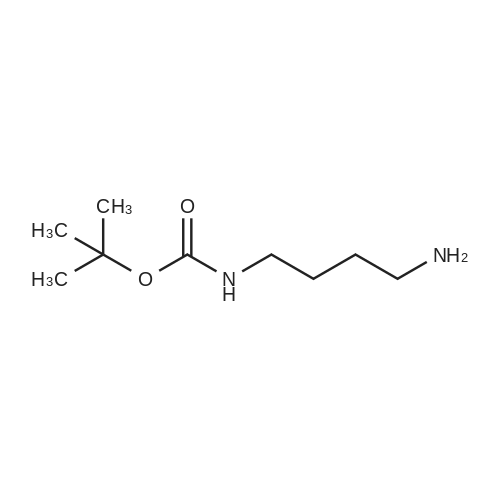
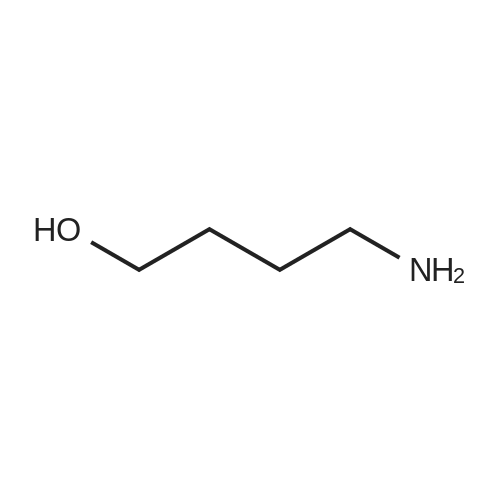
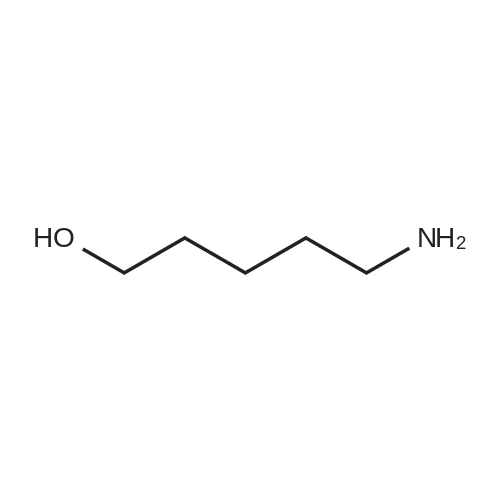
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Chemical Research, Miniprint, 1996, # 8, p. 2143 - 2161
2
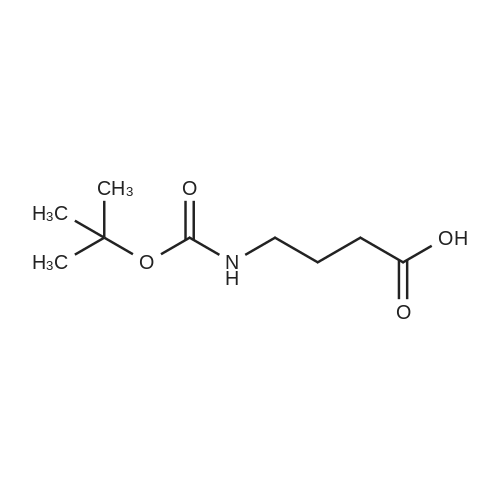
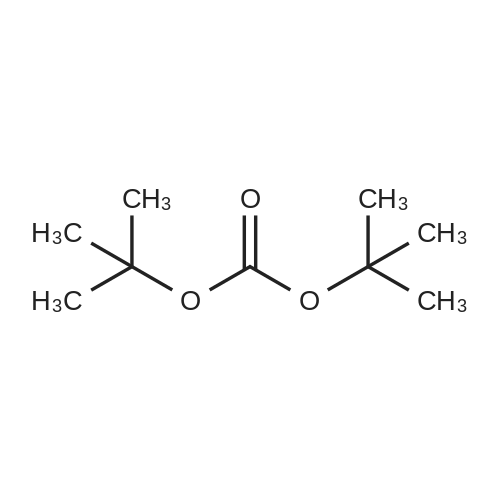
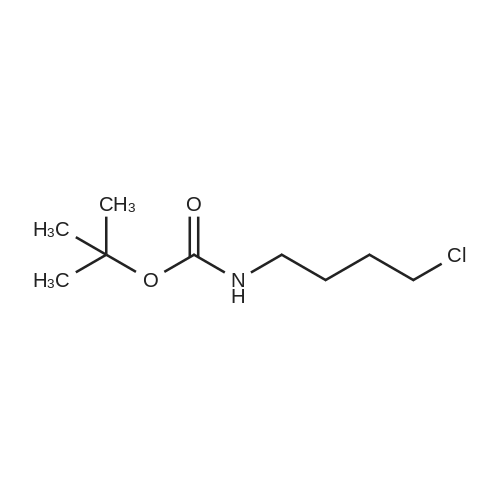
[ 13325-10-5 ]
[ 24424-99-5 ]
[ 75178-87-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
With triethylamine In acetonitrile at 0℃; for 6.5 h; Inert atmosphere
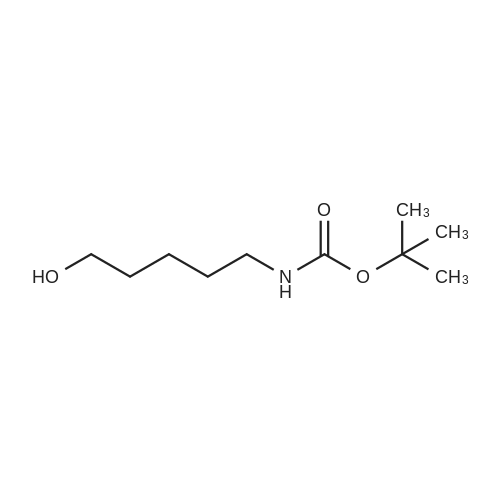
4-amino-1-butanol (0.5 g, 5.61 mmol) was dissolved in acetonitrile (56 ml). The solution was cooled to 0°C and Et3N (1.56 ml, 11.22 mmol, 2 eq.) was added followed by Boc2O (1.346 g, 6.17 mmol). The reaction was followed by TLC and quenched with water (20 ml) after 6.5 h. The solution was extracted with EtOAc (3x30ml), brine was used to help phase separation. The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel (CH2Cl2/MeOH, 95:5) to give the title compound as a colorless oil (1.06 g, quantitative yield)
100%
With triethylamine In methanol at 0 - 20℃;
To the solution of 4-aminobutanol (100 μL) in 2 mL triethylamine/methanol (Et3N/MeOH, v/v, 1:7) di-tert-butyl dicarbonate (278 μL) in 2.5 mL methanol was added dropwise at 0 °C. The solution was stirred at the same temperature overnight and allowed to warm to room temperature during 2 h and kept at ambient temperature for another 6 h. Upon completion of the reaction monitored by TLC visualized by iodine and ninhydin, excess Boc2O and solvent were removed under vacuum. The residue was partitioned between dichloromethane (CH2Cl2) and brine and extracted with CH2Cl2 (3 * 50 mL). The combined organic layers were washed by brine, then dried over MgSO4, filtered and concentrated in vacuo to provide tert-butyl N-(4-hydroxybutyl)carbamate 2 as colorless oil (152 mg, 100percent). 1H NMR (300 MHz, CDCl3) δ 5.02 (s, 1H), 3.63 (t, J = 6.1 Hz, 2H), 3.40 (s, 1H), 3.13 (d, J = 5.9 Hz, 2H), 1.56 (dd, J = 7.2, 4.1 Hz, 4H), 1.44 (s, 9H).
100%
at 20℃; for 3 h;
Butanolamine (5g, 56.09 mmol, 1.0 eq) is introduced in a round-bottomed flask with THF (20 mL) before dropwise addition of di-tert-butylcarbonate (12 mL, 56.09 mmol, 1.0 eq) in THF (10 mL). The reaction mixture is stirred at room temperature for 3 h. After concentration under vacuum, the desired product is obtained as an amorph solid that crystalize upon standing (10 g, quantitative yield).
100%
at 20℃; for 3 h;
To a solution of 4-amino-1 -butanol (2.0 g, 22.5 mmole) in THF at RT was added Boc anhydride (4.90 g, 22.5 mmole). After 3 h, the reaction solution was concentrated under vacuum and the residue purified on silica gel (hexanes/EtOAc, 1 :1 ) to give the title compound (quant.) as a white solid: LCMS (ES) m/z = 190 (M+H)+
96%
With sodium hydroxide In 1,4-dioxane; water at 20℃; for 12 h;
General procedure: To a solution of 2a (or 2b–e, 20 mmol) in a mixture of dioxane (15 mL) and H2O (7 mL), wasadded 5N NaOH (4.8 mL) and a solution of Boc2O (5.0 g, 23 mmol) in dioxane at 0 °C. After stirred atrt overnight, the reaction mixture was concentrated in vacuo. The residue was extracted from 10percentcitric acid with AcOEt, and dried over anhydrous Na2SO4. Evaporation of the solvents gave the pureproduct 3a–e. 3a was obtained as a colorless oil (2.67 g, 83percent).
90.2%
at 0℃; for 4 h;
General procedure: tert-butyl 3-hydroxypropylcarbamate 13a: Boc anhydride(319.2g, 1.46 mol) was added drop wise to 3-aminopropan-1-ol (100.0g, 1.33mol) in methanol ( 1000ml )at 0 °C. Reaction mixture was allowed to stir for 4hr. Reaction mixture was concentrated and diluted with water(1000ml) and ethylacetate (1000ml). Layers were separated.Organic layer was washed with water (250ml), dried oversodium sulfate and concentrated. 202.3g of clear liquid was obtained with 87percent yield. tert-butyl 4-hydroxybutylcarbamate 13b: Followingthe same procedure for 13a, 191.5g of clear liquid with90.2percent yield was obtained. 1H NMR (CDCl3) = 1.44 (s, 9H), 1.48(s, 1 H), 1.52-1.59 (m, 4 H), 3.12-3.16 (m, 2H,),3.66-3.68 (m, 2 H), 4.60-4.69 (br, s, 1 H), 13C NMR (CDCl3)= 26.61, 28.44, 29.74, 40.34, 62.31, 79.21, 156.22.
90.5%
With triethylamine In dichloromethane at 20℃; for 12 h;
4-amino-1-butanol (40) (2.97 g) and triethylamine (5 mL) were added to dichloromethane (60ML), di-tert-butyl dicarbonate (6.61 g) was added under ice-cooling, and the mixture was stirred for 5 minutes. reactionThe solution was returned to room temperature and stirred for 12 hours.Under ice cooling, a 1 N hydrochloric acid aqueous solution was added to the reaction solution and further diluted with dichloromethane. After separating the organic layer, the obtained organic layer was washed with saturated brine and dried with anhydrous sodium sulfate. Filtration and concentration under reduced pressure gave the title compound (41) (5.71 g, yield 90.5percent) as a pale yellow oil.
89%
at 20℃; for 1.5 h;
N-tert-Butoxycarbonyl-4-amino-1-butanol 4-Amino-1-butanol (1.0 g, 11.22 mmol) was dissolved in methanol (10 ml). To this solution, di-tert-butyl carbonate (2.53 g, 11.58 mmol) was added, and the mixture was stirred at room temperature for 1.5 hours. After the reaction was confirmed by TLC to be complete, the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (hexane:acetone=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-butanol (1.88 g, yield: 89percent).
88%
With triethylamine In dichloromethane at 20℃; for 2 h;
To a mixture of 4-aminobutan-l-ol (4.0 g, 45 mmol) and TEA (7.5 mL, 54 mmol) in DCM (200 ml) was added (Boc)20 (10.2 g, 47.2 mmol). The reaction mixture was stirred for 2 h at room temperature and then washed with water (2x150 mL) and the solution of citric acid (100 mL). The organic layer was dried over anhydrous Na2S04 and concentrated to give the product as yellow oil 7.5 g. (yield 88percent).
82%
With sodium hydroxide In 1,4-dioxane; water at 0 - 20℃; for 1.5 h;
4-Aminobutan-1-ol (1.0 g, 11.2 mmol) was suspended in a mixture of dioxane (20 ml), H2O (10 ml) and 1 M NaOH (10 ml) and cooled to 0 °C (ice-water bath) with stirring. Boc2O (2.69 g, 12.34 mmol) was added and stirring was continued at ambient temperature. After 5.5 h additional Boc2O (1.08 g, 4.95 mmol) was added and stirring was continued overnight. Next day additional Boc2O (1.35 g, 6.19 mmol) was added to the suspension. After 1.5 h sulfate buffer (40 ml) was added and the reaction mixture was transferred to a separating funnel and extracted with EtOAc (3 × 30 ml). The combined organic layers were washed with NaHCO3(90 ml) and brine (90 ml), dried (Na2SO4), filtered and concentrated in vacuo. Purification by flash column chromatography (60 percent EtOAc in n-heptane, v/v) gave carbamate 4 (1.62 g, 82percent) as a colourless oil.
54%
With N-ethyl-N,N-diisopropylamine In 1,2-dichloro-ethane at 20℃; for 24 h; Inert atmosphere
A mixture of 4-aminobutan-1-ol (10.0 g, 112.1 mmol, 10.4 mL, 1.0 eq), tert- butoxycarbonyl tert-butyl carbonate (25.7 g, 117.8 mmol, 27.1 mL, 1.05 eq) and DIEA (21.7 g, 168.2 mmol, 29.3 mL, 1.5 eq) in DCE (400 mL) was stirred at 20 °C for 24 h, diluted with water (400 mL) and extracted with DCM (100 mL*3). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2) to give compound 07-4-1 (12.0 g, 60.2 mmol, 54percent yield).1H NMR (CDCl3, 400 MHz): δ 3.67 (d, J=4.85 Hz, 2 H), 3.16 (d, J=5.29 Hz, 2 H), 1.54 - 1.62 (m, 4 H), 1.44 (s, 9 H).
7.54 g
at 20℃; for 24 h; Cooling with ice
Reference Example 35 tert-Butyl 4-hydroxybutylcarbamate (0486) (0487) To a mixture of 4-aminobutanol (3.57 g) and ethyl acetate (9 mL) was dropwise added a mixture of di-tert-butyl dicarbonate (8.73 g) and ethyl acetate (1 mL) under ice-cooling. After stirring at room temperature for 24 hrs., the mixture was concentrated under reduced pressure. The residue was dissolved in ethyl acetate (200 mL), and the mixture was washed with water (50 mL), 1N hydrochloric acid (40 mL), water (30 mL) and saturated brine (30 mL) and dried over anhydrous magnesium sulfate. Concentration under reduced pressure gave the title compound (7.54 g) as a colorless oil. (0488) 1H-NMR(CDCl3): 1.44 (9H, s), 1.47-1.61 (4H, m), 3.07-3.22 (2H, m), 3.61-3.76 (2H, m), 4.62 (1H, bs).
Reference:
[1] Organic Letters, 2009, vol. 11, # 9, p. 2019 - 2022
[2] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 6, p. 2536 - 2543
[3] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 2, p. 1151 - 1155
[4] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 2, p. 191 - 200
[5] Inorganica Chimica Acta, 2016, vol. 452, p. 152 - 158
[6] Patent: WO2006/113837, 2006, A2, . Location in patent: Page/Page column 78
[7] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 8, p. 803 - 808
[8] Chemistry - A European Journal, 2016, vol. 22, # 9, p. 3009 - 3018
[9] Synthesis, 1990, p. 366 - 368
[10] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 23, p. 7050 - 7053
[11] Synthesis, 2011, # 24, p. 3991 - 3996
[12] Tetrahedron Letters, 2008, vol. 49, # 29-30, p. 4491 - 4493
[13] Journal of Medicinal Chemistry, 2009, vol. 52, # 22, p. 7029 - 7043
[14] Tetrahedron Letters, 1998, vol. 39, # 32, p. 5697 - 5700
[15] Bioorganic Chemistry, 2002, vol. 30, # 2, p. 81 - 94
[16] Angewandte Chemie - International Edition, 2012, vol. 51, # 32, p. 8110 - 8113
[17] Molecules, 2013, vol. 18, # 11, p. 13957 - 13978
[18] Journal of Medicinal Chemistry, 2014, vol. 57, # 11, p. 4924 - 4939
[19] Journal of Medicinal Chemistry, 2015, vol. 58, # 13, p. 5287 - 5307
[20] Journal of Medicinal Chemistry, 2006, vol. 49, # 14, p. 4183 - 4195
[21] Farmaco, 1991, vol. 46, # 12, p. 1517 - 1529
[22] Letters in Organic Chemistry, 2013, vol. 10, # 7, p. 518 - 522
[23] Patent: JP2017/71567, 2017, A, . Location in patent: Paragraph 0208-0210
[24] Patent: US2015/353489, 2015, A1, . Location in patent: Paragraph 0070; 0181; 0182
[25] Patent: WO2012/33858, 2012, A2, . Location in patent: Page/Page column 85
[26] Journal of Organic Chemistry, 2010, vol. 75, # 2, p. 518 - 521
[27] Helvetica Chimica Acta, 1996, vol. 79, # 8, p. 2137 - 2151
[28] Journal of Medicinal Chemistry, 2015, vol. 58, # 17, p. 6819 - 6843
[29] Beilstein Journal of Organic Chemistry, 2017, vol. 13, p. 644 - 647
[30] Journal of Labelled Compounds and Radiopharmaceuticals, 2007, vol. 50, # 7, p. 666 - 670
[31] Chemical Communications, 2006, # 20, p. 2156 - 2158
[32] Journal of Medicinal Chemistry, 2014, vol. 57, # 22, p. 9673 - 9686
[33] Patent: WO2017/96045, 2017, A1, . Location in patent: Paragraph 00883
[34] Tetrahedron Letters, 1997, vol. 38, # 33, p. 5741 - 5744
[35] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 21, p. 4828 - 4832
[36] Journal of Medicinal Chemistry, 2004, vol. 47, # 24, p. 6055 - 6069
[37] Journal of Medicinal Chemistry, 2005, vol. 48, # 11, p. 3832 - 3839
[38] Journal of the Chemical Society, Perkin Transactions 2, 2002, # 5, p. 923 - 927
[39] Tetrahedron Letters, 2004, vol. 45, # 38, p. 7081 - 7085
[40] Patent: US5965591, 1999, A,
[41] Patent: WO2003/105845, 2003, A1, . Location in patent: Page 88-89
[42] Patent: EP1602362, 2005, A1, . Location in patent: Page/Page column 50
[43] Patent: EP1607088, 2005, A1, . Location in patent: Page/Page column 53
[44] Biomacromolecules, 2012, vol. 13, # 5, p. 1632 - 1641
[45] Angewandte Chemie - International Edition, 2014, vol. 53, # 3, p. 883 - 887[46] Angew. Chem., 2013, vol. 53, # 3, p. 902 - 906
[47] Tetrahedron Letters, 2015, vol. 56, # 51, p. 7108 - 7111
[48] Patent: US2016/128945, 2016, A1, . Location in patent: Paragraph 0486; 0487; 0488
[49] Biomacromolecules, 2016, vol. 17, # 11, p. 3632 - 3639
[50] Journal of Medicinal Chemistry, 2017, vol. 60, # 16, p. 7067 - 7083
[51] Tetrahedron, 2016, vol. 72, # 41, p. 6492 - 6498
[52] Journal of Medicinal Chemistry, 2018, vol. 61, # 11, p. 4961 - 4977
[53] ChemMedChem, 2018, vol. 13, # 18, p. 1957 - 1971
3
[ 85909-08-6 ]
[ 75178-87-9 ]
Reference:
[1] Angewandte Chemie - International Edition, 2009, vol. 48, # 7, p. 1324 - 1327
[2] Chemistry - An Asian Journal, 2018, vol. 13, # 17, p. 2559 - 2565
4
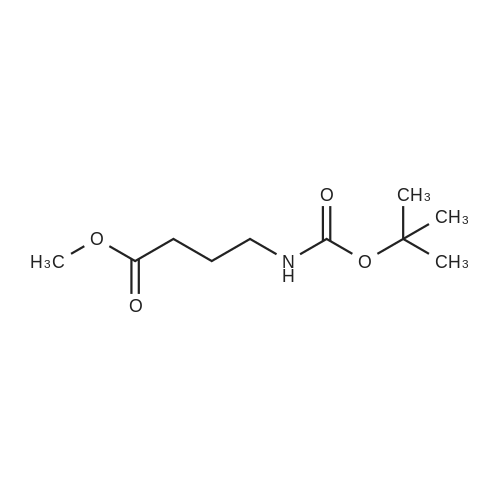
[ 2508-29-4 ]
[ 24424-99-5 ]
[ 75178-87-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 0 - 20℃; for 2.5 h;
The amino alcohol 21b (1.0 equiv, 2.87 mmol, 0.30 mL) was dissolved in 3 mL of CH2Cl2 ([21b] =0.5 mL/L), cooled in an ice bath and Boc2O (1.0 equiv, 2.87 mmol, 0.63 mL) was added. After stirring for 0.5 h at 0 °C, the cooling bath was removed and the mixture was allowed to stir for 2 h at ambient temperature. Subsequently, 3 mL of 1 N HCl solution (aq.) were added, the phases were separated,the organic layer was washed successively with 1 N HCl-, saturated NaHCO3 solution in water and brine/H2O 1:1 (each 3 mL) and dried over MgSO4. After concentration with 2 mL of chloroform under reduced pressure for two times and drying in high vacuum under stirring for 0.5 h delivered the carbamate 12c (2.99 mmol, quant., 567 mg) as a colorless oil.
Reference:
[1] Beilstein Journal of Organic Chemistry, 2014, vol. 10, p. 369 - 383
5
[ 57294-38-9 ]
[ 75178-87-9 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 3, p. 1376 - 1392
[2] Chemical and pharmaceutical bulletin, 1983, vol. 31, # 9, p. 3360 - 3362
[3] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 24, p. 10257 - 10269
6
[ 104700-36-9 ]
[ 75178-87-9 ]
[ 128490-08-4 ]
Reference:
[1] Chemistry Letters, 1986, p. 815 - 818
7
[ 67-56-1 ]
[ 85909-08-6 ]
[ 75178-87-9 ]
[ 85909-04-2 ]
Reference:
[1] Angewandte Chemie - International Edition, 2009, vol. 48, # 7, p. 1324 - 1327
8
[ 85909-08-6 ]
[ 67-63-0 ]
[ 1140511-02-9 ]
[ 75178-87-9 ]
Reference:
[1] Angewandte Chemie - International Edition, 2009, vol. 48, # 7, p. 1324 - 1327
9
[ 13325-10-5 ]
[ 34619-03-9 ]
[ 75178-87-9 ]
Reference:
[1] Journal of the American Chemical Society, 2018, vol. 140, # 20, p. 6278 - 6287
10
[ 24424-99-5 ]
[ 75178-87-9 ]
Reference:
[1] Chemistry - An Asian Journal, 2018, vol. 13, # 17, p. 2559 - 2565
Reference:
[1] Angewandte Chemie - International Edition, 2009, vol. 48, # 7, p. 1324 - 1327
14
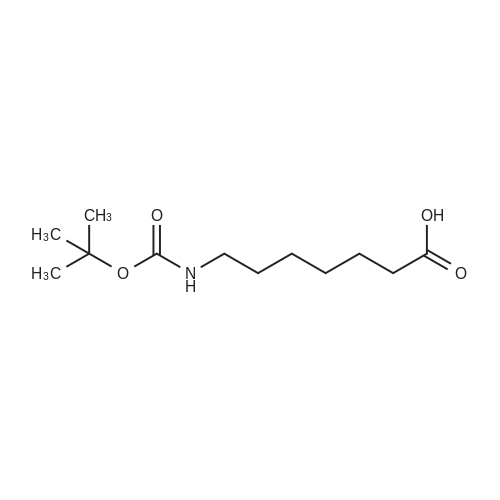
[ 75178-87-9 ]
[ 85909-08-6 ]
Reference:
[1] Chemistry--A European Journal, 2012, vol. 18, # 37, p. 11524 - 11527,4
15
[ 75178-87-9 ]
[ 99207-32-6 ]
Reference:
[1] Patent: JP2017/71567, 2017, A,
16
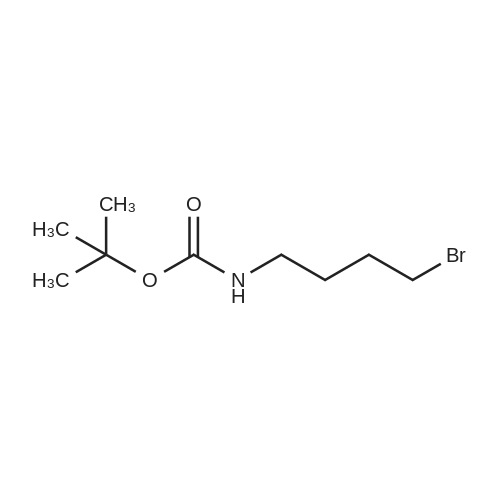
[ 75178-87-9 ]
[ 164365-88-2 ]
Yield
Reaction Conditions
Operation in experiment
100%
With carbon tetrabromide; triphenylphosphine In tetrahydrofuran for 3 h; Inert atmosphere
tert-Butyl (4-hydroxybutyl)carbamate (1.06 g, 5.788 mmol) was dissolved in dry THF (54 ml) followed by addition of Ph3P (2.86 g, 10.92 mmol, 1.9 eq.). Then CBr4 (3.62 g, 10.92 mmol, 1.9 eq.) was slowly added to the mixture. After 3 h the solution was filtered through a celite pad to eliminate the by-products and washed with Et2O. The solvents were removed under high vacuum. The crude product was purified by flash chromatography on silica gel (hexane/EtOAc, 3:1) to give 9 as a white solid at 5°C (1.73 g, quantitative yield)
98%
With carbon tetrabromide; triphenylphosphine In tetrahydrofuran at 0 - 20℃; for 4 h;
To a solution of tert-butyl (4-hydroxybutyl)carbamate (5a) (1.0 g, 5.3 mmol) and PPh3 (2.09 g, 8 mmol) in 20 mL of THF, a solution of CBr4 (2.7 g, 8 mmol) in 10 mL of THF was added dropwise, under stirring, at 0 °C. The mixture was allowed to warm to room temperature and stirred for 4 h. The solvent was evaporated in vacuo, then the residue was purified by silica gel flash chromatography, eluting with hexanes, then hexanes/ethyl acetate, from 95/5 to 8/2, affording 1.31 g (5.2 mmol, 98percent yield) of pure, target product, as a colorless oil.‘H NMR (400 MHz, CDC13): = 4.54 (bs, 1 H); 3.44 (t, 2 H); 3.17-3.14 (m, 2 H); 1.92-1.87 (m, 2 H); 1.69-1.63 (m, 2 H); 1.46 (s, 9 H).
78%
With carbon tetrabromide; triphenylphosphine In dichloromethane at 20℃; for 20 h; Inert atmosphere
A solution of compound 07-4-1 (11.0 g, 58.1 mmol, 1.0 eq) in DCM (200 mL) were added CBr4 (39.5 g, 119.1 mmol, 2.05 eq) and PPh3 (32.9 g, 125.6 mmol, 2.16 eq) at 20°C, and the resulting mixture was stirred at 20°C for 20 h, diluted with water (100 mL) and extracted with DCM (100 mL*3) ,and the mixture was filtered and concentrated under reduced pressure to give a residue which was purified by column chromatography (SiO2) to give compound 07-4-2 (12.0 g, 45.2 mmol, 78percent yield).1H NMR (CDCl3, 400 MHz): δ 3.41 (t, J=6.62 Hz, 2 H), 3.14 (d, J=6.17 Hz, 2 H), 1.83 - 1.94 (m, 2 H), 1.62 (quin, J=7.28 Hz, 2 H), 1.36 - 1.49 (m, 9 H).
63.3%
With carbon tetrabromide; triphenylphosphine In dichloromethane at 0 - 20℃; for 18 h;
To an ice-cold solution of tert-butyl 4-hydroxybutylcarbamate (5 g, 26.41 mmol) in dichloromethane (200 mL) was added triphenylphosphine (10.38 g, 39.61 mmol) followed by carbon tetrabromide (13.15 g, 39.61 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 18 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography (60-120 mesh silica gel) using 10percent ethyl acetate in pet-ether to give tert-butyl 4-bromobutylcarbamate(4.2 g, 63.3 percent) as a light green liquid. XH NMR (400 MHz, CDC13) δ ppm 4.53 (1H, s), 3.45-3.41 (2H, m), 3.18-3.13 (2H, m), 1.93-1.83 (2H, m), 1.68-1.61 (2H, m), 1.47 (9H, s).
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 2, p. 1151 - 1155
[2] ACS Medicinal Chemistry Letters, 2015, vol. 6, # 11, p. 1156 - 1161
[3] Patent: WO2017/53360, 2017, A1, . Location in patent: Paragraph 0154
[4] Patent: WO2017/96045, 2017, A1, . Location in patent: Paragraph 00884
[5] Patent: WO2012/9309, 2012, A1, . Location in patent: Page/Page column 58
[6] Journal of the Chemical Society - Series Chemical Communications, 1995, # 10, p. 1015 - 1016
[7] Journal of the Chemical Society, Perkin Transactions 2, 2002, # 5, p. 923 - 927
17
[ 558-13-4 ]
[ 75178-87-9 ]
[ 164365-88-2 ]
Yield
Reaction Conditions
Operation in experiment
70%
With triphenylphosphine In dichloromethane
7b) tert-Butyl 4-bromobutylcarbamate Carbon tetrabromide (5 g) was added at once to a solution of tert-butyl 4-hydroxybutylcarbamate (1.89 g) obtained in Example 7a) and triphenylphosphine (3.15 g) in methylene chloride (20 ml), and the mixture was further stirred at room temperature for 2 minutes. The reaction mixture was washed by addition of saturated sodium bicarbonate water, followed by further washing with saturated brine. The extract was concentrated, and the residue was purified by silica gel column to obtain the title compound as colorless oil (1.76 g, 70percent). NMR (CDCl3) δ: 1.46 (9H, s), 1.50-2.00 (4H, m), 3.08-3.12 (2H, m), 3.43 (2H, t, J=6.6).
With carbon tetrabromide; triphenylphosphine; In tetrahydrofuran; for 3h;Inert atmosphere;
tert-Butyl (4-hydroxybutyl)carbamate (1.06 g, 5.788 mmol) was dissolved in dry THF (54 ml) followed by addition of Ph3P (2.86 g, 10.92 mmol, 1.9 eq.). Then CBr4 (3.62 g, 10.92 mmol, 1.9 eq.) was slowly added to the mixture. After 3 h the solution was filtered through a celite pad to eliminate the by-products and washed with Et2O. The solvents were removed under high vacuum. The crude product was purified by flash chromatography on silica gel (hexane/EtOAc, 3:1) to give 9 as a white solid at 5C (1.73 g, quantitative yield)
98%
With carbon tetrabromide; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 4h;
To a solution of tert-butyl (4-hydroxybutyl)carbamate (5a) (1.0 g, 5.3 mmol) and PPh3 (2.09 g, 8 mmol) in 20 mL of THF, a solution of CBr4 (2.7 g, 8 mmol) in 10 mL of THF was added dropwise, under stirring, at 0 C. The mixture was allowed to warm to room temperature and stirred for 4 h. The solvent was evaporated in vacuo, then the residue was purified by silica gel flash chromatography, eluting with hexanes, then hexanes/ethyl acetate, from 95/5 to 8/2, affording 1.31 g (5.2 mmol, 98% yield) of pure, target product, as a colorless oil.?H NMR (400 MHz, CDC13): = 4.54 (bs, 1 H); 3.44 (t, 2 H); 3.17-3.14 (m, 2 H); 1.92-1.87 (m, 2 H); 1.69-1.63 (m, 2 H); 1.46 (s, 9 H).
78%
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 20℃; for 20h;Inert atmosphere;
A solution of compound 07-4-1 (11.0 g, 58.1 mmol, 1.0 eq) in DCM (200 mL) were added CBr4 (39.5 g, 119.1 mmol, 2.05 eq) and PPh3 (32.9 g, 125.6 mmol, 2.16 eq) at 20C, and the resulting mixture was stirred at 20C for 20 h, diluted with water (100 mL) and extracted with DCM (100 mL*3) ,and the mixture was filtered and concentrated under reduced pressure to give a residue which was purified by column chromatography (SiO2) to give compound 07-4-2 (12.0 g, 45.2 mmol, 78% yield).1H NMR (CDCl3, 400 MHz): delta 3.41 (t, J=6.62 Hz, 2 H), 3.14 (d, J=6.17 Hz, 2 H), 1.83 - 1.94 (m, 2 H), 1.62 (quin, J=7.28 Hz, 2 H), 1.36 - 1.49 (m, 9 H).
63.3%
With carbon tetrabromide; triphenylphosphine; In dichloromethane; at 0 - 20℃; for 18h;
To an ice-cold solution of tert-butyl 4-hydroxybutylcarbamate (5 g, 26.41 mmol) in dichloromethane (200 mL) was added triphenylphosphine (10.38 g, 39.61 mmol) followed by carbon tetrabromide (13.15 g, 39.61 mmol) at 0 C. The reaction mixture was stirred at room temperature for 18 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography (60-120 mesh silica gel) using 10% ethyl acetate in pet-ether to give tert-butyl 4-bromobutylcarbamate(4.2 g, 63.3 %) as a light green liquid. XH NMR (400 MHz, CDC13) delta ppm 4.53 (1H, s), 3.45-3.41 (2H, m), 3.18-3.13 (2H, m), 1.93-1.83 (2H, m), 1.68-1.61 (2H, m), 1.47 (9H, s).
With N-Bromosuccinimide; triphenylphosphine; In dichloromethane; at 20℃; for 1.5h;Cooling;
ieri-Butyl (4-hydroxybutyl)carbamate (7, 43 g, 230 mmol) was dissolved in DCM (500 mL) and triphenylphosphine (90 g, 340 mmol) and slowly NBS (45 g, 250 mmol) were added while cooling by water. The resulting mixture was stirred at room temperature for 90 minutes. NMR showed full conversion. The mixture was washed with aqueous saturated sodium bicarbonate (sat. 300 mL), dried over Na2S04, and concentrated till one third of the volume. Heptane (500 mL) was added and the remaining DCM was removed in vacuo. Additional heptane (200 mL) was added and the mixture was stirred overnight at room temperature. The mixture was filtered and concentrated to provide crude tert-butyl (4-bromobutyl)carbamate (10, 75 g, 74 %) as a yellow oil. NMR showed 43% PPh3 and 57 % correct product.
With di-isopropyl azodicarboxylate; triphenylphosphine; In dichloromethane; at 20℃; for 1.5h;
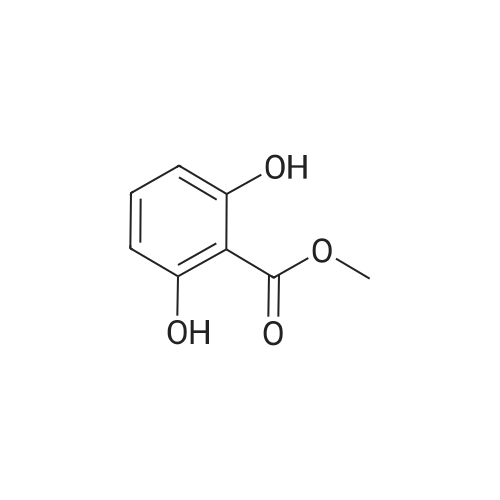
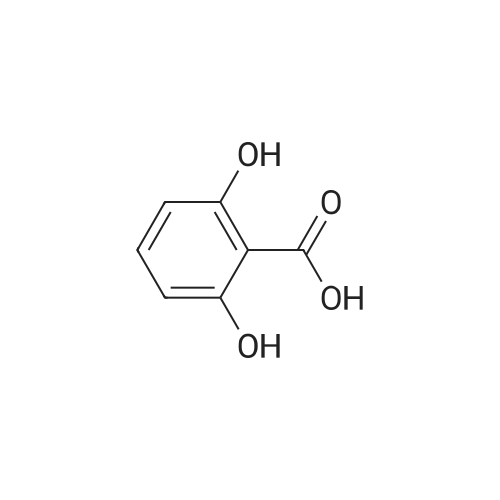
The 2-(4-amino-butoxy)-6-hydroxy- benzoic acid methyl ester was prepared by taking a solution of <strong>[2150-45-0]2,6-dihydroxymethylbenzoate</strong> (1.0 g, 5.95 mmol), 1.5 equivalents N-Boc aminobutanol and 1.5 equivalents of triphenylphosphine (2.34 g, 8.93 mmol) in 40 mL Of CH2Cl2 and adding 1.5 equivalents of DIAD (1.80 g, 8.93 mmol). Stirring continued for 1.5 hours at room temperature. Evaporation to dryness followed by column chromatography (30% EtOAc/hexanes) gave 2- (4-tert-butoxycarbonylamino-butoxy)-6-hydroxy-benzoic acid methyl ester. This compound was cooled to 0 0C in an ice/brine bath. HCl gas was bubbled into the solution for 2 minutes and stirring was continued for 1 hour. Evaporation to dryness followed by precipitation with ether and filtration gave 2-(4-aminobutoxy)-6-hydroxybenzoic acid methyl ester as the HCl salt.
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 20℃; for 18h;
To a solution of methyl 2, 6-dihydroxybenzoate (1.0 g, 5.95 mmol) and 4-(Boc-amino)-1- butanol (1.1 ml_, 5.95 mmol) in 60 mL of THF is added PPh3 (1.7 g, 6.5 mmol) and DEAD (2.97 mL, 6.5 mmol). The solution is stirred at RT for 18 h then the solvent is removed under reduced pressure. The residue is purified by flash chromatography using a gradient of hexane /EtOAc (5:1 to 2:1) to give the product as a colorless liquid.
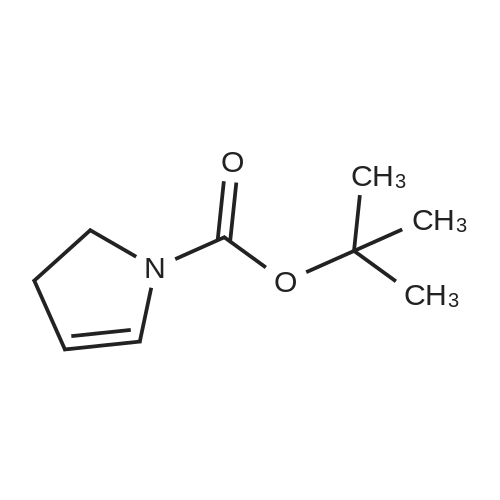
With 2-iodoxybenzoic acid; In dimethyl sulfoxide; at 20℃; for 5.7h;
1-(t-Butyloxycarbonyl)-3, 4-dihydropyrrole.; To a solution of 4- (t- butyloxycarbonylamino) butanol (1. 540 g, 8.1 mmol) in DMSO (15 mL), IBX (2 eq, 4.5 g, 16.2 mmol) was added at rt and reacted for 5.7 hr. The remainder of the workup was similar to that used for Boc-NH-(CH2) 2-CHO. The crude oil was purified by silica gel chromatography in 10% CH30H in CH2C12 and concentrated, leaving the product as a clear, faintly yellow oil: yield 58% ; one spot on TLC, Rf= 0.71 (10% CH30H in CH2Cl2).'H NMR (CDCl3) 8 5.47 (s, 1H), 5.40 (s, 1H), 3.60-3. 20 (m, 2H), 2. 14-1. 74 (m, 2H), 1.45 (s, 9H). MS (ESI+) nilz 170 (M+1).
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 0℃; for 4h;
84.5%
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 20℃; for 5h;
72%
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 20℃; for 2h;
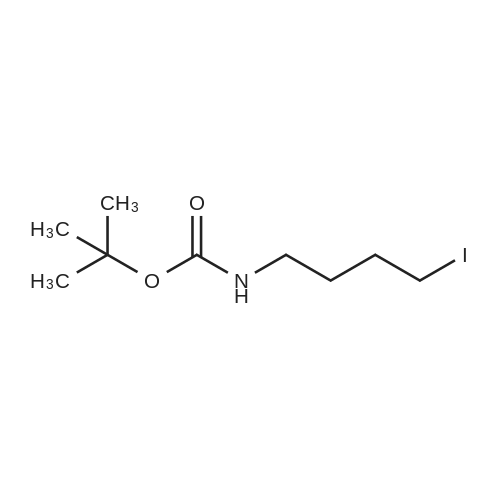
1 N-tert-Butoxycarbonyl-4-amino-1-iodobutane
N-tert-Butoxycarbonyl-4-amino-1-iodobutane Triphenylphosphine (3.3 g, 12.58 mmol) and imidazole (0.86 g, 12.63 mmol) were dissolved in dichloromethane (15 ml), and the solution was stirred at 5 minutes. Then, iodine (3.2 g, 12.61 mmol) was added thereto, and the mixture was stirred for 10 minutes. A dichloromethane (4 ml) solution of N-tert-butoxycarbonyl-4-amino-1-butanol (1.6 g, 8.454 mmol) was added dropwise thereto, and the mixture was stirred at room temperature for 2 hours. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and a 5% aqueous sodium thiosulfate solution was added to the filtrate to remove iodine. The organic layer was washed twice with brine and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified by silica gel column chromatography (hexane:ethyl acetate=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-iodobutane (1.83 g, yield: 72%).
72%
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 20℃; for 2h; Inert atmosphere;
1 N-tert-Butoxycarbonyl-4-amino-1-iodobutane
N-tert-Butoxycarbonyl-4-amino-1-iodobutane Triphenylphosphine (3.3 g, 12.58 mmol) and imidazole (0.86 g, 12.63 mmol) were dissolved in dichloromethane (15 ml), and the solution was stirred at 5 minutes. Then, iodine (3.2 g, 12.61 mmol) was added thereto, and the mixture was stirred for 10 minutes. A dichloromethane (4 ml) solution of N-tert-butoxycarbonyl-4-amino-1-butanol (1.6 g, 8.454 mmol) was added dropwise thereto, and the mixture was stirred at room temperature for 2 hours. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and a 5% aqueous sodium thiosulfate solution was added to the filtrate to remove iodine. The organic layer was washed twice with saturated saline and dehydrated over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified using silica gel column chromatography (hexane:ethyl acetate=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-iodobutane (1.83 g, yield: 72%).
72%
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 20℃; for 2h; Inert atmosphere;
1 N-tert-Butoxycarbonyl-4-amino-1-iodobutane
Triphenylphosphine (3.3 g, 12.58 mmol) and imidazole (0.86 g, 12.63 mmol) were dissolved in dichloromethane (15 ml), and the solution was stirred at 5 minutes. Then, iodine (3.2 g, 12.61 mmol) was added thereto, and the mixture was stirred for 10 minutes. A dichloromethane (4 ml) solution of N-tert-butoxycarbonyl-4-amino-1-butanol (1.6 g, 8.454 mmol) was added dropwise thereto, and the mixture was stirred at room temperature for 2 hours. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and a 5% aqueous sodium thiosulfate solution was added to the filtrate to remove iodine. The organic layer was washed twice with saturated saline and dehydrated over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified using silica gel column chromatography (hexane:ethyl acetate=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-iodobutane (1.83 g, yield: 72%).
72%
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 20℃; for 2h; Inert atmosphere;
1 N-tert-Butoxycarbonyl-4-amino-1-iodobutane
Triphenylphosphine (3.3 g, 12.58 mmol) and imidazole (0.86 g, 12.63 mmol) were dissolved in dichloromethane (15 ml), and the solution was stirred at 5 minutes. Then, iodine (3.2 g, 12.61 mmol) was added thereto, and the mixture was stirred for 10 minutes. A dichloromethane (4 ml) solution of N-tert-butoxycarbonyl-4-amino-1-butanol (1.6 g, 8.454 mmol) was added dropwise thereto, and the mixture was stirred at room temperature for 2 hours. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and a 5% aqueous sodium thiosulfate solution was added to the filtrate to remove iodine. The organic layer was washed twice with saturated saline and dehydrated over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified using silica gel column chromatography (hexane:ethyl acetate=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-iodobutane (1.83 g, yield: 72%).
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 0 - 20℃; for 1h; Inert atmosphere;
General Procedure for Preparation of cis-14
General procedure: To a solution of an aminoalcohol (66.6 mmol) in THF (67 mL) was added di-tert-butyl dicarbonate (15.3 g, 66.6 mmol) at 50 °C under an argon atmosphere. After 3 h stirring, the solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (EtOAc / hexane, 50:50) to give a N-Boc aminoalcohol. Iodine (12.5 g, 49.3 mmol) was added to a solution of N-Boc aminoalcohol (17.6 mmol), PPh3 (6.92 g, 26.3 mmol) and imidazole (1.79 g, 26.3 mmol) in CH2Cl2 (98 mL) at 0 °C. The mixture was stirred at room temperature for 1 h under an argon atmosphere. The reaction mixture was quenched with saturated aqueous Na2S2O3, diluted with CH2Cl2 and extracted with CH2Cl2. The organic layer was washed with H2O and brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (EtOAc/hexane, 10:90) to afford a N-BOC aminoalkyliodide. To a solution of NaH (60% in oil, 0.673 g, 16.8 mmol) in DMF (15.3 mL) was added dropwise a solution of N-hydroxyphthalimide (2.49 mmol) in DMF (10 mL) at 0 °C under an argon atmosphere. After 15 min of stirring, a solution of the iodide (15.3 mmol) in DMF (10 mL) was added dropwise to the solution and stirred at 70 °C for 12 h. After cooled to 0 °C, the reaction was quenched with H2O and filtrated to afford colorless solid, which was recrystallized to give a N-alkoxyphthalimide 13a-d.
With 1H-imidazole; iodine; triphenylphosphine In dichloromethane at 20℃; for 2h; Cooling with ice; Inert atmosphere;
Multi-step reaction with 2 steps
1: 4-dimethylaminopyridine; triethylamine / dichloromethane / 16 h / 25 °C
2: sodium iodide / propan-2-one / 2 h / 60 °C
7b) tert-Butyl 4-bromobutylcarbamate Carbon tetrabromide (5 g) was added at once to a solution of tert-butyl 4-hydroxybutylcarbamate (1.89 g) obtained in Example 7a) and triphenylphosphine (3.15 g) in methylene chloride (20 ml), and the mixture was further stirred at room temperature for 2 minutes. The reaction mixture was washed by addition of saturated sodium bicarbonate water, followed by further washing with saturated brine. The extract was concentrated, and the residue was purified by silica gel column to obtain the title compound as colorless oil (1.76 g, 70%). NMR (CDCl3) delta: 1.46 (9H, s), 1.50-2.00 (4H, m), 3.08-3.12 (2H, m), 3.43 (2H, t, J=6.6).
With triphenylphosphine In tetrahydrofuran; hexane
36.B Methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate
EXAMPLE 36B Methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate To a mixture of tert-butyl 4-hydroxybutylcarbamate (400 mg, 2.1 mmol), 2,6-dihydroxybenzoate (463 mg, 2.7 mmol), and triphenylphosphine (777 mg, 3.0 mmol) under positive nitrogen atmosphere in THF (anhydrous) was added dropwise diethyl azodicarboxylate (433 μL, 2.7 mmol). The mixture was stirred for 16 hour, solvents removed under reduced pressure and the residue was purified on a silica gel chromatography eluding with 15-30% ethyl acetate in hexane to give the titled compound (410 mg, 57%) as a cloroless oil.
With triphenylphosphine; In tetrahydrofuran; hexane;
EXAMPLE 45E Methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate To a round bottom flask was charged with tert-butyl 4-hydroxybutylcarbamate (400 mg, 2.1 rnmol), 463 mg of <strong>[2150-45-0]methyl 2,6-dihydroxybenzoate</strong> (463 mg, 2.7 mmol), and triphenylphosphine (777 mg, 3.0 mmol). The flask was vacuumed and back flushed with nitrogen (3*), capped with a rubber septum, and kept under positive nitrogen atmosphere. THF (anhydrous) (25 mL) was then added, followed by dropwise addition of diethyl azodicarboxylate (433 muL, 2.7 mmol). Solvent were removed under reduced pressure, and the residue purified on a silica gel chromatography eluding with 15-30% ethyl acetate in hexane to give the titled compound (410 mg, 57%) as a colorless oil.
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran; hexane
12.A Methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate
EXAMPLE 12A Methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate To a round bottom flask was charged with tert-butyl 4-hydroxybutylcarbamate (400 mg, 2.1 mmol), 463 mg of 2,6-dihydroxybenzoate (463 mg, 2.7 mmol), and triphenylphosphine (777 mg, 3.0 mmol). The flask was vacuumed and back flushed with nitrogen (3*), capped with a rubber septum, and kept under positive nitrogen atmosphere. THF (anhydrous) was then added, followed by dropwise addition of DEAD (433 μL, 2.7 mmol). Most of the starting material was consumed within the first 30 min. Solvent was then removed in vacuo, and the residue was purified on a silica gel chromatography eluding with 15-30% EtOAc in hexane to give the ether product (410 mg, 57%) as a cloroless oil.
57%
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran; hexane
12.A methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate
EXAMPLE 12A methyl 2-{4-[(tert-butoxycarbonyl)amino]butoxy}-6-hydroxybenzoate To a round bottom flask was charged with tert-butyl 4-hydroxybutylcarbamate (400 mg, 2.1 mmol), 463 mg of 2,6-dihydroxybenzoate (463 mg, 2.7 mmol), and triphenylphosphine (777 mg, 3.0 mmol). The flask was vacuumed and back flushed with nitrogen (3*), capped with a rubber septum, and kept under positive nitrogen atmosphere. THF (anhydrous) was then added, followed by dropwise addition of DEAD (433 μL, 2.7 mmol). Most of the starting material was consumed within the first 30 min. Solvent was then removed in vacuo, and the residue was purified on a silica gel chromatography eluding with 15-30% EtOAc in hexane to give the ether product (410 mg, 57%) as a cloroless oil.
3-(4-(N-t-butoxycarbonylamino)butoxy)-1,2-benzisoxazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
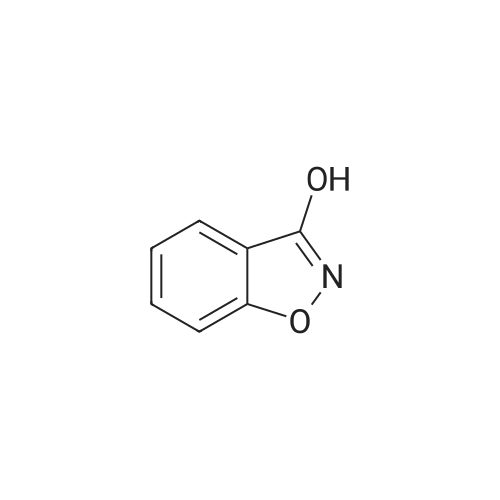
(b) 3-(4-(N-t-Butoxycarbonylamino)butoxy)-1,2-benzisoxazole. The title compound was obtained in 71% yield from <strong>[21725-69-9]3-hydroxy-1,2-benzisoxazole</strong> and 4-(N-t-butoxycarbonylamino)butanol by similar reactions and treatments as in example 1(e). IR spectrum(KBr)numax cm-1: 3321, 1701, 1615, 1539, 1509; NMR spectrum(CDCl3)deltappm: 1.14(9H,s), 1.62-1.74(2H,m), 1.91-1.97(2H,m), 3.15-3.27(2H,brs), 4.46(2H,t,J=6.5 Hz), 4.55-4.70(1H,brs), 7.24-7.66(4H,m).
With methanesulfonyl chloride; triethylamine; In DCM;
Methane sulfonyl chloride (1 eq) was added to a stirring solution of N-Boc propan-1 -ol, N-Boc butan-1-ol or N-Boc pentan-1-ol (1 eq) and EtJM (3 eq) in DCM. Upon completion the reaction was quenched with NaHCO3 solution. The DCM layer was dried over magnesium sulfate and concentrated in vacuo to produce a crude residue which was used crude or purified by flash column chromatography.
1
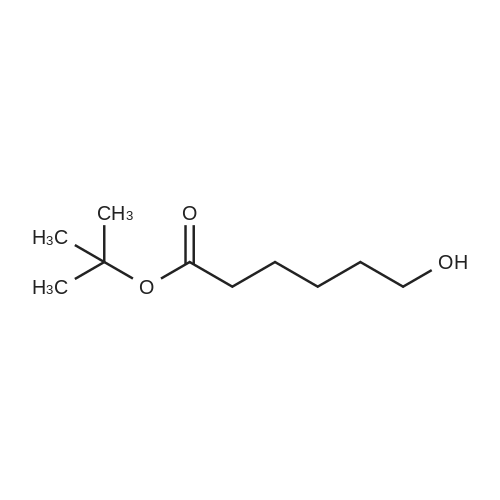
To a solution of tert-butyl 6-hydroxyhexanoate (1) (J.Org.Chem. (1984), 49(12), 2147) (2.6 g, 13.7 mmol) and tert-buty 4-hydroxybutylcarbamate (2) (J. Med. Chem. (2006), 49(14), 4183-4195) (2.6 g, 13.7 mmol) in 20 mL of anhydrous pyridine (20 mL) was added 3.1 mL of diphenylphosphite (85% pure). After being stirred at room temperature overnight, the reaction was concentrated and re-dissolved in ethyl acetate. The solution was washed with 10% citric acid, saturated NaCl and dried over Na2SO4. The crude material obtained after solvent evaporation was chromatographed on silica eluting first with 1 : 1 ethyl acetate: hexane to separate phenol and one of the symmetric by-products and, second, with ethyl acetate to elute the desired phosphite 3. Concentration of the pure product fractions gave 2.05 g of the phosphite 3 as a viscous liquid. H'-NMR (DMSO-d6): ? 6.92 (br t, NH, IH), 6.80 (d, J=692 Hz, PH, IH), 3.97 (m, 4H), 2.93 (q, J=6.6 Hz, 2H), 2.19 (t, J=7 Hz, 2H), 1.59 (m, 4H), 1.49 (m, 4H), 1.40 (s, 9H), 1.38 (s, 9H), 1.36 (m, 2H).
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at -10 - 20℃;Inert atmosphere;
Example 27: Methyl 2-(4-(tert-butoxycarbonylamino)butoxy)-4-fluorobenzoate (1-28)Methyl 4-fluoro-2-hydroxybenzoate (21 mmol), tert-butyl 4-hydroxybutylcarbamate (21 mmol) and triphenyl phosphine (24 mmol) in THF (70 ml) was cooled to -10C. Then, DIAD (43 mmol) was added. The mixture was stirred for 30 min at -10C under N2. The ice-bath was removed and the mixture was stirred overnight at 20C. The solvent was removed under vacuum. The residue was washed with petroleum and diethyl ether. The white solid was filtered and it was confirmed to betriphenylphosphine oxide (RhoRho1¾=0). The filtrate was evaporated to dryness. The residue was purified with flash column (petroleum/ethyl acetate 100:0 to 5: 1). Yield: 8.18 g (purity 90%, yield 98%).
1 N-tert-Butoxycarbonyl-4-amino-1-butanol
N-tert-Butoxycarbonyl-4-amino-1-butanol 4-Amino-1-butanol (1.0 g, 11.22 mmol) was dissolved in methanol (10 ml). To this solution, di-tert-butyl carbonate (2.53 g, 11.58 mmol) was added, and the mixture was stirred at room temperature for 1.5 hours. After the reaction was confirmed by TLC to be complete, the solvent was distilled off under reduced pressure, and the residue was purified using silica gel column chromatography (hexane:acetone=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-butanol (1.88 g, yield: 89%).
89%
In methanol at 20℃; for 1.5h; Inert atmosphere;
1 N-tert-Butoxycarbonyl-4-amino-1-butanol
4-Amino-1-butanol (1.0 g, 11.22 mmol) was dissolved in methanol (10 ml). To this solution, di-tert-butyl carbonate (2.53 g, 11.58 mmol) was added, and the mixture was stirred at room temperature for 1.5 hours. After the reaction was confirmed by TLC to be complete, the solvent was distilled off under reduced pressure, and the residue was purified using silica gel column chromatography (hexane:acetone=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-butanol (1.88 g, yield: 89%).
N-tert-Butoxycarbonyl-4-amino-1-butanol
N-tert-Butoxycarbonyl-4-amino-1-butanol 4-Amino-1-butanol (1.0 g, 11.22 mmol) was dissolved in methanol (10 ml). To this solution, di-tert-butyl carbonate (2.53 g, 11.58 mmol) was added, and the mixture was stirred at room temperature for 1.5 hours. After the reaction was confirmed by TLC to be complete, the solvent was distilled off under reduced pressure, and the residue was purified using silica gel column chromatography (hexane:acetone=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-butanol (1.88 g, yield: 89%).
N-tert-Butoxycarbonyl-4-amino-1-iodobutane
N-tert-Butoxycarbonyl-4-amino-1-iodobutane Triphenylphosphine (3.3 g, 12.58 mmol) and imidazole (0.86 g, 12.63 mmol) were dissolved in dichloromethane (15 ml), and the solution was stirred at 5 minutes. Then, iodine (3.2 g, 12.61 mmol) was added thereto, and the mixture was stirred for 10 minutes. A dichloromethane (4 ml) solution of N-tert-butoxycarbonyl-4-amino-1-butanol (1.6 g, 8.454 mmol) was added dropwise thereto, and the mixture was stirred at room temperature for 2 hours. After the reaction was confirmed by TLC to be complete, the reaction solution was filtered through celite, and a 5% aqueous sodium thiosulfate solution was added to the filtrate to remove iodine. The organic layer was washed twice with saturated saline and dehydrated over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and then, the residue was purified using silica gel column chromatography (hexane:ethyl acetate=9:1) to obtain N-tert-butoxycarbonyl-4-amino-1-iodobutane (1.83 g, yield: 72%).
To an ice-cold solution of triphosgene (13.6 mg, 0.046 mmol) in dry THF (0.5 mL) was added a solution of t-butyl 4-hydroxybutylcarbamate (24.2 mg, 0.128 mmol) and TEA (18.1 muL, 0.13 mmol) in dry THF (1 mL) under argon. After stirring for 1 h at 0 C., the crude chloroformate was slowly added to a solution of <strong>[371935-74-9]PI-103</strong> (25 mg, 0.072 mmol) and TEA (15.1 muL, 0.108 mmol) in NMP (0.5 mL). After several minutes THF was removed under vacuum and NMP (0.5 mL) was added to make the mixture more homogenous. The resulting mixture was stirred overnight at room temperature. Additional chloroformate (from 45 mg BOC-alcohol, prepared as described above) and TEA (15 muL) were added and the reaction mixture was stirred for 40 min at which point LC/MS indicated 95% conversion to the desired product. The reaction mixture was diluted with DCM (150 mL) and then washed with water (2*50 mL) and brine (50 mL). The organic phase was dried over Na2SO4 and concentrated under vacuum. The crude product was purified on silica gel (4 g CombiFlash column, EtOAc:Hex, 0% EtOAc 1 min, then gradient to 80% EtOAc over 16 min) to give 26 mg of a colorless film. Yield 64%. ESI-MS calc for C29H34N5O7 564.3 (M+H+). found 564.1. The BOC-protected carbonate was dissolved in DCM (2 mL) and then treated with 4 M HCl in dioxane (4 mL). The resulting mixture was stirred for 3.5 h and then concentrated under vacuum. The deprotected carbonate was lyophilized from water:CH3CN to afford the title compound as a pale yellow solid (21.9 mg, 96% yield). ESI-MS calc for C24H26N5O5 464.2 (M+H+). found 464.1.
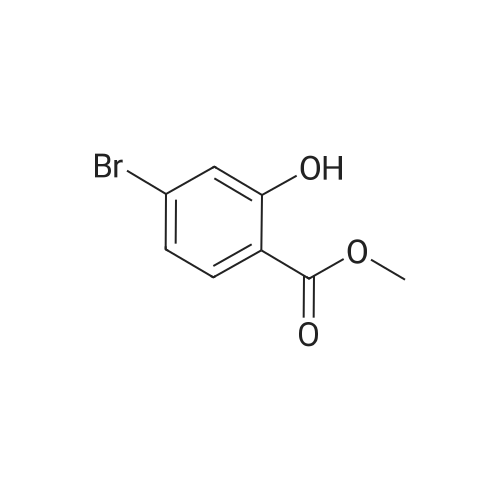
methyl 5-bromo-2-(4-((tert-butoxycarbonyl)amino)butoxy)-4-fluorobenzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 18℃; for 0.5h;
To a stirred mixture of <strong>[4133-72-6]methyl 5-bromo-4-fluoro-2-hydroxybenzoate</strong> (30 g, 0.12 mol), tert-butyl (4-hydroxybutyl)carbamate (32 g, 0.17 mol) and PPh3 (38 g, 0.15 mol) in THF (400 mL) was dropwise added DIAD (30.7 mL, 0.16 mol). The reaction mixture was stirred at 18 C for 0.5 h. After this time the volatiles were removed under reduced pressure to give a crude product. Petroleum ether/EtOAc (6/1) was added until a solid precipitated. The mixture was filtered and the organic layer was concentrated to give the title compound (30 g, 59%) as a white solid. The product was used without further purification in the next step.
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0 - 30℃; for 3h;
77.A Step A. Methyl 5-bromo-2-(4-((tert-butoxycarbonyl)amino)butoxy)-4-(trifluoromethyl)benzoate
A solution of methyl 5-bromo-2-hydroxy-4-(trifluoromethyl)benzoate (6.0 g, 20.1 mmol), tert-butyl (4-hydroxybutyl)carbamate (5.32 g, 28.1 mmol) and PPh3 (6.32 g, 24.08 mmol) in THF (50 mL) at 0 °C was added DIAD (5.1 mL, 26.1 mmol) dropwise via syringe. The reaction was stirred at 30 °C for 3 h. After this time the mixture was concentrated and purified by HPLC (using a Phenomenex Synergi Max-RP, ΙΟμιη 250x50mm column and using water (containing 10 mM NH4HC03)/CH3CN from 20% to 80% as the mobile phase at a flow rate of 115 mL/min) to provide the title compound (2.0 g, 21%) as a white solid. MS (ESI): 470.0 [(M + H) (79Br)]+.
methyl 4-bromo-2-(4-((tert-butoxycarbonyl)amino)butoxy)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
52%
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 28℃; for 2h;
To a solution of <strong>[22717-56-2]methyl 4-bromo-2-hydroxybenzoate</strong> (5 g, 21.6 mmol) in THF (30 mL) was added ieri-butyl (4-hydroxybutyl)carbamate (4.91 g, 26 mmol) and PPh3 (6.81 g, 26 mmol) and the mixture was stirred at 28 C for 10 min. After this time DIAD (5.5 mL, 28.1 mmol) was added dropwise over 1 min and the mixture was stirred at 28 C for 2 h. After this time, the mixture was concentrated and petroleum ether/EtOAc (20 mL, 5/1) was added. The mixture was filtered and the filtrate was concentrated to give the crude product (5 g), which was purified by chromatography on silica gel using DCM/MeOH (100/1 to 10/1) as eluent to give the title compound (4.5 g, 52%) as an oil. MS (ESI): 403.8 [(M + H) (81Br)]+.
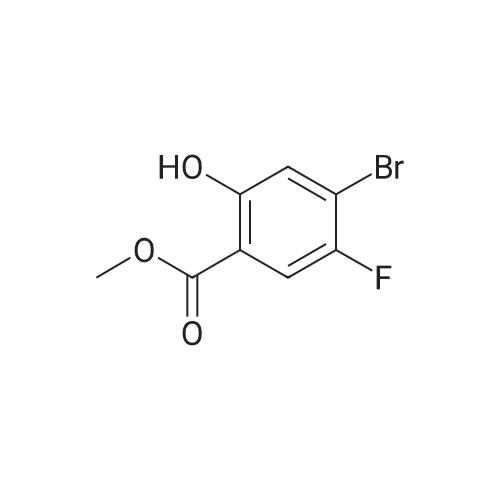
With caesium carbonate In N,N-dimethyl-formamide at 25℃; for 18h;
85.A Step A. Methyl 4-bromo-2-(4-((tert-butoxycarbonyl)amino)butoxy)-5-fluorobenzoate
To a solution of methyl 4-bromo-5-fluoro-2-hydroxybenzoate (360 mg, 1.44 mmol) in DMF (8 mL) was added tert-butyl (4-bromobutyl)carbamate (438 mg, 1.73 mmol) and Cs2C03 (939 mg, 2.88 mmol) and the reaction was stirred at 25 °C for 18 h. After this time, the mixture was poured into water (20 mL) and extracted with EtOAc (3 x 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2S04, filtered and concentrated under vacuum to give the crude product, which was purified by column chromatography on silica using petreoleum ether/EtOAc (10/1 to 5/1) as eluent to give the title compound (500 mg, 83%) as a yellow solid. 1H NMR (400 MHz, DMSO-i) δ ppm 7.59 (d, 7=8.8 Hz, 1H), 7.47 (d, 7=5.2 Hz, 1H), 6.85 - 6.81 (m, 1H), 4.01 - 4.04 (m, 2H), 3.77 (s, 3H), 2.91 - 2.96 (m, 2H), 1.61 - 1.67 (m, 2H), 1.37 - 1.54 (m, 2H), 1.35 (s, 9H).
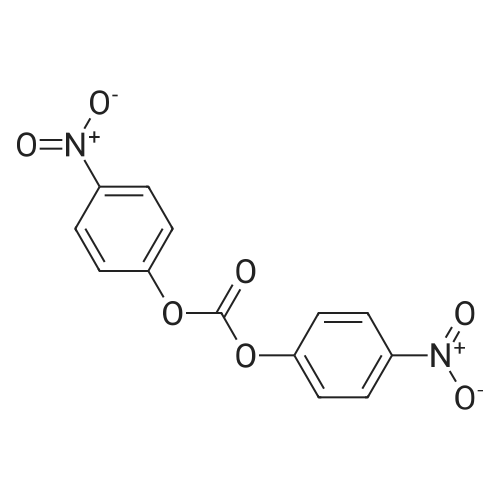
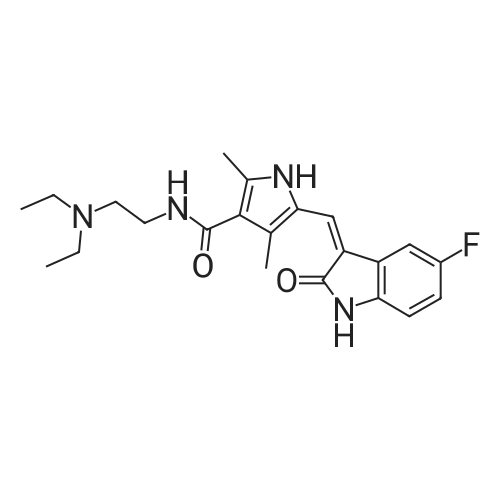
Stage #1: bis-(p-nitrophenyl) carbonate; N-[2-(diethylamino)ethyl]-5-[(5-fluoro-1,2-dihydro-2-oxo-3H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide With triethylamine In dichloromethane at 45℃; for 20h; Inert atmosphere;
Stage #2: (4-hydroxybutyl)carbamic acid tert-butyl ester In dichloromethane at 20℃; for 22h; Inert atmosphere;
101.1 Step 1: Preparation of 4- ((tert-butoxycarbonyl) amino) butyl (Z)-3-((4- ((2- (diethylamino) ethyl) carbamoyl) -3,5-dimethyl-1H-pyrrol-2-yl) methylene) -5-fluoro-2-oxoindoline-1-carboxylate
To a stirred mixture of (Z) -N- (2- (diethylamino) ethyl)-5-((5-fluoro-2-oxoindolin-3-ylidene) methyl) -2,4-dimethyl-1H-pyrrole-3-carboxamide (598 mg, 1.5mmol) and bis (4-nitrophenyl) carbonate (548 mg, 1.8mmol) in DCM (30mL) was added triethylamine (379.5 mg, 3.75mmol). The reaction mixture was heated to 45°C and stirred at this temperature for 20 hours. Then the reaction mixture was cooled to room temperature. Then tert-butyl (4-hydroxybutyl) carbamate (426 mg, 2.25mmol) was added. The reaction mixture was stirred at room temperature for 22 hours. The solution was diluted with DCM and washed with saturated NaHCO 3 solution, water and saturated brine solution. The organic layer was dried over sodium sulfate, filtered and then concentrated under reduced pressure. The crude residue was purified by column chromatography (DCM: Methanol=20: 1~10: 1) to afford the title compound (0.6g, Yield: 65.2%). MS (m/z) : [M+H] + calcd forC 32H 44FN 5O 6, 614.73; found: 614.3. 1H NMR (400 MHz, Chloroform-d) δ 12.93 (s, 1H), 7.81 (dd, J = 8.9, 4.5 Hz, 1H), 7.38 (s, 1H), 7.28 (s, 1H), 7.18 (dd, J = 8.4, 2.6 Hz, 1H), 6.93 (td, J = 9.0, 2.6 Hz, 1H), 4.72 (s, 1H), 4.49 (t, J = 6.6 Hz, 2H), 3.81 (s, 2H), 3.27 -3.03 (m, 8H), 2.65 (s, 3H), 2.58 (s, 3H), 1.90 (t, J = 7.5 Hz, 2H), 1.71 (q, J = 7.3 Hz, 2H), 1.44 (s, 9H), 1.38 (d, J = 9.3 Hz, 6H).
65.2%
Stage #1: bis-(p-nitrophenyl) carbonate; N-[2-(diethylamino)ethyl]-5-[(5-fluoro-1,2-dihydro-2-oxo-3H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide With triethylamine In dichloromethane at 45℃; for 20h; Inert atmosphere;
Stage #2: (4-hydroxybutyl)carbamic acid tert-butyl ester In dichloromethane at 20℃; for 22h; Inert atmosphere;
101.1 Step 1: Preparation of 4- ((tert-butoxycarbonyl) amino) butyl (Z)-3-((4- ((2- (diethylamino) ethyl) carbamoyl) -3,5-dimethyl-1H-pyrrol-2-yl) methylene) -5-fluoro-2-oxoindoline-1-carboxylate
To a stirred mixture of (Z) -N- (2- (diethylamino) ethyl)-5-((5-fluoro-2-oxoindolin-3-ylidene) methyl) -2,4-dimethyl-1H-pyrrole-3-carboxamide (598 mg, 1.5mmol) and bis (4-nitrophenyl) carbonate (548 mg, 1.8mmol) in DCM (30mL) was added triethylamine (379.5 mg, 3.75mmol). The reaction mixture was heated to 45°C and stirred at this temperature for 20 hours. Then the reaction mixture was cooled to room temperature. Then tert-butyl (4-hydroxybutyl) carbamate (426 mg, 2.25mmol) was added. The reaction mixture was stirred at room temperature for 22 hours. The solution was diluted with DCM and washed with saturated NaHCO 3 solution, water and saturated brine solution. The organic layer was dried over sodium sulfate, filtered and then concentrated under reduced pressure. The crude residue was purified by column chromatography (DCM: Methanol=20: 1~10: 1) to afford the title compound (0.6g, Yield: 65.2%). MS (m/z) : [M+H] + calcd forC 32H 44FN 5O 6, 614.73; found: 614.3. 1H NMR (400 MHz, Chloroform-d) δ 12.93 (s, 1H), 7.81 (dd, J = 8.9, 4.5 Hz, 1H), 7.38 (s, 1H), 7.28 (s, 1H), 7.18 (dd, J = 8.4, 2.6 Hz, 1H), 6.93 (td, J = 9.0, 2.6 Hz, 1H), 4.72 (s, 1H), 4.49 (t, J = 6.6 Hz, 2H), 3.81 (s, 2H), 3.27 -3.03 (m, 8H), 2.65 (s, 3H), 2.58 (s, 3H), 1.90 (t, J = 7.5 Hz, 2H), 1.71 (q, J = 7.3 Hz, 2H), 1.44 (s, 9H), 1.38 (d, J = 9.3 Hz, 6H).
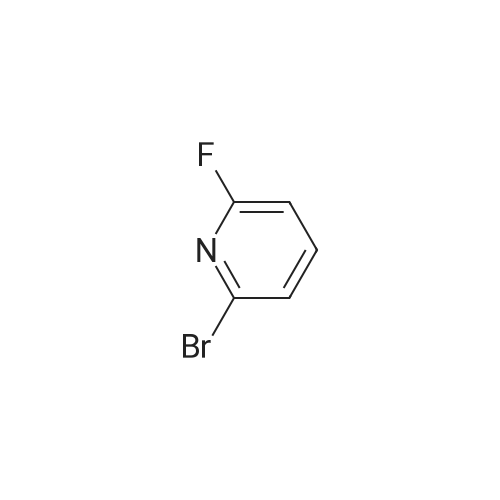
Stage #1: 2-bromo-6-fluoropyridine; (4-hydroxybutyl)carbamic acid tert-butyl ester With sodium hydride In tetrahydrofuran; mineral oil at 0℃;
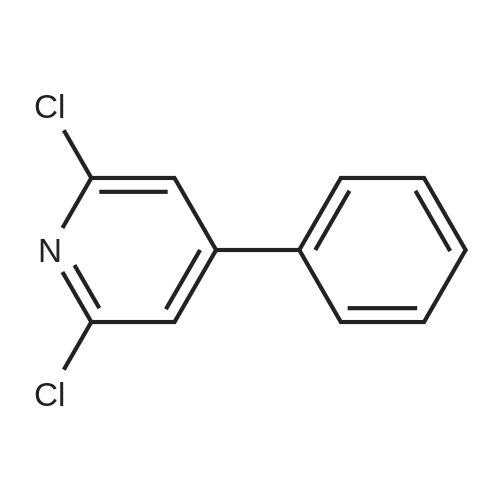
Stage #2: 2,6-dichloro-4-phenylpyridine In tetrahydrofuran; mineral oil at 60℃;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping