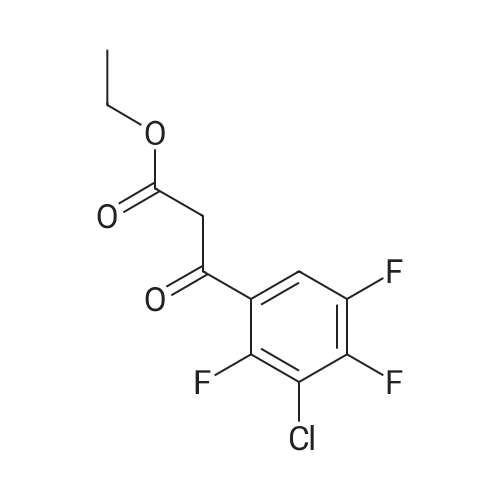
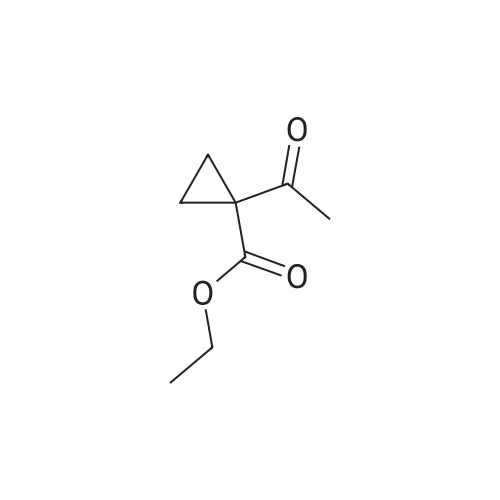
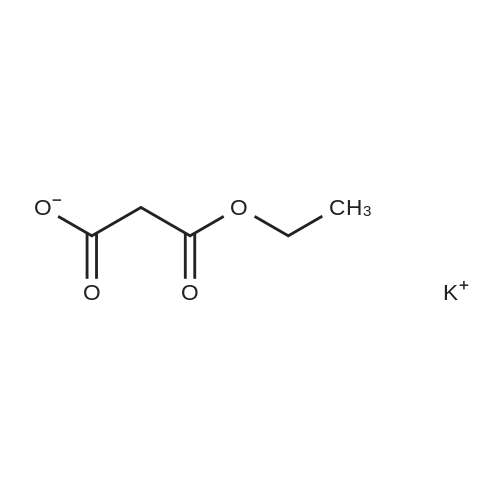
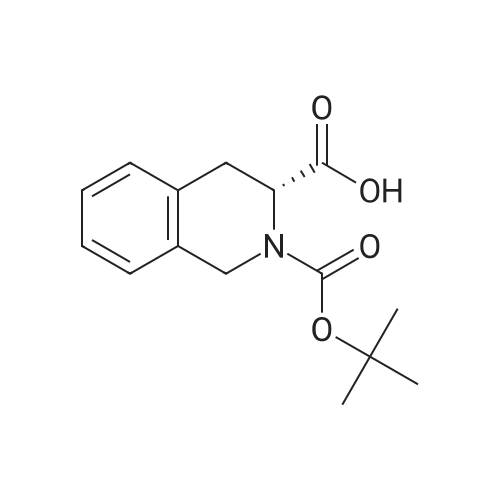
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
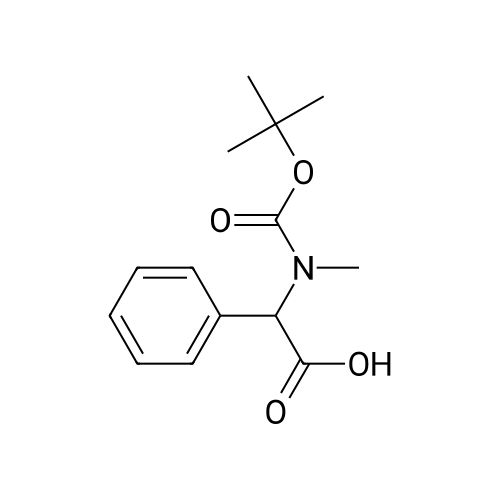
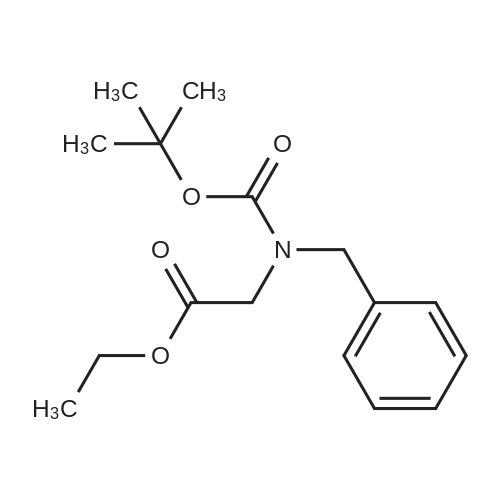
1OA. (Benzyl-tert-butoxycarbonyl-amino^acetic acid: To 2- (benzylamino)acetic acid, HCl (3 g, 14.88 mmol) in DCM (40 mL) was added di-tert- butyl dicarbonate (3.25 g, 14.88 mmol) and TEA (6.22 mL, 44.6 mmol) and stirred for 6 days. The reaction was partitioned with DCM/dil HCl and extracted with DCM (3 x 20 mL). Combined organic layers were washed with water (20 mL), brine (20 mL) and dried (MgSO4) to afford 1OA and containing some triethylamine hydrochloride impurity (5g, 125percent) as a clear colorless oil. 1H NMR (400 MHz,CDCl3) δ 1.38 - 1.46 (d, 9 H), 3.71 (s, 1 H), 3.86 (s, 1 H), 4.54 (d, J=12.38 Hz, 2 H),7.07 - 7.37 (m, 5 H). Used without further purification.
100%
With sodium hydroxide In 1,4-dioxane; water at 0 - 20℃;
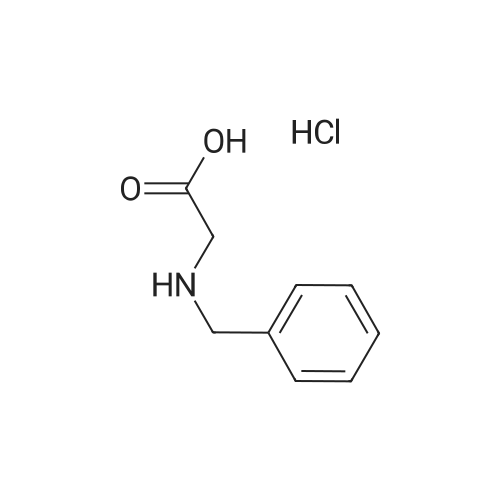
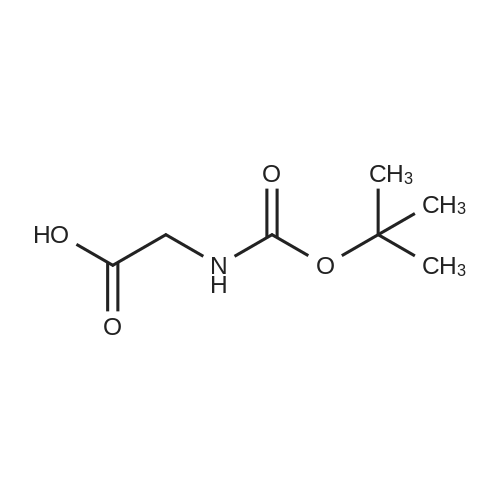
To a stirred solution of N-benzylglycine hydrochloride (402 mg, 2 mmol) in a H2O/dioxane mixture (1/4, 5 mL) were added, at 0 °C, Boc2O (524 mg, 2.4 mmol) and 2 mL of 2 N NaOH solution. The mixture was stirred overnight at room temperature and dioxane was evaporated. The aqueous layer was acidified with a 20percent citric acid solution and extracted by AcOEt. The organic layer was dried over MgSO4 and evaporated to give (Benzyl-tert-butoxycarbonyl-amino)-acetic acid(26a) as a colourless oil. Yield 100percent. 1H NMR (DMSO-d6) δ ppm: 7.23-7.35 (m, 5H), 4.4 (s, 1.1H), 4.38 (s, 0.9H), 3.83 (s, 0.9H), 3.74 (s, 1.1H), 1.38 (s, 1.8H), 1.34 (s, 9.9H); tR,LCMS = 5.4 min, Purity 99percent; MS (ESI-): m/z = 264 (M-H)-.
Reference:
[1] Patent: WO2008/76805, 2008, A2, . Location in patent: Page/Page column 127
[2] European Journal of Medicinal Chemistry, 2015, vol. 90, p. 547 - 567
[3] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1997, # 11, p. 1681 - 1690
[4] Patent: WO2007/58862, 2007, A2, . Location in patent: Page/Page column 65
3
[ 24424-99-5 ]
[ 17136-36-6 ]
[ 76315-01-0 ]
Yield
Reaction Conditions
Operation in experiment
40.70 g
for 6.5 h;
To a suspension of /V-benzylglycine (24.3 g, 147 mmol) in THF:water (1 :1 , 500 mL) was added Boc-anhydride (33.7 g, 154 mmol). After 6.5 h the mixture was diluted with TBME (250 mL) and citric acid (33 g) was added until pH = 4. After 10 min of stirring, the phases were separated and the organic phase was washed with brine (250 ml). The aqueous layer was washed with TBME (2x100 ml) and the combined organic phases were dried over Na2S04, filtered then concentrated in vacuo (45°C), affording the title compound (40.70 g) as a colorless oil, which began to crystallize upon standing. HPLC: 99.7percent by UV, LCMS: Rt = 0.94 min, m/z = 264.3 (M-H), Method LCMS_ 2_MIN_FINAL_ANALYSIS. 1H NMR (600 MHz, DMSO-c/6)* δ 12.62 (br s, 1 H), 7.44-7.09 (m, 5 H), 4.41 (d, J = 8.1 Hz, 2 H) 1 .44-1 .24 (m, 9 H) 3.89-3.67 (m, 2 H). *As a mixture with 0(Boc)2 (ca. 9percent).
Reference:
[1] Journal of the American Chemical Society, 2003, vol. 125, # 35, p. 10664 - 10671
[2] Polish Journal of Chemistry, 2002, vol. 76, # 6, p. 823 - 830
[3] Synthetic Communications, 1994, vol. 24, # 17, p. 2429 - 2435
[4] Acta Crystallographica Section C: Crystal Structure Communications, 1998, vol. 54, # 8, p. 1164 - 1165
[5] Patent: US2010/16316, 2010, A1, . Location in patent: Page/Page column 63
[6] Patent: WO2018/60926, 2018, A1, . Location in patent: Paragraph 00203
4
[ 136159-62-1 ]
[ 76315-01-0 ]
Reference:
[1] Journal of Medicinal Chemistry, 1996, vol. 39, # 7, p. 1472 - 1484
[2] Journal of Medicinal Chemistry, 2005, vol. 48, # 6, p. 1768 - 1780
[3] Tetrahedron, 2010, vol. 66, # 21, p. 3723 - 3729
5
[ 4530-20-5 ]
[ 100-39-0 ]
[ 76315-01-0 ]
Yield
Reaction Conditions
Operation in experiment
0.58% w/w
With hydrogenchloride; NaH In tetrahydrofuran; water
a) tert-Butyl N-(2-hydroxy-2-oxoethyl)-N-(phenylmethyl)carbamate To a cold (-20°) suspension of 60percent NaH (120 g, 3.00 mol) in THF (700 mL) was added (30 min) a solution of Boc-glycine (175 g, 1.00 mol) in THF (300 mL). Thereafter, benzyl bromide was added to the mixture. After 15 min, the cooling bath was removed. The reaction mixture was stirred at room temperature for 1 h and then heated at reflux for 16 h. The reaction mixture was cooled to 0°. H2O (~50 mL) was added dropwise over a 30 min period. After another 30 min, H2O (600 mL) was added. The mixture was washed with hexane (3*500 mL). 1N Aqueous HCl (1.3 L) was added slowly to the aqueous layer, followed by the addition of 4N aqueous HCl (300 mL). The aqueous solution was extracted with EtOAc (1.5 L) and then CH2C12 (3*500 mL). The combined organic layers were serially washed with H2O and brine, dried (MgSO4, Na2SO4, charcoal), filtered through diatomaceous earth and concentrated under reduced pressure to give the title compound (241 g, 91percent yield) as a pale yellow solid: Mp 94-97°; 1H NMR (400 MHz, DMSO-d6) δ10.5 (broad s, 1H), 7.23-7.33 (m, 5H), 4.40 (s, 2H), 3.80, 3.72 (2s, 2H), 1.38, 1.35 (2s, 9H); MS (FAB) m/z 266 (MH)+; Anal. Calcd for C14H19NO4 (and accounting for water content, 0.58percent w/w as determined by Karl Fisher analysis): C, 63.01; H, 7.26; N, 5.25. Found: C, 62.79; H, 7.14; N, 5.13.
Reference:
[1] Polish Journal of Chemistry, 1994, vol. 68, # 5, p. 1047 - 1054
[2] Patent: US6288091, 2001, B1,
6
[ 1070-19-5 ]
[ 7689-50-1 ]
[ 76315-01-0 ]
Yield
Reaction Conditions
Operation in experiment
86%
With sodium hydrogencarbonate In 1,4-dioxane; water; acetic acid
Method B N-Benzylglycine hydrochloride (7.38 g, 36.6 mmol), tert-butyl azidoformate (10.7 mL, 73.2 mmol) and sodium bicarbonate (12.3 g) were reacted in 150 mL dioxane/H2 O-1:1 (v/v) at 45° (24 hr) and then at room temperature (24 h). The reaction suspension was evaporated to dryness and the resulting white residue was dissolved in 200 mL glacial acetic acid/H2 O--1:1(v/v) with cooling to 4°. Water (800 ml) was slowly added with rapid stirring and cooling. The resulting turbid solution was seeded with Boc-N-benzyl-glycine obtained from Method A and allowed to stand in an ice bath for 30 min. The resulting material was gathered by filtration, washed with water followed by petroleum ether (bp 30°-75°) and dried to give 8.34 g (86percent) of crude product as a white solid. Recrystallization from dichloromethane petroleum ether (bp 30°-75°) gave 7.34 g (76percent) of white, crystalline product identical to Boc-N-benzylglycine (Method A) by NMR and melting point (106°).
Reference:
[1] Patent: US5527882, 1996, A,
7
[ 58632-95-4 ]
[ 7689-50-1 ]
[ 76315-01-0 ]
Reference:
[1] Journal of Medicinal Chemistry, 1983, vol. 26, # 2, p. 167 - 174
8
[ 4530-20-5 ]
[ 7732-18-5 ]
[ 100-39-0 ]
[ 76315-01-0 ]
Yield
Reaction Conditions
Operation in experiment
0.58% w/w
With hydrogenchloride; NaH In tetrahydrofuran
a) tert-Butyl N-(2-hydroxy-2-oxoethyl)-N-(phenylmethyl)carbamate
To a cold (-20°) suspension of 60percent NaH (120 g, 3.00 mol) in THF (700 mL) was added (30 min) a solution of Boc-glycine (175 g, 1.00 mol) in THF (300 mL). Thereafter, benzyl bromide was added to the mixture. After 15 min, the cooling bath was removed. The reaction mixture was stirred at room temperature for 1 h and then heated at reflux for 16 h. The reaction mixture was cooled to 0°. H2 O (~50 mL) was added dropwise over a 30 min period. After another 30 min, H2 O (600 mL) was added. The mixture was washed with hexane (3*500 mL). 1N Aqueous HCl (1.3 L) was added slowly to the aqueous layer, followed by the addition of 4N aqueous HCl (300 mL). The aqueous solution was extracted with EtOAc (1.5 L) and then CH2 Cl2 (3*500 mL). The combined organic layers were serially washed with H2 O and brine, dried (MgSO4, Na2 SO4, charcoal), filtered through diatomaceous earth and concentrated under reduced pressure to give the title compound (241 g, 91percent yield) as a pale yellow solid: Mp 94-97°; 1 H NM (400 MHz, DMSO-d6) δ10.5 (broad s, 1H), 7.23-7.33 (m, 5H), 4.40 (s, 2H), 3.80, 3.72 (2s, 2H), 1.38, 1.35 (2s, 9H); MS (FAB) m/z 266 (MH)+; Anal. Calcd for C14 H19 NO4 (and accounting for water content, 0.58percent w/w as determined by Karl Fisher analysis): C, 63.01; H, 7.26; N, 5.25. Found: C, 62.79; H, 7.14; N, 5.13.
1OA. (Benzyl-tert-butoxycarbonyl-amino^acetic acid: To 2- (benzylamino)acetic acid, HCl (3 g, 14.88 mmol) in DCM (40 mL) was added di-tert- butyl dicarbonate (3.25 g, 14.88 mmol) and TEA (6.22 mL, 44.6 mmol) and stirred for 6 days. The reaction was partitioned with DCM/dil HCl and extracted with DCM (3 x 20 mL). Combined organic layers were washed with water (20 mL), brine (20 mL) and dried (MgSO4) to afford 1OA and containing some triethylamine hydrochloride impurity (5g, 125%) as a clear colorless oil. 1H NMR (400 MHz,CDCl3) delta 1.38 - 1.46 (d, 9 H), 3.71 (s, 1 H), 3.86 (s, 1 H), 4.54 (d, J=12.38 Hz, 2 H),7.07 - 7.37 (m, 5 H). Used without further purification.
100%
With sodium hydroxide; In 1,4-dioxane; water; at 0 - 20℃;
To a stirred solution of <strong>[7689-50-1]N-benzylglycine hydrochloride</strong> (402 mg, 2 mmol) in a H2O/dioxane mixture (1/4, 5 mL) were added, at 0 C, Boc2O (524 mg, 2.4 mmol) and 2 mL of 2 N NaOH solution. The mixture was stirred overnight at room temperature and dioxane was evaporated. The aqueous layer was acidified with a 20% citric acid solution and extracted by AcOEt. The organic layer was dried over MgSO4 and evaporated to give (Benzyl-tert-butoxycarbonyl-amino)-acetic acid(26a) as a colourless oil. Yield 100%. 1H NMR (DMSO-d6) delta ppm: 7.23-7.35 (m, 5H), 4.4 (s, 1.1H), 4.38 (s, 0.9H), 3.83 (s, 0.9H), 3.74 (s, 1.1H), 1.38 (s, 1.8H), 1.34 (s, 9.9H); tR,LCMS = 5.4 min, Purity 99%; MS (ESI-): m/z = 264 (M-H)-.
With sodium hydroxide; In 1,4-dioxane; water; at 20℃;
EXAMPLE 47; N- {(1.5,25)- 1 -Benzyl-2-hydroxy-2-[(45)- 1 ,2,2-trimethyl-5-oxoimidazolidin-4-yl]ethyl} -N-[( IR)-I -(4- fluorophenyl)ethyl]-5-[methyl(methylsulfonyl)amino]isophthalamide; Step 1; N-Benzyl-N-(fer/-butoxycarbonyl)glycme; To a solution of <strong>[7689-50-1]N-benzylglycine hydrochloride</strong> (5.00 g, 24.80 mmol) in dioxane (100 mL) and water (25 mL) were added di-tert-butyldicarbonate (5.95 g, 27.28 mmol) and aqueous IN NaOH solution (50 mL), and the reaction was stirred at rt overnight. The solvents were removed in vacuo, and the resulting residue was partitioned between EtOAc and aqueous 10% KHSO4 solution. The layers were separated and the aqueous was extracted twice more with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to afford the product as a white solid. lH NMR (400 MHz, CDCl3) delta 7.36-7.22 (m, 5H), 4.53 (d, J= 14.4 Hz, 2H), 3.96 (s, IH), 3.83 (s, IH), 1.48 (s, 9H); ES MS [M-IOO (loss of BOC)+1] = 166.
(S)-2-[2-({(S)-2-[2-(Benzyl-tert-butoxycarbonyl-amino)-acetylamino]-3-methyl-pentyl}-naphthalen-1-ylmethyl-amino)-acetylamino]-4-methylsulfanyl-butyric acid[ No CAS ]
<i>N</i>4-benzyl-<i>N</i>1-(1-cyclohexylmethyl-2,3-dihydroxy-5-methyl-hexyl)-2-cyclopropylmethyl-<i>N</i>4-dimethylcarbamoylmethyl-succinamide[ No CAS ]
2-(2-amino-thiazol-4-ylmethyl)-<i>N</i>4-benzyl-<i>N</i>1-(1-cyclohexylmethyl-2,3-dihydroxy-5-methyl-hexyl)-<i>N</i>4-dimethylcarbamoylmethyl-succinamide[ No CAS ]
<i>N</i>4-benzyl-<i>N</i>1-(1-cyclohexylmethyl-2,3-dihydroxy-5-methyl-hexyl)-2-cyclopropylmethyl-<i>N</i>4-(2-morpholin-4-yl-2-oxo-ethyl)-succinamide[ No CAS ]
<i>N</i>4-benzyl-<i>N</i>1-(1-cyclohexylmethyl-2,3-dihydroxy-5-methyl-hexyl)-2-cyclopropylmethyl-<i>N</i>4-[methyl-(2-pyridin-2-yl-ethyl)-carbamoyl]-methyl}-succinamide[ No CAS ]
With sodium hydrogencarbonate; In 1,4-dioxane; water; acetic acid;
Method B N-Benzylglycine hydrochloride (7.38 g, 36.6 mmol), tert-butyl azidoformate (10.7 mL, 73.2 mmol) and sodium bicarbonate (12.3 g) were reacted in 150 mL dioxane/H2 O-1:1 (v/v) at 45 (24 hr) and then at room temperature (24 h). The reaction suspension was evaporated to dryness and the resulting white residue was dissolved in 200 mL glacial acetic acid/H2 O--1:1(v/v) with cooling to 4. Water (800 ml) was slowly added with rapid stirring and cooling. The resulting turbid solution was seeded with Boc-N-benzyl-glycine obtained from Method A and allowed to stand in an ice bath for 30 min. The resulting material was gathered by filtration, washed with water followed by petroleum ether (bp 30-75) and dried to give 8.34 g (86%) of crude product as a white solid. Recrystallization from dichloromethane petroleum ether (bp 30-75) gave 7.34 g (76%) of white, crystalline product identical to Boc-N-benzylglycine (Method A) by NMR and melting point (106).
(5S,10S,11S,14S)-11-benzyl-5,14-di-tert-butyl-3,6,13,16-tetraoxo-8-(4-(pyridin-2-yl)benzyl)-2,17-dioxa-4,7,8,12,15-pentaazaoctadecan-10-yl 2-(benzyl(tert-butoxycarbonyl)amino)acetate[ No CAS ]






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping