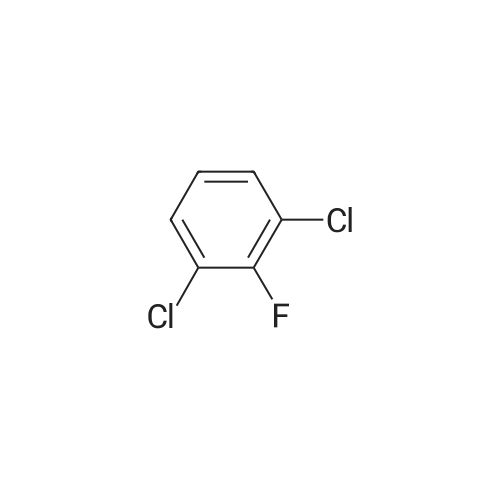
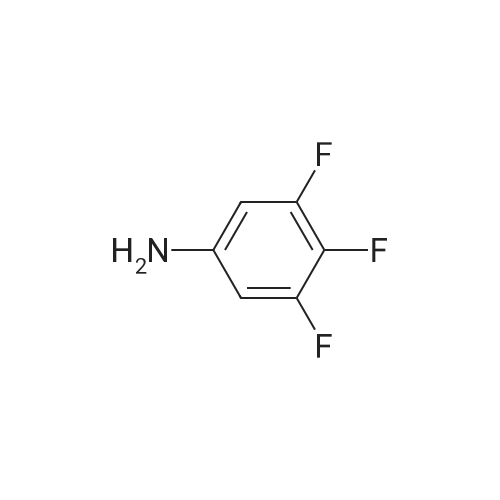
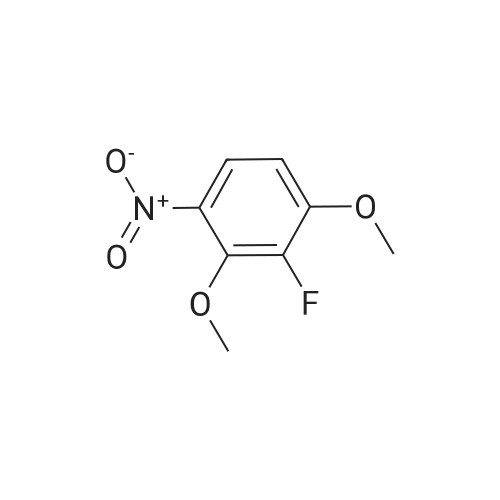
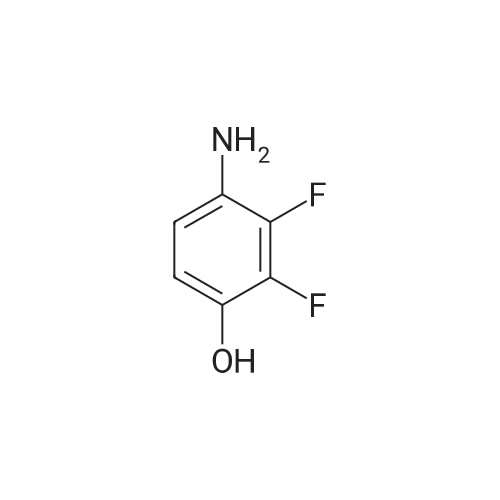
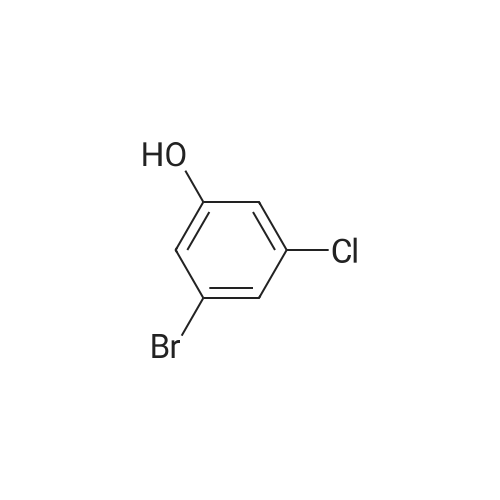
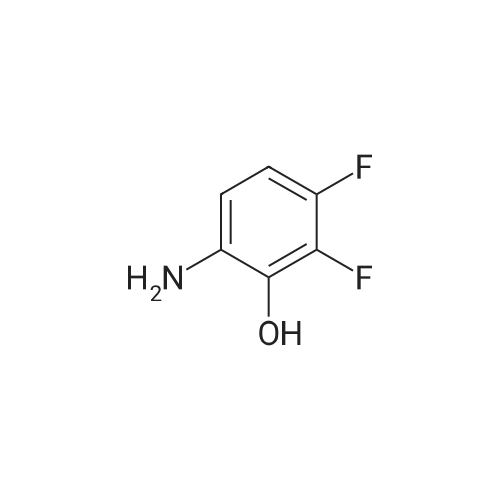
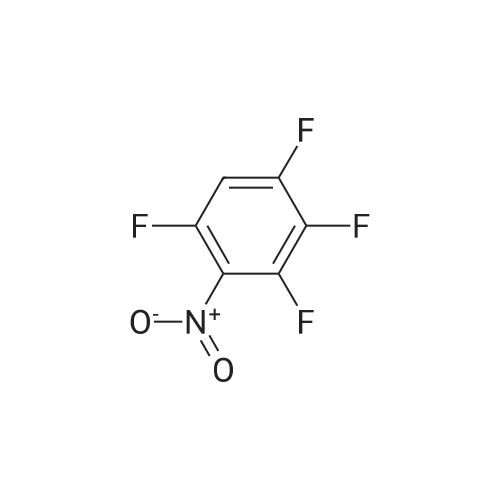
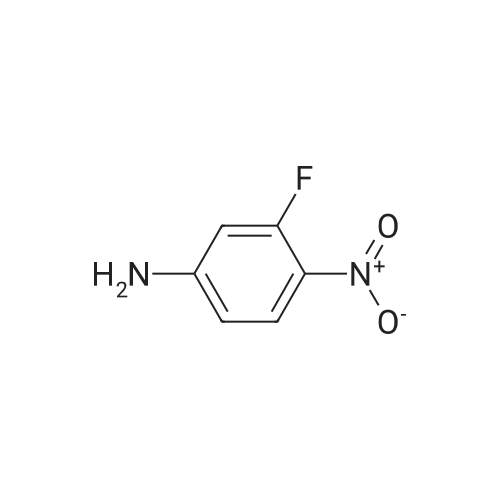
[ 17700-09-3 ]
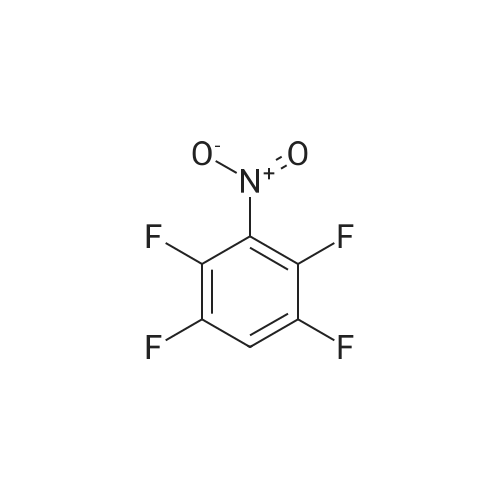
[ 771-69-7 ]
Yield
Reaction Conditions
Operation in experiment
99.7%
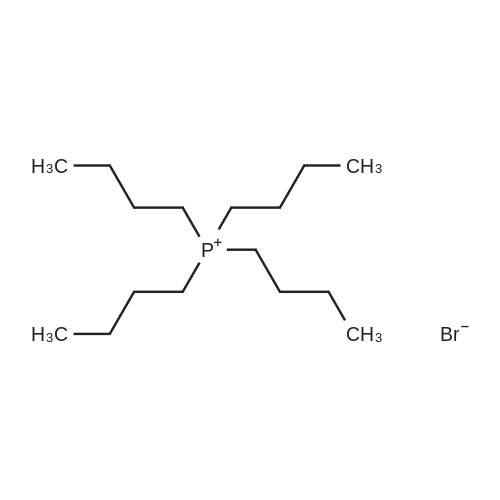
Stage #1: With potassium fluoride; tetrabutylammomium bromide In dimethyl sulfoxide at 75 - 80℃; for 4 h; Stage #2: With potassium fluoride; tetrabutyl ammonium fluoride In dimethyl sulfoxide at 75 - 180℃; for 12 h;
(2) Add 240 g of DMSO in an anhydrous reaction flask,120g 2,3,4-trichloronitrobenzene, open stirring,The temperature was raised to 75 to 80 ° C under reduced pressure, and after stirring for 2 hours, 76.8 g of KF was added,12g TBAB, dehydrated under reduced pressure 75 ~ 80 for 2 hours,Until the distillation head no drops so far;(3) The reaction system was warmed to 180 ,The reaction was started, the progress of the reaction was followed by GC,After 10 hours, the content of 2-fluoro-3,4-dichioronitrobenzene and 2,3-dichloro-4-fluoronitrobenzene were both less than 0.2percentFor the reaction end, cooled to 70 ~ 75 ,Filtration, the filtrate vacuum distillation and then put into anhydrous reaction device, plus 46g KF,12g TBAF, decompression 75 ~ 80 dehydration for 2 hours, the distillation head without water droplets, set the reaction temperature 120 , ultrasonic power: 20KHZ, the reaction,The progress of the reaction was followed by GC, the reaction was completed after 2 hours, cooled to 70-75 ° C, filtered,Distillation of the filtrate separates the DMSO from the product. After testing,The yield of 2,3,4-trifluoronitrobenzene was 99.7percent.
Reference:
[1] Patent: CN107325001, 2017, A, . Location in patent: Paragraph 0028-0029; 0031-0034; 0036-0039; 0041-0044; 0046
5
[ 2268-05-5 ]
[ 771-69-7 ]
Yield
Reaction Conditions
Operation in experiment
85.5%
Stage #1: at 20 - 25℃; for 2.5 h; Stage #2: for 5 h;
n a 250 ml four-necked flask, 64 g of 98percent concentrated sulfuric acid was charged(0.606 mol) of 2,6-dichlorofluorobenzene was added dropwise to a stirred solution of 33 g of 98percent concentrated sulfuric acid at 20 to 25 ° CMixed with 39 g of 98percent concentrated nitric acid, 2.5 hThe addition is complete.After warming up to 40 ~ 45 ° C and heat 1.5 hours. After the end of the washing, to neutral, organic subtractionPressure dehydration after use.In a 250 ml four-necked flask285gSulfolane and87g (1.5mol) KF, after dehydration by adding the last step nitration products,In 195 ± 5 ° C heat 5h, vacuum distillation of the solvent and then distillation 2,3,4-trifluoronitrobenzene 91. 7g,The content was 99.8percent and the yield was 85.5percent
Reference:
[1] Patent: CN102249881, 2016, B, . Location in patent: Paragraph 0015; 0057-0059
[2] Journal of the American Chemical Society, 1959, vol. 81, p. 94,95, 97
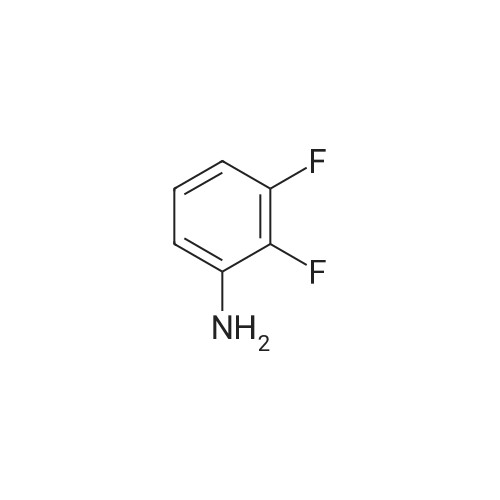
6
[ 393-79-3 ]
[ 771-69-7 ]
Reference:
[1] Tetrahedron Letters, 1989, vol. 30, # 51, p. 7199 - 7202
[2] Journal of the American Chemical Society, 1959, vol. 81, p. 94,95, 97
7
[ 393-79-3 ]
[ 3115-68-2 ]
[ 771-69-7 ]
Reference:
[1] Patent: US5502260, 1996, A,
8
[ 163733-96-8 ]
[ 771-69-7 ]
Reference:
[1] Russian Journal of Organic Chemistry, 1995, vol. 31, # 2, p. 200 - 206[2] Zhurnal Organicheskoi Khimii, 1995, vol. 31, # 2, p. 226 - 232
9
[ 95-50-1 ]
[ 771-69-7 ]
Reference:
[1] Patent: CN102249881, 2016, B,
10
[ 3209-22-1 ]
[ 771-69-7 ]
Reference:
[1] Patent: CN102249881, 2016, B,
11
[ 2106-49-2 ]
[ 771-69-7 ]
Reference:
[1] Patent: CN102249881, 2016, B,
12
[ 1489-53-8 ]
[ 771-69-7 ]
Reference:
[1] Russian Journal of Organic Chemistry, 1995, vol. 31, # 2, p. 200 - 206[2] Zhurnal Organicheskoi Khimii, 1995, vol. 31, # 2, p. 226 - 232
Reference:
[1] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
[2] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
[3] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
17
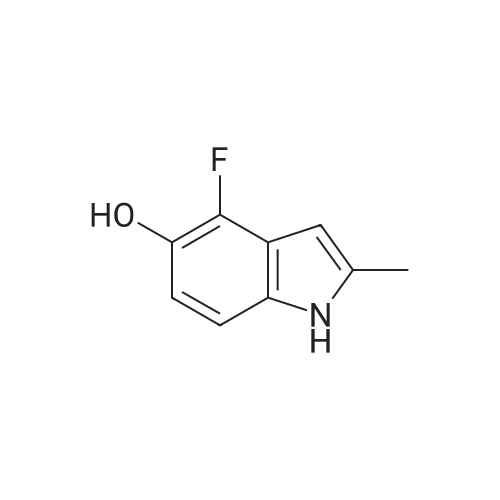
[ 771-69-7 ]
[ 4519-40-8 ]
Reference:
[1] Patent: JP2015/227293, 2015, A,
[2] Patent: JP2015/227293, 2015, A,
[3] Patent: JP2015/227293, 2015, A,
[4] Patent: JP2015/227293, 2015, A,
18
[ 771-69-7 ]
[ 3862-73-5 ]
Reference:
[1] Journal of the American Chemical Society, 1959, vol. 81, p. 94,95, 97
[2] Advanced Synthesis and Catalysis, 2011, vol. 353, # 8, p. 1306 - 1316
[3] Science, 2013, vol. 342, # 6162, p. 1073 - 1076
[4] Green Chemistry, 2015, vol. 17, # 2, p. 898 - 902
[5] ACS Catalysis, 2015, vol. 5, # 3, p. 1526 - 1529
[6] Patent: CN104163764, 2016, B, . Location in patent: Paragraph 0050-0053
[7] ChemCatChem, 2017, vol. 9, # 19, p. 3743 - 3751
[8] European Journal of Organic Chemistry, 2018, vol. 2018, # 2, p. 209 - 214
[9] Applied Catalysis A: General, 2018, vol. 559, p. 127 - 137
19
[ 771-69-7 ]
[ 369-34-6 ]
[ 6921-22-8 ]
[ 3862-73-5 ]
Reference:
[1] Angewandte Chemie - International Edition, 2013, vol. 52, # 11, p. 3203 - 3207[2] Angew. Chem., 2013, vol. 125, # 11, p. 3285 - 3289,5
20
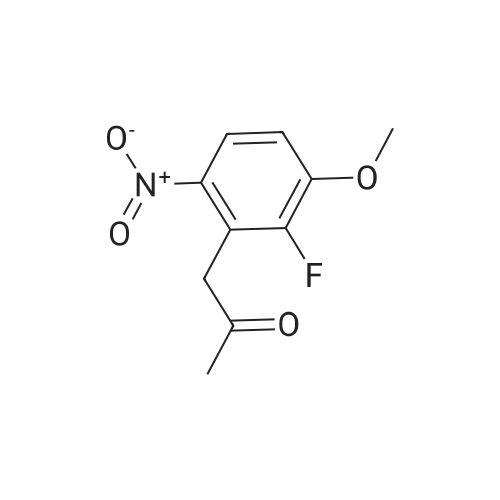
[ 771-69-7 ]
[ 82419-36-1 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1984, vol. 32, # 12, p. 4907 - 4913
21
[ 771-69-7 ]
[ 82419-35-0 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1984, vol. 32, # 12, p. 4907 - 4913
22
[ 771-69-7 ]
[ 82419-26-9 ]
Yield
Reaction Conditions
Operation in experiment
86%
With potassium hydroxide; sulfuric acid In water
EXAMPLE 1 1.77 kg (10 mol) of 2,3,4-trifluoronitrobenzene are added to 4 l of water. The mixture is then warmed to 40° C. and vigorously stirred. 389.4 g (22 mol) of 31.7percent strength potassium hydroxide solution are then added dropwise in such a way that the temperature remains between 40° and 55° C. After about 4 h, the reaction is complete (GC checking). The reaction mixture is brought to pH 3 using 70percent strength sulfuric acid at 70° C. Steam is then passed into the solution, the product passing over with it and being isolated after cooling to 10° C. 1.51 kg of 2,3-difluoro-6-nitrophenol having a purity >99.9percent (GC) are obtained after drying (melting point 63.5° C.), which corresponds to a yield of 86percent of theory. The mixture can also be acidified with, for example, 85percent strength phosphoric acid instead of with the 70percent strength sulfuric acid.
38.8g
With potassium hydroxide In dimethyl sulfoxide at 15 - 17℃; for 3 h;
a. To a 1000 mL flask was added 45.2 g of trifluoronitrobenzene and99.5g dimethyl sulfoxide, stirring;b, adjust the temperature to 15 ~ 17 start dropping125 g mass fraction of 32percent KOH solution,Dropping process control temperature 15 ~ 17 ;c, after the drop in the 15 ~ 17 stirring reaction 3h;d, after the end of the reaction at 14 ~ 16 To the reaction system was added 290 g of dilute hydrochloric acid with a mass fraction of 7.6percente, after stirring for 1 h,Filter cake to 50g water / times washing, washing a total of three times;f, dried to give 38.8 g of 2,3-difluoro-6-nitrophenol;
Reference:
[1] Patent: US5292967, 1994, A,
[2] Chemical and Pharmaceutical Bulletin, 1984, vol. 32, # 12, p. 4907 - 4913
[3] Patent: US4382892, 1983, A,
[4] Patent: CN106632444, 2017, A, . Location in patent: Paragraph 0032; 0033; 0034; 0035; 0036; 0037; 0038-0041
23
[ 771-69-7 ]
[ 124-41-4 ]
[ 82419-26-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
24
[ 771-69-7 ]
[ 82419-34-9 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1984, vol. 32, # 12, p. 4907 - 4913
25
[ 771-69-7 ]
[ 151414-46-9 ]
Reference:
[1] Patent: US2003/144267, 2003, A1,
26
[ 771-69-7 ]
[ 124-41-4 ]
[ 155020-44-3 ]
Yield
Reaction Conditions
Operation in experiment
99%
at 4 - 20℃; for 0.5 h;
A freshly prepared solution of sodium methoxide [0.58 g (25 mmol) of sodium dissolved in 3 niL of anhydrous methanol] was added dropwise to a solution of 2 g (11.2 mmol) of l52,3-trifluoro-4-nitrobenzene in 30 mL of anhydrous methanol under nitrogen at +40C. The resulting mixture was stirred overnight at room temperature and quenched with 1 M aqueous citric acid (0.1 eq) and methanol was evaporated under reduced pressure. The residue was taken up with ether, washed with 1 M citric acid, brine and dried over anhydrous magnesium sulfate and filtered off. The filtrate was evaporated under reduced pressure to give 2.2 g (99percent yield) of the crude product, which was purified by crystallisation from hexane. H1 NMR (CDCl3) 3.95 (s, 3H, OMe); 4.06 (d, 3H5 J= 1.6 Hz); 6.72 (dd, IH, CH5 J = 9.4 Hz5 J = 7.5 Hz) 7.72 (dd, IH, CH, J = 9.4 Hz, J = 2.23 Hz).
80%
at 0 - 20℃;
Example 25 N-(2-cvano- 1 -(7-fluoro- 1 -hydroxy- 1.3 -dihydrobenzo [c] [ 1.2"|oxaborol-6-yloxy)propan-2-v l)-4-(trifluoromethoxy)benzamide To a stirring solution of l,2,3-trifluoro-4-nitrobenzene (80 g, 0.45 mmol) in MeOH (800 mL) is slowly added MeONa (54 g, 0.99 mmol) in portions at 0°C. The resulting mixture is stirred at rt overnight. TLC showed the reaction is completed. The solvent is evaporated and the residue is washed with water, acidified with diluted HC1 solution to pH=7.0 and extracted with EtOAc. The separated organics is dried and concentrated to give a residue, which is purified by silica gel chromatography (PE:EtOAc=100:l to 30:1) to give the desired product (72 g, 80percent yield).
Reference:
[1] Journal of Organic Chemistry, 1997, vol. 62, # 19, p. 6469 - 6475
[2] Patent: WO2006/84338, 2006, A1, . Location in patent: Page/Page column 90
[3] Journal of the American Chemical Society, 2011, vol. 133, # 9, p. 2989 - 2997
[4] Patent: WO2014/149793, 2014, A1, . Location in patent: Page/Page column 85
27
[ 67-56-1 ]
[ 771-69-7 ]
[ 155020-44-3 ]
Yield
Reaction Conditions
Operation in experiment
89%
for 48 h; Reflux
Step 1:To a solution of 1,2,3-trifluoro-4-nitrobenzene(5.00 g, 28.2 mmol) in anhydrous MeOH (100 mL) was added NaOMe (6.86 g, 12.7 mmol). The reaction mixture was heated at reflux for 48 h, adjusted to pH 6 with 2 M citric acid, and extracted with EtOAc (2 × 400 mL). The organic layers were combined, washed with brine (50 mL), dried over MgSO4, and concentrated under reduced pressure to give 2-fluoro-1,3-dimethoxy-4-nitrobenzene as a yellow powder (5.10 g, 89percent).1H-NMR(CDCl3):δ3.97 (s, 3H, CH3), 4.07 (d, 3H,J= 1.1 Hz, CH3), 6.74 (dd, 1H,J= 9.3 & 7.6 Hz, Ph-H), 7.74 (dd, 1H,J= 9.4 & 2.1 Hz, Ph-H).
Reference:
[1] European Journal of Medicinal Chemistry, 2017, vol. 139, p. 762 - 772
28
[ 771-69-7 ]
[ 155020-44-3 ]
Reference:
[1] Patent: US6162931, 2000, A,
29
[ 771-69-7 ]
[ 124-41-4 ]
[ 66684-59-1 ]
[ 66684-60-4 ]
[ 155020-44-3 ]
Reference:
[1] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
[2] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
[3] Journal of Medicinal Chemistry, 1994, vol. 37, # 9, p. 1362 - 1370
30
[ 771-69-7 ]
[ 103068-40-2 ]
Reference:
[1] Journal of Organic Chemistry, 1997, vol. 62, # 19, p. 6469 - 6475
[2] Journal of the American Chemical Society, 2011, vol. 133, # 9, p. 2989 - 2997
Reference:
[1] Organic Process Research and Development, 2014, vol. 18, # 1, p. 89 - 102
[2] Chinese Chemical Letters, 2015, vol. 26, # 9, p. 1165 - 1168
33
[ 771-69-7 ]
[ 288385-99-9 ]
Reference:
[1] Organic Process Research and Development, 2014, vol. 18, # 1, p. 89 - 102
[2] Patent: EP2940031, 2015, A1,
34
[ 771-69-7 ]
[ 649736-31-2 ]
Reference:
[1] Organic Process Research and Development, 2014, vol. 18, # 1, p. 89 - 102
35
[ 771-69-7 ]
[ 211693-73-1 ]
Yield
Reaction Conditions
Operation in experiment
91.2%
With ammonium hydroxide In ethanol at 20℃; for 8 h;
1,2,3-trifluoro-4-nitrobenzene 8.85g of (50 mmol) was dissolved in ethanol 50 ml. To this solution 25percent aqueous ammonia 13.62g (the ammonia 200mmol equivalent) was added, followed by stirring at room temperature for 8 hours. Then, the ethanol was distilled off, and filtered. The resulting crude crystals were washed with water, then dried under reduced pressure. 2,3-difluoro-6-nitroaniline 7.94g (45.6mmol, 91.2percent yield). 1H-NMR analysis results of the resulting compound were as follows.
73%
With ammonia In methanol at 70℃; for 1.5 h; Microwave irradiation
Ten batches, each of 2.00 g (11.3 mmol) of l,2,3-trifluoro-4-nitrobenzene and 4.2 ml (29.6 mmol) of 7N methanolic ammonia solution, were reacted sequentially for 90 min at 700C in a single-mode microwave. The resultant solutions were combined and the volatile components were removed in a rotary evaporator. The residue was taken up in ethyl acetate, washed with water and dried over sodium sulfate. The solvent was removed by distillation at reduced pressure and the residue was recrystallized from methanol/water (1:1). We obtained 14.4 g of the target compound (73percent of theor., based on the total amount of l,2,3-trifluoro-4-nitrobenzene used).GC-MS (method 6): R, = 4.09 min; MS (EIpos): m/z = 174 [M]+.IH-NMR (400 MHz, DMSO-D6): δ [ppm] = 6.72 (m, IH), 7.54 (sbr, 2H), 7.93 (m, IH). 10.00 g (56.471 mmol) of 2,3,4-trifiuornitrobenzene was dissolved in a 2M methanolic ammonia solution and heated in an autoclave for 2 h at 700C. After cooling, the solvent was removed in a rotary evaporator and the residue was taken up in ethyl acetate. The organic phase was washed with water, dried over sodium sulfate and concentrated in a rotary evaporator. The resultant solid was recrystallized from 120 ml methanol/water (v/v = 1:1). We obtained 7.21 g (73percent of theor.) of the target compound.GC-MS (method 6): R, = 4.10 min; MS (EIpos): m/z = 174 [M]+. IH-NMR (400 MHz, DMSO-D6): δ [ppm] = 6.72 (dt, IH), 7.53 (sbr, 2H), 7.93 (ddd, IH).
35.6%
With ammonia In methanol at 70℃; for 1.5 h; Microwave irradiation
A solution of 1,2,3-trifluoro-4-nitrobenzene (1) (1.00 g, 5.64 mmol, 1.00 equiv) in methanolic ammonia (1.5 mL) was taken in microwave vial and heated to 70° C. for 90 min in the microwave. The solvent was evaporated under vacuum to give crude; which was purified by silica gel column chromatography (EtOAc/Hexane 1:49) to furnish compound 2 (0.350 g, 35.6percent) as yellow solid. TLC: 10percent EtOAc/Hexane (Rf: 0.10); 1H NMR (500 MHz, DMSO-d6): δ 7.94-7.91 (m, 1H), 7.51 (s, 2H), 6.75-6.70 (m, 1H); LC-MS: m/z=173 (M+-1) at RT 3.15 (99.8percent purity)
Reference:
[1] Patent: JP2015/227293, 2015, A, . Location in patent: Paragraph 0033; 0034
[2] Patent: WO2010/20363, 2010, A1, . Location in patent: Page/Page column 96-97, 119-120
[3] Patent: US2013/287686, 2013, A1, . Location in patent: Paragraph 0135; 0136
With sodium hypophosphite hydrate; 1% platinum on charcoal; hydrogen; at 65 - 80℃; under 6750.68 Torr; for 2.75h;Autoclave;
Under the premise of Example 1, the catalyst application experiment (ie, catalyst recycling) was carried out: (1) 200 g of 2,3,4-trifluoronitrobenzene was added, and 0.2 g of a 1% Pt/C catalyst and 1 g of sodium hypophosphite were added to the autoclave, and nitrogen and hydrogen were each replaced three times.(2) The stirring was started, the temperature was raised to 65 C, hydrogen gas was introduced at 0.9 Mpa, and the reaction was started to control the reaction temperature to 80 C. (3) During the reaction, the instantaneous reaction rate of the reaction was measured and expressed by the pressure drop of hydrogen per minute, and the reaction rate was 0.13 MPa/min. (4) The reaction time was 2 hours and 45 minutes. At the end of the reaction, the reaction pressure was increased, the reaction temperature was lowered, and the reaction rate was zero.(5) separating the catalyst from the reaction solution by means of suction filtration, the reaction liquid is simply separated into water, and the final product 2,3,4-trifluoroaniline is obtained by distillation under reduced pressure, and the conversion rate of the reaction is 100%, and the purity of the product is obtained. 99.78%, defluorination 0.101%, product yield 98.52%.
With 2 wt% Pd/C; hydrogen; at 120℃; under 7500.75 Torr;
Examples 27 to 34 investigated the effect of carbon-supported large-particle palladium catalysts on the synthesis of halogenated aromatic amines by solvent-free hydrogenation of different halogenated aromatic nitro compounds. In a 500 ml reactor, 200 g of different halogenated aromatic nitro compounds were added, 2 g of a 2 wt% palladium catalyst containing a large particle size in Example 5, shutting down the reactor; first with nitrogen replacement reactor inside the air three times, and then replaced with hydrogen three times, And then heated to 120 C, and the hydrogen pressure rose to 1MPa, open stirring to 1000r/min; to maintain the reaction temperature and pressure until the end of the reaction; cooling cooling, remove the reactor liquid, filter separation The catalyst was used and the water in the filtrate was separated by phase separation to give the desired product Halogenated aromatic amine. The reaction product was analyzed by gas chromatography. The results are shown in Table 4.
With hydrazine hydrate; In ethanol; at 30℃; for 0.833333h;Autoclave;
General procedure: The catalytic reduction of nitro aromatics was conducted in a 25 mlTelfon-lined stainless steel autoclave with magnetic stirring. In a typicalprocess, 6 mmol nitroarene, and desired amounts of reducing agent andsolvents were introduced in the reactor. The autoclave was transferredinto a water bath at the set temperature with an accuracy of better than0.2 C for ?0.5 h. Then, 10 mg catalyst was added into the reactionmixture and started the reduction at a stirring rate of 450 rpm. After thereaction, the catalyst was rapidly separated by ltration. n-Decane(500 muL) as standard was added into the ltrate and was dried withanhydrous Na 2 SO 4 . The products were analyzed by gas chromato-graphy-mass spectrometry (GC-MS) (Shimadzu GCMS-QP2010 Plus)and GC (Varian CP-3800) with a capillary column (column VF-1 ms,15 m, 0.25 mm, 0.25 mum) and a ame ionization detector (FID).
Step 2, with a thermometer,Blender,Adding solvent to the three-necked flask of the reflux condenserNitrification, PTC and dried KF,Stir under heating, react for 7 h, cool and filter,DMF is recovered at atmospheric pressure, followed by steam distillation. The distillate was extracted with ethyl acetate and dried over anhydrous Na2SO4.After recovering ethyl acetate under normal pressure,The distillation was carried out under reduced pressure to collect 92-93 C fractions.That is, the reaction temperature is 160 to 165 C.The yield is about 85%.
With potassium hydroxide; sulfuric acid; In water;
EXAMPLE 1 1.77 kg (10 mol) of 2,3,4-trifluoronitrobenzene are added to 4 l of water. The mixture is then warmed to 40 C. and vigorously stirred. 389.4 g (22 mol) of 31.7% strength potassium hydroxide solution are then added dropwise in such a way that the temperature remains between 40 and 55 C. After about 4 h, the reaction is complete (GC checking). The reaction mixture is brought to pH 3 using 70% strength sulfuric acid at 70 C. Steam is then passed into the solution, the product passing over with it and being isolated after cooling to 10 C. 1.51 kg of 2,3-difluoro-6-nitrophenol having a purity >99.9% (GC) are obtained after drying (melting point 63.5 C.), which corresponds to a yield of 86% of theory. The mixture can also be acidified with, for example, 85% strength phosphoric acid instead of with the 70% strength sulfuric acid.
With potassium hydroxide; In water; dimethyl sulfoxide;
EXAMPLE 1 Production of starting material 20 g of 2,3,4-trifluoronitrobenzene was dissolved in 150 ml of dimethyl sulfoxide, and to this mixture a solution of 10% potassium hydroxide was added dropwise while keeping the temperature at 18 to 20 C. Then, the mixture was stirred for 2 hours at room temperature and one liter of water was added to this reaction mixture and the mixture was shaken with chloroform. The water layer was acidified with hydrochloric acid and was extracted with chloroform. The extract was washed with water and was dried, then chloroform layer was concentrated. The residue was purified by silica gel column chromatography to provide 5.8 g of 2,3-difluoro-6-nitrophenol as yellow oil.
38.8g
With potassium hydroxide; In dimethyl sulfoxide; at 15 - 17℃; for 3h;
a. To a 1000 mL flask was added 45.2 g of trifluoronitrobenzene and99.5g dimethyl sulfoxide, stirring;b, adjust the temperature to 15 ~ 17 start dropping125 g mass fraction of 32% KOH solution,Dropping process control temperature 15 ~ 17 ;c, after the drop in the 15 ~ 17 stirring reaction 3h;d, after the end of the reaction at 14 ~ 16 To the reaction system was added 290 g of dilute hydrochloric acid with a mass fraction of 7.6%e, after stirring for 1 h,Filter cake to 50g water / times washing, washing a total of three times;f, dried to give 38.8 g of 2,3-difluoro-6-nitrophenol;
A freshly prepared solution of sodium methoxide [0.58 g (25 mmol) of sodium dissolved in 3 niL of anhydrous methanol] was added dropwise to a solution of 2 g (11.2 mmol) of l52,3-trifluoro-4-nitrobenzene in 30 mL of anhydrous methanol under nitrogen at +40C. The resulting mixture was stirred overnight at room temperature and quenched with 1 M aqueous citric acid (0.1 eq) and methanol was evaporated under reduced pressure. The residue was taken up with ether, washed with 1 M citric acid, brine and dried over anhydrous magnesium sulfate and filtered off. The filtrate was evaporated under reduced pressure to give 2.2 g (99% yield) of the crude product, which was purified by crystallisation from hexane. H1 NMR (CDCl3) 3.95 (s, 3H, OMe); 4.06 (d, 3H5 J= 1.6 Hz); 6.72 (dd, IH, CH5 J = 9.4 Hz5 J = 7.5 Hz) 7.72 (dd, IH, CH, J = 9.4 Hz, J = 2.23 Hz).
80%
In methanol; at 0 - 20℃;
Example 25 N-(2-cvano- 1 -(7-fluoro- 1 -hydroxy- 1.3 -dihydrobenzo [c] [ 1.2"|oxaborol-6-yloxy)propan-2-v l)-4-(trifluoromethoxy)benzamide To a stirring solution of l,2,3-trifluoro-4-nitrobenzene (80 g, 0.45 mmol) in MeOH (800 mL) is slowly added MeONa (54 g, 0.99 mmol) in portions at 0C. The resulting mixture is stirred at rt overnight. TLC showed the reaction is completed. The solvent is evaporated and the residue is washed with water, acidified with diluted HC1 solution to pH=7.0 and extracted with EtOAc. The separated organics is dried and concentrated to give a residue, which is purified by silica gel chromatography (PE:EtOAc=100:l to 30:1) to give the desired product (72 g, 80% yield).
n a 250 ml four-necked flask, 64 g of 98% concentrated sulfuric acid was charged(0.606 mol) of <strong>[2268-05-5]2,6-dichlorofluorobenzene</strong> was added dropwise to a stirred solution of 33 g of 98% concentrated sulfuric acid at 20 to 25 CMixed with 39 g of 98% concentrated nitric acid, 2.5 hThe addition is complete.After warming up to 40 ~ 45 C and heat 1.5 hours. After the end of the washing, to neutral, organic subtractionPressure dehydration after use.In a 250 ml four-necked flask285gSulfolane and87g (1.5mol) KF, after dehydration by adding the last step nitration products,In 195 ± 5 C heat 5h, vacuum distillation of the solvent and then distillation 2,3,4-trifluoronitrobenzene 91. 7g,The content was 99.8% and the yield was 85.5%
(R)-3,4-difluoro-2-(2-benzyloxypropoxy)nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium hydroxide; potassium carbonate In toluene at 0℃; for 1h;
6 Example 6 (R)-3,4-Difluoro-2-(2-benzyloxypropoxy)nitrobenzene
Under ice cooling, potassium hydroxide (5.40 g) and potassium carbonate (3.33 g) were suspended in toluene (180 mL). To the resulting suspension, a solution of (R)-2-benzyloxy-1-propanol (4.0 g), which had been obtained as in Example 2, and 2,3,4-trifluoronitrobenzene (4.13 g) in toluene (40 mL) was added, followed by stirring at the same temperature for 1 hour. Water was added to the reaction mixture, followed by extraction with toluene. The organic layer was washed with water and then dried over anhydrous magnesium sulfate. After the solvent was distilled off, the resulting residue was subjected to chromatography on a silica gel column to afford the title compound as a yellow oil (7.55 g).1H-NMR (CDCl3) δ:1. 29 (3H, d, J = 6. 4Hz), 3. 93-4. 03 (2H, m), 4. 53-4. 65 (2H, m), 6. 90-6. 99 (1H, m), 7. 23-7. 35 (5H, m), 7. 60-7. 66 (1H, m)
With sodium t-butanolate In toluene at 0℃; for 1h;
7 Example 7 (R)-3,4-Difluoro-2-(2-benzyloxypropoxy)nitrobenzene
Under ice cooling, sodium tert-butoxide (63.6 mg) was suspended in toluene (0.5 mL). To the resulting suspension, (R)-2-benzyloxy-1-propanol (100 mg) which had been obtained as in Example 2 was added. The thus-obtained suspension was then added under ice cooling to a solution of 2,3,4-trifluoronitrobenzene (103.5 mg) in toluene (0.5 mL), followed by stirring for 1 hour. Water was added to the reaction mixture, followed by extraction with toluene. The organic layer was washed with water and then dried over anhydrous magnesium sulfate. After the solvent was distilled off, the resulting residue was subjected to chromatography on a silica gel column to afford the title compound as a yellow oil (161.2 mg).1H-NMR spectrum data of the compound were in conformity with those of the compound in Example 6.
With sodium t-butanolate; In tetrahydrofuran; at 0℃; for 2h;
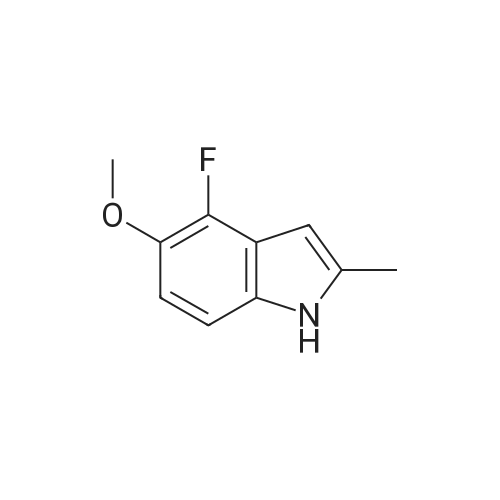
Solid sodium tert-butoxide was added to a ice cold solution of <strong>[2380-84-9]7-hydroxyindole</strong> (1.23 g, 9.24 mmol; Synthetic Communications 2003 33:507) in anhydrous THF (145 mL) under a nitrogen atmosphere. The mixture was stirred for 10 min, and 2,3,4-trifluoronitrobenzene (1.06 mL, 9.24 mmol) was added dropwise. The brown solution was stirred for 2 h, and then added to a saturated aqueous solution of NH4Cl (150 mL). The aqueous layer was extracted with EtOAc (3×100 mL), and the combined organic fractions were washed with H2O (100 mL), brine (75 mL), and dried over anhydrous MgSO4. The solvents were evaporated, and the remaining oil was purified by flash chromatography on silica gel (0% to 30% EtOAc/hexanes) to afford 2.26 g (84%) of 27.
84%
Solid sodium tert-butoxide was added to a ice cold solution of 4-hydroxyindole (1.23 g, 9.24 mmol; Synthetic Communications 2003 33:507) in anhydrous THF (145 mL) under a nitrogen atmosphere. The mixture was stirred for 10 m, and 2,3,4-trifluoronitrobenzene (1.06 mL, 9.24 mmol) was added dropwise. The brown solution was stirred for 2 h, and then added to a saturated aqueous solution of NH4Cl (150 mL). The aqueous layer was extracted with EtOAc (3×100 mL), and the combined organic fractions were washed with H2O (100 mL), brine (75 mL), and dried over anhydrous MgSO4. The solvents were evaporated, and the remaining oil was purified by flash chromatography on silica gel (0% to 30% EtOAc/hexanes) to afford 2.26 g (84%) of 109.
With sodium; boron tribromide; In methanol; dichloromethane; water;
i 3,5-Difluoro-6-nitrophenol To a stirred solution of 2,3,4-trifluoronitrobenzene (5 g, 28.23 mmol) in dry methanol (70 ml) was added a solution of sodium (0.68, 29.46) in dry methanol (30 ml). The solution was stirred until all starting material was consumed (~2 h). After concentration water was added and the solution was extracted with ether, dried over Na2SO4, filtered and concentrated to a yellow residue (4.65 g). To the solution of the yellow residue in dichloromethane (140 ml) was added boron tribromide (1M in dichloromethane, 40 ml) and stirred at room temperature overnight. Water was then added and the solution stirred for further 30 min. The organic phase was separated and the water phase was extracted with ether. The combined organic phase were dried over Na2SO4, filtered and concentrated in vacuo to give a brownish residue. The residue was taken up into ether and washed with 2M sodium hydroxide and water. The water and sodium hydroxide washings were combined and neutralized with 6M HCl and extracted with ether, dried over Na2SO4 and evaporated to give a yellow residue which was purified by flash chromatography on silica gel with EtOAc:Heptan; 1:2 as eluent to give the desired product 2 g, 11.42 mmol. GC-MS: m/z 175(M+) 1H-NMR (400 MHz, CD3OD) deltappm, 6.63-6.68(1H, m), 6.60-6.67(1H, dt)
A. 2,3-Difluoro-6-nitroaniline 1,2,3-Trifluoronitrobenzene (20.2 g, 114.1 mmol, Aldrich, Milwaukee) and methanolic ammonia (150 mL of a 2.0 molar solution, Aldrich, Milwaukee) were placed in a sealed tube and were heated to 70 C. for 2 h, after which time the solvents were removed in vacuo. The remaining residue was dissolved in ethyl acetate and was extracted with water, saturated aqueous sodium chloride, dried over magnesium sulfate, filtered and the solvents were removed in vacuo to leave a yellow solid. The title compound was recrystallized from water-methanol to afford yellow needles (16.6 g, 84%); 1H NMR (DMSO-d6) delta: 7.94 (m, 1H, Ar-H), 7.56 (br s, 2H, NH2), 6.72 (m, 1H, Ar-H).
Example 2 Preparation of 2,4-Dimethoxy-3-fluoronitrobenzene (2) Beginning with 2,3,4-trifluoronitrobenzene (5.53 g, 31.2 mmol, Aldrich), Compound 2 is obtained as 6.30 g (99%) as a pale yellow crystalline solid. An analytical sample is obtained by crystallization from light petroleum ether/dichloromethane: m.p. 59-61 C.; 1 H NMR (CDCl3) 7.72 (dd, 1H), 6.71 (dd, 1H), 4.08 (s, 3H), 3.95 (s, 3H); 19 F NMR (CDCl3) 149.1 (d). Anal. calc. for C8 H8 NO4 F: C, 47.77; H, 4.01; N, 6.96. Found: C, 47.64; H, 4.05; N, 6.80.
anhydrous ethylene glycol dimethyl ether[ No CAS ]
trimethyl(ethoxypolyoxypropylmethyl ether)ammonium chloride[ No CAS ]
[ 393-79-3 ]
[ 3115-68-2 ]
[ 771-69-7 ]
Yield
Reaction Conditions
Operation in experiment
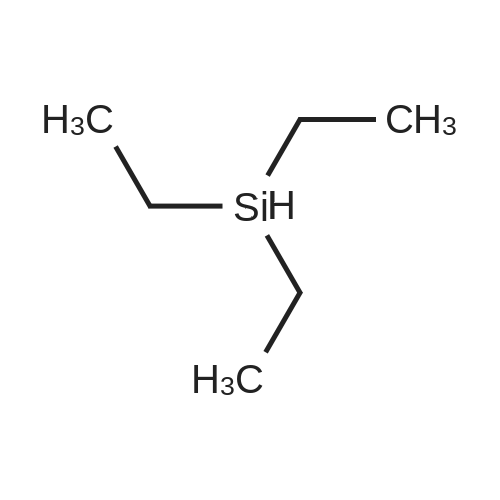
With potassium fluoride; In 5,5-dimethyl-1,3-cyclohexadiene;
EXAMPLE 7 2,3,4-Trifluoronitrobenzene In a 2.5 liter flange flask fitted with a distillation bridge and anchor stirrer, 488 g (8.4 mol) of potassium fluoride, 40.0 g (0.06 mol) of trimethyl(ethoxypolyoxypropyl methyl ether)ammonium chloride, 20.0 g (0.04 mol) of polyethylene glycol dimethyl ether 500 and 20.4 g (0.06 mol) of tetrabutylphosphonium bromide were introduced at 110 C. into the melt of 840 g (4 mol) of <strong>[393-79-3]2,4-dichloro-3-fluoronitrobenzene</strong>. Subsequently, 60 g (0.57 mol) of xylene were added and the reaction suspension was azeotropically dried by application of a vacuum of 30 mbar and heating to 130 C. After the xylene had been distilled off, the distillation bridge was replaced by a reflux condenser, the reaction suspension was heated to 150 C. and stirred for 21 hours at this temperature. Subsequently, the reaction suspension was cooled to 25 C. and filtered with suction (25 C.). The salts separated off were washed twice with a total of 400 g of xylene and the combined organic phases were fractionated. 516.8 g (73% of theory) of 2,3,4-trifluoronitrobenzene were isolated. Amount of 2,3,4-trifluoronitrobenzene formed, according to GC analysis: after 5 hours, 14 GC area-%; after 21 hours, 75 GC area-%.
(R)-2-[2-(1-ethoxyethoxy)propoxy]-3,4-difluoronitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
In tetrahydrofuran; diethyl ether; water
31 (R)-2-[2-(1-Ethoxyethoxy)propoxy]-3,4-difluoronitrobenzene
Example 31 (R)-2-[2-(1-Ethoxyethoxy)propoxy]-3,4-difluoronitrobenzene 0.4 G of 60% sodium hydride, previously washed with pentane, was suspended in 20 ml of tetrahydrofuran. Next, a mixture of 2.96 of (R)-2-(1-ethoxyethoxy)-1-propanol and 10 ml of tetrahydrofuran was added to the stirred suspension. After reacting for 0.5 hour the clear solution was added slowly to 1.79 g of 2,3,4-trifluoronitrobenzene dissolved in 10 ml of tetrahydrofuran and the reaction mixture was stirred for another 0.5 hour. The solvent was removed under reduced pressure, and diethyl ether and water were added to the residue. The organic layer was separated, washed with water, dried over anhydrous sodium sulfate, filtered and evaporated to dryness. The crude residue was purified by chromatography on silica gel (eluent: hexane/diethyl ether/triethylamine: 7/3/0.1). Finally, 2.5 g of title product were isolated as a mixture of diastereomers (ratio: 2/1). Yield: 82%. 1H NMR (CDCl3, ppm) delta = 1.17 (tr, 3H), 1.20 (tr, 3H), 1.29 (m, 6H), 3.57 (m, 2H), 4.0-4.4 (m, 3H), 4.82 (q, 1H), 4.89 (q, 1H), 7.01 (m, 1H), 7.66 (m, 1H).
6.7%
With potassium hydroxide; potassium carbonate In water; toluene
93 (R)-2-[2-(1-Ethoxyethoxy)propoxy]-3,4-difluoronitrobenzene
Example 93 (R)-2-[2-(1-Ethoxyethoxy)propoxy]-3,4-difluoronitrobenzene A stirred suspension of 6.72 g of powdered potassium hydroxide and 4.14 g of powdered potassium carbonate in 80 ml of toluene was cooled to -5°C, after which a solution of 5.31 g of 2,3,4-trifluoronitrobenzene and 5.18 g of (R)-2-(1-ethoxyethoxy)-1-propanol in 45 ml of toluene was added slowly, while maintaining the temperature at 0°C. The mixture was stirred for 0.5 hour. Next, water was added and the organic phase was separated. The aqueous layer was extracted twice with toluene and the combined organic layers were washed with water several times till pH=8. The organic phase was evaporated to dryness and the residue was purified by chromatography on silica gel (eluent: hexane/diethyl ether/triethylamine: 2/1/0.01) to afford 8.0 g of title compound, as a 1:1 mixture of diastereomers. Yield: 87%. 1H NMR (CDCl3, ppm) delta = 1.18 (tr, 3H), 1.21 (tr, 3H), 1.30 (m, 6H), 3.58 (m, 2H), 4.1-4.4 (m, 3H), 4.82 (q, 1H), 4.86 (q, 1H), 7.00 (m, 1H), 7.66 (m, 1H). In addition, 0.7 g of a more polar by-product was obtained, which was identified as (R,R)-2,4-bis[2-(1-ethoxyethoxy)propoxy]-3-fluoronitrobenzene. Yield: 6.7%. 1H NMR (CDCl3, ppm) delta = 1.20 (m, 6H), 1.33 (m, 12H), 3.52 and 3.68 (2x m, 4H), 4.01-4.16 (m, 6H), 4.86 (m, 2H), 6.76 (m, 1H), 7.71 (m, 1H).
With sodium t-butanolate; In tetrahydrofuran; at 0 - 20℃;
step 1-Sodium tert-butoxide (0.76 g, 1 equiv) was added in one portion to a solution of <strong>[626-41-5]3,5-dibromophenol</strong> (40, 2.0 g, 7.9 mmol) in dry THF (16 mL). The solution was cooled to 0° C., and 2,3,4-trifluoronitrobenzene (32, 0.91 mL, 1 equiv) was added dropwise via syringe. The solution was warmed to RT, stirred overnight, and poured into water. The mixture was extracted with EtOAc, and the combined organics were washed with water, brine, and dried over magnesium sulfate. Filtration and evaporation afforded 3.26 g (100percent) of 41a as an oil that slowly crystallized.
With N-ethyl-N,N-diisopropylamine In acetonitrile at -10 - 20℃;
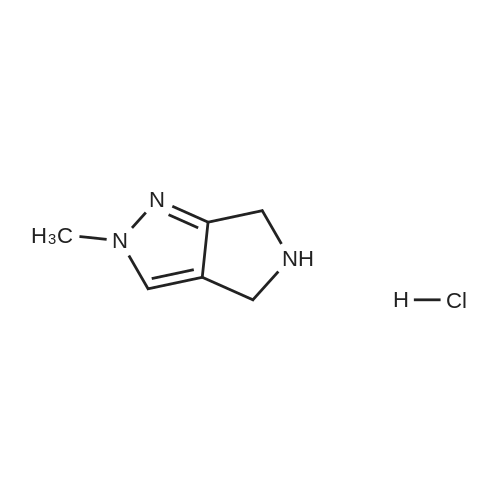
24
Example 24; Compound of Structure Scheme for Compound of Example 24: Intermediate 42. DIEA (3.8 mL) was added dropwise with stirring to 2-methyl-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole hydrochlororide (1.0 g, 7.04 mmol; prepared as described in JP 6073056) and 2,3,4-trifluoronitrobenzene (1.5 g, 8.45 mmol) in MeCN (100 mL) at -10° C. The mixture was allowed to warm up to r.t. and stirred for 6 h. Solvent was removed under vacuum, and the residue was taken into EtOAc (60 mL), washed with water (40 mL×3), brine (40 mL), and dried (Na2SO4). Solvent was removed under vacuum, and the product was purified by column chromatography (gradient 17% to 75% petroleum ether/ethyl acetate). Yellow solid. 1H NMR (400 MHz): 7.54 (m, 1H), 7.27 (d, J=6.4 Hz, 1H), 6.95 (m, 1H), 4.54 (s, 2H), 4.49 (s, 2H), 3.85 (s, 3H).
With N-ethyl-N,N-diisopropylamine In acetonitrile at -10 - 20℃; for 6h;
24
DIEA (3.8 mL) was added dropwise with stirring to 2-methyl-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole hydrochloride (1.0 g, 7.04 mmol; prepared as described in JP 6073056) and 2,3,4-trifluoronitrobenzene (1.5 g, 8.45 mmol) in MeCN (100 mL) at -10° C. The mixture was allowed to warm up to r.t. and stirred for 6 h. Solvent was removed under vacuum, and the residue was taken into EtOAc (60 mL), washed with water (40 mL×3), brine (40 mL), and dried (Na2SO4). Solvent was removed under vacuum, and the product was purified by column chromatography (gradient 17% to 75% petroleum ether/ethyl acetate). Yellow solid. 1H NMR (400 MHz): 7.54 (m, 1H), 7.27 (d, J=6.4 Hz, 1H), 6.95 (m, 1H), 4.54 (s, 2H), 4.49 (s, 2H), 3.85 (s, 3H).
With ammonium hydroxide; In ethanol; at 20℃; for 8h;
1,2,3-trifluoro-4-nitrobenzene 8.85g of (50 mmol) was dissolved in ethanol 50 ml. To this solution 25% aqueous ammonia 13.62g (the ammonia 200mmol equivalent) was added, followed by stirring at room temperature for 8 hours. Then, the ethanol was distilled off, and filtered. The resulting crude crystals were washed with water, then dried under reduced pressure. 2,3-difluoro-6-nitroaniline 7.94g (45.6mmol, 91.2% yield). 1H-NMR analysis results of the resulting compound were as follows.
73%
With ammonia; In methanol; at 70℃; for 1.5h;Microwave irradiation;
Ten batches, each of 2.00 g (11.3 mmol) of l,2,3-trifluoro-4-nitrobenzene and 4.2 ml (29.6 mmol) of 7N methanolic ammonia solution, were reacted sequentially for 90 min at 700C in a single-mode microwave. The resultant solutions were combined and the volatile components were removed in a rotary evaporator. The residue was taken up in ethyl acetate, washed with water and dried over sodium sulfate. The solvent was removed by distillation at reduced pressure and the residue was recrystallized from methanol/water (1:1). We obtained 14.4 g of the target compound (73% of theor., based on the total amount of l,2,3-trifluoro-4-nitrobenzene used).GC-MS (method 6): R, = 4.09 min; MS (EIpos): m/z = 174 [M]+.IH-NMR (400 MHz, DMSO-D6): delta [ppm] = 6.72 (m, IH), 7.54 (sbr, 2H), 7.93 (m, IH). 10.00 g (56.471 mmol) of 2,3,4-trifiuornitrobenzene was dissolved in a 2M methanolic ammonia solution and heated in an autoclave for 2 h at 700C. After cooling, the solvent was removed in a rotary evaporator and the residue was taken up in ethyl acetate. The organic phase was washed with water, dried over sodium sulfate and concentrated in a rotary evaporator. The resultant solid was recrystallized from 120 ml methanol/water (v/v = 1:1). We obtained 7.21 g (73% of theor.) of the target compound.GC-MS (method 6): R, = 4.10 min; MS (EIpos): m/z = 174 [M]+. IH-NMR (400 MHz, DMSO-D6): delta [ppm] = 6.72 (dt, IH), 7.53 (sbr, 2H), 7.93 (ddd, IH).
35.6%
With ammonia; In methanol; at 70℃; for 1.5h;Microwave irradiation;
A solution of 1,2,3-trifluoro-4-nitrobenzene (1) (1.00 g, 5.64 mmol, 1.00 equiv) in methanolic ammonia (1.5 mL) was taken in microwave vial and heated to 70 C. for 90 min in the microwave. The solvent was evaporated under vacuum to give crude; which was purified by silica gel column chromatography (EtOAc/Hexane 1:49) to furnish compound 2 (0.350 g, 35.6%) as yellow solid. TLC: 10% EtOAc/Hexane (Rf: 0.10); 1H NMR (500 MHz, DMSO-d6): delta 7.94-7.91 (m, 1H), 7.51 (s, 2H), 6.75-6.70 (m, 1H); LC-MS: m/z=173 (M+-1) at RT 3.15 (99.8% purity)
Stage #1: aniline With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 0.166667h; Inert atmosphere;
Stage #2: 2,3,4-trifluoronitrobenzene In tetrahydrofuran at -78℃; for 0.5h;
Stage #3: With water In tetrahydrofuran
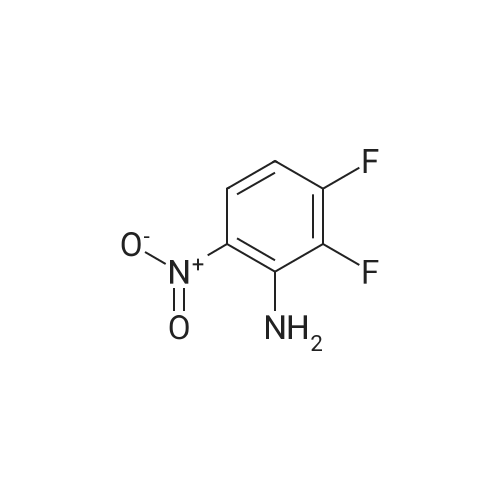
6-Fluoro-3-nitro-2-phenylaminobenzonitrile LiHMDS (1.0M in THF, 38 mL, 38.0 mmol) was added dropwise to a stirred solution of aniline (1.86 g, 19.9 mmol) in anhydrous THF (30 mL) under a nitrogen atmosphere at -78° C. After 10 min stirring at -78° C., a solution of 2,6-difluoro-3-nitrobenzonitrile (3.5 g, 19.0 mmol) in THF (15 mL) was added and stirring at -78° C. continued for 30 min. The crude mixture was quenched with water and diluted with EtOAc. The resulting emulsion was filtered through a pad of Celite and the organic fraction separated, washed with brine, dried (MgSO4) and concentrated in vacuo. The resulting residue was purified by column chromatography (Si-PCC, gradient 0-100% EtOAc in cyclohexane) to afford the title compound (2.7 g, 55%). 1H NMR (CDCl3, 400 MHz): δ 9.94 (1H, br s), 8.51 (1H, dd, J=9.50, 5.88 Hz), 7.51-7.21 (5H, m), 6.68 (1H, dd, J=9.50, 7.46 Hz).
To a suspension of sodium f-butoxide in THF (600 mL) was added 3-bromo-5- chlorophenol (34.4 g, 166 mmol) at 0 °C, and the mixture stirred at rt for 15 min. The mixture was cooled to 0 °C and 1 ,2,3-trifluoro-4-nitrobenzene (28.0 g, 158 mmol) was added dropwise. The mixture was stirred for 3 h at rt and concentrated to dryness. The residue was dissolved in ethyl acetate, washed with water and brine, dried over magnesium sulfate, filtered and concentrated to give 51.4 g of the title compound. 1H <n="38"/>NMR (DMSO-d6) .sect. 8.07 - 8.19 (m, 1 H), 7.64 - 7.75 (m, 1 H), 7.45 - 7.51 (m, 1 H), 7.35 - 7.41 (m, 1 H), 7.30 (s, 1 H).
With sodium t-butanolate; In tetrahydrofuran; at 0 - 20℃; for 0.25h;
To a suspension of sodium f-butoxide in THF (600 ml.) was added 3-bromo-5- chlorophenol (34.4 g, 166 mmol) at 0 0C, and the mixture stirred at rt for 15 min. The <n="36"/>mixture was cooled to 0 0C and 1 ,2,3-trifluoro-4-nitrobenzene (28.0 g, 158 mmol) was added dropwise. The mixture was stirred for 3 h at rt and concentrated to dryness. The residue was dissolved in ethyl acetate, washed with water and brine, dried over magnesium sulfate, filtered and concentrated to give 51.4 g of the title compound. 1H NMR (DMSOd6) delta 8.07 - 8.19 (m, 1 H), 7.64 - 7.75 (m, 1 H), 7.45 - 7.51 (m, 1 H), 7.35 - 7.41 (m, 1 H), 7.30 (s, 1 H).
With potassium carbonate In N,N-dimethyl-formamide at 0 - 20℃;
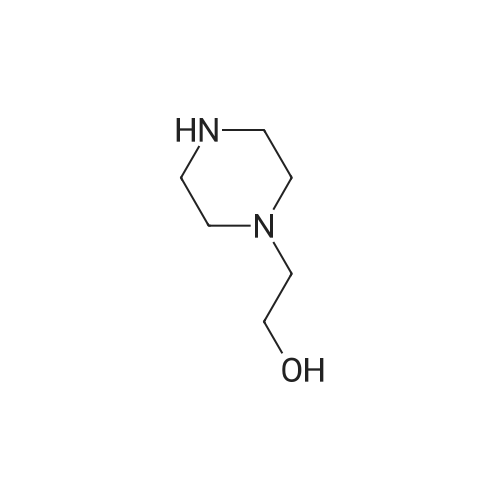
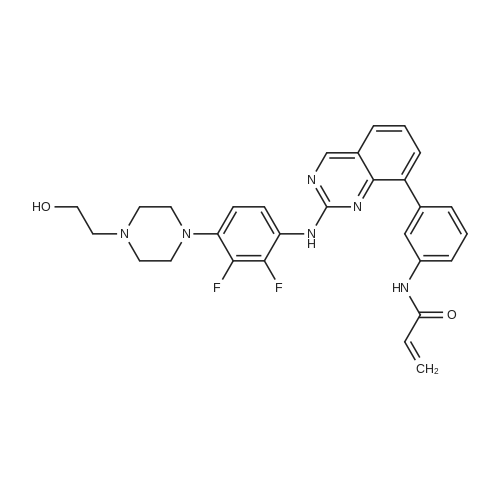
153 Preparation of N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2CO3 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro- 4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
67.5%
With potassium carbonate In N,N-dimethyl-formamide at 0 - 20℃;
1 Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2- hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4- nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
75 %
With potassium carbonate In N,N-dimethyl-formamide at 0 - 20℃; Inert atmosphere;
8.1 Step 1.
To a solutio ene (5.00 g, 28.2 mmol, 3.25 mL, 1.0 eq), 2-(piperazin-1-yl)ethan-1-ol (3.68 g, 28.2 mmol, 3.47 mL, 1.0 eq) in DMF (40 mL) was added K2CO3 (7.80 g, 56.5 mmol, 2.0 eq) at 0 °C. The mixture was stirred at 20 °C for 12 h. This reaction mixture was poured into 200 mL ice-water. The resulting solid was collected by filtration, washed with cold water three times, and concentrated under reduce pressure to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-1-yl)ethan-1-ol (6.10 g, 21.2 mmol, 75% yield) as a yellow solid.1H NMR: (400 MHz, CDCl3) δ 7.89 - 7.80 (m, 1H), 6.64 - 6.69 (m, J = 1.9, 7.9, 9.5 Hz, 1H), 3.67 (s, 2H), 3.41 - 3.34 (m, 4H), 2.72 - 2.67 (m, 4H), 2.65 - 2.61 (m, 2H).
Step 1:To a solution of 1,2,3-trifluoro-4-nitrobenzene(5.00 g, 28.2 mmol) in anhydrous MeOH (100 mL) was added NaOMe (6.86 g, 12.7 mmol). The reaction mixture was heated at reflux for 48 h, adjusted to pH 6 with 2 M citric acid, and extracted with EtOAc (2 × 400 mL). The organic layers were combined, washed with brine (50 mL), dried over MgSO4, and concentrated under reduced pressure to give 2-fluoro-1,3-dimethoxy-4-nitrobenzene as a yellow powder (5.10 g, 89%).1H-NMR (CDCl3):delta3.97 (s, 3H, CH3), 4.07 (d, 3H,J= 1.1 Hz, CH3), 6.74 (dd, 1H,J= 9.3 & 7.6 Hz, Ph-H), 7.74 (dd, 1H,J= 9.4 & 2.1 Hz, Ph-H).
Stage #1: 4-nitro-1,2,3-trichlorobenzene With potassium fluoride; tetrabutylammomium bromide In dimethyl sulfoxide at 75 - 80℃; for 4h;
Stage #2: With potassium fluoride; tetrabutyl ammonium fluoride In dimethyl sulfoxide at 75 - 180℃; for 12h;
4.2; 4.3 Example 4
(2) Add 240 g of DMSO in an anhydrous reaction flask,120g 2,3,4-trichloronitrobenzene, open stirring,The temperature was raised to 75 to 80 ° C under reduced pressure, and after stirring for 2 hours, 76.8 g of KF was added,12g TBAB, dehydrated under reduced pressure 75 ~ 80 for 2 hours,Until the distillation head no drops so far;(3) The reaction system was warmed to 180 ,The reaction was started, the progress of the reaction was followed by GC,After 10 hours, the content of 2-fluoro-3,4-dichioronitrobenzene and 2,3-dichloro-4-fluoronitrobenzene were both less than 0.2%For the reaction end, cooled to 70 ~ 75 ,Filtration, the filtrate vacuum distillation and then put into anhydrous reaction device, plus 46g KF,12g TBAF, decompression 75 ~ 80 dehydration for 2 hours, the distillation head without water droplets, set the reaction temperature 120 , ultrasonic power: 20KHZ, the reaction,The progress of the reaction was followed by GC, the reaction was completed after 2 hours, cooled to 70-75 ° C, filtered,Distillation of the filtrate separates the DMSO from the product. After testing,The yield of 2,3,4-trifluoronitrobenzene was 99.7%.
92%
With potassium fluoride In water; dimethyl sulfoxide at 20℃;
3 Example 1-1, a method for preparing aryl (heterocyclic) fluoride, including the following steps:
General procedure: Reaction solution A is made up of 22mL water, 23mL dimethyl sulfoxide, 8.82g potassium fluoride, stirred and dissolved at room temperature. At this time, the mass content of water in reaction solution A is about 39.2%; reaction solution B consists of 200mL dimethyl sulfoxide Sulfoxide, 18.27g p-chloronitrobenzene, stirred and dissolved at room temperature.
With sulfuric acid; dihydrogen peroxide; bromine; acetic acid In water Cooling;
8.7; 9.7; 10.7
Add 100 grams of water to the flask,200 grams of acetic acid, 200 grams of sulfuric acid,147 grams of 2,3,4-trifluoroaniline obtained in Example 7 were added with stirring,80 grams of bromine were added dropwise successively under cooling,112 grams of 30% hydrogen peroxide,After the reaction is completed, pour it into the sodium bisulfite aqueous solution,Separate at rest to obtain crude 2,3,4-trifluoro-6-bromoaniline.Add 318 grams of nitrososulfuric acid to the flask,Keep it at 10-15 degrees,Slowly add crude 2,3,4-trifluoro-6-bromoaniline under stirring,After the addition, the reaction was incubated for 6 hours to obtain a clear diazonium solution.Add 150 grams of water to another flask,50 grams of sulfuric acid, 114 grams of sodium hypophosphite,2 grams of cuprous oxide, slowly add the above diazonium solution under stirring,Carry out deamination reaction, stir and react for one hour after adding,Carry out steam distillation, the distillate is separated into oil layer rectification,169 g of 3,4,5-trifluorobromobenzene was obtained. The total yield of the two steps is 80%.
5-(2,3-difluoro-4-nitrophenoxy)-2-fluoroaniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%
With potassium carbonate; In dimethyl sulfoxide; at 90℃; for 3h;
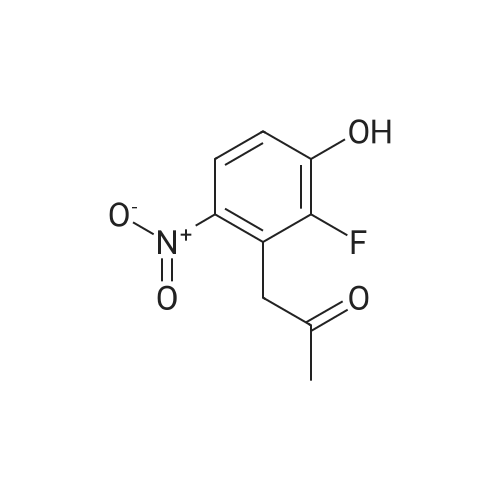
To <strong>[62257-16-3]3-amino-4-fluorophenol</strong> (456 mg, 3.953 mmol) and 1,2,3-trifluoro-4-nitrobenzene at room temperature(700mg, 3.953mmol) in DMSO (10ml) was added K2CD3 (1.49g, 10.78mmol).Stir the solution at 90CAfter stirring for 3 hours, the solution was extracted with ethyl acetate, washed with saturated sodium chloride, dried over anhydrous sodium sulfate, and the filtrate was concentrated under reduced pressure.The obtained residue was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 7:1-6:1), and the obtained solution was reducedIt was concentrated under pressure to obtain 440 mg of yellow solid with a yield of 43%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping