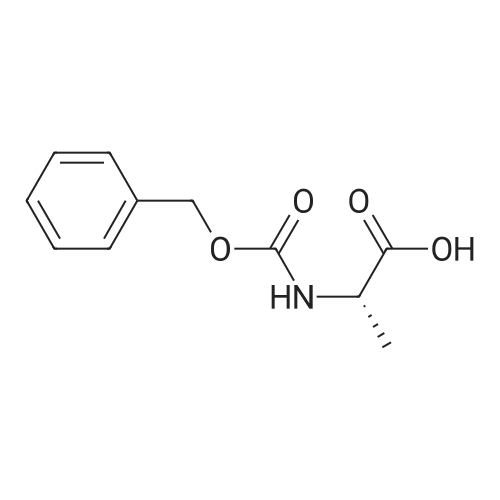
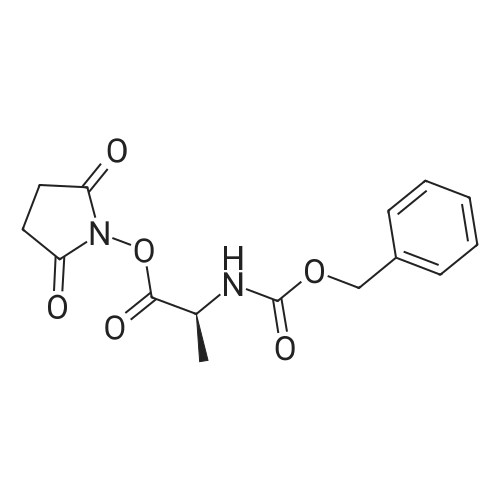
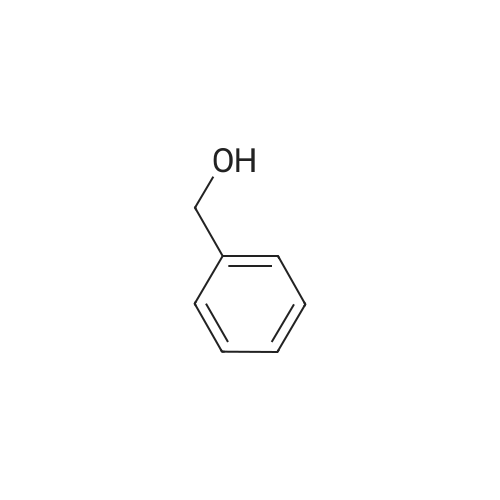
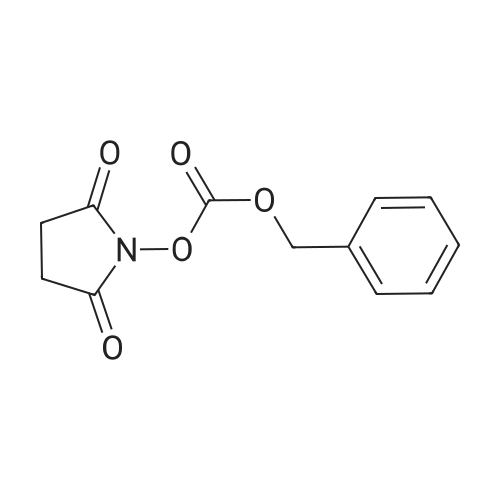
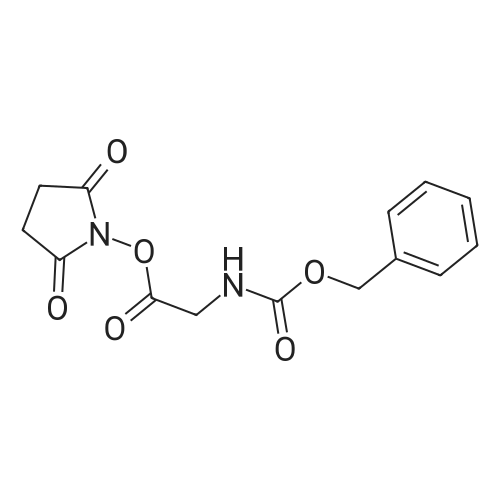
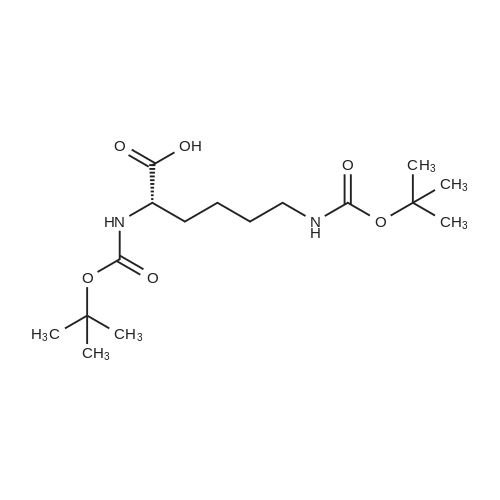
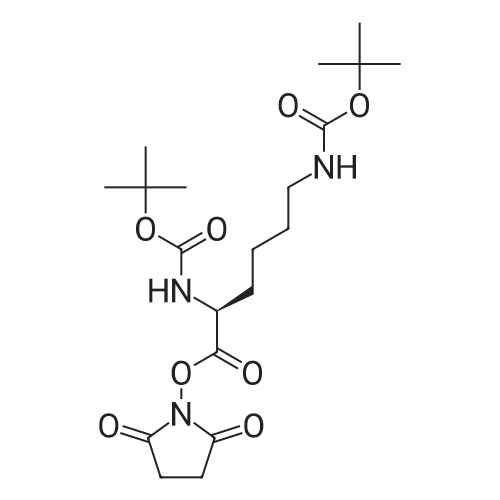
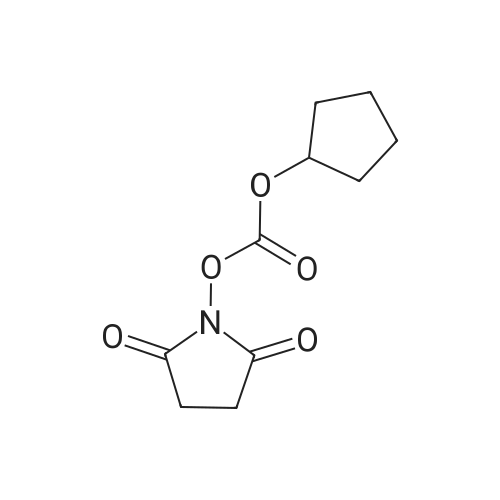
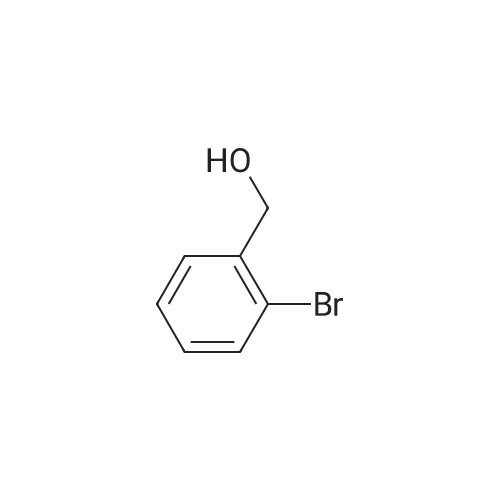
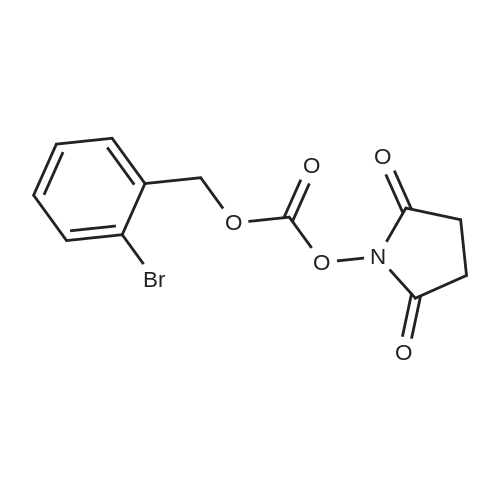
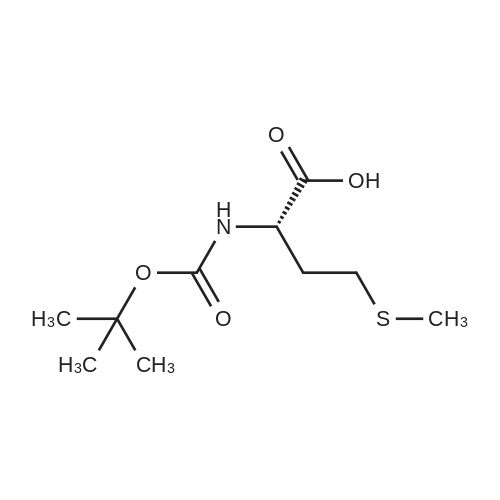
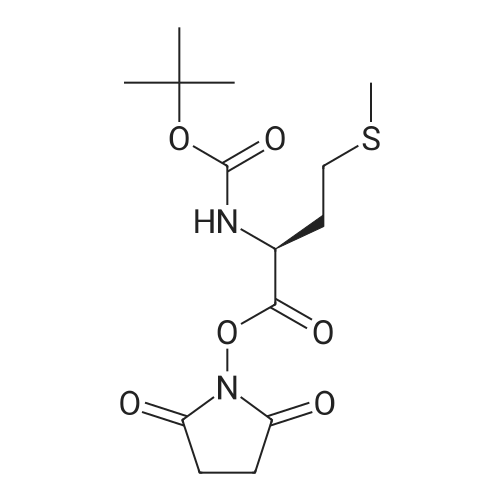
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
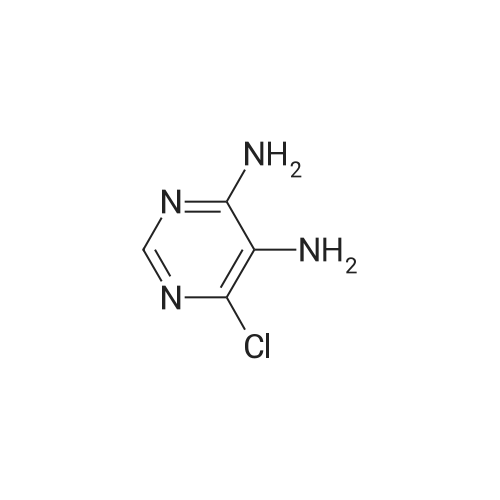
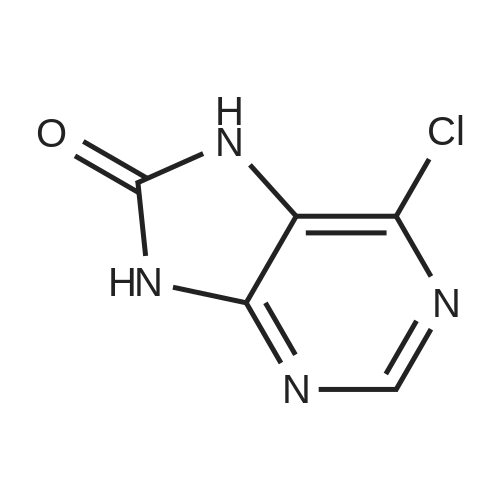
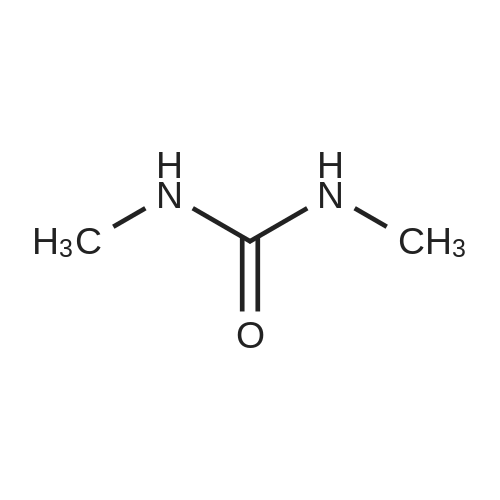
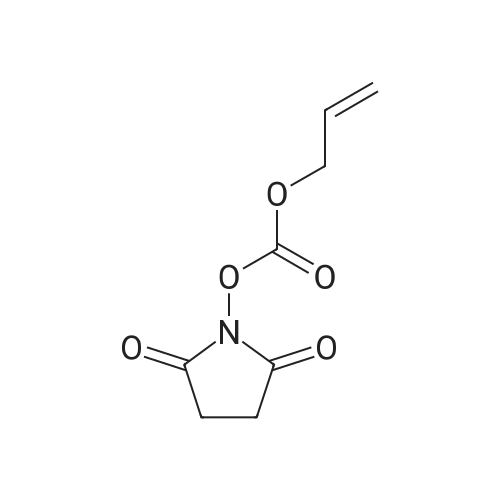
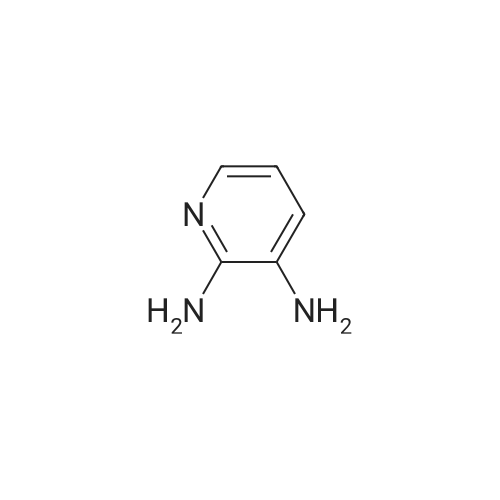
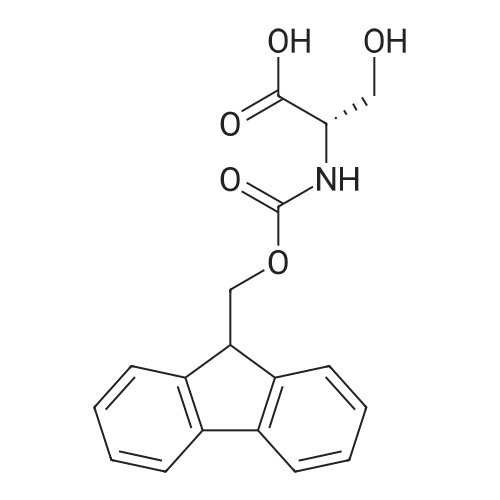
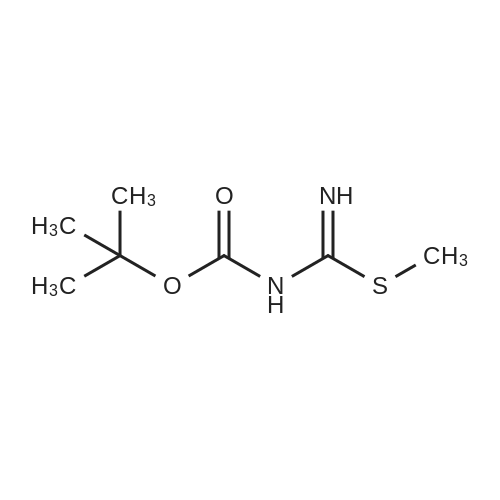
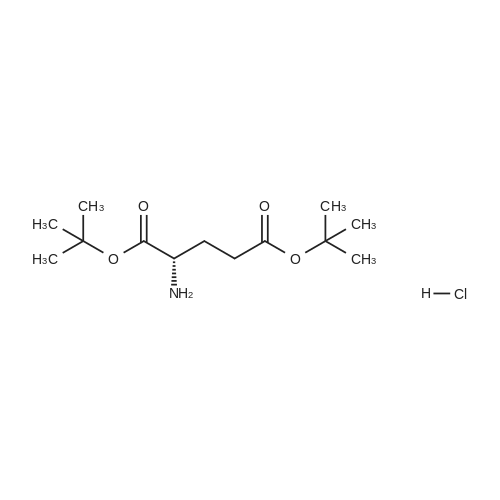
Synthesis 20-1 -A; 6-Chloro-7H-purin-8(9H)-one; NXrNH2 A mixture of 4-chloropyrimidine-2,3-diamine (36 mg, 0.25 mmol) and di(/V-succinimidyl)carbonate (128 mg, 0.50 mmol) in acetonitrile (10 mL) was refluxed for 16 hours. The solids formed were collected, washed with acetonitrile (2 x 5 mL) and dried, to give the title compound as a light yellow powder (33 mg, 78percent).1H NMR (500 MHz, de-DMSO) δ 8.35 (1 H, s), 11.90 (2H, s, broad); LC-MS (2) R1 1.96 min; m/z (ESI) 171 [MH+].
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 20, p. 5559 - 5562
5
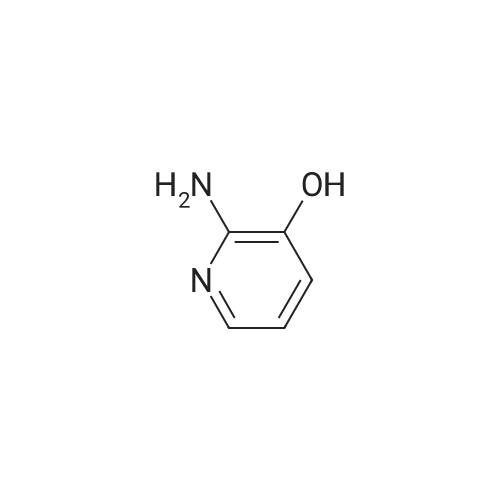
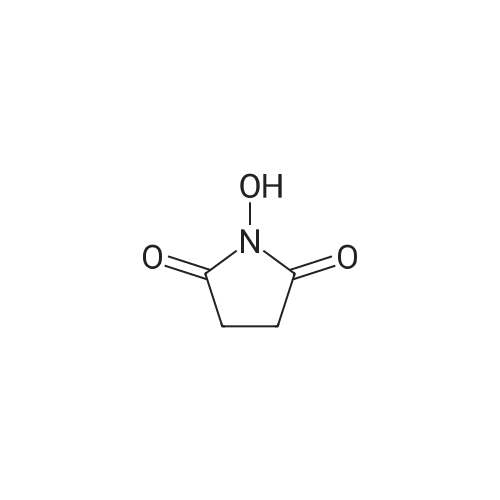
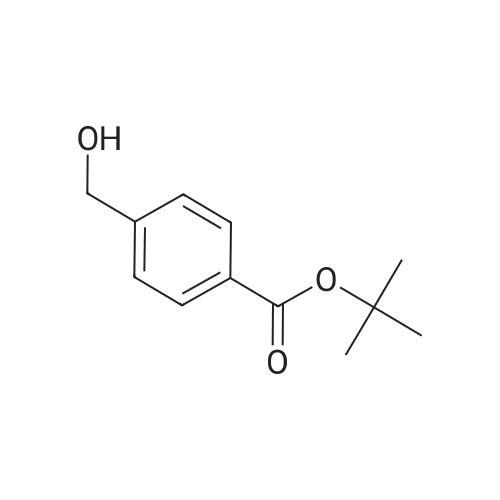
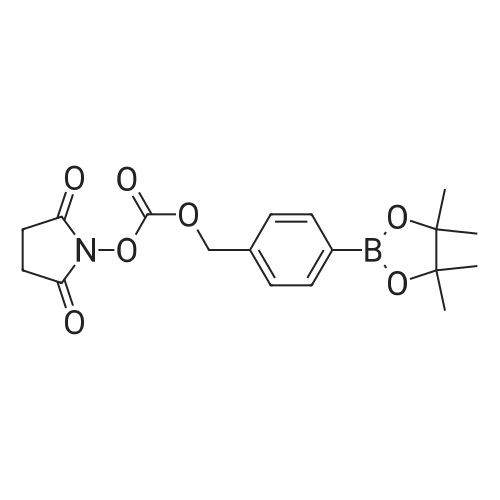
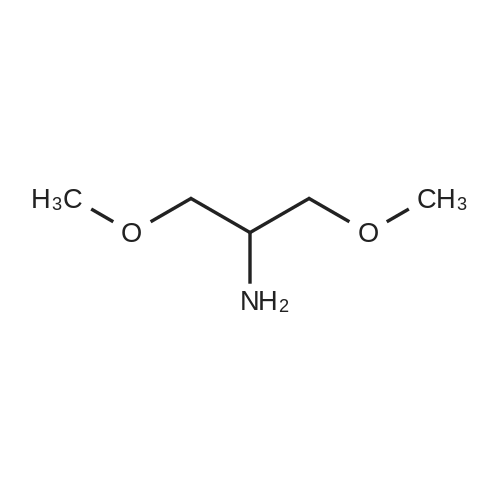
[ 74124-79-1 ]
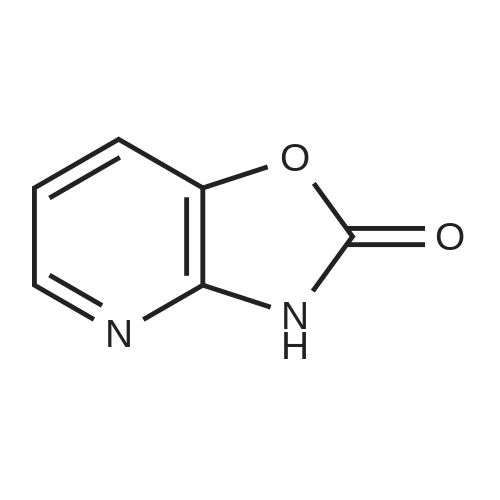
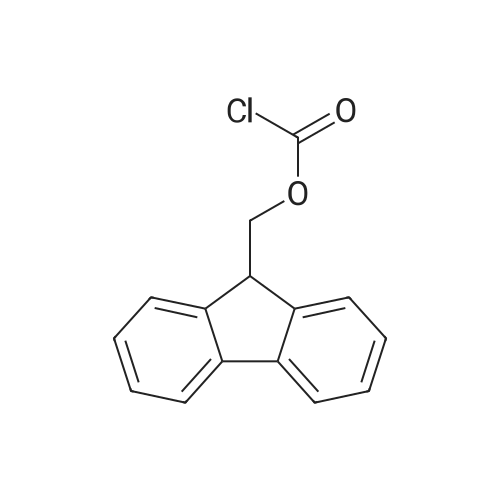
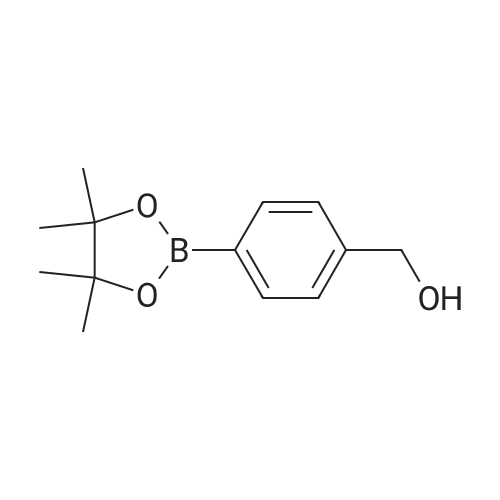
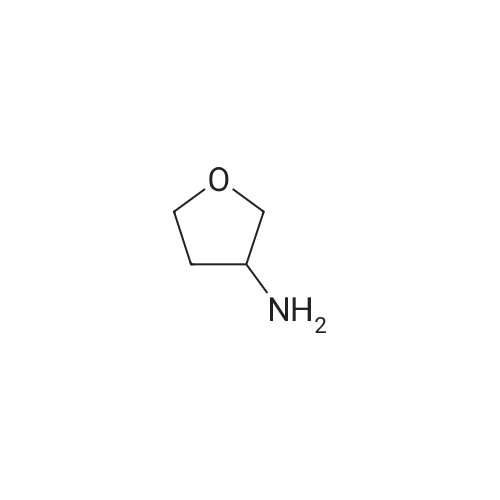
[ 1142-20-7 ]
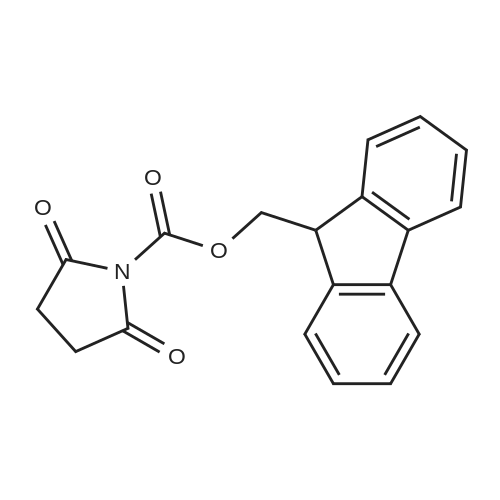
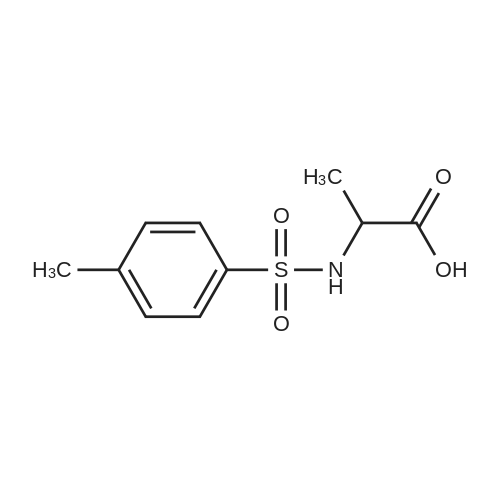
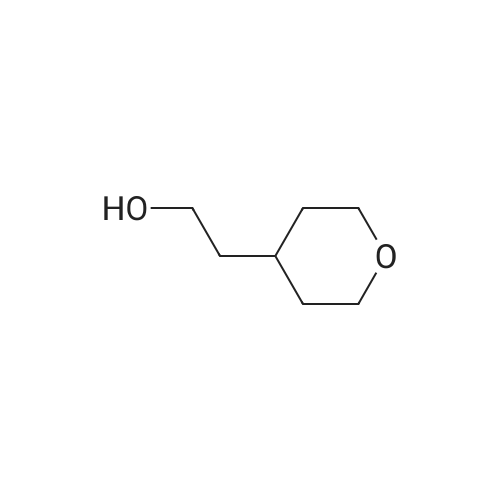
[ 3401-36-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 20, p. 5559 - 5562
6
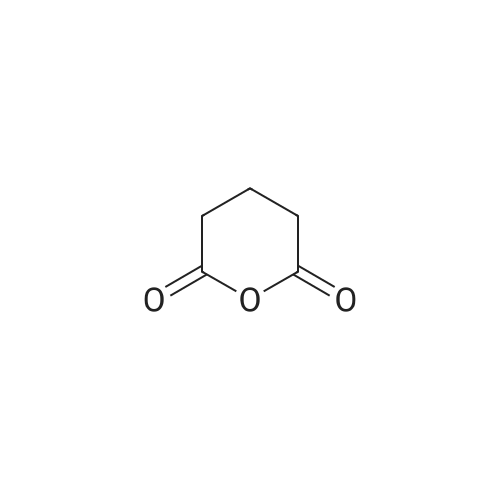
[ 74124-79-1 ]
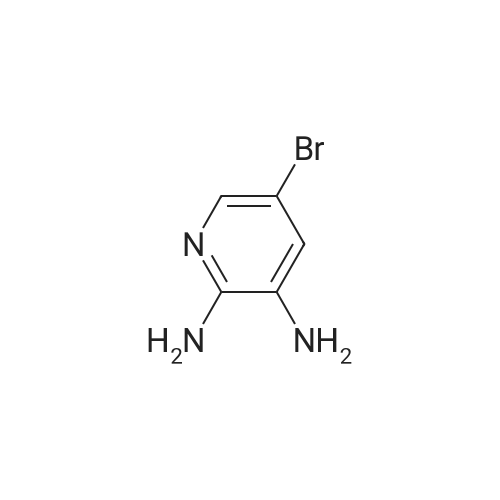
[ 100-51-6 ]
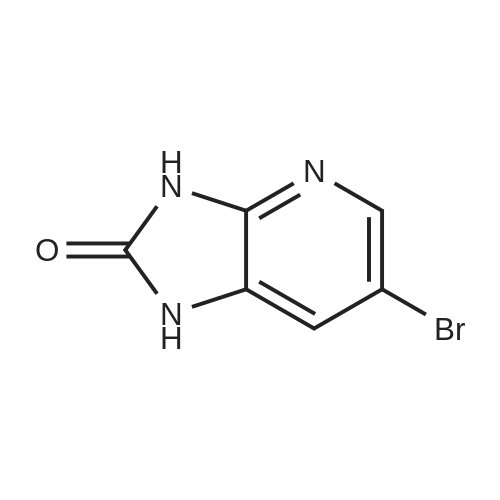
[ 13139-17-8 ]
Reference:
[1] European Journal of Medicinal Chemistry, 1999, vol. 34, # 7-8, p. 625 - 638
[2] Patent: WO2008/64218, 2008, A2, . Location in patent: Page/Page column 72; 83-85; 86-87; 98-99; 125
7
[ 6066-82-6 ]
[ 28920-43-6 ]
[ 74124-79-1 ]
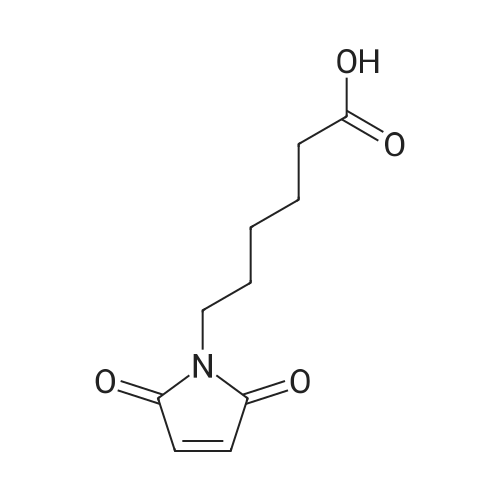
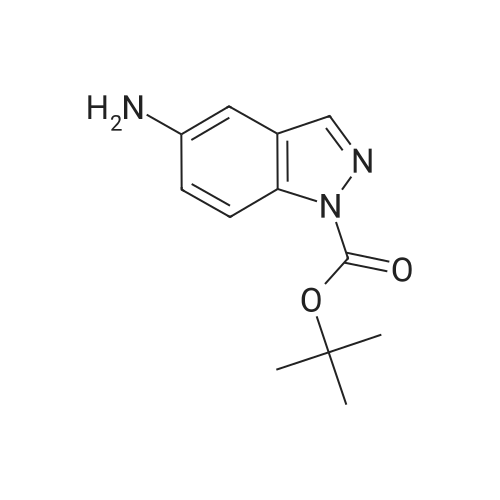
[ 102774-86-7 ]
Yield
Reaction Conditions
Operation in experiment
95 kg
With tributyl-amine In tetrahydrofuran at -2 - 5℃; Large scale
From the reactor solid feed port one - time investment HosuL43. 8Kg (l. 25KmOl),With stirring,So that the kettle liquid at -2 ° C ~ 0 ° C.The tributylamine300Kg (l. 62 kP)And tetrahydrofuran450Kg into the high slot,After mixing,In 10 ~ llhr slowly added to the reactor in the reaction,The amount of added per hour for the 50 ~ 57Kg;And keep the kettle temperature not to exceed 5 ° C,Because of the exothermic reaction,After adding tributylamine in tetrahydrofuran solution,The reaction vessel was allowed to warm to room temperature and the reaction was continued for 7.5 h.A large amount of white solid crystals were observed to be precipitated by DSC from the reaction vessel.(3) filtered DSC crude: the reactor material in lhr evenly introduced into a fully enclosed centrifuge in N2Under the protection of throwing dry, and then twice the introduction of about 0 ° C tetrahydrofuran solvent 100Kg in the centrifuge rinse, throw dry,The DSC wet goods 120Kg. The wet goods in a vacuum oven at 40 ~ 45 ° C drying 7 ~ 8hr to constant weight, get out of the white crystalline powder 95Kg; HPLC analysis of 99. 1percent. The resulting filtrate was centrifuged to proceed to the next step. (3) filtered to obtain DSC crude: the reactor material in 1hr evenly introduced into a fully enclosed centrifuge, in the N2Under the protection of throwing dry, and then twice the introduction of about 0 ° C tetrahydrofuran solvent 100Kg in the centrifuge rinse, throw dry, get DSC wet goods 120Kg.The wet product in a vacuum oven at 40 ~ 45 dry 7 ~ 8hr to constant weight, get out of the white crystalline powder 95Kg; HPLC analysis of 99.1percent.The resulting filtrate was centrifuged to proceed to the next step.(4) The centrifugal filtrate in step (3) is combined with two washing solvents and introduced into a 2000L distilling apparatus. The tetrahydrofuran solvent is removed at a vacuum control temperature of 0 to 5 DEG C, and the inside temperature of the kettle is kept at 40 to 42 DEG C .After the tetrahydrofuran was removed, 230 kg of dichloroethane was introduced, stirred at 40-50 ° C for 0.5 hr, and washed with 160 kg of water 3 times. The phases were separated and the dichloromethane was introduced into a 500 L autoclave.
With sodium hydrogencarbonate In N,N-dimethyl-formamide at 0℃; for 1 h; Inert atmosphere
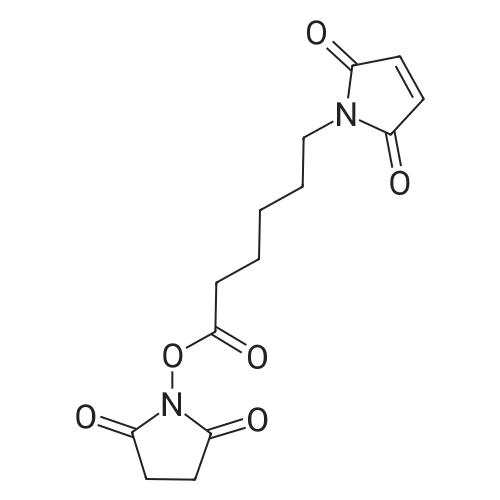
6-Maleimidohexanoic acid N-hydroxysuccinimide ester 4. Sodium bicarbonate (0.0512 g, 0.483 mmol ) was added to a solution of 6-maleimidocaproic acid (0.211 g, 1.000 mmol) in DI water (10 mL). After the reagents were dissolved in DI water, the DI water was evaporated by blowing with air. The resulting solid was dissolved in anhydrous DMF (3 mL) and the solution was cooled to 0 °C. Disuccinimide carbonate (0.282 g, 1.10 mmol) was added into the solution and the reaction was stirred at 0 °C for 1 h. CH2C12 (25 mL) was added into reaction which was then washed with water (3 x 10 mL). The resulting organic layer was dried over Na2S04, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: MeOH; solvent B: CH2C12; gradient: 0percentA / 100percentB (1 CV), 0percentA / 100percentB→ 2percentA / 98percentB (10 CV), 2percentA / 98percentB (2 CV); flow rate: 25 mL/min; monitored at 210 and 280 nm] to afford ester 4 (0.215 g, 0.697 mmol, 70percent yield) as a light yellow liquid. [000194] NMR (500 MHz, CDC13) 66.69 (2H, s), 3.53 (2H, t, J= 7.1 Hz), 2.83 (4H, s), 2.60 (2H, t, .7=7.4 Hz), 1.78 (2H,p, .7=7.5 Hz), 1.63 (2H,p, .7=7.4 Hz), 1.41 (2H,p,J=7.6 Hz). [000195] 13CNMR(125 MHz, CDC13) δ 170.9, 169.2, 168.5, 134.2,37.5,30.8,28.1,25.9,25.7,24.1.
Reference:
[1] Journal of the American Chemical Society, 1994, vol. 116, # 14, p. 6101 - 6106
[2] Patent: WO2017/66668, 2017, A1, . Location in patent: Paragraph 000193-000195
With N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 40℃; for 1 h;
Lauric acid (1.2 g, 5.99 mmol, 1 eq) is dissolved in N, N-dimethylformamide (DMF, 70 ml). N, N-disuccinimidyl carbonate (DSC, 4.6 g, 17.97 mmol, 3 eq) and N, N-diisopropylethylamine Hunig base, 10.2 ml, 59.91 mmol, 10 eq), and the mixture was stirred at 40 ° C for 1 hour. After completion of the reaction, the compound was lyophilized to obtain Compound 2-1 (2.1 g, 100percent)
With triethylamine In acetonitrile at 20℃; for 3 h;
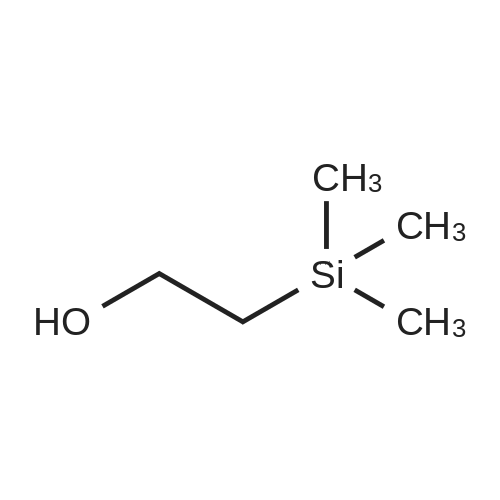
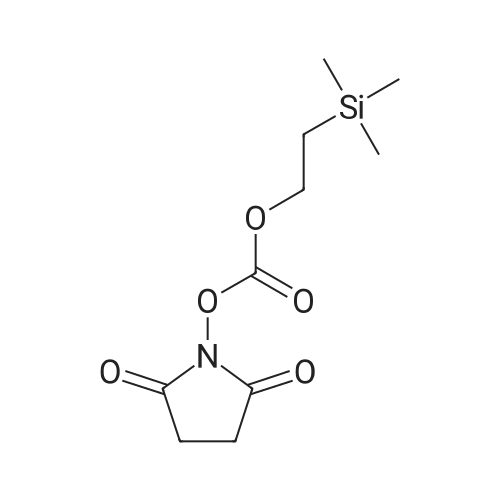
Compound 41; TEA (21.9 mL, 155.6 mmol, 3.0 eq.) was added to 2-trimethylsilanyl- ethanol (7.4 mL, 51.88 mmol, 1.0 eq.) in 260 mL MeCN, followed by di- succinimidyl carbonate (20 g, 1.5 eq.). The reaction mixture was stirred at room temperature for 3 hours. The reaction mixture was concentrated and extracted using EtOAc/ saturated NaHCO3. The organic layer was concentrated after dried over Na2SO4. Ether (100 mL) was added to the residue to form a precipitate. The precipitate was filtered and dried to give Compound 41 as a white solid (11.1 g, 84percent). 1H NMR (300 MHz, CDCl3): δ 4.42 (t, 3 H), 2.82 (s, 4 H), 1.16 (t, 2 H), 0.1 (s, 9 H).
82%
With triethylamine In acetonitrile at 25℃; for 16 h;
Example 5; methyl (l-l- ( {2- [ (2, 36)-3-amino-2-hydroxy-4-phenylbutyl]-2- benzylhydrazino} carbonyl)-2, 2-dimethylpropylcarbamate; Example 5A; 1- (f [2- (trimethylsilyl) ethoxy] carbonyl} oxy)-2, 5-pyrrolidinedione; Trimethylsilylethanol (7.4 mL, 52 mmol) was dissolved in acetonitrile (260 mL) and treated with disuccinimoyl carbonate (20 g, 1.5 equivalents) and triethylamine (33 mL, 3 equivalents) at 25°C for 16h. The solvents were evaporated, and the residue was partitioned between ethyl acetate and saturated sodium bicarbonate, the organic layer was separated and washed with brine, dried over sodium sulfate, filtered and concentrated. The crude residue was triturated with ether to form a solid which was filtered and dried to give 11.12 g (82percent) of the title compound.
82%
With triethylamine In acetonitrile
EXAMPLE 5A 1-([2-(trimethylsilyl)ethoxy]carbonyl}oxy)-2,5-pyrrolidinedione Trimethylsilylethanol (7.4 mL, 52 mmol) was dissolved in acetonitrile (260 mL) and treated with disuccinimoyl carbonate (20 g, 1.5 equivalents) and triethylamine (33 mL, 3 equivalents) at 25° C. for 16 h. The solvents were evaporated, and the residue was partitioned between ethyl acetate and saturated sodium bicarbonate, the organic layer was separated and washed with brine, dried over sodium sulfate, filtered and concentrated. The crude residue was triturated with ether to form a solid which was filtered and dried to give 11.12 g (82percent) of the title compound.
With pyridine In tert-butyl methyl ether at 25 - 40℃; for 62 h;
26.3 g of tert-butyl methyl ether,35.09 g (135 mmol) of di (N-succinimidyl) carbonate,And 20.00 g (the content of the compound (II-1): 73.0percent) of the compound (II-1) and the compound (III-2) obtained in the same manner as in Example 2 were mixed at room temperature And the temperature was raised to 40 ° C. 11.89 g (150 mmol) of pyridine was added dropwise to the obtained solution at 40 ° C., and the mixture was stirred at 40 ° C. for 22 hours. The whole amount of the solution was cooled to 20 ° C., 73.0 g of 2-propanol was added dropwise at 20 ° C. to precipitate compound (I-1), cooled to 0 to 5 ° C. and then cooled to 0 to 5 ° C. for 19 hours Followed by stirring.The precipitated compound (I-1) was filtered and washed to obtain 27.83 g of compound (I-1) (content: 97.8percent, yield 90percent, enantiomeric excess of compound (I-1) Rate> 99.9percent ee). 30.0 g of tert-butylmethyl ether, 11.87 g of the compound (II-0)(The diastereomer ratio of the compound (II-1) to the 3S, 3aS, 6aR-OH form: 91.1 / 8.9 containing 84.3percent diastereomer) and the enzyme (CHIRAZYME L-2c, -flyo, manufactured by Roche Diagnostics) was added to the mixture at a temperature of 25 ° C. 3.31 g (38.4 mmol) of vinyl acetate was added dropwise to the mixture,After stirring at 25 ° C. for 40 hours, insoluble matter was filtered off.The filtrate was concentrated to obtain 12.73 g of a mixture containing compound (II-1) and compound (III-2) (yield of compound (II-1): 90percent 3S, 3aS, 6aR-OH isomer: 100.0 / 0.0). (II-1) and the compound (III-2) obtained in the same manner as in Example 2, 26.3 g of tert-butyl methyl ether, 35.09 g (135 mmol) of di (N-succinimidyl carbonate) And 20.00 g of the compound (II-1) in an amount of 73.0percent) were mixed at room temperature, and the mixture was heated to 40 ° C. To the resulting solution, 11.89 g (150 mmol) of pyridine was added dropwise at 40 ° C. And the mixture was stirred for 22 hours at 40 ° C. The total amount of the solution was cooled to 20 ° C. and 73.0 g of 2-propanol was dropwise added at 20 ° C. to precipitate the compound (I-1) and cooled to 0 to 5 ° C. , And the mixture was stirred for 19 hours at 0 to 5 ° C. The precipitated compound (I-1) was filtered,(Yield: 97.8percent, yield: 90percent, enantiomer excess of compound (I-1)> 99.9percent ee) was obtained by filtration and washing with water to obtain 27.83 g of compound (I-1).
Reference:
[1] Patent: JP2016/150901, 2016, A, . Location in patent: Paragraph 0016; 0017; 0023; 0024
[2] Journal of Organic Chemistry, 2004, vol. 69, # 23, p. 7822 - 7829
[3] European Journal of Organic Chemistry, 2016, vol. 2016, # 10, p. 1874 - 1880
[4] Journal of Medicinal Chemistry, 2005, vol. 48, # 6, p. 1813 - 1822
[5] Patent: WO2010/23322, 2010, A1, . Location in patent: Page/Page column 29
[6] Patent: US2008/85918, 2008, A1, . Location in patent: Page/Page column 18; Sheet 3
[7] Patent: WO2011/92687, 2011, A1, . Location in patent: Page/Page column 31
[8] Patent: WO2016/207907, 2016, A1, . Location in patent: Page/Page column 23
[9] Organic Process Research and Development, 2017, vol. 21, # 1, p. 98 - 106
[10] Patent: WO2007/126812, 2007, A2, . Location in patent: Page/Page column 41
[11] Patent: WO2008/133734, 2008, A2, . Location in patent: Page/Page column 28-29
α-methyl-6-nitropiperonylol succinimidyl carbonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
With triethylamine; In acetonitrile; at 20℃;
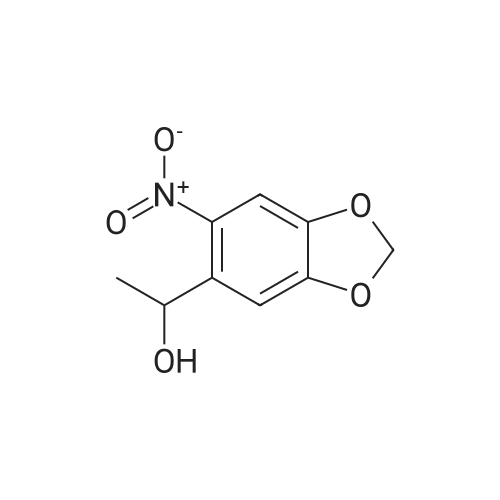
l-(3,4-(Methylenedioxy)-6-nitrophenyl)ethanol was synthesized as reported (Lusic, H.; Deiters, A., Synthesis 2006, 2147-2150) and 200 mg (0.947 mmol) were dissolved in 5 ml of dry CH3CN. To the solution were added N.N'-disuccinimidyl carbonate (485 mg, 1.894 mmol) and TEA (0.396 ml, 2.840 mmol). The reaction was stirred at rt overnight. The solvent was removed under reduced pressure and product was directly purified by column chromatography on Si02 (eluted with hexanes/ethyl acetate 5: 1), delivering 296 mg (89percent yield) of NPOC-NHS as a light yellow solid. 3/4 NMR (300 MHz, CDC13) delta 7.49 (s, 1H), 7.09 (s, 1H), 6.39 (q, J = 6.4 Hz, 1H), 6.14-6.12 (m, 2H), 2.79 (s, 4H), 1.73 (d, J = 6.4 Hz); 13C NMR (100 MHz, CDC13) delta 168.7, 153.0, 150.8, 148.0, 141.6, 133.2, 105.9, 105.6, 103.5, 76.5, 25.6, 22.3; MS calcd C14H12N2Na09 375.04405, found 375.1.
4-[(2S)-2-amino-3-methylbutanoyl]-1'-cyclopropylcarbonyl-hexahydropyrrolo-(3'aS,6'aR)-spiro[cyclobutane-1,3'-pyrrolo[3,2-b]pyrrol]-2(1H)-one hydrochloride[ No CAS ]
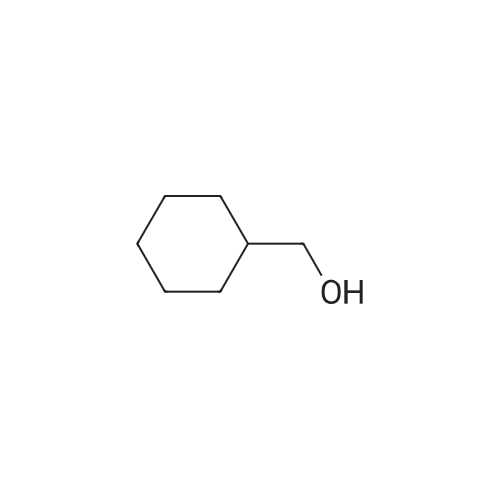
Stage a Synthesis of the activated alcohol N-succinimidyl carbonic acid cyclohexylmethyl ester 5.38 g (10.5 mmoles)of di(N-succinimidyl)carbonate is solubilized in 60 ml of methylene chloride. 2.16 ml (17.55 mmoles) of cyclohexylmethanol solubilized in 10 ml of methylene chloride and 4.92 ml (35.1 mmoles) of triethylamine are added at ambient temperature. The reaction mixture is left under stirring overnight. Then, the reaction mixture is concentrated to dryness before extraction with ethyl acetate and washing with a saturated solution of sodium bicarbonate. The organic phase obtained is dried over magnesium sulphate before being concentrated to dryness under reduced pressure (2 kPa). 4.4 g of activated alcohol is obtained which is used as it is in the following stage. (quantitative yield) TLC: Rf=0.84 (silica gel, eluent: ethyl acetate -triethylamine (90-10). 1H-NMR (CDCl3): delta 1.02 (m, 2H, CH2-CH2--CH2-CH2); 1.25 (m, 4H, CH2--CH2--CH2); 1.75 (m, 5H, CH2-CH2--CH2-CH2 and CH2--CH--CH2); 2.85 (s, 4H, CO---CO); 4.15 (d, 2H, OCOO--cyclohexyl)
With pyridine; triethylamine; methylamine; In ethanol; water; acetonitrile;Product distribution / selectivity;
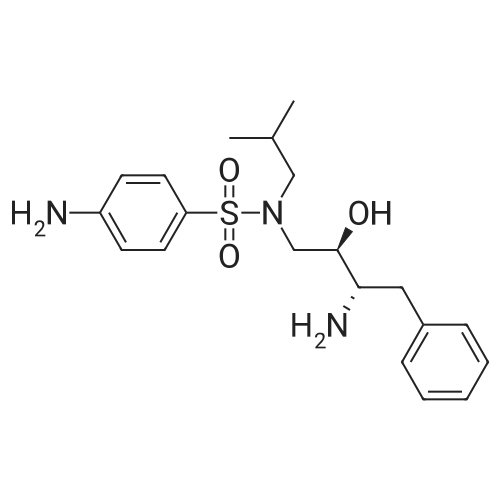
Example 9: Preparation of 3aS. 6aR-hexahvdrofuro [2. 3-bl]furan-3-yl (1S, 2R)-3- [[4-aminophenvl) suulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate ethanolate. 100 mmol (3R, 3aS, 6aR)-hexahydrofuro [2, 3-b] furan-3-ol in acetonitrile were added onto 110 mmol of disuccimidylcarbonate (95%) in acetonitrile. Following 300 mmol pyridine was added and stirred. The mixture was cooled and treated with a suspension of 95 mmol of 4-Amino-N-( (2R, 3S)-3-amino-2-hydroxy-4-phenylbutyl)-N- (isobutyl)- benzene sulfonamide in acetonitrile, followed by 100 mmol of triethylamine 20 mmol methylamine, 41% aqueous solution in water were added and the mixture was warmed 80 g solvent were distilled off, MTBE was added and the reaction mixture was washed with 10% Na2C03-solution, with a mixture of sodium sulfate in sulfuric acid and again with 10% Na2CO3-solution. Solvent was evaporated and ethanol was added. Another portion of solvent was distilled off. The temperature was kept around 40-45 C and crystallization was initiated by seeding. After stirring the mixture was cooled, stirred for another 90 min stirred, cooled and again stirred for 60 min. The precipitate was filtered and washed with ethanol. The wet product was dried in vacuo at 40 C. 43.5 g of (3R, 3aS, 6aR)-hexahydrofuro [2, 3-b] furan-3-yl (lS, 2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate were suspended in ethanol abs. and dissolved. The clear solution was cooled and seeding was applied. Crystallization occurred while cooling the mixture. Stirring was continued for another 60 min, followed by cooling, stirring and filtering off the product, which was washed with cold ethanol abs. The wet product was dried in vacuo at 40 C. Yield: 48.1 g = 81 %.
71%
With triethylamine; methylamine; In ethanol; water; ethyl acetate; acetonitrile;
Example 7: Preparation of (3R3aS. 6aR1-hexahydrofuro f2. 3-b] furan-3-vl (1S. 2R)-3- [[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate ethanolate. 100 mmol (3R, 3aS, 6aR)-hexahydrofuro [2,3-b] furan-3-ol in ethyl acetate were added onto 120 mmol of disuccimidylcarbonate (95%) in acetonitrile. Following, a solution of 140 mmol triethylamine in ethylacetate was added and stirred. The mixture was cooled and treated with a suspension of 92 mmol of 4-Amino-N-((2R, 3S)-3-amino-2- hydroxy-4-phenylbutyl)-N-(isobutyl) benzene sulfonamide in ethyl acetate. 20 mmol methylamine, 41% aqueous solution in ethanol were added and the mixture was warmed. The reaction was washed twice with 10% Na2CO3-solution and with water. Solvent was evaporated and ethanol was added. Another portion of solvent was distilled off. The temperature was kept around 40-45 C and crystallization was initiated by seeding. After stirring the mixture was cooled, stirred for another 90 min stirred, cooled and again stirred for 60 min. The precipitate was filtered and washed with ethanoL The wet product was dried in vacuo at 40 C. 43.5 g of (3R, 3aS, 6aR) - hexahydrofuro [2, 3-b] furan-3-yl(1S,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate were suspended in ethanol abs. and dissolved. The clear solution was cooled and seeding was applied. Crystallization occurred while cooling the mixture. Stirring was continued for another 60 min, followed by cooling, stirring and filtering off the product, which was washed with cold ethanol abs. The wet product was dried in vacuo at 40 C. Yield: 42. 1 g = 71 %.
To a mixture of (3R,3aS,6aR)-hexahdrofuro [2,3-b] furan-3-ol (25 gm) and acetonitrile (180 ml) was added disuccinimidyl carbonate (56 gm) and pyridine (46 gm) at 25 to 30C. The mixture was stirred for 1 hour at 25 to 30C and cooled to 0C. A solution of 4-amino-N-((2R,3S)-3-amino-2-hydroxy-4- phenylbutyl)-N-(isobutyl)benzene sulfonamide (70 gm) in acetonitrile (300 ml) was added to the reaction mass at 0 to 5C for 30 minutes. To the reaction mass was added triethylamine (19 gm) and monomethylamine (3 gm) at 0 to 5C, the temperature was slowly raised to 25 to 30C and stirred for 22 hours. Distilled off the solvent completely under vacuum at 45C to obtain a residue and to the residue was added ethyl acetate (250 ml). The ethyl acetate layer was washed with 10% sodium bicarbonate (100 ml), 2% sulfuric acid (100 ml), 10% sodium sulfate (100 ml) and 10% sodium chloride solution (100 ml). The layer was dried over sodium sulfate. The layer was treated with carbon and distilled off the solvent under vacuum at below 45C to obtain 85 gm of darunavir.
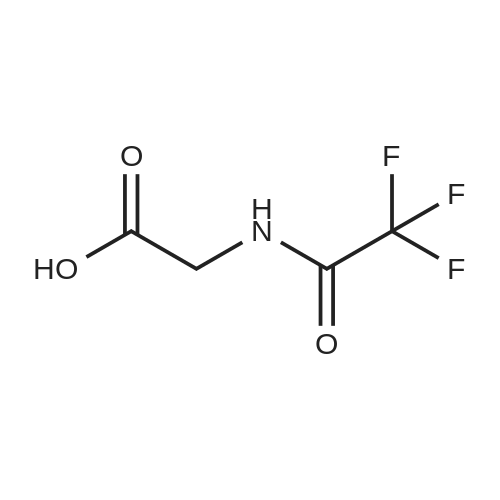
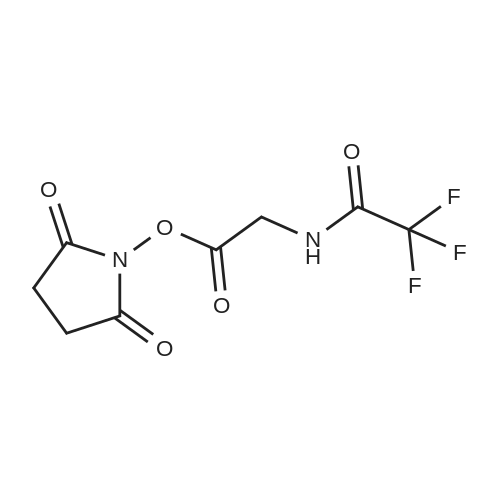
Disuccinimidyl carbonate (300 mg, 1.17 mmoles) was added to a solution of 13 (200 mg, 1.17 mmoles) and pyridine (94.6 muL, 1.17 mmoles) in acetonitrile (1.0 mL). The reaction mixture was stirred at room temperature for 3 h, during which the solution became clear and evolved gas. The solution was added to ethyl acetate (10 mL), washed with 1N HCl (2 x 5 mL) and saturated aqueous NaHCO3 (2 x 5 mL), dried over MgSO4, and concentrated in vacuo to yield a white solid (232 mg, 865 mumoles, 74%). LRMS: (ES+) calcd for C8H7N2O5F3 (M + NH4): 286; found: 286. 1H NMR: spectrum is consistent with the predicted structure.
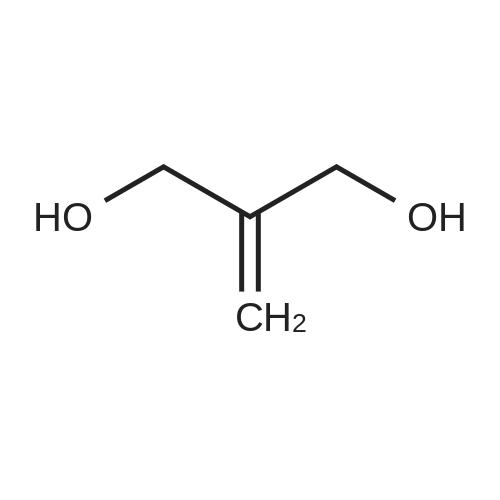
A. N-hydroxysuccinimidyl-1-methoxypropane-3-carbonate A solution of 355 mg of <strong>[3513-81-3]2-methylene-1,3-propanediol</strong> in acetonitrile (30 mL) was added sequentially, at ambient temperature, 65 mg of sodium hydride and 0.25 mL iodomethane. The mixture was stirred for 12 h and concentrated in vacuo. The residue was then taken up in 15 mL of acetonitrile and treated sequentially, at ambient temperature under an atmosphere of nitrogen, with 1.3 g of N,N-disuccinimidyl carbonate and 1.6 mL of triethylamine. After stirring for 14 h, the reaction mixture was concentrated in vacuo and the residue was diluted CH2 Cl2, washed with saturated sodium bicarbonate solution and saturated brine, dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography with EtOAc as eluant to give 95 mg of the title compound. (1 H)-NMR (CDCl3) consistent with structure.
With triethylamine; In acetonitrile;
A. N-hydroxysuccinimidyl-1-methoxypropane-3-carbonate A solution of 355 mg of <strong>[3513-81-3]2-methylene-1,3-propanediol</strong> in acetonitrile (30 mL) was added sequentially, at ambient temperature, 65 mg of sodium hydride and 0.25 mL iodomethane. The mixture was stirred for 12 h and concentrated in vacuo. The residue was then taken up in 15 mL of acetonitrile and treated sequentially, at ambient temperature under an atmosphere of nitrogen, with 1.3 g of N,N-disuccinimidyl carbonate and 1.6 mL of triethylamine. After stirring for 14 h, the reaction mixture was concentrated in vacuo and the residue was diluted CH2 Cl2, washed with saturated sodium bicarbonate solution and saturated brine, dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography with EtOAc as eluant to give 95 mg of the title compound. (1 H)-NMR (CDCl3) consistent with structure.
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 3h;
[0616] To a room temperature suspension of Fmoc-MeVal-OH (3.03 g, 8.57 mmol) and N,N'-disuccimidyl carbonate (3.29 g, 12.86 mmol) in CH2Cl2 (80 mL) is added DIEA (4.48 mL, 25.71 mmol). This reaction mixture is allowed to stir for 3 hr, and then poured into a separation funnel where the organic mixture is extracted with 0.1 M HCl (aq). The crude organic residue is concentrated under reduced pressure, and the product is isolated by flash column chromatography on silica gel using a 20-100% ethyl acetate/hexanes linear gradient {e.g., a total of 2.18 g of pure Fmoc-MeVal-OSu (4.80 mmoles, 56% yield) can be recovered).
56%
With N-ethyl-N,N-diisopropylamine; In dichloromethane; at 20℃; for 3h;
[0561] To a room temperature suspension of Fmoc-MeVal-OH (3.03 g, 8.57 mmol) andN,N'-disuccimidyl carbonate (3.29 g, 12.86 mmol) in CH2Cl2 (80 mL) is added DIEA (4.48 mL, 25.71 mmol). This reaction mixture is allowed to stir for 3 hr, and then poured into a separation funnel where the organic mixture is extracted with 0.1 M HCl (aq). The crude organic residue is concentrated under reduced pressure, and the product is isolated by flash column chromatography on silica gel using a 20-100% ethyl acetate/hexanes linear gradient. A total of 2.18 g of pure Fmoc-MeVal-OSu (4.80 mmoles, 56% yield) is EPO <DP n="172"/>recovered. To a room temperature suspension of Fmoc-MeVal-OSu (2.18 g, 4.84 mmol) in DME (13 niL) and THF (6.5 mL) is added a solution of L-citrulline (0.85 g, 4.84 mmol) and NaHCO3 (0.41 g, 4.84 mmol) in H2O (13 mL). The suspension is allowed to stir at room temperature for 16 hr, then it is extracted into ter?-BuOH/CHCl3/H2O, acidified to pH=2-3 with 1 M HCl. The organic phase is separated, dried and concentrated under reduced pressure. The residue is triturated with diethyl ether resulting in 2.01 g of Fmoc-MeVal-Cit-COOH which is used without further purification.
56%
With N-ethyl-N,N-diisopropylamine; In dichloromethane; for 3h;
To a room temperature suspension of Fmoc-MeVal-OH (3.03 g, 8.57 mmol) and N,N?-disuccimidyl carbonate(3.29 g, 12.86 mmol) in CH2Cl2 (80 mL) was added DIEA (4.48 mL, 25.71 mmol). This reaction mixture was allowed tostir for 3.0 hr, and then poured into a separation funnel where the organic mixture was extracted with 0.1 M HCl (aq).The crude organic residue was concentrated under reduced pressure, and the product was isolated by flash columnchromatography on silica gel using a 20-100% ethyl acetate/hexanes linear gradient. A total of 2.18 g of pure Fmoc-MeVal-OSu (4.80 mmoles, 56% yield) was recovered.
(+/-)-3-(6-methoxy-pyridin-3-yl)-2-[4-(2-oxo-1,4-dihydro-2H-quinazolin-3-yl)-piperidine-1-carbonyl]-amino}-propionic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
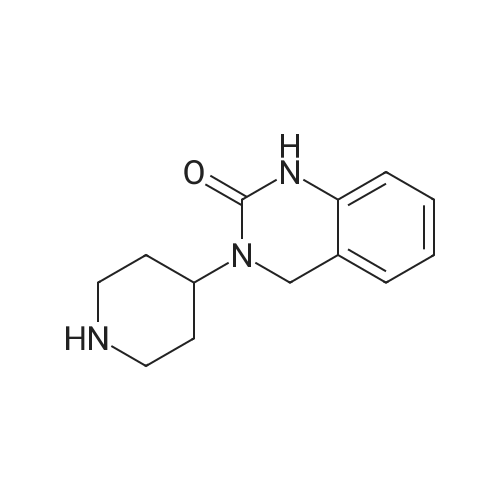
To a solution of [2-AMINO-3- (6-METHOXY-PYRIDIN-3-YL)-PROPIONIC] acid methyl ester (130 mg) and diisopropylethylamine (0.3 mL) in methylene chloride (2 mL, [0C)] was added N, N'-disuccinimidyl carbonate (158 mg). After 30 min, 3-piperidin- 4-yl-3, [4-DIHYDRO-LH-QUINAZOLIN-2-ONE] (120 mg) in methylene chloride [(1] mL) was added via canula. The reaction was warmed to room temperature and stirred overnight. The reaction was concentrated and purified by prep HPLC to give 160mg (55%). Mass spec.: 468.19 [(MH) +.]
160 mg (55%)
With N-ethyl-N,N-diisopropylamine; In dichloromethane;
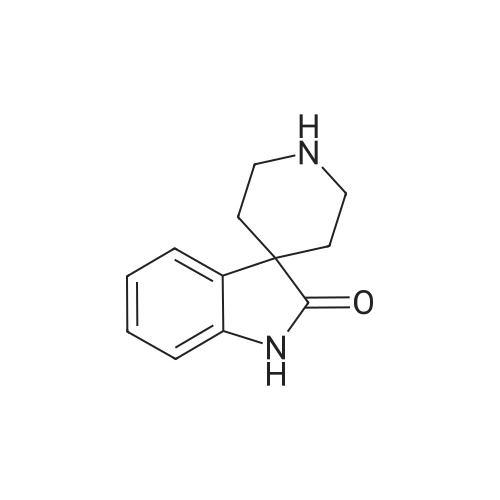
(+-)-3-(6-Methoxy-pyridin-3-yl)-2-[4-(2-oxo-1,4-dihydro-2H-quinazolin-3-yl)-piperidine-1-carbonyl]-amino}-propionic acid methyl ester To a solution of 2-amino-3-(6-methoxy-pyridin-3-yl)-propionic acid methyl ester (130 mg) and diisopropylethylamine (0.3 mL) in methylene chloride (2 mL, 0 C.) was added N,N'-disuccinimidyl carbonate (158 mg). After 30 min, <strong>[79098-75-2]3-piperidin-4-yl-3,4-dihydro-1H-quinazolin-2-one</strong> (120 mg) in methylene chloride (1 mL) was added via canula. The reaction was warmed to room temperature and stirred overnight. The reaction was concentrated and purified by prep HPLC to give 160 mg (55%). Mass spec.: 468.19 (MH)+.
Synthesis 20-1 -A; 6-Chloro-7H-purin-8(9H)-one; NXrNH2 A mixture of 4-chloropyrimidine-2,3-diamine (36 mg, 0.25 mmol) and di(/V-succinimidyl)carbonate (128 mg, 0.50 mmol) in acetonitrile (10 mL) was refluxed for 16 hours. The solids formed were collected, washed with acetonitrile (2 x 5 mL) and dried, to give the title compound as a light yellow powder (33 mg, 78%).1H NMR (500 MHz, de-DMSO) delta 8.35 (1 H, s), 11.90 (2H, s, broad); LC-MS (2) R1 1.96 min; m/z (ESI) 171 [MH+].
A solution OF3-METHYL-4-NITROANILINE (231 mg, 1.52 mmol) and triethylamine (0.42 mL, 3.04 mmol) in dichloromethane (10 mL) was added slowly to a suspension of disuccinylcarbonate (389 mg, 1.52 mmol) in dichloromethane (10 mL) during which time the suspension became homogeneous. The reaction was stirred until all starting aniline was consumed, then treated with the aniline prepared from 4-fluorobenyl bromide using procedure described in example 1 (300 mg, 1.17 mmol) and stirred at rt overnight. The reaction mixture was concentrated to dryness, then diluted with DMF and heated to 90C for 4 hrs at which time it was cooled to rt, diluted with water which was then extrated twice with ethyl acetate and the combined organic layers then dried over magnesium sulfate. After concentration, the crude product was purified by silica gel chromatography. The resulting yellow solid was then reduced as described in Example 2 affording 10 mg of the desired aniline (10% yield, 2 steps). ES+ MS showed 491 m/z, the correct mass for the product NMR (400 MHz, CDC13) showed 8 = 1.40 (s, 9 H), 2.20 (s, 3 H), 4.83 (s, 2 H), 6.73 (M, 1 H), 6.86 (M, 7 H), 7.13 (M, 2 H), 7.99 (M, 2 H), 10.08 (s, 1 H).
With triethylamine; In dichloromethane; at 20℃; for 2.16667 - 2.25h;
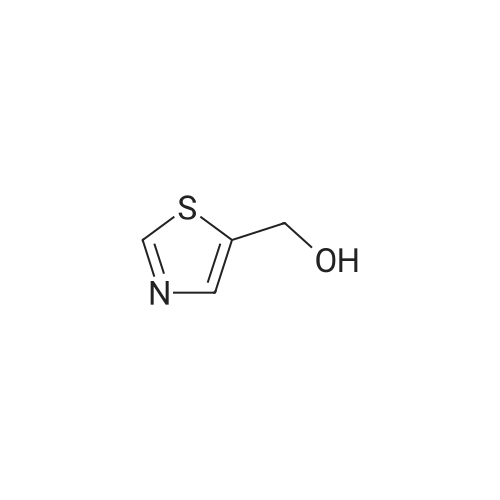
A mixture of 1.15 g of <strong>[38585-74-9]thiazol-5-yl-methanol</strong> (1-1) and 1.2 g triethylamine (TEA) in 25 ml of dichloromethane (DCM) was stirred at room temperature under an atmosphere of nitrogen. 2.56 g of N,N'-disuccinimidyl carbonate was then added and the resulting mixture was stirred for 10-15 minutes. The solution was stirred for an additional 2 hours. The resulting intermediate (1-2) was used directly in the subsequent reaction with the amine (1-3). Instead of amines also salts thereof can be used.
With triethylamine; In acetonitrile; at 20℃; for 3h;
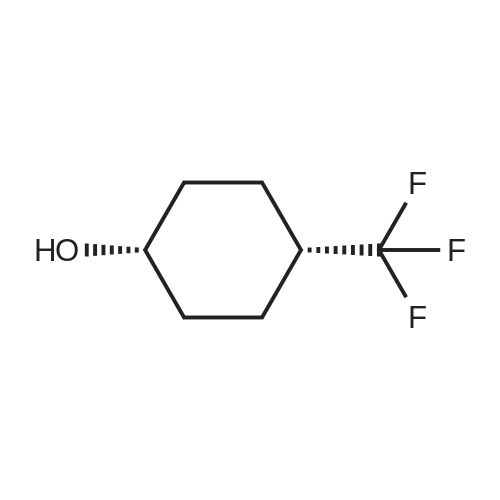
Example 12Preparation of Compounds 162 and 167 To a solution of 4-trifluorophenylcyclohexanol (0.225 g, 1.34 mmol) dissolved inCH3CN (4.0 mL) was added triethylamine (0.56 mL, 4.02 mmol), then disuccinimidyl carbonate (0.307 g, 1.61 mmol). To the resulting mixture was added dropwise a solution of the HCl salt of compound 2 (0.15 g, 0.41 mmol) in CH2Cl2 (3.0 mL) and triethylamine (0.1 mL). The resulting reaction was allowed to stir for about 3 hours at room temperature, then was diluted with CH2Cl2, washed with water and the organics were concentrated in vacuo. The resulting residue was purified using flash column chromatography (20% acetone/hexanes) to provide the separate isomeric compounds 162 and 167 (0.183 g, combined yield of 85%) with the isomer 167 as the major product.
(+/-)-3-(2-methoxy-pyrimidin-5-yl)-2-[4-(2-oxo-1,4-dihydro-2H-quinazolin-3-yl)-piperidine-1-carbonyl]-amino}-propionic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
36%
To a solution of [2-AMINO-3-(2-METHOXY-PYRIMIDIN-5-YL)-PROPIONIC] acid methyl ester (125 mg) and diisopropylethylamine (0.3 mL) in methylene chloride (2 mL, [0C)] was added N, N'-disuccinimidyl carbonate (155 mg). After 30 min, 3- [PIPERIDIN-4-YL-3,] 4-dihydro-lH-quinazolin-2-one (120 mg) in methylene chloride (2 [ML)] was added via canula. The reaction was warmed to room temperature and stirred overnight. The reaction was concentrated and purified by prep HPLC to give 99mg (36%). Mass spec.: 469.10 [(MH)] [+.]
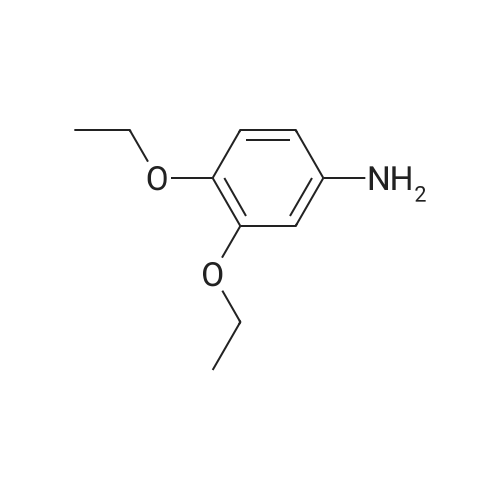
To a solution (6 ML) of 1-[5-amino-2-(benzhydryloxy)phenyl]-2-methylpropan-1-one (200 mg, 0.579 mmol) in acetonitrile were added diisopropylethyl amine (0.116 ML, 0.695 mmol) and N,N'-disuccinimidyl carbonate (178 mg, 0.695 mmol) under ice-cooling, and the mixture was stirred under ice-cooling for 1 hour.. To the reaction solution were added diisopropylethyl amine (0.116 ML, 0.695 mmol) and <strong>[39052-12-5]3,4-diethoxyaniline</strong> (126 mg, 0.695 mmol) under ice-cooling, and the mixture was stirred under ice-cooling for 1 hour and at room temperature for 12 hours.. The reaction solution was poured into water and was extracted with ethyl acetate.. The extracted solution was washed with water, and was dried with anhydrous magnesium sulfate.. The solvent was distilled off under reduced pressure, and the residue was purified by silicagel column chromatography (hexane:ethyl acetate = 2:1), to obtain the titled compound as a solid. 236 mg (73.8percent) 1H-NMR (CDCl3) delta; 1.04 (6H, d, J = 6.4 Hz), 1.39 to 1.46 (6H, m), 3.51 to 3.57 (1H, m), 3.99 to 4.11 (4H, m), 6.21 (1H, s), 6.64 to 7.58 (18H, m)
Example 6-3 [Show Image] To Compound 1 (154 mg) were added acetonitrile (5 ml)-THF (5 ml), then N,N'-disuccinimidyl carbonate (323 mg) and N,N'-dimethylaminopyridine (6 mg), and the mixture was stirred at room temperature for 20 hours. Then, thereto was added Compound 2 (178 mg), and the mixture was stirred at room temperature for 24 hours. To the reaction mixture was added 5percent aqueous potassium carbonate solution, and the mixture was extracted with ethyl acetate. The extract layers were combined, washed with brine, dried over anhydrous sodium sulfate, and then concentrated under reduced pressure. The resulting residue was dissolved in methanol (5 ml), and thereto were added water (2.5 ml) and potassium carbonate (346 mg) with stirring under ice cooling. The mixture was stirred at room temperature for 2.5 hours. To the reaction mixture was added water, and the mixture was extracted with ethyl acetate. The extract layers were combined, washed with brine, dried over anhydrous sodium sulfate, and then the solvent was distilled away under reduced pressure. The residue was purified by silica gel column chromatography (eluent: methanol-ethyl acetate 3:97 to 12:88 gradient) to give Compound 3 (91 mg) as a colorless oil. MS (APCI) 424 [M+H]+
Into a three-necked flask equipped with a magnetic stirrer and placed under N2 is introduced tert-butyl (2S)-2-[(3,4-dichlorophenyl)(hydroxy)methyl]piperidine-1-carboxylate (1.9 g, 5.2 mmol) dissolved in acetonitrile (30 mL). N,N'-Disuccinimyl carbamate (2.05 g, 8 mmol) and triethylamine (2.19 mL, 15.6 mmol) are then added and the reaction medium is stirred for 4 hours at RT. After concentrating the reaction medium by evaporation under RP, the residue thus obtained is taken up in saturated aqueous sodium hydrogen carbonate solution and the aqueous phase is extracted with EtOAc (3.x.30 mL). The organic phase is washed with aqueous NaCl solution, dried over MgSO4 and concentrated by evaporation under RP. The residue obtained is diluted in DCM (15 mL). This solution is then added dropwise to solution, prepared beforehand and placed in a one-necked flask, of <strong>[129488-10-4]5-amino-N-tert-butoxycarbonyl-1H-indazole</strong> (1.45 g, 6.2 mmol), DCM (40 mL) and triethylamine (1.1 mL, 7.8 mmol). The reaction medium is stirred at RT overnight. 40 mL of DCM and 30 mL of saturated aqueous sodium hydrogen carbonate solution are then added. After separation of the phases by settling, the organic phase is washed with aqueous NaCl solution, dried over MgSO4, filtered and concentrated by evaporation under RP. The residue is purified by chromatography on silica gel eluted with a 3/1 cyclohexane/EtOAc mixture. tert-Butyl (2S)-2-[(S)-(3,4-dichlorophenyl)[(1-(tert-butoxycarbonyl)-1H-indazol-5-yl)carbamoyl]oxy}methyl]piperidine-1-carboxylate (0.215 g) is thus obtained the form of a colourless lacquer and tert-butyl (2S)-2-[(R)-(3,4-dichlorophenyl)[(1-(tert-butoxycarbonyl)-1H-indazol-5-yl)carbamoyl]oxy}methyl]piperidine-1-carboxylate (0.434 g) is obtained in the form of a white foam. (M-H)-=617.
Into a one-necked flask equipped with a magnetic stirrer is introduced tert-butyl (2S)-2-[3-(ethoxycarbonyl)phenyl](hydroxy)methyl}piperidine-1-carboxylate (4.7 g, 12.8 mmol) dissolved in acetonitrile (85 mL) with N,N'-disuccinimidyl carbonate (13.1 g, 51 mmol). Triethylamine (8.95 mL, 64 mmol) is then added and the reaction medium is stirred for 4 hours at a temperature in the region of 20° C. The reaction medium is concentrated to dryness and the evaporation residue is taken up in saturated aqueous sodium hydrogen carbonate solution (50 mL) and extracted with twice 40 mL of EtOAc. A persistent insoluble material is removed by filtration of the organic phases through a sinter funnel. The filtrate is dried over MgSO4, filtered and concentrated to dryness under RP to give the activated intermediate. Into a second one-necked flask equipped with a magnetic stirrer is introduced tert-butyl 5-amino-indazole-1-carboxylate (3 g, 12.8 mmol) with DCM (125 mL) and triethylamine (2.7 mL, 19.1 mmol). Into this solution is poured the activated intermediate dissolved in DCM (40 mL) over about 10 minutes. The reaction medium is stirred in the region of 20° C. for 16 hours. The medium is hydrolysed with saturated aqueous sodium hydrogen carbonate solution (80 mL), the phases are separated by settling and the aqueous phase is re-extracted with DCM (30 mL). The combined organic extracts are dried over MgSO4, filtered and concentrated to dryness under RP. The garnet-coloured oil isolated is chromatographed on 420 g of silica gel 60, particle size 15-40 mum, contained in a column 5 cm in diameter, eluting with a 7/3v/v cyclohexane/EtOAc mixture, under an excess pressure of 0.6 bar of argon. The evaporation of the fractions gives 1.53 g of tert-butyl 5-[([(2S)-1-(tert-butoxycarbonyl)piperid-2-yl][3-(ethoxycarbonyl)phenyl]methoxy}carbonyl)amino]-1H-indazole-1-carboxylate in the form of a white-coloured foam. (M-H)-=621. 1H NMR (DMSO, 400 MHz): 70percent-30percent mixture of isomers, delta (ppm) from 0.98 to 2.00 (m, 27H); 2.98 (m, 1H); 3.90 (broad m, 1H); 4.33 (q, J=7.5 Hz, 2H); 4.50 (broad m, 1H); 6.09 (d, J=10.0 Hz, 0.7H); 6.23 (broad d, J=9.0 Hz, 0.3H); 7.50 (t, J=7.5 Hz, 0.7H); 7.58 (m, 1.3H); 7.68 (broad d, J=7.5 Hz, 0.7H); 7.75 (broad d, J=7.5 Hz, 0.3H); from 7.85 to 8.00 (m, 3H); 8.04 (broad s, 0.7H); 8.08 (broad s, 0.3H); 8.32 (s, 0.7H); 8.34 (s, 0.3H); 9.82 (broad m, 0.3H); 10.1 (s, 0.7H).
Example 1 :Preparation of crystalline hydrochloride salt of darunavirTo a mixture of (3R,3aS,6aR)-hexahydrofuro [2,3-b] furan-3-ol (66 gm) and acetonitrile (300 ml) was added disuccinimidyl carbonate (1 10 gm) at 25 to 30C. The reaction mass was cooled to 10C under nitrogen atmosphere and then added pyridine (93 gm) for 30 minutes. The temperature of the reaction mass was raised to 30C and stirred for 1 hour 30 minutes at 30C. The reaction mass further cooled to -10C under nitrogen atmosphere. A solution of 4-amino-N-((2R,3S)-3-amino-2-hydroxy-4- phenylbutyl)-N-(isobutyl)benzene sulfonamide (135 gm) in acetonitrile (330 ml) was added to the reaction mass at -10 to -15C for 45 minutes. To the reaction mass was added triethylamine (39 gm) and monomethylamine (5.8 gm) at -10C, and the temperature was slowly raised to 20 to 25C and stirred for 1 hour. The reaction mass distilled off the solvent completely removed under vacuum at below 50C to obtain a residue. To the residue was added dichloromethane (1000 ml). The dichloromethane layer was washed with 10% sodium bicarbonate (500 ml), 2% sulfuric acid (500 ml), 10% sodium sulfate (500 ml) and 10% sodium chloride solution (500 ml). The layer was treated with carbon and then added concentrated hydrochloric acid (40 ml). The reaction mass was stirred for 1 hour at 25 to 30C and filtered. The solid obtained was dried under vacuum at 60 to 65C for 6 hours to obtain 140 gm of crystalline hydrochloride salt of darunavir.
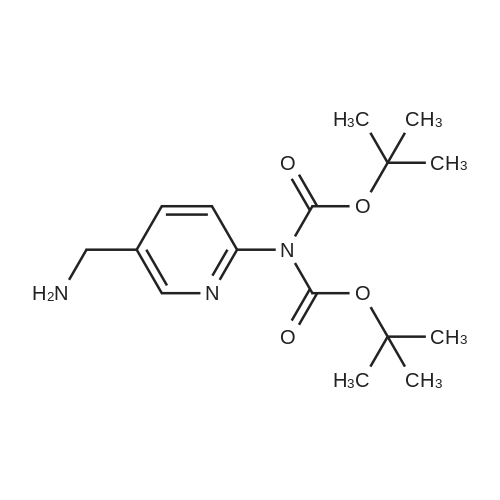
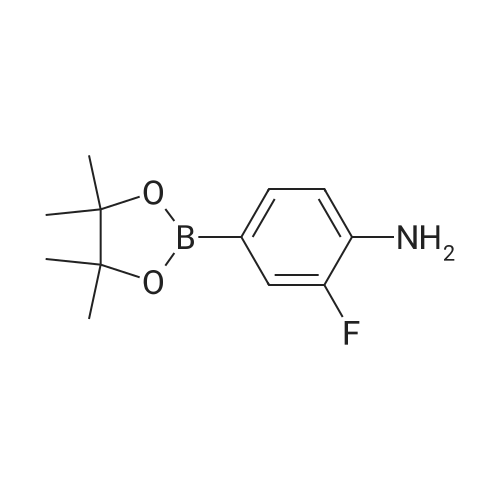
2.2. Di(tert-butyl){5-[([2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]carbamoyl}amino)methyl]pyridin-2-yl}imidodicarbonate 5.0 g (21.09 mmol) of <strong>[819058-34-9]2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline</strong> and 3.09 g (25.31 mmol) of DMAP (4-dimethylaminopyridine) are dissolved in 500 ml of THF. 6.48 g (23.31 mmol) of DSC are added and the mixture is stirred at AT for 18 h. 8.81 ml (63.27 mmol) of triethylamine and 8.18 g (23.31 mmol) of di(tert-butyl) [5-(aminomethyl)pyridin-2-yl]imidodicarbonate are added. The mixture is stirred at AT for 5 h. The solvent is evaporated and the residue is taken up in DCM. The organic phase is washed with water and than with a saturated NaCl solution. It is dried over sodium sulphate, filtered and evaporated. The residue is purified by flash chromatography. 12 g of product composed of a 50/50 mixture of pinacolic ester and of boronic acid are obtained. LCMS (LS) MH+ 587, rt=6.17 min, and MH+ 505, rt=4.97 min.
With triethanolamine; In acetonitrile; at 20℃; for 2h;Inert atmosphere;
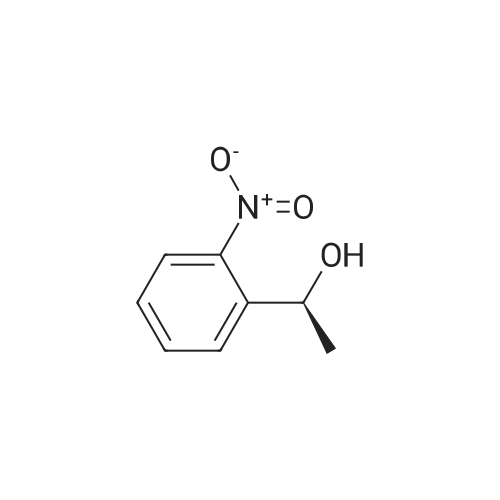
To a solution of1-(2-nitrophenyl) ethanol (1.70 g, 10 mmol) in dry acetonitrile (15 mL)containing TEA (3.5 mL) was added di(N-succinimidyl) carbonate (3.60 g, 14mmol) under nitrogen. The reaction mixture was stirred for 2 hours at roomtemperature. After removal of solvents in vacuo, the residue was purified bysilica gel column, eluted with PE:EA (3:1 to 1:1) to give 1-(2-nitrophenyl)ethylN-succinimidyl carbonate as a light yellow solid (2.60 g, 8.5 mmol, yield 85percent). 1H NMR (400 MHz, CDCl3) delta 8.04-7.48 (m, 4H), 6.40 (m,1H), 2.79 (s, 4H), 1.79 (d, 2H). 13C NMR (100 MHz, CDCl3)delta 168.4, 150.6, 147.2, 135.7, 134.2, 129.2, 126.9, 124.7, 75.9, 25.3, 22.0
N-(3-(1H-imidazol-1-yl)propyl)-2-((4-fluorobenzyl)amino)-1,3-thiazole-5-carboxamide[ No CAS ]
N1-(4-fluorobenzyl)-N1-(5-[3-(1H-imidazol-1-yl)propyl]carbamoyl}-1,3-thiazol-2-yl)piperidine-1,3-dicarboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: Example 6B (0.018 g, 0.05 mmol) was dissolved in acetonitrile (0.5 ml), and treated with potassium carbonate (0.021 g, 0.15 mmol) and 1 -(2-chloroethyl)pyrrolidine (0.017 g, 0.1 mmol). The reaction mixture was heated via microwave at 180 °C for 30 minutes, concentrated in vacuo, and submitted to reverse-phase HPLC (as described in Example 6C) to provide the title compound.
N-(3-(1H-imidazol-1-yl)propyl)-2-((4-fluorobenzyl)amino)-1,3-thiazole-5-carboxamide[ No CAS ]
[ 78531-29-0 ]
2-[(1,3-dimethoxypropan-2-yl)carbamoyl](4-fluorobenzyl)amino}-N-[3-(1H-imidazol-1-yl)propyl]-1,3-thiazole-5-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: Example 6B (0.018 g, 0.05 mmol) was dissolved in acetonitrile (0.5 ml), and treated with potassium carbonate (0.021 g, 0.15 mmol) and 1 -(2-chloroethyl)pyrrolidine (0.017 g, 0.1 mmol). The reaction mixture was heated via microwave at 180 C for 30 minutes, concentrated in vacuo, and submitted to reverse-phase HPLC (as described in Example 6C) to provide the title compound.
N-(3-(1H-imidazol-1-yl)propyl)-2-((4-fluorobenzyl)amino)-1,3-thiazole-5-carboxamide[ No CAS ]
2-[(4-fluorobenzyl)(tetrahydrofuran-3-ylcarbamoyl)amino]-N-[3-(1H-imidazol-1-yl)propyl]-1,3-thiazole-5-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: Example 6B (0.018 g, 0.05 mmol) was dissolved in acetonitrile (0.5 ml), and treated with potassium carbonate (0.021 g, 0.15 mmol) and 1 -(2-chloroethyl)pyrrolidine (0.017 g, 0.1 mmol). The reaction mixture was heated via microwave at 180 °C for 30 minutes, concentrated in vacuo, and submitted to reverse-phase HPLC (as described in Example 6C) to provide the title compound.
(E)-cyclooct-4-en-1-yl (2,5-dioxopyrrolidin-1-yl) glutarate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
51%
Synthesis of trans-cyclooctene NHS ((E)-cyclooct-4-en-1-yl (2,5-dioxopyrrolidin-1-yl) glutarate) DMAP (6.1 mg, 0.05 mmol) was added to a stirred solution of (E)-cyclooct-4-enol (5.0 mg, 0.040 mmol) in toluene (1.0 mL), followed by glutaric anhydride (6.0 mg, 0.05 mmol). The resulting reaction solution was heated to 100° C. and stirred at this temperature for 18 hours. After TLC indicated that the reaction had finished the solvent was evaporated and the residue was dissolved in CH2Cl2, followed by addition of N,N'-disuccinimidyl carbonate (13.0 mg, 0.05 mmol). After stirring at room temperature for 30 minutes, the reaction solution was evaporated and the residue was purified by preparative TLC (Hexanes:Ethyl Acetate (EtOAc)=2:1) to afford 7.0 mg product as a colorless liquid, in 51percent yield. 1H NMR (500 MHz, CDCl3) delta 1.59-1.71 (2H, m), 1.89-2.05 (6H, m), 2.30-2.40 (6H, m), 2.68 (t, J=10 Hz, 2H), 2.83 (4H, bs), 4.42-4.45 (1H, t, m), 5.46-5.60 (2H, m); 13C (100 MHz, CDCl3) 20.05, 25.80, 30.28, 31.18, 32.72, 33.30, 34.46, 38.81, 41.10, 80.64, 133.27, 135.13, 168.32, 169.27, 171.95; HRMS [M+Na]+ m/z calc. for [C17H23NO6Na]+ 360.1418, found 360.1419.
2,5-dioxopyrrolidin-1-yl (4-((2-(diethylamino)ethyl)carbamoyl)phenyl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetonitrile; for 5h;Inert atmosphere;
In a first method, 2.6 g of procaine amide A was added and dissolved in 47 g of dry acetonitrile in a dry 100 mL Erlenmeyer flask equipped with a stir bar.In a 1 L separable flask equipped with a dropping funnel and a stir bar, 3.2 g of N, N-disuccinimidyl carbonate (DSC) B was dissolved in 417 g of dry acetonitrile and the system was purged with N 2.The solution of procainamide was then transferred to a dropping funnel and added dropwise to the DSC solution over 1 hour. The solution was then stirred for 4 hours.At this point the solvent was removed and the 2,5-dioxopyrrolidin-1-yl (4 - ((2- (diethylamino) ethyl) carbamoyl) phenyl) carbamate C was dried at room temperature under high vacuum.
(2R,5R)-1,6-diphenylhexane-2,5-diamine dihydrochloride[ No CAS ]
[ 1247119-33-0 ]
Yield
Reaction Conditions
Operation in experiment
80.7%
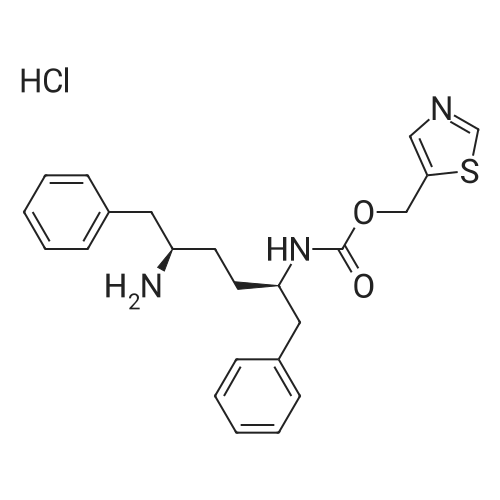
50 mL of acetonitrile was added to the dried reaction flask, cooled to 0 C., and then N,N?-disuccinimidyl carbonate (DSC) 28.2 g was added, and 12.7.0 g containing <strong>[38585-74-9]5-hydroxymethylthiazole</strong> was slowly added dropwise. A solution of 50 mL of acetonitrile was added and the system was stirred at 0C for 1 hour until used. In another reaction flask(2R,5R)-1,6-diphenyl-2,5-hexanediamine dihydrochloride 34.1 g,25.0 g of pyridine was added to 200 mL of acetonitrile, cooled to 0 C., and the above DSC prepared solution was dropped.After one hour of dropwise addition, the reaction system was gradually warmed to room temperature (25 C) and stirred for 8 h;TLC tests the reaction of the raw materials.Concentrate to remove most of the acetonitrile, then add 1 mol/L sodium hydroxide aqueous solution 150mL,Extract with ethyl acetate three times, and combine the organic phase with water,Saturated sodium chloride solution was washed, dried over anhydrous magnesium sulfate, and the solvent was concentrated.The resulting residue was dissolved in isopropanol and added to a solution of isopropyl alcohol in hydrochloric acid to form a salt.A large number of white solids appear, filtered, rinsed, and dried to give a white powder productN-((1R,4R)-4-Amino-5-phenyl-1-benzylpentyl)carbamic acid 5-thiazolylmethyl ester hydrochloride (I) 36.0 g, yield 80.7%.
6-(4-aminophenyl)-2-cyclopentyl-5-methyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-[4-(1-cyclopentyl-4-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
To a solution of Intermediate 9, 60 mg (0.22 minol), and 4-dimethylaminopyridine, 32 mg (0.26 minol), in tetrahydrofuran, 25 mL, was added N,N-disuccinimidyl carbonate, 67.9 mg, (0.26 minol). The reaction was stirred for 1 hour then triethylamine, 0.092 mL (0.66 minol), and 2,3-dihydro-IH-pyrrolo[3,4-c]pyridine hydrochloride (6000-50-6), 41.3 mg (0.26 minol), was added and the reaction was left to stir at room temperature for 18 hours. Water was added and the mixture was extracted with dichloromethane. The combined organics were dried over solid sodium sulfate and concentrated under vacuum. Purification by flash chromatography on silica gel 60 (eluent: ethyl acetate-heptane 0:1, 1:1, 1:0 and methanol-ethyl acetate 1:9) gave thedesired product, 62.8 mg (68percent).1H NMR (300 MHz, CDCI3): 6 [ppm] = 1.16 (d, 3H), 1.57-2.00 (m, 8H), 2.46 (d, IH), 2.64 (dd, IH), 3.27 (m, IH), 4.90 (d, 4H), 5.21 (m, IH), 6.40 (s, IH), 7.30 (d, IH), 7.52 (d, 2H), 7.76 (d, 2H), 8.58 (d, IH), 8.63 (s, IH).UPLC-MS (Method 4): R 1.93 min., 100percent. MS (ESIpos): mz[M+H]418.
6-(4-aminophenyl)-5-methyl-2-(2,2,2-trifluoroethyl)-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-{4-[4-methyl-6-oxo-1-(2,2,2-trifluoroethyl)-1,4,5,6-tetrahydropyridazin-3-yl]phenyl}-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
20%
A solution of 100mg of Intermediate 11(0.35 minol, 1.00 eq) in DMF (10 mL) was treated with108 mg of N,N?-disuccinimidyl carbonate (0.42 minol, 1.20 eq) and 51.4 mg of 4-dimethylaminopyridine (0.42 minol, 1.20 eq) and was left over night at room temperature. Asuspension of 81 .2 mg of 2,3-dihydro-1 H-pyrrolo[3,4-c]pyridine dihydrochloride (0.42 minol, 1 .20eq) and 667 pL of triethylamine (4.78 minol, 3.60 eq) in DMF (5 mL) was added. The reactionmixture was left for 3 days at room temperature. The mixture was poured into water. The aqueous phase was three times extracted with ethyl acetate. The combined organic layers were washed with brine, dried over Na2504 and the solved was removed under reduced pressure. The residue was purified by preparative reverse phase HPLC to yield the desired productExample 11(30 mg, 20percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.09 (d, 3H), 2.40-2.47 (m, IH), 2.79-2.87 (m, IH),3.42-3.54 (m, IH), 4.26-4.38 (m, IH), 4.37-4.37 (m, IH), 4.80-4.83 (m, 4H), 7.41 -7.47 (m, I H), 7.65 - 7.72 (m, 2H), 7.72 - 7.79 (m, 2H), 8.50 (d, I H), 8.61 (s, I H), 8.66 (s, I H).U PLC-MS (Method 2): R= 0.81 min; MS (ESipos): mz [M÷H] 432.
6-(4-aminophenyl)-5-methyl-2-(tetrahydro-2H-pyran-4-yl)-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-{4-[4-methyl-6-oxo-1-(tetrahydro-2H-pyran-4-yl)-1,4,5,6-tetrahydropyridazin-3-yl]phenyl}-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
11%
A solution of 177 mg of Intermediate 14 (0.31 minol, 50percent purity 1.00 eq) in DMF (19 mL) wastreated with 94.7 mg of N,N?-disuccinimidyl carbonate (0.37 minol, 1.20 eq) and 45.2 mg, of 4-dimethylaminopyridine (0.37 minol, 1.20 eq). The mixture was left over night at room temperature. A suspension of 71.4 mg of 2,3-dihydro-IH-pyrrolo[3,4-c]pyridine dihydrochloride (0.37 minol, 1.20 eq) and 155 pL of triethylamine (1.11 minol, 3.60 eq) on DMF (3 mL) wasadded. The reaction mixture was stirred 3 days at room temperature. The mixture was poured into water and extracted three times with ethyl acetate. The combined organic layers were washed with brine and dried over Na2SO4. The aqueous and the organic phase were evaporated under reduced pressure. Both residues were purified by preparative HPLC to yield in total 15.0mg of the desired product Example 21(0.03 minol, 11percent).1H-NMR (400MHz, DMSO-d6): oe [ppm]= 1.05 (d, 3H), 1.49- 1.61 (m, 2H), 1.80- 1.93 (m, IH),2.02 - 2.16 (m, I H), 2.29 - 2.37 (m, I H), 2.68 - 2.76 (m, I H), 3.88 - 3.99 (m, 2H), 4.70 - 4.80 (m,IH), 4.91 (d, 4H), 7.64 - 7.71 (m, 3H), 7.74 - 7.79 (m, 2H), 8.64 (d, IH), 8.72 (s, IH), 8.76 (s,I H). Three protons are not visible.U PLC-MS (Method 2): R 0.71 min; MS (ESIpos): mz [M÷H] 434.
6-(4-aminophenyl)-5-methyl-2-phenyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-[4-(4-methyl-6-oxo-1-phenyl-1,4,5,6-tetrahydropyridazin-3-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
21%
A solution of 100 mg of Intermediate 23(0.36 minol, 1.00 eq) in DMF (11 mL) was treated with110 mg of N,N?-disuccinimidyl carbonate (0.43 minol, 1.20 eq) and 53.4 mg of 4-dimethylaminopyrdine (0.44 minol, 1.20 eq). The mixture was stirred over night at roomtemperature. A solution of 82.9 mg of 2,3-dihydro-1 H-pyrrolo[3,4-c]pyridine dihydrochloride (0.43minol, 1 .20 eq) and 2.99 mL of triethylamine (21.47 minol, 30 eq) in DMF (2 mL) was added. The mixture was stirred over night. THE was removed from under reduced pressure. The remaining solution was poured into water. The solid was removed by filtration, the filtrate was taken to dryness and the residue was purified by preparative reverse phase HPLC to yield 35.0mg of the desired product (21percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.07 (d, 3H), 2.17 (s, 6H), 2.22-2.36 (m, IH), 2.40-2.48 (m, 2H), 2.64 - 2.70 (m, I H), 3.37 - 3.44 (m, I H), 3.59 - 3.75 (m, I H), 3.91 - 4.08 (m, I H),4.83 (d, 4H), 7.44 (d, IH), 7.63 - 7.71 (m, 2H), 7.71 - 7.79 (m, 2H), 8.50 (d, IH), 8.65 (s, IH),8.61 (s, IH).UPLC-MS (Method 1): R0.83 min; MS (ESIpos): mz[M+H]426.
6-(4-aminophenyl)-2-[2-(dimethylamino)ethyl]-5-methyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-(4-{1-[2-(dimethylamino)ethyl]-4-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl}phenyl)-1,3- dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
13%
A solution of 100mg of Intermediate 24 (0.36 minol, 1.00 eq) in DMF (11 mL) was treated with112 mg of N,N?-disuccinimidyl carbonate (0.44 minol, 1.20 eq) and 53.4 mg of 4-dimethylaminopyrdine (0.44 minol, 1.20 eq). The mixture was stirred over night at roomtemperature. A solution of 84.4 mg of 2,3-dihydro-1 H-pyrrolo[3,4-c]pyridine dihydrochloride (0.44minol, 1 .20 eq) and 3.05 mL of triethylamine (21.87 minol, 30 eq) in DMF (2 mL) was added. The mixture was stirred over night. THE was removed from under reduced pressure. The remaining solution was poured into water. The solid was removed by filtration, the filtrate was taken to dryness and the residue was purified by preparative reverse phase HPLC to yield 20.0mg of the desired product (13percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.07 (d, 3H), 2.17 (s, 6H), 2.22-2.36 (m, IH), 2.40-2.48 (m, 2H), 2.64 - 2.70 (m, I H), 3.37 - 3.44 (m, I H), 3.59 - 3.75 (m, I H), 3.91 - 4.08 (m, I H),4.83 (d, 4H), 7.44 (d, IH), 7.63 - 7.71 (m, 2H), 7.71 - 7.79 (m, 2H), 8.50 (d, IH), 8.65 (s, IH),8.61 (s, IH).UPLC-MS (Method 1): R0.47 min; MS (ESIpos): mz[M+2H]421
6-(4-aminophenyl)-2-[4-(difluoromethoxy)benzyl]-5-methyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-(4-{1-[4-(difluoromethoxy)benzyl]-4-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl}phenyl)-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
12%
A solution of 200 mg of Intermediate 25 (0.56 minol, 1.00 eq) in DMF (8 mL) was treated with171 mg of N,N?-disuccinimidyl carbonate (0.67 minol, 1.20 eq) and 82.0 mg of 4- dimethylaminopyrdine (0.67 minol, 1.20 eq). The mixture was stirred for 3 days at room temperature. A suspension of 129 mg of 2,3-dihydro-IH-pyrrolo[3,4-c]pyridine dihydrochloride (0.67 minol, 1.20 eq) and 279 pL of triethylamine (2.00 minol, 3.6 eq) in DMF (2 mL) was added. The mixture was stirred over night. The resulting suspension was filtered, the solid was removedand the filtrate was taken to dryness. The residue was purified by reverse phase preparative HPLC to yield 34.0mg of the desired product (12percent).1H-NMR (400MHz, DMSO-d6): oe [ppm]= 1.04 (d, 3H), 2.30 -2.40 (m, IH), 2.75 -2.86 (m, IH),3.38-3.48 (m, IH), 4.78 -4.87 (m, 5H), 4.99 (s, IH), 7.00- 7.22 (m, 3H), 7.34 -7.40 (m, 2H),7.44 (d, I H), 7.62 - 7.68 (m, 2H), 7.69 - 7.75 (m, 2H), 8.50 (d, I H), 8.63 (d, 2H).UPLC-MS (Method 2): Rt0.92 min; MS (ESIpos): mz [M÷H] 506.
N-[4-(4-oxo-3,4-dihydrophthalazin-1-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.5%
A solution of 100.0 mg of 4-(4-aminophenyl)-2-methylphthalazin-1(2H)-one (by S. Demirayak etal. in Eur. J. Med. Chem. 2004, 39, 1089?1095, 0.42 minol, 1.00 eq) in DMF (12 mL) wastreated with 129.6mg of N,N?-disuccinimidyl carbonate (0.51 minol, 1.20 eq) and 61.8mg of 4-dimethylaminopyridine (0.51 minol, 1.20 eq). The mixture was left of night at room temperature.A suspension of 97.7 mg of 2,3-dihydro-1 H-pyrrolo[3,4-c]pyridine dihydrochloride (0.51 minol,1.20 eq) and 1.76 mL of triethylamine (12.79 minol, 30 eq) in DMF (2 mL) was added and themixture was again stirred over night. The suspension was filtered, the filtrate was taken to dryness and the residue was purified by preparative reverse phase HPLC to obtain 2.5 mg of the desired material (1.5percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 4.86 (d, 4H), 7.39 - 7.60 (m, 3H), 7.76 (d, 3H), 7.83 -8.00 (m, 2H), 8.24-8.40 (m, IH), 8.52 (d, IH), 8.67 (s, IH), 8.63 (s, IH), 12.80 (s, IH).UPLC-MS (Method 2): R0.80 min; MS (ESIpos): mz [M÷H] 384.
6-(4-aminophenyl)-5-methyl-2-[2-(morpholin-4-yl)ethyl]-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-(4-{4-methyl-1-[2-(morpholin-4-yl)ethyl]-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl}phenyl)-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
11%
A solution of 150 mg of Intermediate 34 (0.47 minol, 1.00 eq) in DMF (5 mL) was treated with145 mg of N,N?-disuccinimidyl carbonate (0.57 minol, 1.20 eq) and 181 mg of 4-dimethylaminopyrdine (0.57 minol, 1.20 eq). The mixture was stirred over night at roomtemperature. A suspension of 109 mg of 2,3-dihhydro-IH-pyrrolo[3,4-c]pyridine dihydrochloride(0.57 minol, 1.20 eq) and 2.00 mL of triethylamine in DMF (2 mL) was added. The mixture was stirred for 3 days. The solid was removed by filtration, the filtrate was taken to dryness and the residue was purified by preparative reverse phase HPLC to yield 24.2 mg of the desired product(11percent).1H-NMR(400MHz, DMSO-d6): oe [ppm]= 1.09-1.13 (m, 3H), 2.30 (dd, IH), 2.35 -2.45 (m, 4H),2.65-2.75 (m, IH), 3.36-3.43 (m, 4H), 3.50 (t, 4H), 3.59-3.68 (m, IH), 4.12 (dt, IH), 4.82 (d,4H), 7.44 (d, I H), 7.64 - 7.69 (m, 2H), 7.71 - 7.77 (m, I H), 8.50 (d, I H), 8.63 (d, 2H).UPLC-MS (Method 2): R = 0.49 min; MS (ESIpos): mz [M÷H] 463.
N-[4-(1,4-dimethyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
A solution of 108 mg of Intermediate 4 (0.50 minol, 1.00 eq) in THE (18 mL) was treated with154 mg N,N?-disuccinimidyl carbonate (0.60 minol, 1.20 eq) and 73.3 mg of 4-dimethylaminopyrindine. The reaction mixture was stirred at room temperature over night. Asuspension of 116mg 2,3-dihydro-IH-pyrrolo[3,4-c]pyridine dihydrochloride (0.6 minol, 1.20 eq)and 251 pL of triethylamine (1.80 minol, 3.60 eq) was added. The reaction mixture was stirred at room temperature and additionally 250 pL triethylamine were added. After stirring for 3 days the precipitate was filtered off and discarded. The filtrate was taken to dryness. The remaining residue was purified by preparative reverse phase HPLC to provide 150 mg of the desiredproduct Example 4 (83percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.07 (d, 3H), 2.30 (dd, IH), 2.65-2.75 (m, IH), 3.32(s, 3H), 4.83 (d, 4H), 7.41 -7.46 (m, IH), 7.64-7.69 (m, 2H), 7.71 -7.76 (m, 2H), 8.14 (s, IH),8.50 (d, I H), 8.62 (d, 2H). One proton under the water protons.LC-MS (Method 2): R = 0.62 min; MS (ESIpos): mz = 364 [M÷H]
6-(6-aminopyridin-3-yl)-2,5-dimethyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-[5-(1,4-dimethyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)pyridin-2-yl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
21%
To a solution of Intermediate 83 45 mg (0.21 minol) in acetonitrile, 6mL, was added N,Ndisuccinimidyl carbonate, 63.38 mg (0.25 minol). The reaction was stirred at room temperature for 18 hours in a sealed tube. To a suspension of 2,3-dihydro-IH-pyrrolo[3,4-c]pyridinehydrochloride, 32 mg (0.21 minol) in acetonitrile, I mL, was added triethylamine, 0.057 mL (0.41 minol), the slurry was transferred to the tube and the reaction was left to stir at room temperature for 18 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The combined organics were dried over solid sodium sulfate and concentrated under vacuum.Purification by MDAP (Eluent: Acetonitrile: 0.1percent NH4OH 5:90, 40:60) gave 102.4, 15.9 mg (21percent) as a white solid.1H NMR (300 MHz, CDCI3): 6 [ppm] = 1.23 (d, 3H), 2.50 (d, IH), 2.70 (dd, IH), 3.27 (m, IH),3.47 (s, 3H), 4.92 (d, 4H), 7.32 (m, IH), 8.14 (m, IH), 8.22 (m, IH), 8.58-8.64 (m, 3H).UPLC-MS (Method 4): R= 1.31min., 99percent ES (ESIpos) [M÷H] 365.
(5S)-6-(4-aminophenyl)-2,5-dimethyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-{4-[(4S)-1,4-dimethyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl]phenyl}-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
A solution of 500 mg of Intermediate 6 (2.30 minol, 1.00 eq) in THE (60 mL) was treated with707 mg of N,N?-disuccinimidyl carbonate (2.76 minol, 1.20 eq) and 338 mg of 4-dimethylaminopyridine (2.76 minol, 1.20 eq). The mixture was stirred for 3 days at roomtemperature. A suspension of 533 mg 2,3-dihydro-IH-pyrrolo[3,4-c] pyrimidine dihydrochloride(2.76 minol, 1.20 eq) and 1.16 mL of triethylamine (8.29 minol, 3.60 eq) in DMF (3 mL) wasadded. Additionally 5 mL of DMF were added to the mixture and it was stirred again over night at room temperature. The precipitate was filtered off and discarded. The filtrate was poured into water. The resulting suspension was stirred over night, the precipitate collected by filtration, washed with water and dried to provide the desired product Example 5 (732 mg, 83percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.07 (d, 3H), 2.30 (dd, IH), 2.70 (dd, IH), 3.35-3.45 (m, I H), 4.82 (d, 4H), 7.44 (d, I H), 7.63 - 7.70 (m, 2H), 7.71 - 7.76 (m, 2H), 8.50 (d, I H), 8.62 (d,I H). The methyl group is under the water protons.LC-MS (Method 2): R = 0.60 min; MS (ESIpos): mz = 364 [M÷H].Optical rotation (Method 5): [a] = + 35750 (c = 1.00, DMSO).
6-(4-aminophenyl)-5-isopropyl-2-methyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-[4-(4-isopropyl-1-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
To a solution of 6-(4-aminophenyl)-5-isopropyl-2-methyl-4, 5-dihydropyridazin-3(2H )-one Intermediate 91, 50 mg (0.20 minol) and 4-dimethylaminopyridine, 30 mg (0.25 minol) in tetrahydrofuran, 5 mL, N,N-disuccinimidyl carbonate, 63 mg (0.25 minol) was added. After 2 hours at room temperature 2,3-dihydro-1 H-pyrollo[3,4-c]pyridine dihydrochloride, 59 mg (6000- 50-6, 0.31 minol) and triethylamine, 0.29 mL, (2.04 minol) dissolved in N,N dimethylformide (3mL) were added and the mixture was stirred for a further 16 hours. The reaction mixture was diluted with a saturated solution of aminonium chloride and extracted with ethyl acetate. The combined organic layers were dried over solid sodium sulfate, filtered and concentrated undervacuum. The crude compound was purified by reverse phase chromatography (BIOTAGE SP4, 30 g Biotage cartridge) using acetonitrile and water containing 10min aminonium bicarbonate pH 10 buffer (3:97 to 100:0) to give Example 78, 31 mg (42percent) as a colourless solid.1H NMR (400 MHz, CDCI3): 6 [ppm] = 0.89 (m, 6H), 1.96 (m, IH), 2.56 (dd, IH), 2.70 (d, IH),3.04 (m, IH), 3.41 (s, 3H), 4.89 (d, 4H), 6.44 (s, IH), 7.33 (m, IH), 7.51 (d, 2H), 7.73 (d, 2H),8.59 (m, IH), 8.65 (m, IH).UPLC (Method 3): R = 0.61 min., 95percent. MS (ESIpos): mz = [M÷H] 392.
N-[4-(4-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
A solution of 406 mg of Intermediate 7 (2.00 minol, 1.00 eq) in THE (30 mL) was treated with615 mg of N,N?-disuccinimidyl carbonate (2.40 minol, 1.20 eq) and 293 mg of 4-dimethylaminopyridine (2.40 minol, 1 .20 eq). The mixture was stirred for three days at roomtemperature. A suspension of 463 mg of 2,3-dihydro-IH-pyrrolo[3,4-c]pyridine dihydrochloride(2.40 minol, 1.20 eq) and 1.00 mL of triethylamine (7.2Ominol, 3.60 eq) in DMF (2 mL) was added and the resulting suspension as again stirred over night. The mixture was poured into water, the precipitate was collected by filtration and was washed with water to provide after trituration with ethanol 440 mg of the desired product (63percent).1H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.07 (d, 3H), 2.17-2.26 (m, IH), 2.62-2.71 (m, IH),3.35-3.42 (m, IH), 4.82 (d, 4H), 7.41 -7.46 (m, IH), 7.61 -7.74 (m, 4H), 8.50 (d, IH), 8.61 (d,2H), 10.85 (s, IH).LC-MS (Method 1): R = 0.57 min; MS (ESIpos): mz = 350 [M÷H].
6-(4-aminophenyl)-2-isopropyl-5-methyl-4,5-dihydropyridazin-3(2H)-one[ No CAS ]
[ 6000-50-6 ]
N-[4-(1-isopropyl-4-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)phenyl]-1,3-dihydro-2H-pyrrolo[3,4-c]pyridine-2-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
25%
A solution of 100mg of Intermediate 22 (0.41 minol, 1.00 eq) in DMF (12 mL)was treated with125 mg of N,N?-disuccinimidyl carbonate (0.49 minol, 1.20 eq) and 59.7 mg of 4-dimethylaminopyrdine (0.49 minol, 1.20 eq). The mixture was stirred over night at roomtemperature. A solution of 94.4 mg of 2,3-dihydro-I H-pyrrolo[3,4-c]pyridine dihydrochloride (0.49minol, 1 .20 eq) and 1.70 mL of triethylamine (12.37 minol, 30 eq) in DMF (2 mL) was added.The mixture was stirred over night. THE was removed from under reduced pressure. The remaining solution was poured into water. The solid was removed by filtration, the filtrate was taken to dryness and the residue was purified by preparative reverse phase HPLC to yield 40.3 mg of the desired product (25percent).H-NMR (400MHz, DMSO-d6): oe [ppm] = 1.04 (d, 3H), 1.15 (d, 3H), 1.24 (d, 3H), 2.29 (dd, IH),2.68 (dd, IH), 3.21 - 3.42 (m, IH), 4.83 (d, 4H), 4.90 (quin, IH), 7.44 (d, IH), 7.60 - 7.72 (m,2H), 7.72 - 7.83 (m, 2H), 8.50 (d, I H), 8.62 (d, 2H).UPLC-MS (Method 1): RO.78 min; MS (ESIpos): mz [M÷2H] 393.
tert-butyl 4-((1S,2S)-1-(4-(N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)sulfamoyl)phenoxy)-6-chloro-4-cyano-2,3-dihydro-1H-inden-2-yl)piperazine-1-carboxylate[ No CAS ]
C70H94Cl2N12O16S2[ No CAS ]
tert-butyl 4-((1S,2S)-1-(4-(N-(19-((4-(((1S,2S)-2-(4-(tert-butoxycarbonyl)piperazin-1-yl)-6-chloro-4-cyano-2,3-dihydro-1H-inden-1-yl)oxy)phenyl)sulfonamido)-10-oxo-3,6,14,17-tetraoxa-9,11-diazanonadecyl)sulfamoyl)phenoxy)-6-chloro-4-cyano-2,3-dihydro-1H-inden-2-yl)piperazine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%; 22%
The amine 1NT-M5E is treated with disuccinimidyl carbonate (DSC) or similar activating agent (others including Iota, -carbonyldiimidazole, p- nitrophenylchloroformate, etc) in DMF, followed by addition of the desired diamine, with (lA',4s)-cyclohexane-l,4-diamine shown. Bis(2,5-dioxopyrrolidin-l-yl)carbonate (85 mg, 0.33 mmol, 1.1 eq) and tert-butyl 4-((lS,2S)-2-(4-(N-(2-(2-(2- aminoe1hoxy)e1hoxy)e1hyl)sulfamoyl)phenoxy)-4-cWoro-6-cj'ano- l-yl)piperazine-l-carboxylate (200 mg, 0.3 mmol) were stirred in DMF (1 mL) for 1.5 hours before a solution of (ls,4s)-cyclohexane-l,4-diamine (15.5 mg, 0.135 mmol, 0.45 eq) in DMF (0.2 mL) was added. The mixture was stirred for 2 h at 60 C. LCMS showed significant amount of monourea side-product, example 155. The DMF was removed under vacuum, residue dissolved in 4: 1 MeCN: H20, filtered, and purified by prep-HPLC with the following conditions: Column, Atlantis Prep T3 OBD, 50*250 mm, 10 urn; mobile phase, water (0.1% TFA) and CH3CN (25.0% CH3CN up to 80.0% in 60 min); Detector, UV 214 nm. Product eluted ~ 62% MeCN. Boc-protected cyclohexyl diamine product: 143 mg (61%); LCMS: ret time 3.3 min. MS (m/z): [M/2+H]+ 747.4. The product was deprotected in the following step. Step A: Bis(2,5-dioxopyrrolidin-l-yl)carbonate (85 mg, 0.33 mmol, 1.1 eq) and tert-butyl 4-((lS,2S)-2-(4-(N-(2-(2-(2-ammoemoxy)emoxy)e (2130) cy ano-2,3 -dihy dro- 1 H-inden- 1 -y l)piperazine- 1 -carboxy late (200 mg, 0.3 mmol) were stirred in DMF (1 mL) for 1.5 hours before a solution of (ls,4s)-cyclohexane-l,4-cuamine (15.5 mg, 0.135 mmol, 0.45 eq) in DMF (0.2 mL) was added. The mixture was stirred for 2 h at 60 C. LCMS showed significant amount of monourea side-product, example 155. The DMF was removed under vacuum, residue dissolved in 4:1 MeCN: H20, filtered, and purified by prep-HPLC with the following conditions: Column, Atlantis Prep T3 OBD, 50*250 mm, 10 urn; mobile phase, water (0.1% TFA) and CH3CN (25.0% CH3CN up to 80.0% in 60 min); Detector, UV 214 nm. Product eluted ~ 62% MeCN. Cyclohexyl diamine product: 143 mg (61%); LCMS: ret time 3.3 min. MS (m/z): [M/2+H]+ 747.4. Symmetric urea product: 47 mg (22%); LCMS: ret time 3.4 min MS (m/z): [M/2+H]+ 677.3. The products were each deprotected in the following step.
(S)-2-((cyclopropoxycarbonyl) amino)-3,3-dimethylbutanoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To asolution of <strong>[16545-68-9]cyclopropanol</strong> (0.4 ml, 6.37 mmol) in CH3CN (18 mL) at 0C, was added bis(2,5-dioxopyrrolidin-1-yl) carbonate (DSC) (3.26 g, 12.74 mmol) followed by Et3N (2.66 ml, 19.11 mmol). The reaction mixture was warmed up to 40C and stirred overnight. After cooling to room temperature, the reaction was concentrated under reduced pressure and the residue triturated with DCM, the solid filtered, and the filtrated was purified by silica column chromatography (10% - 100%EtOAc/hexanes). The product (663 mg, 3.33 mmol) was dissolved5 in THF (5 mL) and L-tert-leucine methyl ester hydrochloride (0.91 g, 5 mmol) and Et3N (1.39 ml, 0.01 mol) were added, the reaction was warmed up to 40C for 18h, then at room temperature for 48 h, diluted with EtOAc, washed with water. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure and the residue (760 mg, 3.31 mmol) was dissolved in a mixture of Methanol (4 mL) / water (2 mL), lithium hydroxide, monohydrate10 (0.56 g, 0.01 mol) was added. After 16 h, the mixture was concentrated, diluted with EtOAc, washed with brine, the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford A5 1HNMR (400 MHz, Chloroform-d) oe 4.19 (d, J 9.6 Hz, 1H), 1.02 (s, 11H), 0.68 (d, J= 4.8 Hz, 5H).
With triethylamine; In N,N-dimethyl-formamide; at 20℃; for 2.25h;
To a suspension of compound 9 (1 eq., 520 mg, 1.45 mmol) in DMF (25.5 mL) weresubsequently added N,N-disuccinimidyl carbonate (2.5 eq., 931 mg, 3.64 mmol) and TEA(2 eq., 294 mg, 0.404 mL, 2.91 mmol). After 15 minutes the precipitate solubilized, stirringcontinued for 2h at room temperature. lOx volume of Et20 was then added and theobtained precipitate was filtered to yield the crude product, which was recrystallized froma minimum amount of ACN to yield compound 10 as a white solid. The structure of 10 wasconfirmed by ESI-MS analysis (Method 1).ESI-MS m/z: 455.5 [M+H]
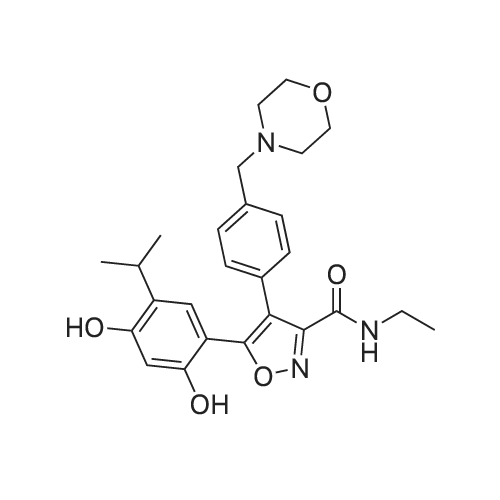
5-carboxy-2-(6-(dimethylamino)-3-(dimethyliminio)-3H-xanthen-9-yl)benzoate[ No CAS ]
C108H153N23O19S2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
25 pmol of NH2-Fx-r-Fx-r-Fx-r-K(Mtt) on resin was reacted with S-trityl-2- mercaptoproprionic acid (4 eq), PyBOP (4 eq), and DIPEA (8 eq) in 1 mL DMF. The peptide was washed (2 x DMF/MeOH/DCM) and deprotected with trifluoroacetic (0146) acid:triisopropylsilane:DCM (5:3:92, 2 x 15 minutes). The peptide washed and equilibrated in acetonitrile:water (5: 1 ). Cysteamine (20 eq) was dissolved in 1 mL acetonitrile:water (5: 1 ) and added to the reaction mixture followed by iodine (10 eq). The reaction was stirred for 30 minutes. The peptide was washed (2 x DMF:MeOH:DCM) and reacted with 5- Carboxytetramethylrhodamine (2 eq), HBTU (2 eq), and DIPEA (4 eq) in 0.5 mL DMF for 2 hours. The peptide was washed, cleaved from resin using trifluoroacetic (0147) acid:triisopropylsilane:water (95:2.5:2.5) and precipitated in ether at -20°C for 1 hour. The precipitate was purified by HPLC and lyophilized. 5-(2,4-Dihydroxy-5-isopropylphenyl)-N-ethyl- 4-(4-(morpholinomethyl)phenyl)isoxazole-3-carboxamide (Luminespib, 3 eq, Adooq Bioscience, Irvine CA) was reacted with Nu,Nu'-Disuccinimidyl carbonate (3 eq, Sigma-Aldrich) and 4- (Dimethylamino)pyridine (12 eq, Sigma-Aldrich) in 0.4 mL DMF for 1 hour. The peptide was dissolved in 0.1 mL DMF and added to the reaction mixture and the solution was left stirring overnight. The peptide was precipitated in ether and purified by HPLC. The earlier eluting isomer was purified and tested due to its higher relative abundance. The solution was frozen in dry ice as the compound eluted from the column and lyophilized. The peptide was identified by ESI mass spectrometry, expected m/z = 2140.12, found m/z = 2140.12. The peptide was quantified using the 5-Carboxytetramethylrhodamine absorbance at 547 nm with an extinction coefficient of 92000 M"1cm"1.
25 muetaetaomicronIota of NH2-Fx-r-Fx-r-Fx-r on resin was reacted with S-trityl-2- mercaptoproprionic acid (4 eq), PyBOP (4 eq), and DIPEA (8 eq) in 1 mL DMF. The peptide was washed (2 x DMF/MeOH/DCM), cleaved from resin using trifluoroacetic (0139) acid:triisopropylsilane:water (95:2.5:2.5) and precipitated in ether at -20°C for 1 hour. The precipitate was purified by RP-HPLC, dried under vacuum and dissolved in 0.5 mL (0140) acetonitrile:water (5:1). 2-mercpatoethanol (20 eq, Sigma-Aldrich) was added to the reaction mixture followed by iodine (10 eq) and the reaction was stirred for 30 minutes. The peptide was purified by HPLC and lyophilized. 5-(2,4-Dihydroxy-5-isopropylphenyl)-N-ethyl-4-(4- (morpholinomethyl)phenyl)isoxazole-3-carboxamide (Luminespib, 3 eq, Adooq Bioscience, Irvine CA) was reacted with Nu,Nu'-Disuccinimidyl carbonate (3 eq, Sigma-Aldrich) and 4- (Dimethylamino)pyridine (12 eq, Sigma-Aldrich) in 0.4 mL DMF for 1 hour. The peptide was dissolved in 0.1 mL DMF and added to the reaction mixture and the solution was left stirring overnight. The peptide was precipitated in ether and purified by HPLC. Two isomers were identified during HPLC purification, likely due to attachment to either of the two resorcinol hydroxyls. The earlier eluting isomer was purified and tested due to its higher relative abundance. The solution was frozen in dry ice as the compound eluted from the column and lyophilized. The peptide was identified by ESI mass spectrometry, expected m/z = 1599.88, found m/z = 1599.88. The peptide was quantified via absorbance spectrophotometry using a SpectraMax M5 spectrophotometer. The absorbance profile of Compound 5 was found to be shifted as compared to Luminespib itself, therefore the peptide was quantified by cleavage in 25 mM TCEP in PBS pH 7.4 for 10 minutes, then measuring free Luminespib absorbance at 305 nm with an extinction coefficient of 8520 M"1cm"1. TCEP was not found to affect the extinction coefficient of Luminespib.
To a solution of DSC (1.73 g, 6.76 mmol) in DMF (31.6 ml), a,y-di-tertbutyl L-glutamate (2 g, 6.76 mmol) was added in portions at 0C. After 50 minutes, TEA (937 p1, 6.76 mmol) was added. After complete conversion, a-tert-butyl-y-benzyl L-glutamate (2.23 g, 6.76 mmol) and TEA (1.87 ml, 13.52 mmol) were added at 000. The reaction mixture was stirred overnight at room temperature. DMF was removed in vacuo and the residue was dissolved in MTBE (100 ml). The organic layer was washed with 15% citric acid solution (2x100 ml), water (2xlOOml), saturated NaHCO3 solution (2x1 00 ml) and water (80 ml) in sequence. The organic layer was dried over MgSO4, filtered and concentrated. The resulting yellowish oil was purified by chromatography on silica gel column (0-33% gradient of ethyl acetate (EtOAc) in hexane) to provide the urea HOP 30.21 75 as colorless syrup (3.02 g, 77%).[00153] MS (ESI+): m/zfound: 579.17 calc.: 579.72 [M+H] found: 601.35 calc.:601.70 [M-?-Na] found: 1180.35 calc.: 1180.41 [2M-?-Na].
(4-{2-[(S)-amino(cyclopentyl)methyl]-4-fluoro-1H-benzimidazol-5-yl}tetrahydrofuran-3-yl)(3,3-difluoroazetidin-1-yl)methanone[ No CAS ]
3,3-difluorocyclobutyl N-[(S)-cyclopentyl{5-[4-(3,3-difluoroazetidine-1-carbonyl)-tetrahydrofuran-3-yl]-4-fluoro-1H-benzimidazol-2-yl}methyl]carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
15%
To a solution of <strong>[637031-88-0]3,3-difluorocyclobutanol</strong> (20 mg, 0.18 mmol) in DCM (1 mL) were added triethylamine (40 muL, 0.3 mmol) and N,N-disuccinimidyl carbonate (75 mg, 0.29 mmol). The reaction mixture was stirred at r.t. for 30 minutes, then a solution of Intermediate 222 (60 mg, 0.14 mmol) in DCM (1 mL) was added dropwise. After stirring for 80 minutes, the reaction mixture was partitioned between DCM and water. The organic layers were separated, dried over Na2SO4, and concentrated in vacuo. The residue was purified by preparative HPLC to give the title compound (12 mg, 15%) as a white solid. dH (400 MHz, DMSO-d6) 7.35 (d, J 8.4 Hz, 1H), 7.26 (dd, J 8.4, 6.4 Hz, 1H), 4.64 (d, J 9.4 Hz, 1H), 4.48 (q, J 11.7 Hz, 2H), 4.32-4.20 (m, 3H), 4.04 (dt, J 11.7, 7.6 Hz, 2H), 3.94 (t, J 8.0 Hz, 1H), 3.86 (t, J 10.9 Hz, 1H), 3.38-3.33 (m, 1H), 2.95 (dtd, J 17.9, 13.3, 12.6, 6.6 Hz, 2H), 2.73-2.45 (m, 3H), 1.91 (dd, J 12.7, 5.8 Hz, 1H), 1.74-1.40 (m, 6H), 1.27 (d, J 11.9 Hz, 1H). LCMS (Method 6): [M+H]+ m/z 557, RT 1.66 minutes.
(1r,4r)-N1-(3-aminopropyl)-N4-(5-chloro-4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)pyrimidin-2-yl)cyclohexane-1,4-diamine[ No CAS ]
(Z)-cyclooct-4-en-1-yl(3-(((1r,4r)-4-((5-chloro-4-(5-(cyclopropylmethyl)-1-methyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)cyclohexyl)amino)propyl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
7%
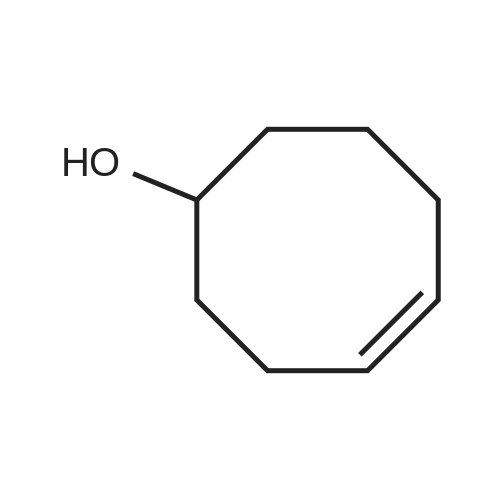
Compound AA8. A mixture of <strong>[4277-34-3](Z)-cyclooct-4-en-1-ol</strong> (42 mg, 330 mumol, 1.5 eq.), N,N?-disuccinimidyl carbonate (DSC) (101 mg, 396 mumol, 1.8 eq.), and TEA (40 mg, 396 mumol, 1.8 eq.) in MeCN (0.5 mL) was degassed and purged with N2 for 3 times. After stirred at 25 C for 4 h under N2, the mixture was added into a mixture of compound AA7 (91.98 mg, 220 mumol, 1 eq.) and TEA (40 mg, 396 mumol, 1.8 eq.) in DMF (0.5 mL) dropwise at 25 C under N2. After stirred at 25 C for 0.5 h under N2, the reaction mixture was filtered and the filtrate was concentrated in vacuo. The mixture was purificated by prep-HPLC (HCl) to afford compound AA8 (8.9 mg, 16 mumol, 7% yield) as a yellow oil. 1H NMR (CD3OD, 400 MHz) delta 8.47 (br s, 1H), 8.42 (s, 1H), 5.75-5.50 (m, 2H), 4.70 (br s, 1H), 4.15-4.00 (m, 1H), 3.96 (s, 3H), 3.29-3.18 (m, 4H), 3.14-2.97 (m, 2H), 2.62-2.49 (m, 2H), 2.44-2.32 (m, 1H), 2.25 (br d, J = 13.2 Hz, 2H), 2.21-2.13 (m, 2H), 2.10-2.00 (m, 1H), 1.95-1.81 (m, 4H), 1.73 (br dd, J = 4.6, 9.9 Hz, 1H), 1.68-1.34 (m, 7H), 1.10 (br s, 1H), 0.54 (br d, J = 7.3 Hz, 2H), 0.30 (br s, 1H).33
tert-butyl 3-((((2,5-dioxopyrrolidin-1-yl)oxy)carbonyl)oxy)-3-methylazetidine-1- carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 20℃; for 18h;
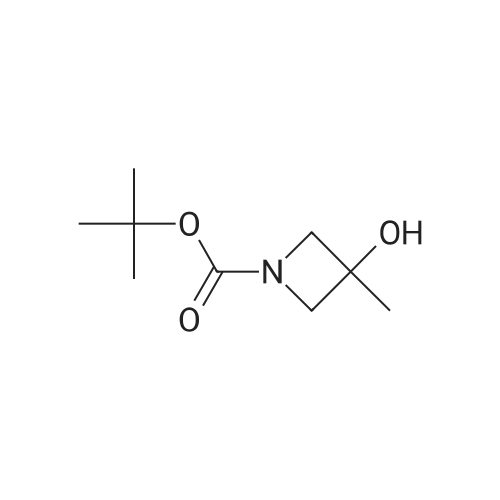
A solution of tert-butyl 3-hydroxy-3-methylazetidine-l-carboxylate (1.09 g, 5.81 mmol) in ACN (50 mL) was added bis(2,5-dioxopyrrolidin-l-yl) carbonate (2.98 g, 11.6 mmol) and DIPEA (2.02 mL, 11.6 mmol). The reaction was allowed to stir at rt for 18 h and was then concentrated and used without further purification.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping