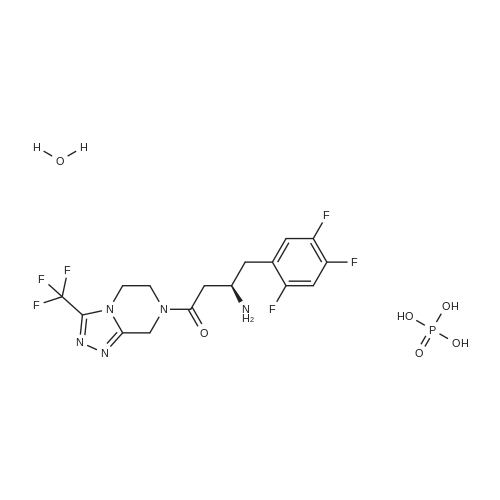
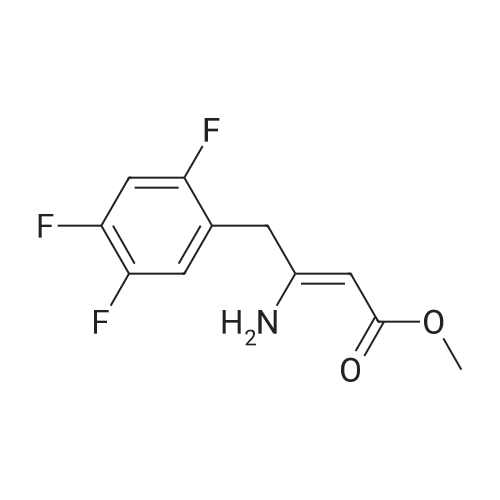
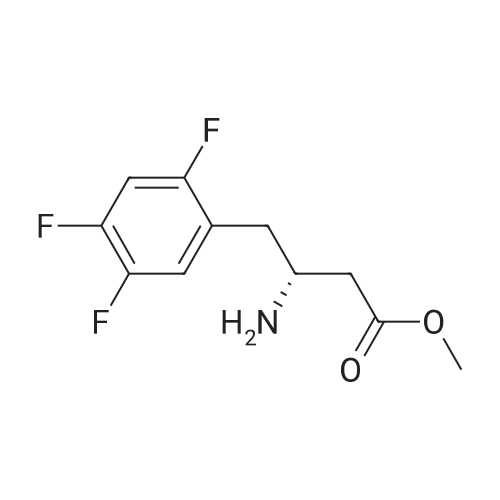
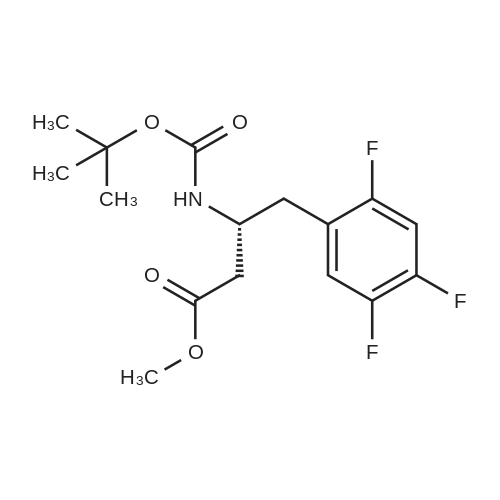
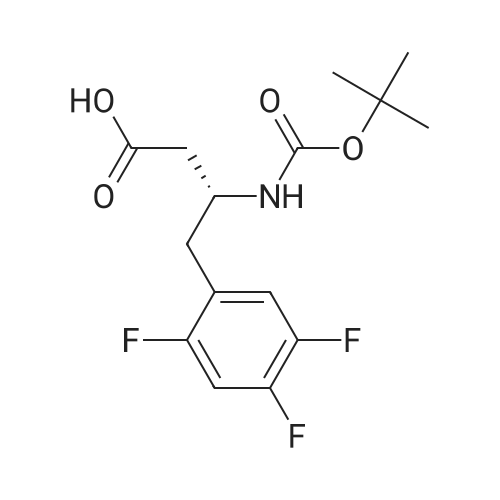
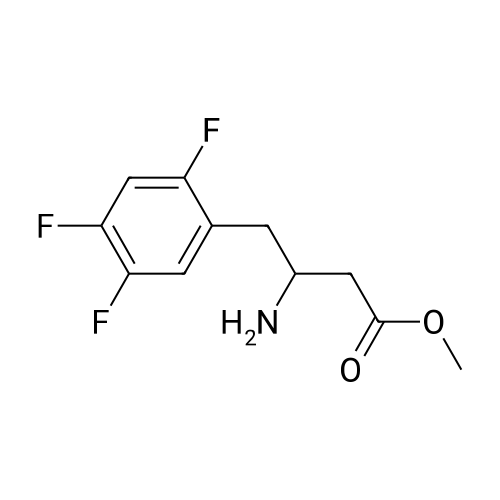
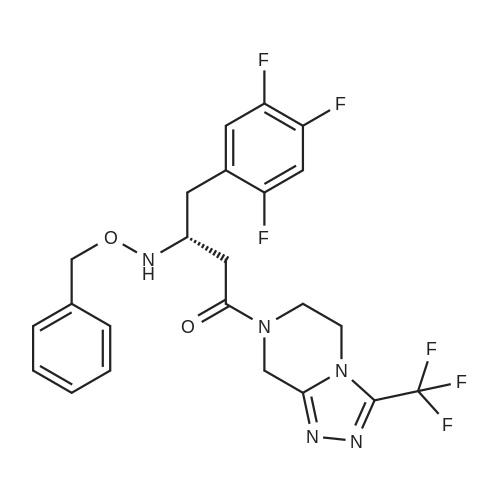
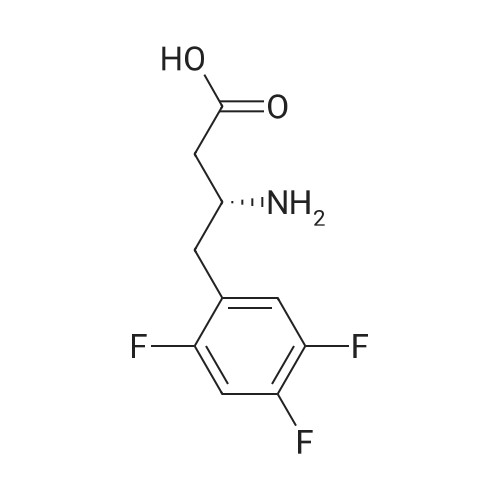
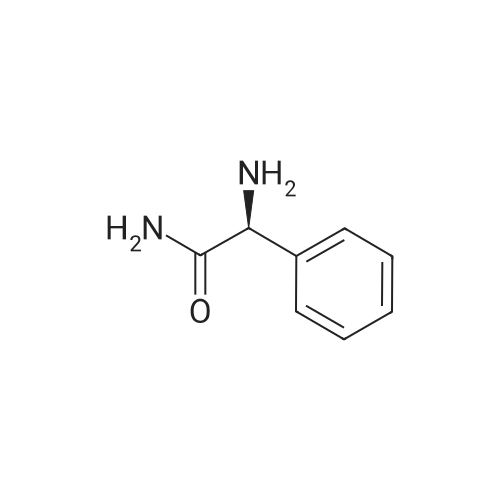
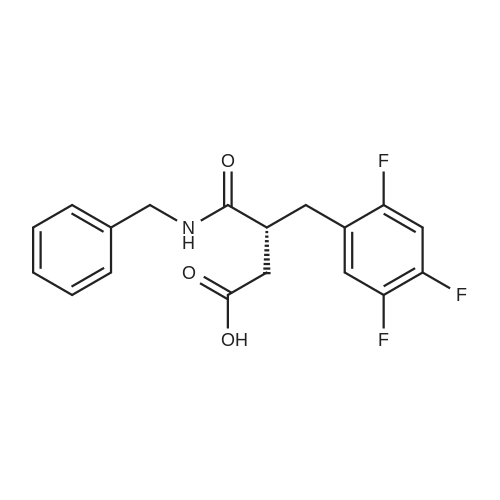
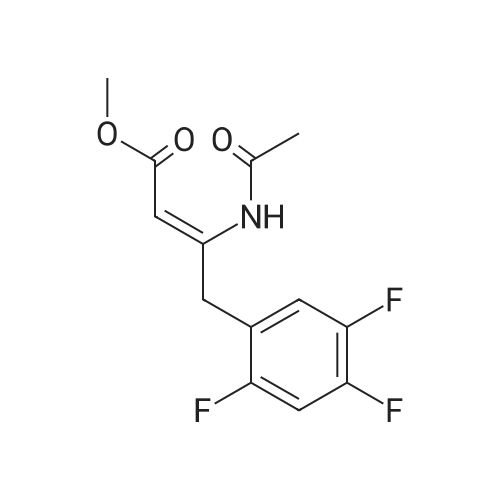
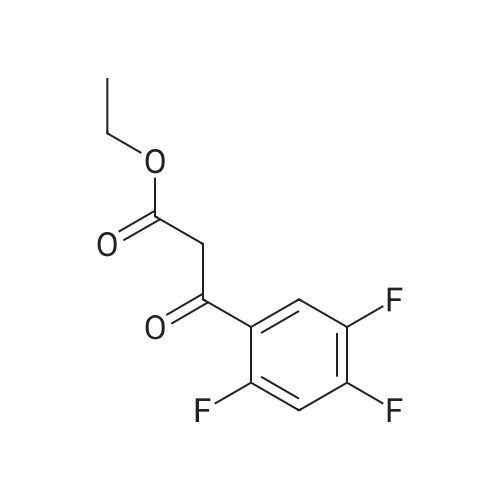
| 91% |
Stage #1: With iodine; magnesium In tetrahydrofuran at 45 - 50℃; Inert atmosphere
Stage #2: With copper(l) iodide In tetrahydrofuran at -20 - 5℃; Inert atmosphere |
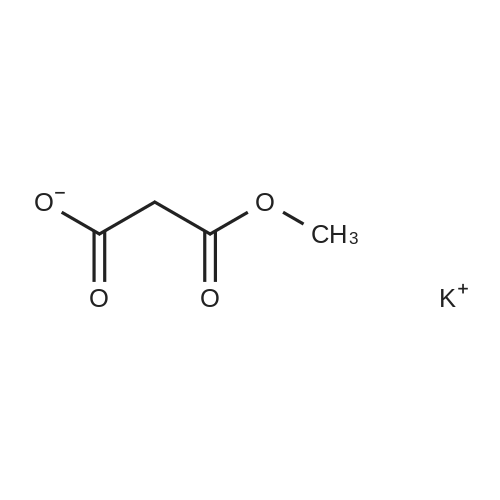
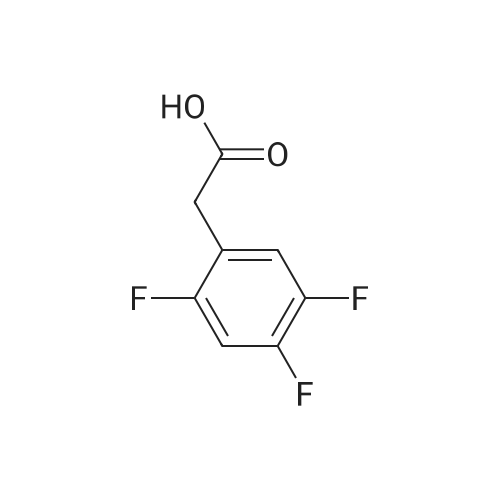
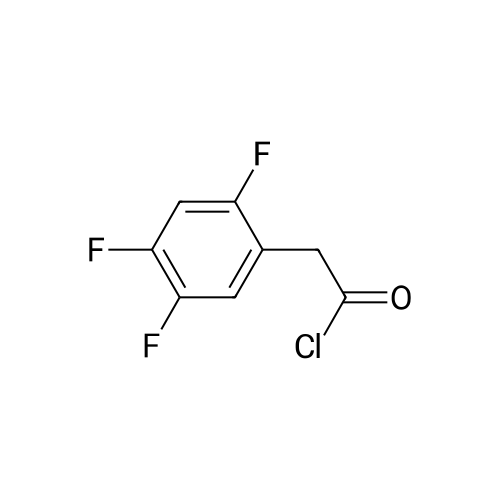
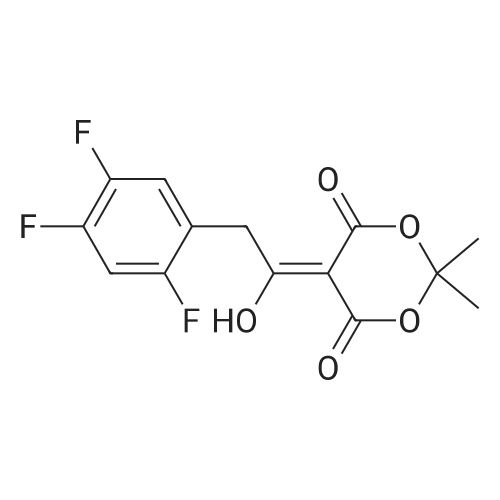
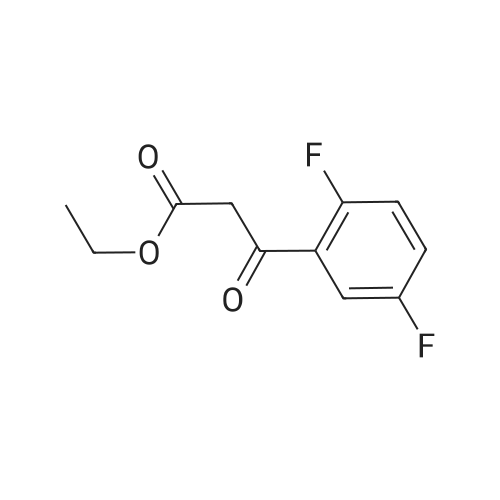
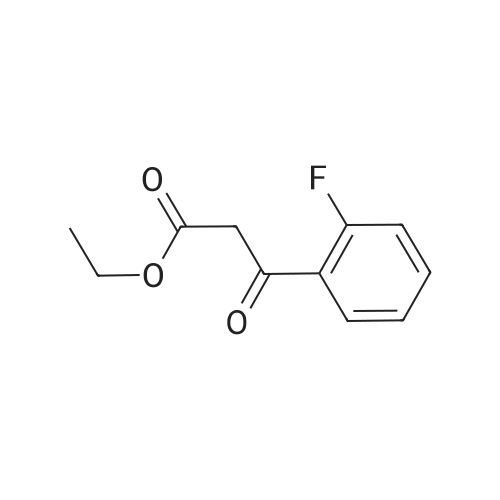
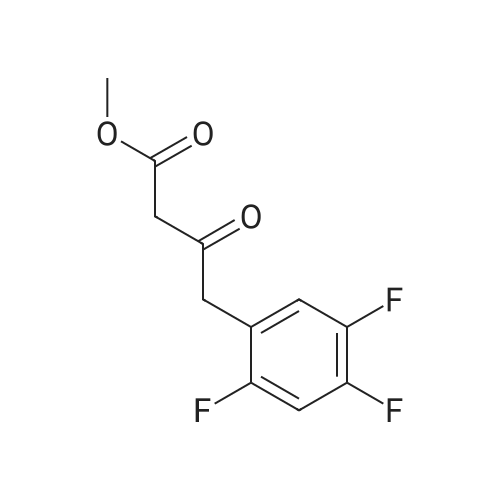
Synthesis of Grignard reagent (1) Mix 50 g of methyl bromoacetate with 200 ml of tetrahydrofuran and stir until clear. spare.(2) In a 1 L glass flask, 8.6 g of magnesium turnings and 200 ml of tetrahydrofuran are sequentially added, and the mixture is stirred thoroughly to start mixing;The flask was filled with nitrogen gas was isolated, 10ml of methyl bromoacetate tetrahydrofuran solution into the flask, heatWarmed to 45-50°C ; to reach the specified temperature, stop stirring, adding 0.5g elemental iodine, triggering the reaction, the bottom of the system gasBubble overflow. To be a large number of bubbles overflow, stir slowly start to stir the system from dark red to colorless and transparent, the systemTemperature is significantly increased. After the initiation of normal speed recovery, and began dropping the remaining methyl bromoacetate tetrahydrofuran solution, bodyThe temperature is maintained at a slight reflux (about 40-45 ° C) for about 1-1.5 hours and the addition is complete. After the drop was completed micro-reflux reversedShould be 2 hours, End of the reaction to give a Grignard reagent (2-2), spare. Synthesis of methyl 2,4,5-trifluorophenylacetoacetate (1) The prepared grignard reagent (2-2) was cooled to 0-5 ° C, 12.4g of cuprous iodide was added to the flask, and further cooled to -20--15 ° C.(2) 75 g of 2- (2,4,5-trifluorophenyl) acetyl chloride was mixed in 200 ml of tetrahydrofuran for use.(3) After reaching the specified temperature,A solution of 2- (2,4,5-trifluorophenyl) acetyl chloride in tetrahydrofuran was slowly added dropwise and the temperature was raised well before the dropwise addition. The reaction was controlled at -15 - -20 ° C. Did not increase the latter part of the heating temperature was about 2-2.5 hours was added dropwise, the solution is yellow-green. After the addition was completed, the mixture was stirred for 2 hours. Stop cooling, naturally warmed to room temperature, stirring was continued for 12 hours. Remove nitrogen protection,Open the stopper, the flask was added 70ml concentrated hydrochloric acid and 210ml of pure water mixed solution,System temperature has significantly increased and the bubble overflow, appropriate heating up to 30-40°C ,Fully stirred (about 3h) to the system the remaining consumption of magnesium shaving to give a yellow clear solution.Stop stirring, static stratification, collecting the upper organic phase,The aqueous phase was extracted twice more with 200 ml of ethyl acetate, the organic phases were combined,The solvent was distilled off under reduced pressure at 55-60 ° C. Fast dry, add the right amount of anhydrous ethanol with dry,Concentration will be iodine was brought out, the color of concentrated solution.300ml absolute ethanol was drawn into the rotary flask, mixed well, drawn into a 2L reactor,Open heat to 50-60°C , incubated for 30 minutes. Stop heating900ml of pure water was poured into the reaction vessel, and the system was dispersed for about 10 minutes.Stir for 30 minutes. Filter, filter cake washed with pure water. Drum air at 55-60 ° C for 24 hours.60.7 g of a white solid was obtained. Yield: 91percent, purity: 99.1percent. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping