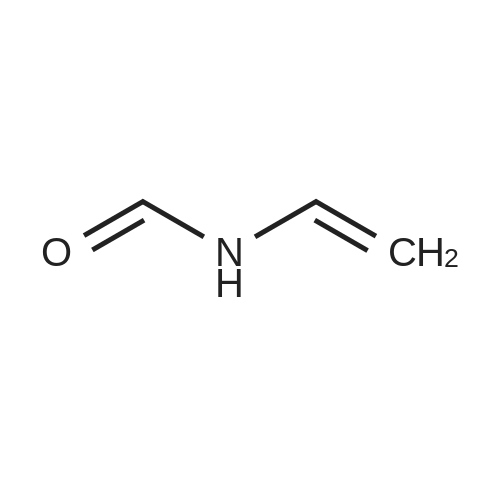
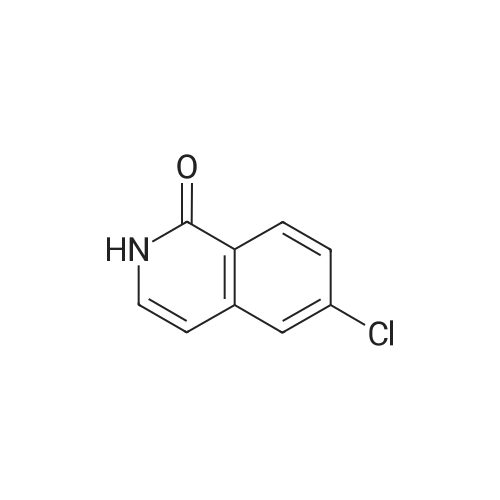
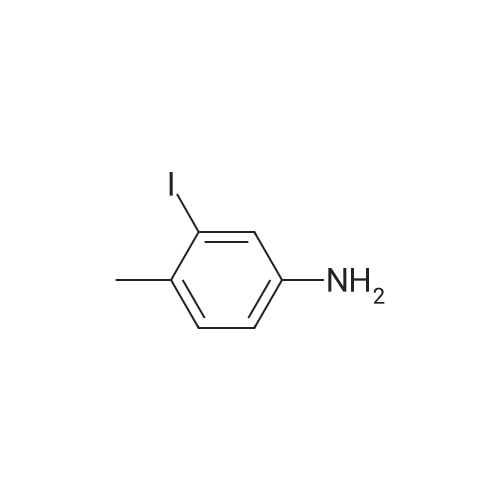
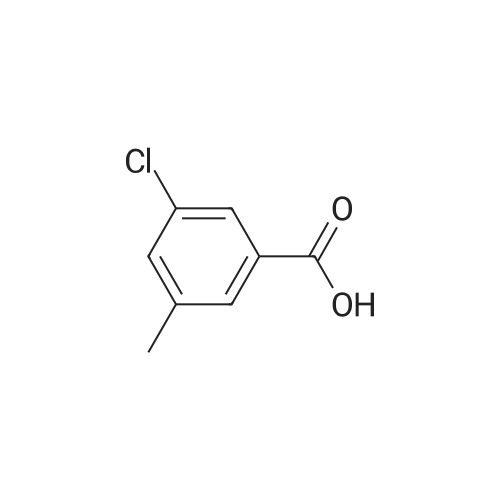
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
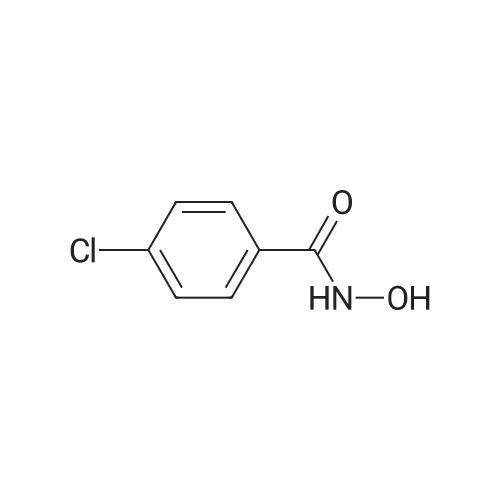
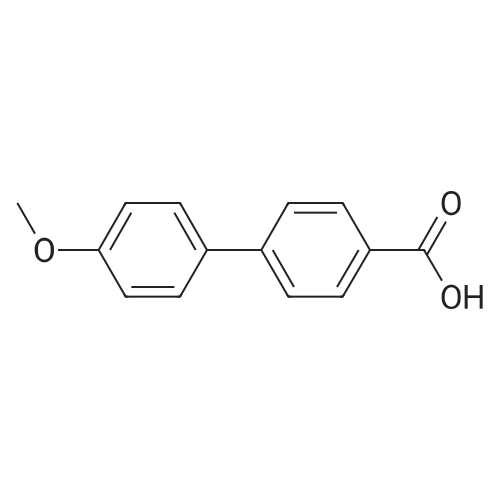
[1] Chemical and Pharmaceutical Bulletin, 1985, vol. 33, # 4, p. 1351 - 1359
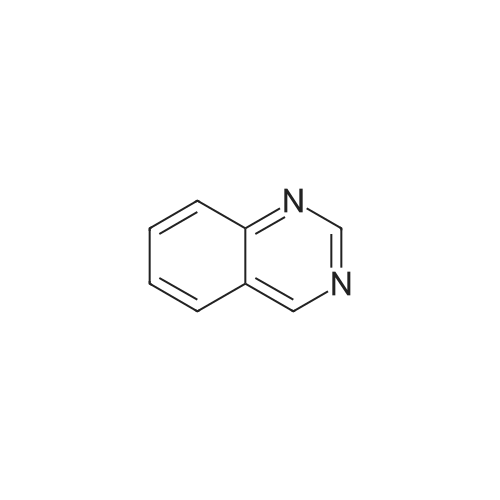
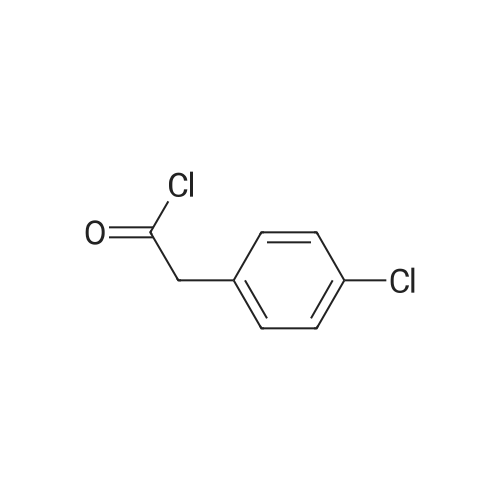
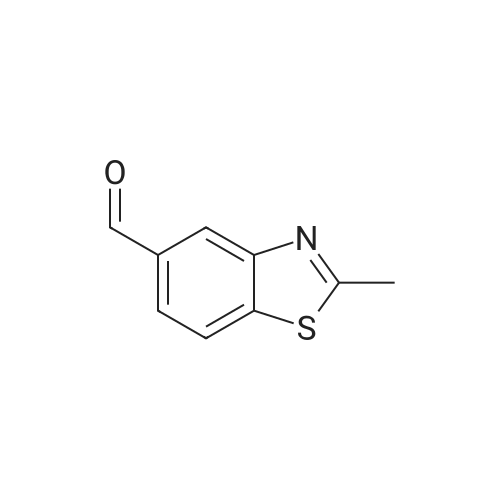
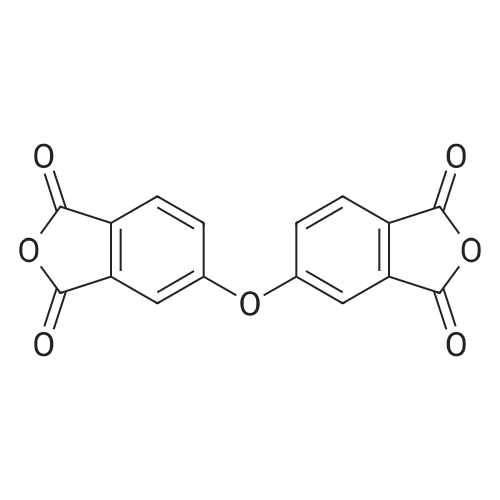
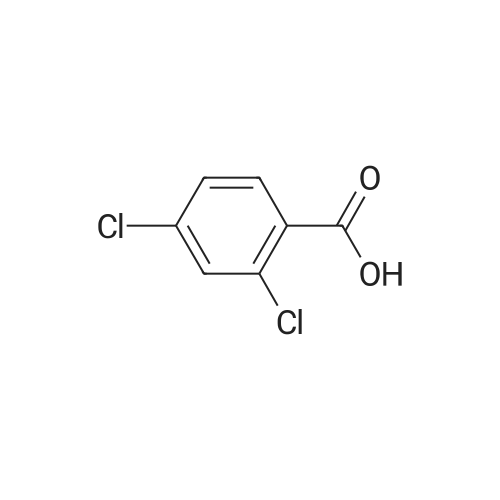
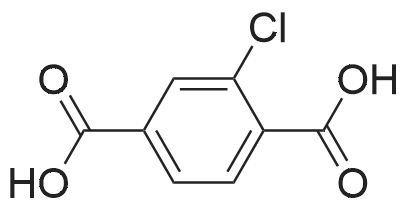
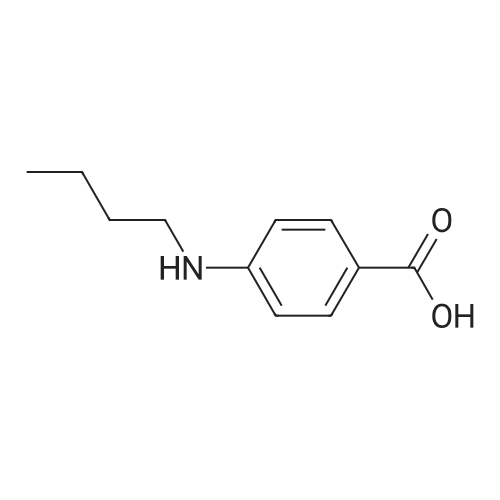
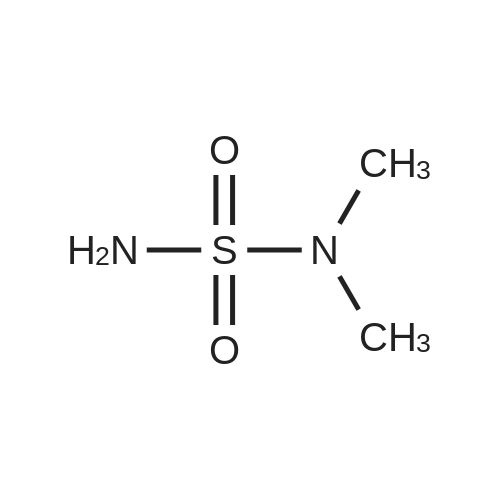
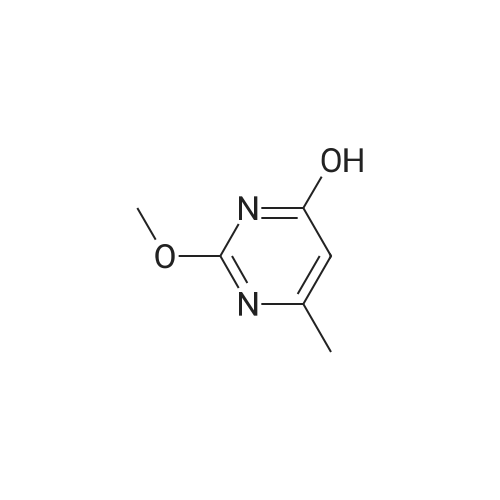
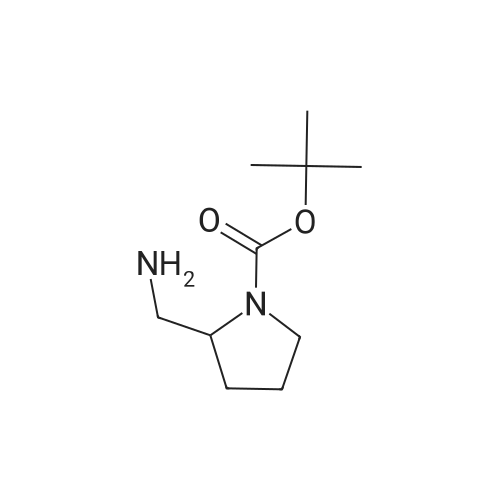
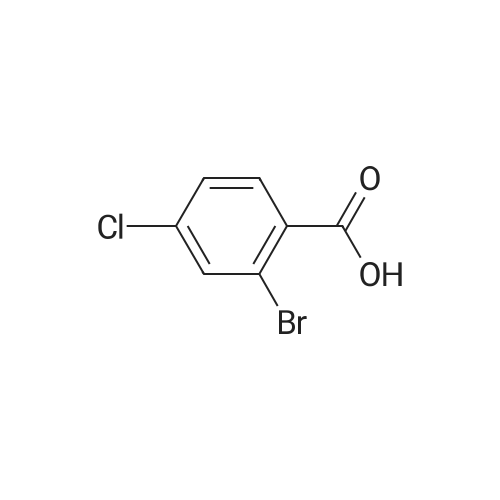
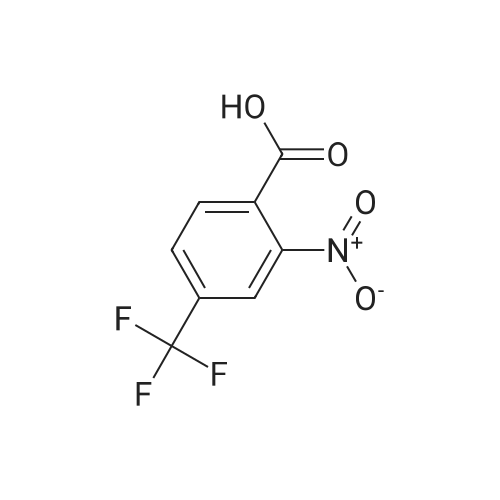
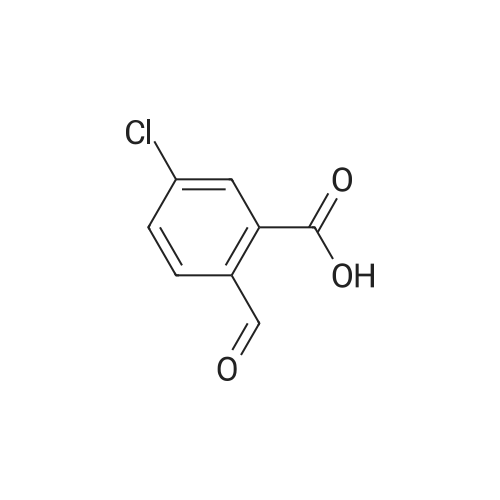
2
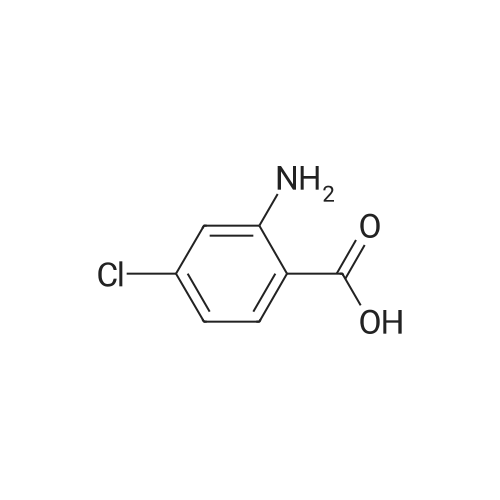
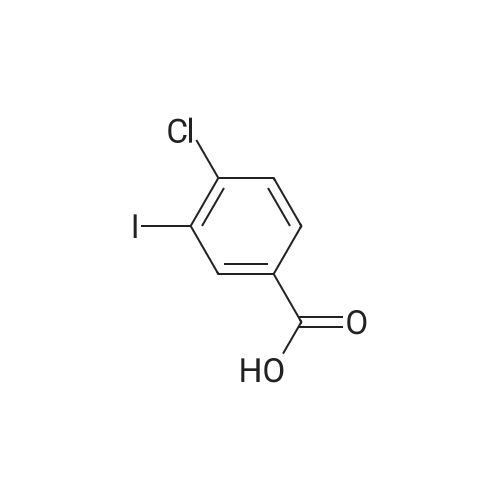
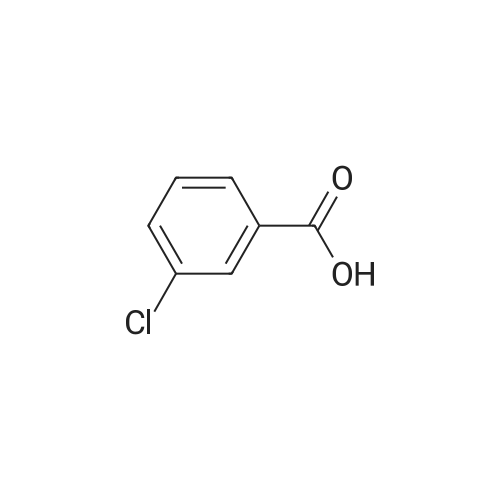
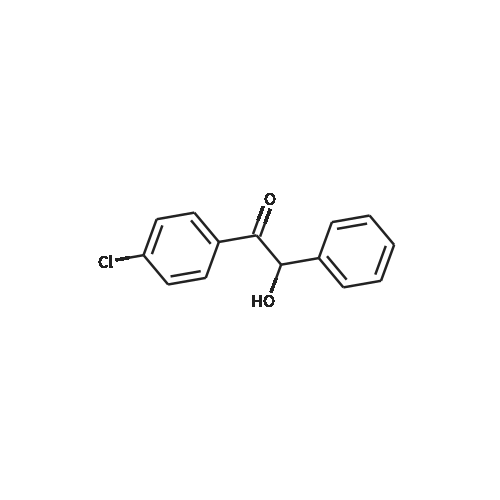
[ 74-11-3 ]
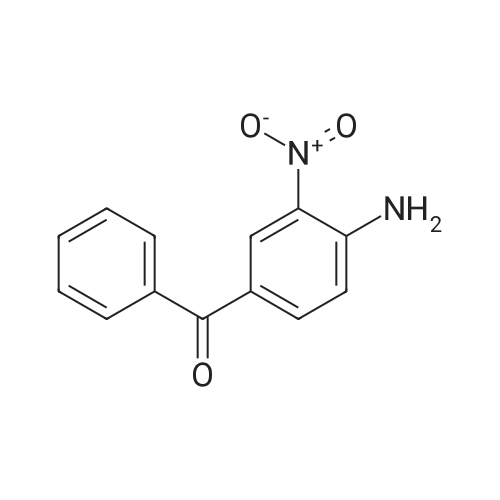
[ 31374-18-2 ]
Reference:
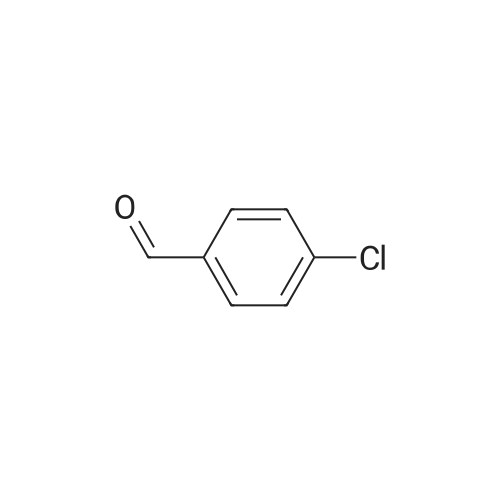
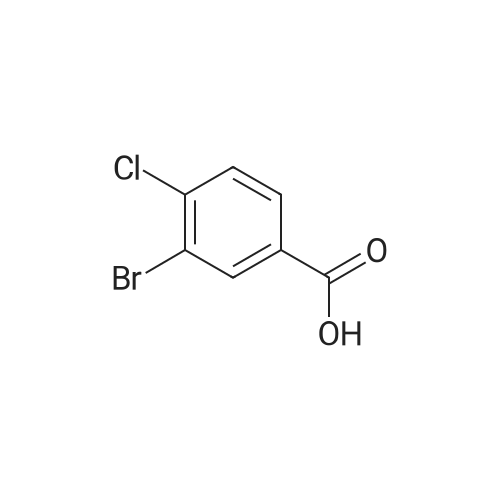
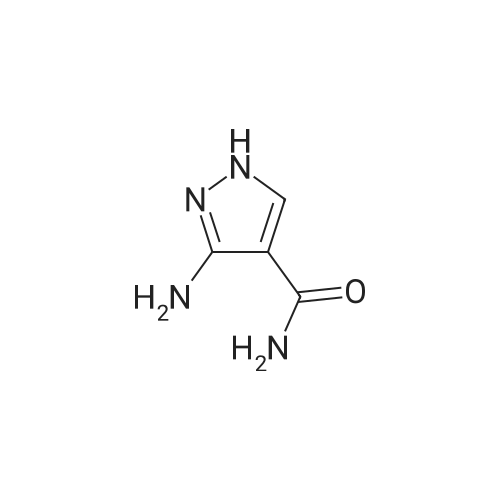
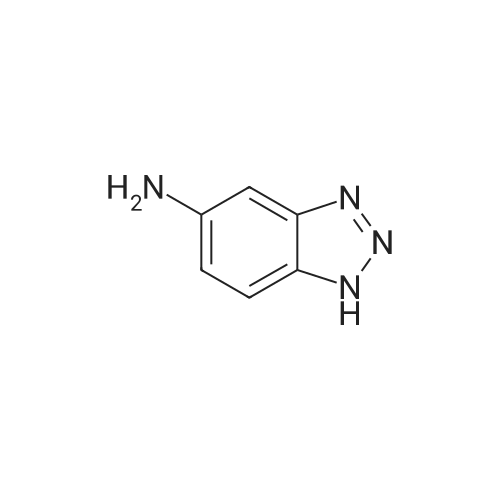
[1] European Journal of Medicinal Chemistry, 2018, vol. 147, p. 227 - 237
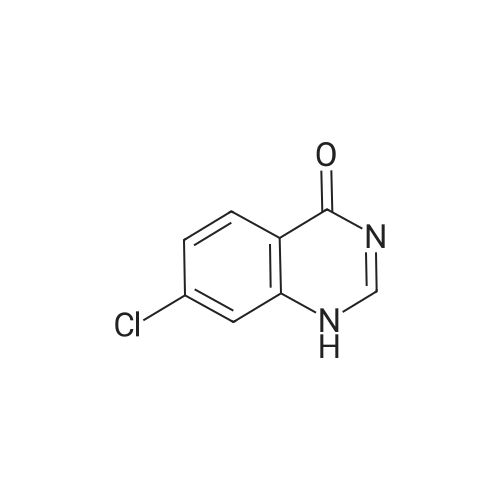
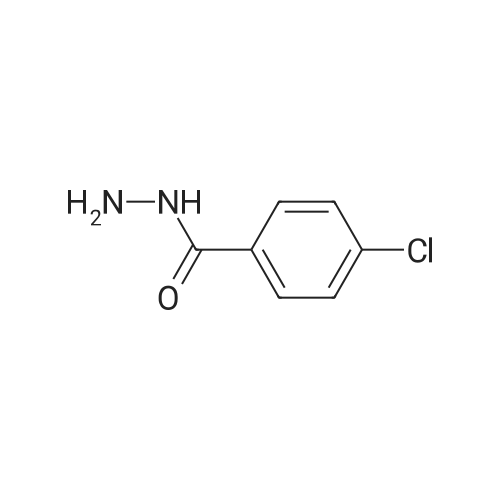
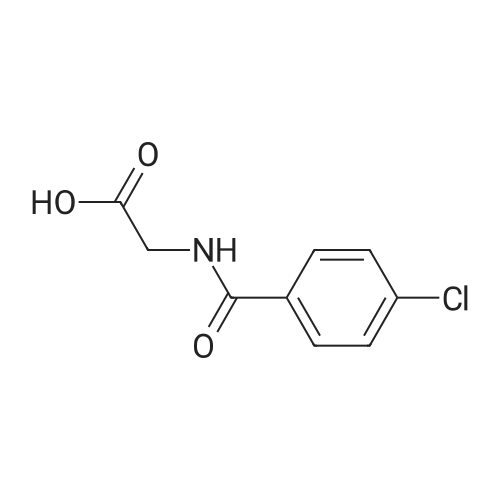
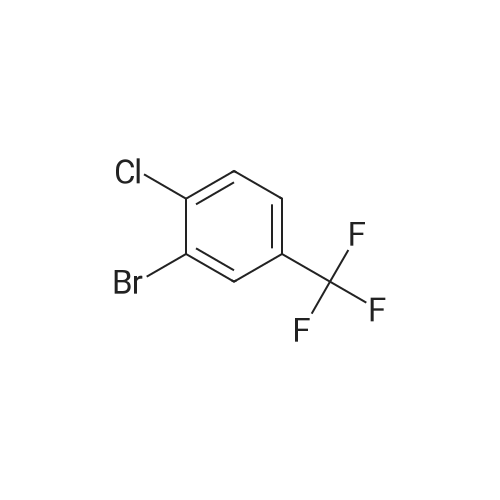
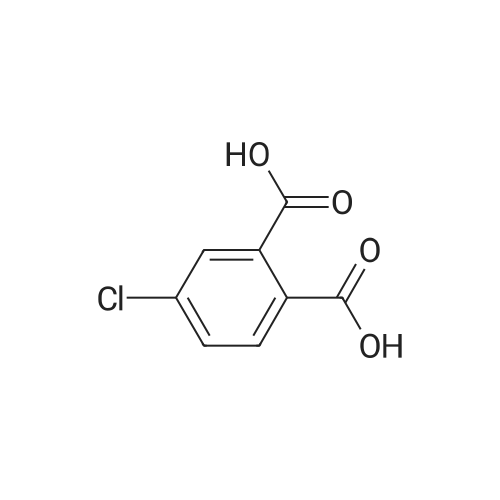
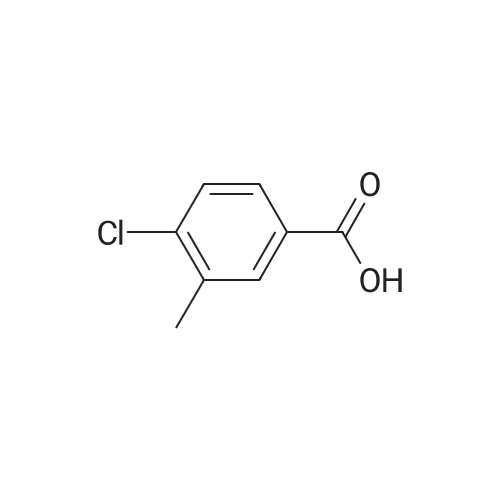
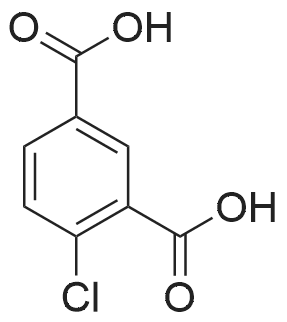
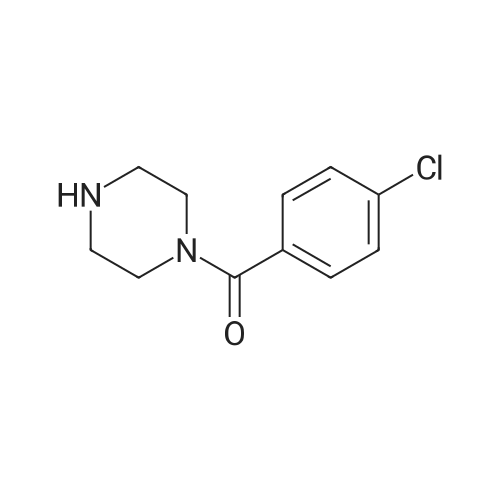
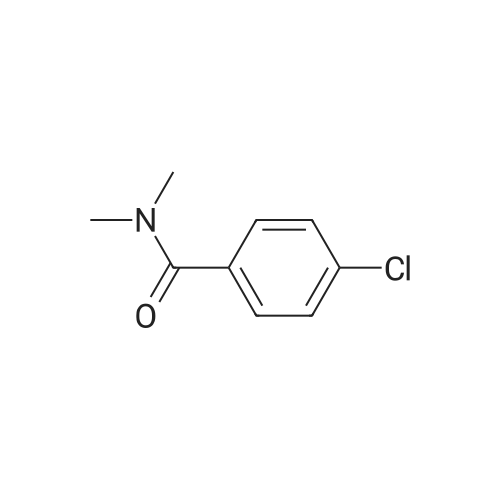
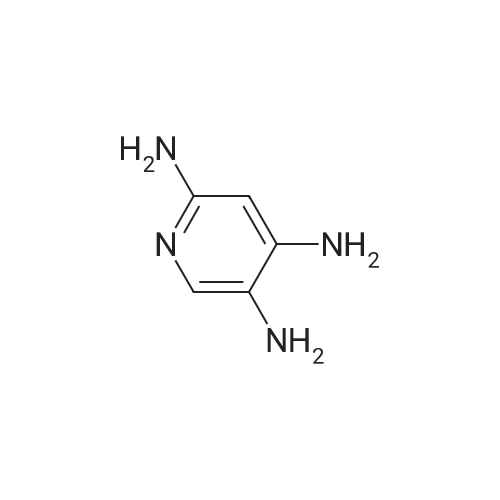
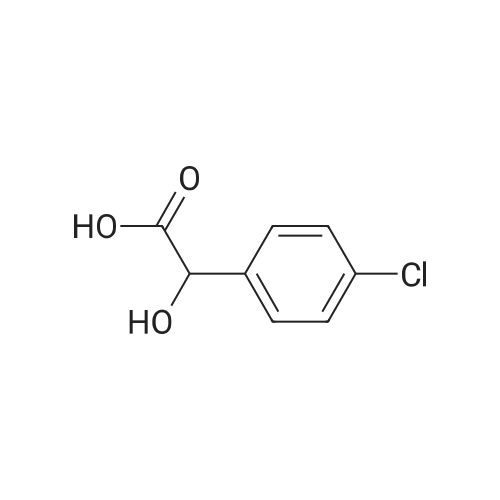
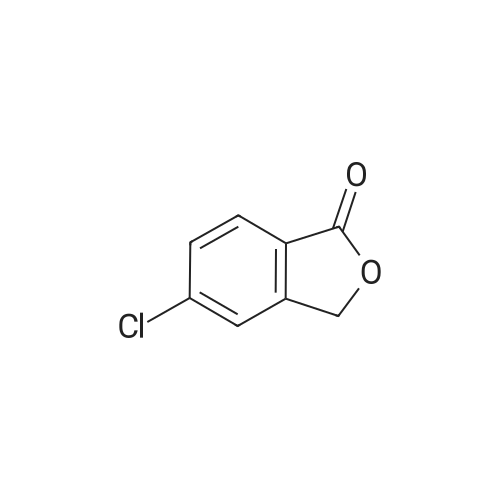
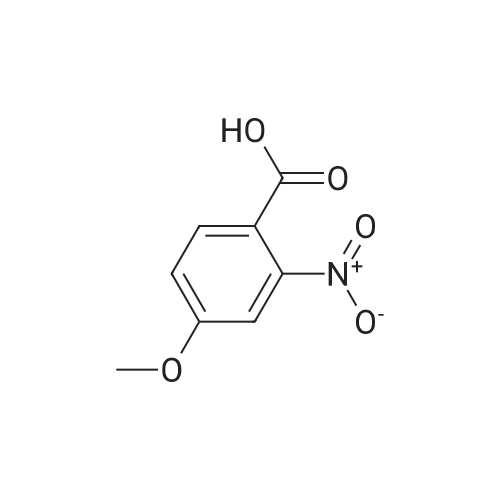
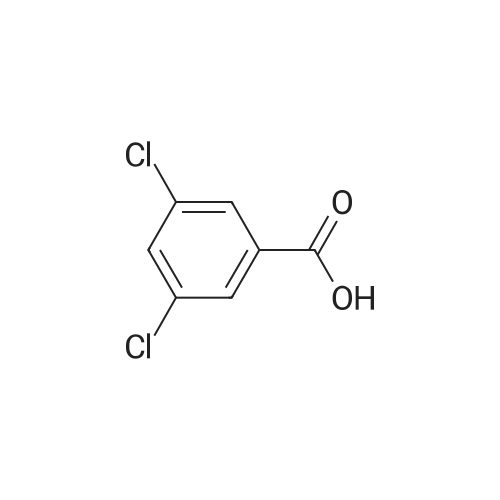
3
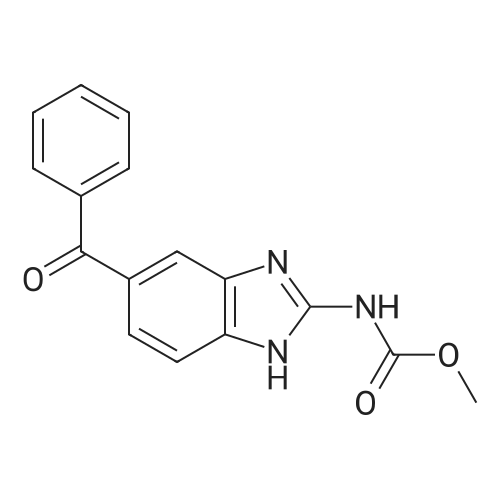
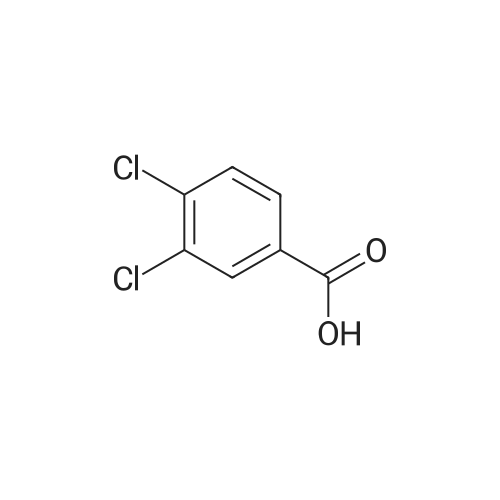
[ 74-11-3 ]
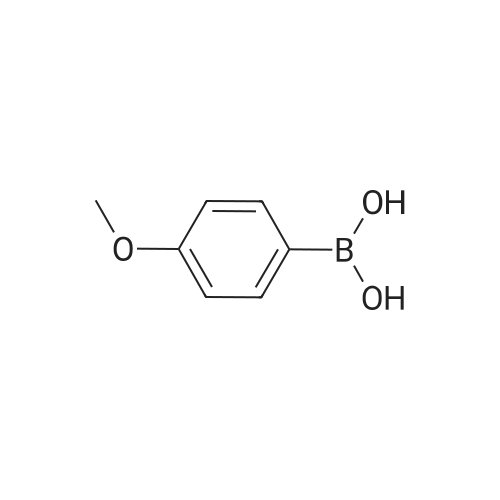
[ 536-40-3 ]
Yield
Reaction Conditions
Operation in experiment
74%
Stage #1: With thionyl chloride In methanol at 50℃; for 4 h; Cooling with ice Stage #2: With hydrazine hydrate In ethanol for 4 h; Reflux
Dissolve 4-chlorobenzoic acid (3. 2g, 20mmol) in methanol (30mL) and slowly add 2mL of thionyl chloride in an ice bath. Heat at 50 ° C for 4h (15mLX3), spin-dried for use; the previous step in 30mL ethanol, 80percent hydrazine hydrate added 4mL, reflux reaction heating (15mLX3), the mixture was cooled to room temperature, 4h, cooling, precipitation of white solid, filter drying, a white solid 2. 5g, yield 74percent.
Reference:
[1] European Journal of Medicinal Chemistry, 2017, vol. 138, p. 396 - 406
[2] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2010, vol. 49, # 4, p. 526 - 531
[3] Journal of Chemical Research - Part S, 2003, # 12, p. 768 - 769
[4] Patent: CN106146487, 2016, A, . Location in patent: Paragraph 0167; 0168; 0169
[5] Journal of Pharmacy and Pharmacology, 2001, vol. 53, # 2, p. 267 - 272
[6] Heterocyclic Communications, 2002, vol. 8, # 6, p. 601 - 606
[7] Phosphorus, Sulfur and Silicon and the Related Elements, 2006, vol. 181, # 9, p. 2079 - 2087
[8] European Journal of Medicinal Chemistry, 2006, vol. 41, # 7, p. 841 - 846
[9] Journal of Organic Chemistry, 2004, vol. 69, # 19, p. 6449 - 6454
[10] Patent: US2009/312374, 2009, A1, . Location in patent: Page/Page column 5
[11] Journal of Heterocyclic Chemistry, 2010, vol. 47, # 4, p. 838 - 845
[12] Molecules, 2010, vol. 15, # 12, p. 9046 - 9056
[13] Journal of Chemical Research, 2010, vol. 34, # 12, p. 680 - 683
[14] Journal of Chemical Research, 2011, vol. 35, # 4, p. 234 - 237
[15] Monatshefte fur Chemie, 2010, vol. 141, # 4, p. 479 - 484
[16] Journal of Chemical Research, 2011, vol. 35, # 6, p. 364 - 367
[17] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 16, p. 5031 - 5038
[18] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 21, p. 6293 - 6301
[19] Journal of Molecular Structure, 2011, vol. 1003, # 1-3, p. 52 - 61
[20] Journal of Molecular Structure, 2012, vol. 1011, p. 121 - 127
[21] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 24, p. 7246 - 7250
[22] Chinese Journal of Chemistry, 2011, vol. 29, # 10, p. 2153 - 2156
[23] Arzneimittel-Forschung/Drug Research, 2011, vol. 61, # 8, p. 452 - 457
[24] Journal of Heterocyclic Chemistry, 2018, vol. 55, # 4, p. 863 - 870
[25] Letters in Drug Design and Discovery, 2012, vol. 9, # 3, p. 276 - 281
[26] Molecules, 2012, vol. 17, # 5, p. 5095 - 5107
[27] Journal of Chemical Research, 2012, vol. 36, # 7, p. 383 - 386
[28] Phosphorus, Sulfur and Silicon and the Related Elements, 2012, vol. 187, # 11, p. 1401 - 1408
[29] Bulletin of the Korean Chemical Society, 2012, vol. 33, # 12, p. 3943 - 3949
[30] Australian Journal of Chemistry, 2012, vol. 65, # 10, p. 1413 - 1419,7
[31] Australian Journal of Chemistry, 2012, vol. 65, # 10, p. 1413 - 1419
[32] Chemical Communications (Cambridge, United Kingdom), 2012, vol. 48, # 92, p. 11307 - 11309,3
[33] Journal of Agricultural and Food Chemistry, 2012, vol. 60, # 47, p. 11649 - 11656
[34] Medicinal Chemistry Research, 2012, vol. 21, # 11, p. 3646 - 3655
[35] Medicinal Chemistry Research, 2013, vol. 22, # 3, p. 1305 - 1312
[36] Journal of the Brazilian Chemical Society, 2013, vol. 24, # 1, p. 115 - 120
[37] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 8, p. 2286 - 2297
[38] Monatshefte fur Chemie, 2013, vol. 144, # 6, p. 825 - 849
[39] Medicinal Chemistry Research, 2013, vol. 22, # 6, p. 2755 - 2767
[40] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 17, p. 5395 - 5406
[41] Bulletin of the Korean Chemical Society, 2012, vol. 33, # 12, p. 4180 - 4184
[42] Medicinal Chemistry Research, 2013, vol. 22, # 10, p. 4980 - 4991
[43] Indian Journal of Heterocyclic Chemistry, 2011, vol. 21, # 1, p. 41 - 44
[44] Medicinal Chemistry Research, 2013, vol. 22, # 11, p. 5344 - 5348
[45] European Journal of Medicinal Chemistry, 2014, vol. 71, p. 199 - 218
[46] Chemical Biology and Drug Design, 2013, vol. 82, # 5, p. 546 - 556
[47] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 1, p. 192 - 194
[48] European Journal of Medicinal Chemistry, 2014, vol. 74, p. 73 - 84
[49] Tetrahedron, 2014, vol. 70, # 12, p. 2190 - 2194
[50] Journal of the Brazilian Chemical Society, 2014, vol. 25, # 1, p. 104 - 111
[51] European Journal of Medicinal Chemistry, 2014, vol. 80, p. 167 - 174
[52] Medicinal Chemistry Research, 2014, vol. 23, # 4, p. 1661 - 1671
[53] Molecules, 2014, vol. 19, # 3, p. 3436 - 3439
[54] Dyes and Pigments, 2014, vol. 108, p. 32 - 40
[55] Archives of Pharmacal Research, 2014, vol. 37, # 7, p. 852 - 861
[56] Synthetic Communications, 2014, vol. 44, # 18, p. 2724 - 2737
[57] Journal of the Chemical Society of Pakistan, 2014, vol. 36, # 3, p. 503 - 511
[58] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 22, p. 5154 - 5156
[59] European Journal of Medicinal Chemistry, 2014, vol. 87, p. 175 - 185
[60] New Journal of Chemistry, 2015, vol. 39, # 1, p. 453 - 460
[61] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 3, p. 481 - 484
[62] Journal of Heterocyclic Chemistry, 2015, vol. 52, # 2, p. 352 - 357
[63] European Journal of Medicinal Chemistry, 2015, vol. 96, p. 330 - 339
[64] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 15, p. 3052 - 3056
[65] Medicinal Chemistry Research, 2014, vol. 24, # 6, p. 2514 - 2528
[66] Phosphorus, Sulfur and Silicon and the Related Elements, 2015, vol. 190, # 7, p. 1045 - 1055
[67] Chemical Communications, 2015, vol. 51, # 76, p. 14365 - 14368
[68] Medicinal Chemistry Research, 2015, vol. 24, # 12, p. 4166 - 4180
[69] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 17, p. 6014 - 6024
[70] Transition Metal Chemistry, 2015, vol. 40, # 6, p. 665 - 671
[71] RSC Advances, 2015, vol. 5, # 118, p. 97089 - 97101
[72] Asian Journal of Chemistry, 2015, vol. 27, # 10, p. 3605 - 3608
[73] Asian Journal of Chemistry, 2016, vol. 28, # 3, p. 639 - 643
[74] Zeitschrift fur Naturforschung - Section B Journal of Chemical Sciences, 2015, vol. 70, # 8, p. 609 - 616
[75] Bioorganic Chemistry, 2016, vol. 65, p. 126 - 136
[76] European Journal of Medicinal Chemistry, 2016, vol. 120, p. 134 - 147
[77] Phosphorus, Sulfur and Silicon and the Related Elements, 2016, vol. 191, # 6, p. 904 - 907
[78] Patent: WO2016/108249, 2016, A1,
[79] Journal of Heterocyclic Chemistry, 2016, vol. 53, # 5, p. 1651 - 1654
[80] Bioorganic Chemistry, 2016, vol. 69, p. 48 - 63
[81] Oriental Journal of Chemistry, 2016, vol. 32, # 4, p. 2155 - 2161
[82] Journal of the Brazilian Chemical Society, 2016, vol. 27, # 11, p. 1998 - 2010
[83] Letters in Drug Design and Discovery, 2016, vol. 13, # 9, p. 968 - 981
[84] Journal of the Chemical Society of Pakistan, 2016, vol. 38, # 5, p. 864 - 881
[85] Patent: CN106008390, 2016, A,
[86] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 1, p. 213 - 220
[87] Chemical Biology and Drug Design, 2017, vol. 89, # 1, p. 47 - 60
[88] Chinese Journal of Chemistry, 2016, vol. 34, # 12, p. 1236 - 1244
[89] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2016, vol. 55B, # 2, p. 207 - 212
[90] Arkivoc, 2016, vol. 2017, # 2, p. 87 - 106
[91] Oriental Journal of Chemistry, 2017, vol. 33, # 1, p. 335 - 345
[92] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 2, p. 1423 - 1429
[93] ChemMedChem, 2017, vol. 12, # 12, p. 972 - 985
[94] European Journal of Medicinal Chemistry, 2017, vol. 138, p. 140 - 151
[95] Chemical Biology and Drug Design, 2017, vol. 90, # 2, p. 236 - 243
[96] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 20, p. 5652 - 5661
[97] Medicinal Chemistry Research, 2017, vol. 26, # 10, p. 2260 - 2271
[98] Medicinal Chemistry Research, 2017, vol. 26, # 12, p. 3367 - 3374
[99] Oriental Journal of Chemistry, 2017, vol. 33, # 2, p. 971 - 978
[100] Oriental Journal of Chemistry, 2017, vol. 33, # 6, p. 2930 - 2936
[101] Pharmaceutical Chemistry Journal, 2018, vol. 51, # 10, p. 907 - 917
[102] RSC Advances, 2018, vol. 8, # 12, p. 6306 - 6314
[103] European Journal of Medicinal Chemistry, 2018, vol. 151, p. 705 - 722
[104] Bioorganic and Medicinal Chemistry, 2018, vol. 26, # 8, p. 1971 - 1985
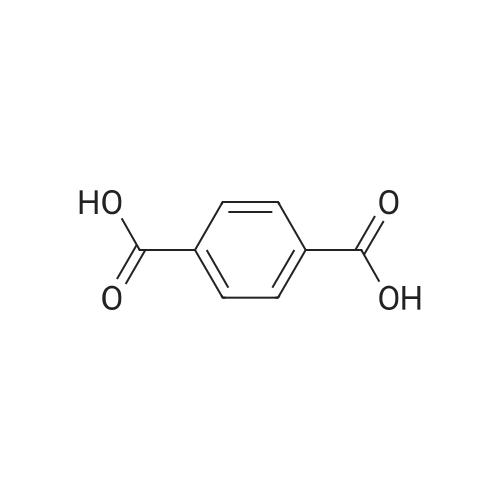
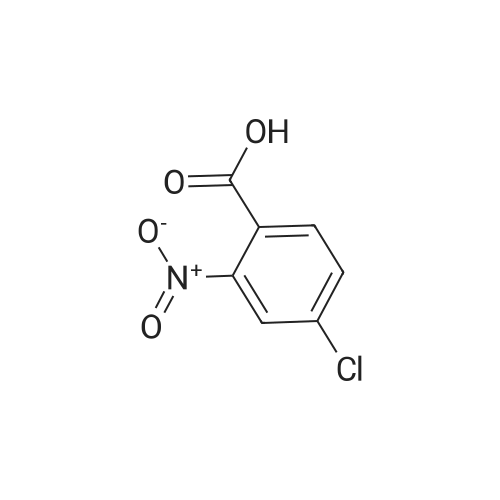
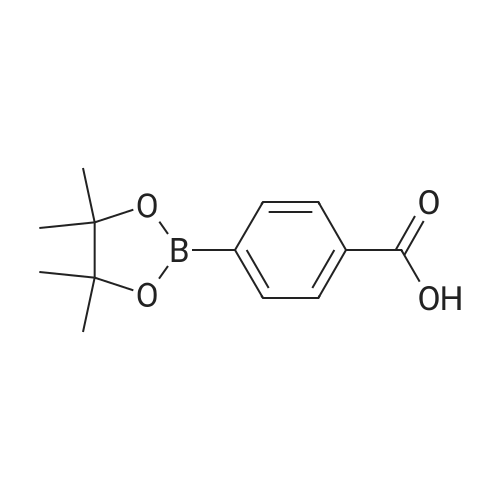
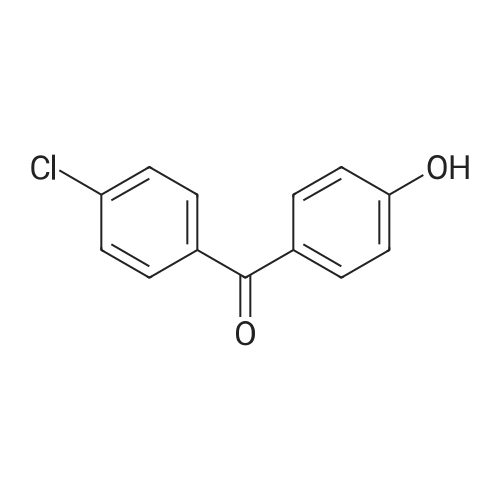
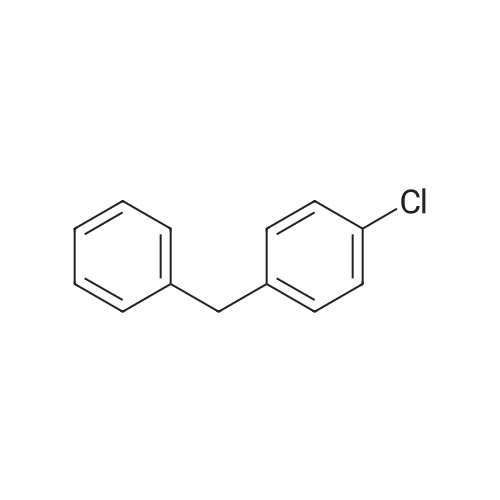
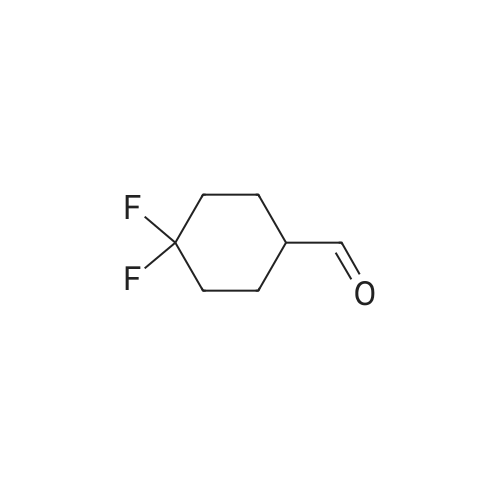
4
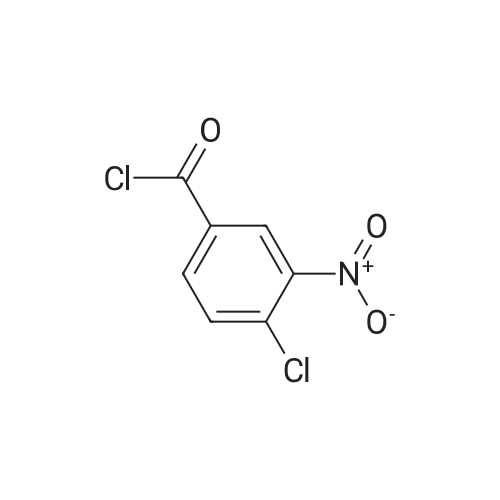
[ 74-11-3 ]
[ 25026-34-0 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2000, vol. 10, # 15, p. 1723 - 1727
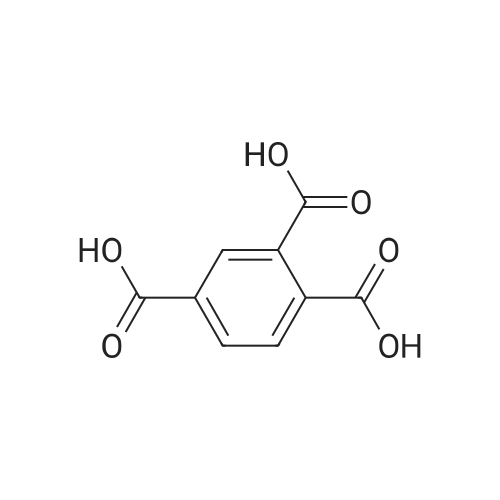
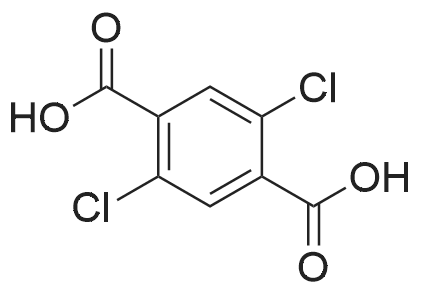
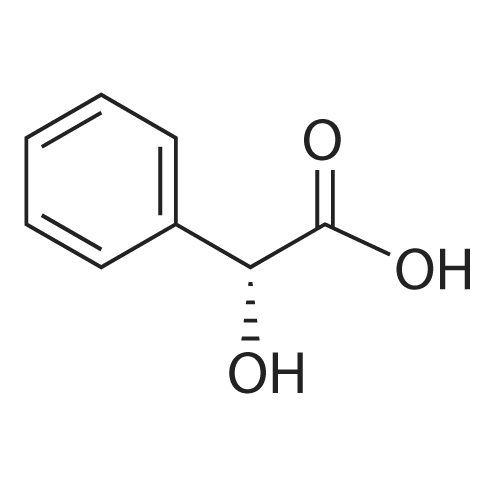
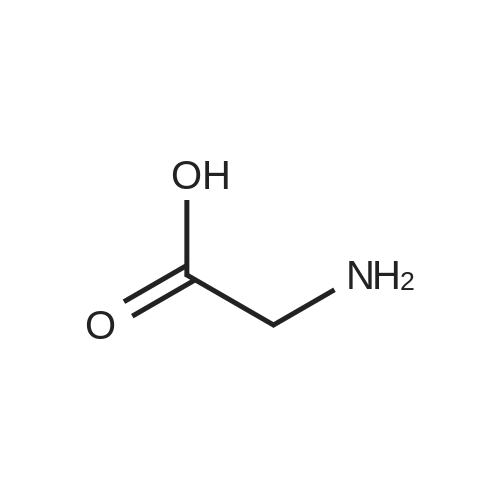
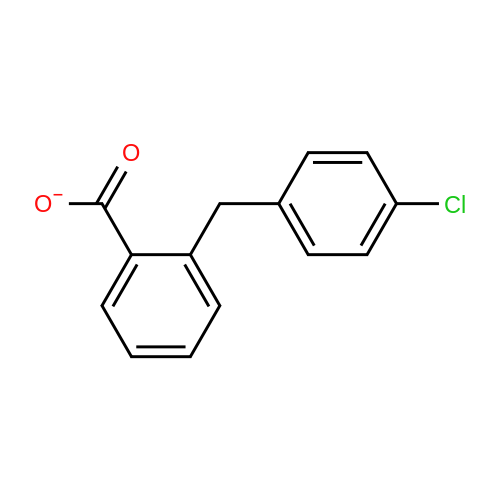
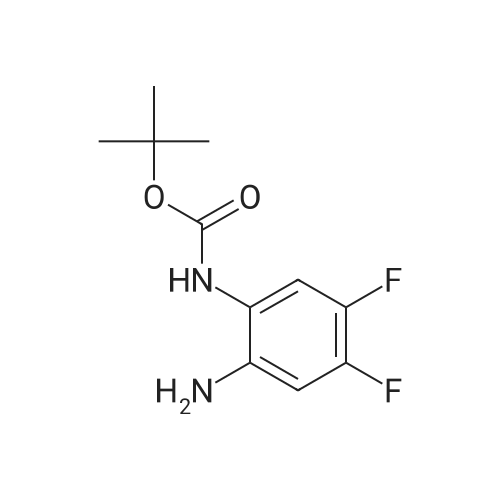
5
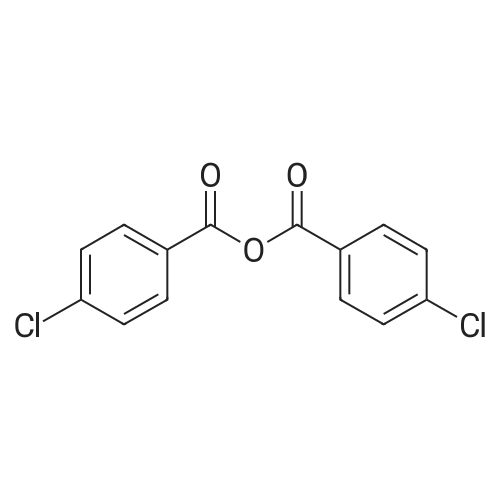
[ 74-11-3 ]
[ 118-97-8 ]
Reference:
[1] Journal of Organic Chemistry, 1983, vol. 48, # 7, p. 1056 - 1059
[2] Russian Chemical Bulletin, 2007, vol. 56, # 6, p. 1216 - 1226
[3] Chemische Berichte, 1906, vol. 39, p. 1344
[4] Justus Liebigs Annalen der Chemie, 1909, vol. 366, p. 89
[5] Acta Chemica Scandinavica (1947-1973), 1950, vol. 4, p. 392
[6] Journal of Organic Chemistry, 2014, vol. 79, # 11, p. 5134 - 5144
[7] Chemical Communications, 2018, vol. 54, # 73, p. 10296 - 10299
6
[ 74-11-3 ]
[ 89-77-0 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2018, vol. 147, p. 227 - 237
7
[ 74-11-3 ]
[ 31431-39-7 ]
Reference:
[1] Patent: WO2016/127168, 2016, A2,
8
[ 74-11-3 ]
[ 38818-50-7 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1900, vol. 19, p. 55
9
[ 74-11-3 ]
[ 790-41-0 ]
Yield
Reaction Conditions
Operation in experiment
99%
With iodine; triethylamine; triphenylphosphine In dichloromethane at 0 - 20℃;
General procedure: To a solution of iodine (0.1573 g, 0.62 mmol) in CH2Cl2 (2 mL) was added with triphenylphosphine (0.1626 g, 0.62 mmol) in one portion at 0 oC. Carboxylic acid (0.41 mmol)was subsequently added into the mixture, followed by addition of triethylamine (0.17 mL, 1.23mmol) at 0 oC. The reaction mixture was allowed to warm up to room temperature and stirred until completion of the reaction (typically within 10 min). The crude mixture was concentrated under reduced pressure and then purified by column chromatography (CC) using 5-10percent ethylacetate in hexane to give the desired anhydride product.
86%
With potassium carbonate; p-toluenesulfonyl chloride In acetonitrile at 20℃; for 1 h;
General procedure: TsCl (0.5 mmol, 0.10 g) was added to the mixture of carboxylic acid (1 mmol) and K2CO3 (1.5 mmol, 0.20 g) in dry CH3CN (5 mL), the reaction mixture was stirred at roomtemperature for the appropriate time (20-180 min, Table 2) until the TsCl was no longer detectable by TLC. After the completion of the reaction, CH2Cl2 or Et2O (2×10 mL) was added to the reaction mixture, stirred, filtered, and the organic layer was dried over CaCl2. The evaporation of the solvent afforded a highly pure product.
Reference:
[1] Tetrahedron Letters, 2016, vol. 57, # 3, p. 325 - 328
[2] Synthesis, 1981, # 8, p. 616 - 620
[3] Tetrahedron Letters, 1986, vol. 27, # 41, p. 4937 - 4940
[4] Synthesis, 1981, # 3, p. 218 - 220
[5] Synthetic Communications, 1983, vol. 13, # 6, p. 471 - 488
[6] Synthetic Communications, 2001, vol. 31, # 3, p. 395 - 399
[7] Advanced Synthesis and Catalysis, 2016, vol. 358, # 11, p. 1725 - 1730
[8] Letters in Organic Chemistry, 2017, vol. 14, # 6, p. 431 - 439
[9] Journal of the Iranian Chemical Society, 2010, vol. 7, # 2, p. 455 - 460
[10] Archiv der Pharmazie, 2018, vol. 351, # 6,
[11] Journal of Organic Chemistry, 1994, vol. 59, # 10, p. 2913 - 2914
[12] Journal of the American Chemical Society, 1918, vol. 40, p. 427
[13] Polish Journal of Chemistry, 1978, vol. 52, p. 307 - 314
[14] Synthesis, 1982, # 4, p. 288 - 291
[15] Synthesis, 1982, # 4, p. 288 - 291
[16] Journal of Medicinal Chemistry, 2007, vol. 50, # 9, p. 2249 - 2253
[17] Synthesis, 2007, # 22, p. 3489 - 3496
[18] Chemical Papers, 2015, vol. 69, # 3, p. 479 - 485
[19] Bioorganic and Medicinal Chemistry, 2018, vol. 26, # 4, p. 891 - 902
[20] Bioorganic Chemistry, 2018, vol. 81, p. 191 - 202
[21] Medicinal Chemistry, 2018, vol. 14, # 7, p. 660 - 673
10
[ 15163-60-7 ]
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] Chemical Papers, 2015, vol. 69, # 3, p. 479 - 485
11
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] J. Gen. Chem. USSR (Engl. Transl.), 1985, vol. 55, # 12, p. 2393 - 2396[2] Zhurnal Obshchei Khimii, 1985, vol. 55, # 12, p. 2692 - 2696
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2007, vol. 55, # 1, p. 156 - 158
14
[ 77339-54-9 ]
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] J. Gen. Chem. USSR (Engl. Transl.), 1985, vol. 55, # 12, p. 2393 - 2396[2] Zhurnal Obshchei Khimii, 1985, vol. 55, # 12, p. 2692 - 2696
15
[ 64-17-5 ]
[ 74-11-3 ]
[ 7335-27-5 ]
[ 790-41-0 ]
Reference:
[1] Synthesis, 2007, # 22, p. 3489 - 3496
16
[ 122-01-0 ]
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 2002, vol. 75, # 1, p. 137 - 148
[2] New Journal of Chemistry, 2017, vol. 41, # 3, p. 931 - 939
17
[ 108-24-7 ]
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] Journal of the American Chemical Society, 1952, vol. 74, p. 4110
18
[ 541-41-3 ]
[ 74-11-3 ]
[ 7335-27-5 ]
[ 790-41-0 ]
Reference:
[1] Journal of Organic Chemistry, 1985, vol. 50, # 5, p. 560 - 565
19
[ 674-82-8 ]
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] Patent: US2476859, 1948, ,
20
[ 2078-12-8 ]
[ 74-11-3 ]
[ 790-41-0 ]
Reference:
[1] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1987, vol. 26, # 1-12, p. 407 - 411
21
[ 74-11-3 ]
[ 53449-14-2 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2018, vol. 147, p. 227 - 237
With iodine; triethylamine; triphenylphosphine; In dichloromethane; at 0 - 20℃;
General procedure: To a solution of iodine (0.1573 g, 0.62 mmol) in CH2Cl2 (2 mL) was added with triphenylphosphine (0.1626 g, 0.62 mmol) in one portion at 0 oC. Carboxylic acid (0.41 mmol)was subsequently added into the mixture, followed by addition of triethylamine (0.17 mL, 1.23mmol) at 0 oC. The reaction mixture was allowed to warm up to room temperature and stirred until completion of the reaction (typically within 10 min). The crude mixture was concentrated under reduced pressure and then purified by column chromatography (CC) using 5-10% ethylacetate in hexane to give the desired anhydride product.
86%
With potassium carbonate; p-toluenesulfonyl chloride; In acetonitrile; at 20℃; for 1h;
General procedure: TsCl (0.5 mmol, 0.10 g) was added to the mixture of carboxylic acid (1 mmol) and K2CO3 (1.5 mmol, 0.20 g) in dry CH3CN (5 mL), the reaction mixture was stirred at roomtemperature for the appropriate time (20-180 min, Table 2) until the TsCl was no longer detectable by TLC. After the completion of the reaction, CH2Cl2 or Et2O (2×10 mL) was added to the reaction mixture, stirred, filtered, and the organic layer was dried over CaCl2. The evaporation of the solvent afforded a highly pure product.
With dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;
General procedure: General procedure for synthesis of anhydride (12a-12p) Mixture of substituted acid derivative (0.02?mol) and dicyclohexylcarbodiimide (0.01?mol) was dissolved in dichloromethane (50?ml) and stirred at room temperature for 3-4?h. After then solvent was separated (12a-12p), the reaction mixture was filtered to remove the precipitated dicyclohexylurea and the filtrate was evaporated to get the oily product (12a-12p).
With dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;
General procedure: To 50 ml of methylene chloride (DCM), substituted acid derivative (0.02 mol) and dicyclohexylcarbodiimide (DCC) (0.01 mol) was added and stirred at room temperature for 3-4 h. After then the solvent was separated (2a-2n), the reaction mixture was filtered to remove precipitate of dicyclohexylurea and the filtrate was collected, washed three times with 10% sodium bicarbonate and three time with water to make it free from alkali and dried over anhydrous magnesium sulfate. The dried extract was filtered, and the solvent was removed under reduced pressure. The residue was dissolved in ethanol and water was added until turbidity appeared. It was then kept in freezer overnight. The oily precipitate obtained was solidified and collected (2a-2n) [40-42].
With dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;
General procedure: Substituted aromatic acid (0.02 M) and dicyclohexylcarbodiimide (0.01 M) were dissolved in 50 ml methylene chloride. The reaction mixture was stirred at room temperature for 3-4 h. The precipitated dicyclohexylurea was removed by filtration. The solvent was evaporated and the oily product obtained was collected and used without purification.
With pyridine; In toluene; at 0℃; for 2h;Reflux; Inert atmosphere;
General procedure: Step 1 In a three-necked 100 mL flask under N2-atmosphere, the corresponding benzoic acid (32 mmol) was suspended in anhydrous toluene (21 mL, 1.5 M). A few drops of anhydrous DMF (126 muL, 1.6 mmol, 0.05 equiv) and SOCl2 (3.0 mL, 41.5 mmol, 1.3 equiv) were added atr.t. sequentially. The mixture was heated to 70 C and stirred at this temperature for 1 hour, then cooled to r.t. and the excess SOCl2 was purged from the system by flushing with N2 for 15 min. The solution of the benzoyl chloride was used without purification in the next step. Step 2 In a three-necked flask under N2-atmosphere, the corresponding benzoic acid (32 mmol, 1.0 equiv) was suspended in anhydrous toluene (21 mL, 1.5 M). Anhydrous pyridine (3.1 mL, 38.3 mmol, 1.2 equiv) was added to the mixture and the resulting solution was cooled to0 C. A solution of the corresponding benzoyl chloride from Step 1 was added at such rate that the temperature was kept under 20 C, while a white precipitate formed. The resulting suspension was heated at reflux for 2 h then cooled to r.t. The precipitate (Pyr·HCl) was filtered, washed with toluene, and the filtrate was evaporated. The residue was dissolved in CH2Cl2 (100 mL), washed with 10% HCl (10 mL) then washed with cc. NaHCO3 (3 × 10 mL) and brine (10 mL). The organic layer was dried over Na2SO4, filtered and evaporated to dryness. Pure anhydrides were obtained in 91-98% yield; NMR data were consistent with the reported values.
2-[[(6-bromo-benzo[1,3]dioxol-5-yl)-cyclohexylcarbamoyl-methyl]-(4-chloro-benzoyl)-amino]-methyl}-pyrrolidine-1-carboxylic acid <i>tert</i>-butyl ester[ No CAS ]
EXAMPLE 12 4-Chlorophthalic anhydride (30 g, 160 mmol) and 1,2,4-trichlorobenzene (30 g) were heated to reflux (approximately 224 C.) and tetraphenyl phosphonium bromide (Hokko Chemical, 0.30 g, 0.7 mmol) and 4-chlorobenzoic acid (Aldrich, 0.06 g, 0.4 mmol) were added. Dry potassium carbonate (7.95 g, 58 mmol) was then added in portions over one hour. The reaction was monitored by GC and showed 62.9% conversion to 4,4'-oxydiphthalic anhydride (ODPAN) 3 hours after addition of carbonate was completed.
With copper; In water; N,N-dimethyl-formamide; at 150℃; for 28h;Autoclave;
To a 200 ml pressure reactionvessel was added p-chlorobenzoic acid 15.7g (1.0eq), 50 ml of water, N, Ndimethylformamide5 ml, n butylamine 21.9g (3.0eq), copper powder 0.3g (0.05eq), addedafter the temperature was raised to 150 degrees for 28 hours, and cooled to 25 degrees,with 10% sodium hydroxide to adjust PH = 10-12, filtered and the filtrate concentratedhydrochloric acid to adjust pH = 1-2, collected by filtration cake was dried in vacuo togive 4-amino-n-butyric acid 16.6g.
e) N-Benzyl-2-[2-(4-chloro-phenyl)-5,6-difluoro-benzoimidazol-l-yll-2-(4,4-difluoro- cyclohexyl) -acetamide; To a solution of 3.8 g (15.56 mmol) (2-amino-4,5-difluoro-phenyl)-carbamic acid tert- butyl ester (Example 73, intermediate g) in 38 ml methanol, 2.54 g (17.12 mmol) 4,4- difluorocyclohexanone (commercially available) were added. After stirring for 5 min. at room temperature the solution was treated with 2.44 g (15.58 mmol) p-chlorobenzoic acid, followed by an addition of 1.9 ml (15.56 mmol) benzyl isocyanide (commercially available). To the viscous slurry 12 ml methanol were added and the reaction for 22.5 h. Then 19.45 ml (77.80 mmol) 4M hydrochloric acid in dioxane were added dropwise over 5 min. After 4.5 h another 19.45 ml (77.80 mmol) 4M hydrochloric acid in dioxane were added. After 24 h, the suspension was poured onto 300 ml aqueous saturated sodium bicarbonate solution and the phases were separated. The aqueous layer was extracted three times with ethyl acetate and the organic layers were washed with brine, dried over magnesium sulfate, filtered, and evaporated. The residue was purified by silica gel chromatography using a MPLC system (CombiFlash Companion, Isco Inc.) eluting with a gradient of heptane : ethyl acetate (100 : 0 to 50 : 50). Light yellow solid (76%). MS (Turbo Spray): m/z = 530.2 [M+H].).
With di-tert-butyl peroxide; copper(II) bis(trifluoromethanesulfonate); In 1,2-dichloro-ethane; at 130℃; for 12h;Sealed tube;
General procedure: A 50 mL sealed tube (with a Teflon high pressure valve) equipped with a magnetic stir bar was charged with Cu(OTf)2 (0.05 mmol), followed by carboxylic acid (0.5 mmol), formamide (2.0 mmol), tert-butyl peroxide (DTBP, 1 mmol), and DCE (1 mL). After the reaction mixture was stirred at 130 C for 12 h, it was allowed to cool to ambient temperature. The reaction mixture was diluted with ethyl acetate, and then filtered through a small pad of Celite. The filtrate was washed with saturated aqueous NaHCO3 (5 mL) and brine (5 mL, twice). The organic phase was dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica gel preparative TLC to give the corresponding product.
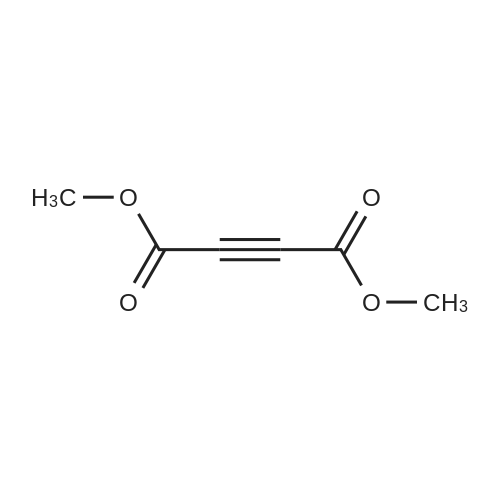
General procedure: At room temperature, organic carbonyl acid 3 (R-COOH, 0.5 mmol) was added into a reaction tube equipped with a small magnet. Then a solution of tertiary amine 1 (R1CH2-NR2R3, 0.5 mmol ) in DCM (2.5 mL) was added dropwise in 2 min. After the mixture was stirred at room temperature for a few minutes, 1 equivalents of dimethyl acetylenedicarboxylate (DMAD, 2) was added. The reaction was stirred overnight at room temperature, and then monitored by TLC with silica gel coated plates. After being stirred for 14 h, the solvent was removed and the residue was purified by a flash column chromatography with silica gel with ethyl acetate/hexane (1:25-30) as eluent to give the desired products 4, 5, and 7. Most of compounds are known and confirmed by NMR, ESI-MS, IR.
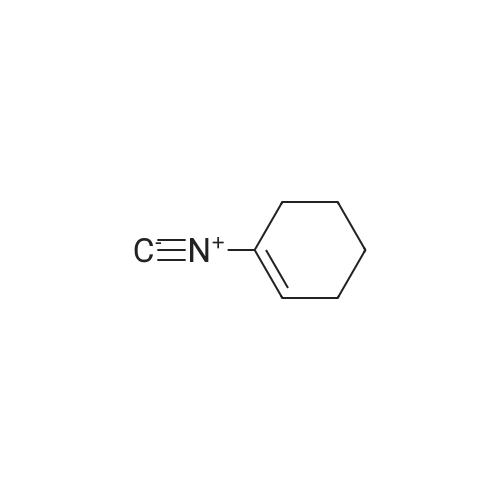
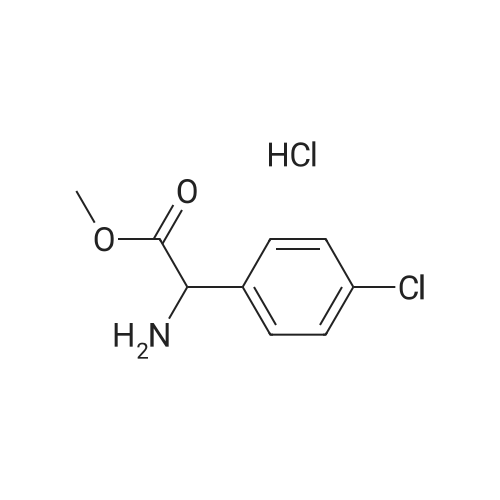
General procedure: Powdered substituted 2-nitrobenzoic acid (10 mmol)was added to a well stirred solution of KOH (10 mmol) in CH3OH (10 mL). The resulting suspension was stirred at room temperature for 10 min and then cooled to 0 C and treated with methyl 2-amino-2-(4-chlorophenyl)acetate hydrochloride (10 mmol) or 2-amino-2-(4-fluorophenyl)acetate hydrochloride (10 mmol) (3), a solution of 1-isocyanocyclohexene (4) (11 mmol) in CH3OH (2 mL) and a solution of p-chlorobenzaldehyde or 4-fluorobenzaldehyde (5) (10 mmol) in CH3OH (2 mL), in the order given. The cooling bath was removed and the reaction mixture was stirred at room temperature for 1 day. Removal of the solvent under reduced pressure left a residue which was stirred with H2O (10 mL) and CH2Cl2 (80 mL). The organic layer was dried (Na2SO4) and evaporated to dryness. The residue was stirred with boiling hexanes (50 mL) for 5 min. The supernatant liquid was discarded while still warm. An analogous treatment was performed with boiling H2O (50 mL x 2). The resulting solid product was stirred with AcOH (60 mL). The resulting solution was heated at 50 C and treated under vigorous stirring with iron powder (40 mmol) in one portion. When the exothermic reaction had subsided, the reaction mixture was heated at 70 C for 1 h and then allowed to cool and stirred with CH2Cl2 (50 mL) and H2O (50 mL). The resulting suspension was filtered to remove the unreacted iron and the filtrate transferred to a separating funnel. The organic layer was washed with H2O (50 mL), NaHCO3 (aq. 2%, 50 mL), H2O (50 mL) and then separated, dried (Na2SO4), and evaporated to dryness. The residue was purified by column chromatography (n-hexane-ethyl acetate, 5:1) togive 7a-r, yield: 45-52%.
General procedure: Powdered substituted 2-nitrobenzoic acid (10 mmol)was added to a well stirred solution of KOH (10 mmol) in CH3OH (10 mL). The resulting suspension was stirred at room temperature for 10 min and then cooled to 0 C and treated with methyl 2-amino-2-(4-chlorophenyl)acetate hydrochloride (10 mmol) or 2-amino-2-(4-fluorophenyl)acetate hydrochloride (10 mmol) (3), a solution of 1-isocyanocyclohexene (4) (11 mmol) in CH3OH (2 mL) and a solution of p-chlorobenzaldehyde or 4-fluorobenzaldehyde (5) (10 mmol) in CH3OH (2 mL), in the order given. The cooling bath was removed and the reaction mixture was stirred at room temperature for 1 day. Removal of the solvent under reduced pressure left a residue which was stirred with H2O (10 mL) and CH2Cl2 (80 mL). The organic layer was dried (Na2SO4) and evaporated to dryness. The residue was stirred with boiling hexanes (50 mL) for 5 min. The supernatant liquid was discarded while still warm. An analogous treatment was performed with boiling H2O (50 mL x 2). The resulting solid product was stirred with AcOH (60 mL). The resulting solution was heated at 50 C and treated under vigorous stirring with iron powder (40 mmol) in one portion. When the exothermic reaction had subsided, the reaction mixture was heated at 70 C for 1 h and then allowed to cool and stirred with CH2Cl2 (50 mL) and H2O (50 mL). The resulting suspension was filtered to remove the unreacted iron and the filtrate transferred to a separating funnel. The organic layer was washed with H2O (50 mL), NaHCO3 (aq. 2%, 50 mL), H2O (50 mL) and then separated, dried (Na2SO4), and evaporated to dryness. The residue was purified by column chromatography (n-hexane-ethyl acetate, 5:1) togive 7a-r, yield: 45-52%.
dl-α-4-fluorophenylglycine methyl ester hydrochloride[ No CAS ]
[ 74-11-3 ]
C31H22ClF4N3O6[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: Powdered substituted 2-nitrobenzoic acid (10 mmol)was added to a well stirred solution of KOH (10 mmol) in CH3OH (10 mL). The resulting suspension was stirred at room temperature for 10 min and then cooled to 0 C and treated with methyl 2-amino-2-(4-chlorophenyl)acetate hydrochloride (10 mmol) or 2-amino-2-(4-fluorophenyl)acetate hydrochloride (10 mmol) (3), a solution of 1-isocyanocyclohexene (4) (11 mmol) in CH3OH (2 mL) and a solution of p-chlorobenzaldehyde or 4-fluorobenzaldehyde (5) (10 mmol) in CH3OH (2 mL), in the order given. The cooling bath was removed and the reaction mixture was stirred at room temperature for 1 day. Removal of the solvent under reduced pressure left a residue which was stirred with H2O (10 mL) and CH2Cl2 (80 mL). The organic layer was dried (Na2SO4) and evaporated to dryness. The residue was stirred with boiling hexanes (50 mL) for 5 min. The supernatant liquid was discarded while still warm. An analogous treatment was performed with boiling H2O (50 mL x 2). The resulting solid product was stirred with AcOH (60 mL). The resulting solution was heated at 50 C and treated under vigorous stirring with iron powder (40 mmol) in one portion. When the exothermic reaction had subsided, the reaction mixture was heated at 70 C for 1 h and then allowed to cool and stirred with CH2Cl2 (50 mL) and H2O (50 mL). The resulting suspension was filtered to remove the unreacted iron and the filtrate transferred to a separating funnel. The organic layer was washed with H2O (50 mL), NaHCO3 (aq. 2%, 50 mL), H2O (50 mL) and then separated, dried (Na2SO4), and evaporated to dryness. The residue was purified by column chromatography (n-hexane-ethyl acetate, 5:1) togive 7a-r, yield: 45-52%.
General procedure: Powdered substituted 2-nitrobenzoic acid (10 mmol)was added to a well stirred solution of KOH (10 mmol) in CH3OH (10 mL). The resulting suspension was stirred at room temperature for 10 min and then cooled to 0 C and treated with methyl 2-amino-2-(4-chlorophenyl)acetate hydrochloride (10 mmol) or 2-amino-2-(4-fluorophenyl)acetate hydrochloride (10 mmol) (3), a solution of 1-isocyanocyclohexene (4) (11 mmol) in CH3OH (2 mL) and a solution of p-chlorobenzaldehyde or 4-fluorobenzaldehyde (5) (10 mmol) in CH3OH (2 mL), in the order given. The cooling bath was removed and the reaction mixture was stirred at room temperature for 1 day. Removal of the solvent under reduced pressure left a residue which was stirred with H2O (10 mL) and CH2Cl2 (80 mL). The organic layer was dried (Na2SO4) and evaporated to dryness. The residue was stirred with boiling hexanes (50 mL) for 5 min. The supernatant liquid was discarded while still warm. An analogous treatment was performed with boiling H2O (50 mL x 2). The resulting solid product was stirred with AcOH (60 mL). The resulting solution was heated at 50 C and treated under vigorous stirring with iron powder (40 mmol) in one portion. When the exothermic reaction had subsided, the reaction mixture was heated at 70 C for 1 h and then allowed to cool and stirred with CH2Cl2 (50 mL) and H2O (50 mL). The resulting suspension was filtered to remove the unreacted iron and the filtrate transferred to a separating funnel. The organic layer was washed with H2O (50 mL), NaHCO3 (aq. 2%, 50 mL), H2O (50 mL) and then separated, dried (Na2SO4), and evaporated to dryness. The residue was purified by column chromatography (n-hexane-ethyl acetate, 5:1) togive 7a-r, yield: 45-52%.
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In N,N-dimethyl-formamide; at 20℃; for 12h;
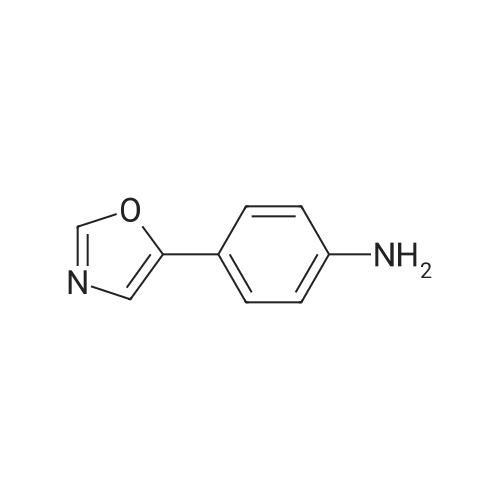
General procedure: All aryl or heteroaryl carboxylic acids and the corresponding 4-(2-alkyl/cyclopropyloxazol-5-yl) anilines cited in this study are commercially available. To a solution of appropriate benzoic acid A (1.0 mmol) and 2-substituted oxazol-5-yl aniline B (1.0 mmol), HATU (570 mg, 1.5 mmol) in anhydrous DMF (3.5 mL) and diisopropylethylamine (DIEA) (387 mg, 3.0 mmol) were added. The reaction mixture was stirred at room temperature for 12 h and then poured into 5 mL of ice-cold water. The resulting precipitates were filtered and washed with water to give to the crude product, which was further purified by preparative HPLC to give the substituted N-(4-(oxazol-5-yl)phenyl)benzamide in good yield.
General procedure: To a solution of 2-Methylbenzoic acid (1.36 g, 10 mmol) in DCM (50 mL), Et3N (1.12 g, 11 mmol) and TBTU (3.60 g,11 mmol) were added in turn. After 20 min, compound b (n =4, 1.17 g, 5 mmol) and Et3N (0.50 g, 5 mmol) were added. The reaction solution was stirred at room temperature for 8 h.Then, the solvent was evaporated with the residue being takenup in EtOAc (50 mL). the EtOAc solution was washed with saturated citric acid (3 × 20 mL), NaHCO3 (3 × 20 mL), and brine (3 × 20 mL), dried over MgSO4, and evaporated under vacuum. The desired compound c (1.50 g, 76 percent yield) was derived by crystallization in EtOAc/petroleum ether (1/4) as white powder. ESI-MS: m/z: 397.8 [M+H]+.
General procedure: To the solution of (naproxen, 230mg 1.0mmol) in 8mL of acetonitrile, DMAP (18mg, 0.15mmol) and EDCI (190mg, 1.0mmol) were added with stirring at room temperature for 0.5h. To the reaction mixture <strong>[25984-63-8]4-hydroxythiobenzamide</strong> (153mg, 1.0mmol) was added and stirred for 4hat room temperature. After filtration, the filtrate was evaporated under reduced pressure to remove the solvent. The oily residue thus obtained was dissolved in trichloromethane; the organic layer was washed with brine, with NaCl 5%, and then dried on anhydrous Na2SO4, filtered and the solvent evaporated. The crude product was chromatographed on a silica gel (CHCl3/CH3OH 30:1), and 148.8mg pale yellow solid was obtained.
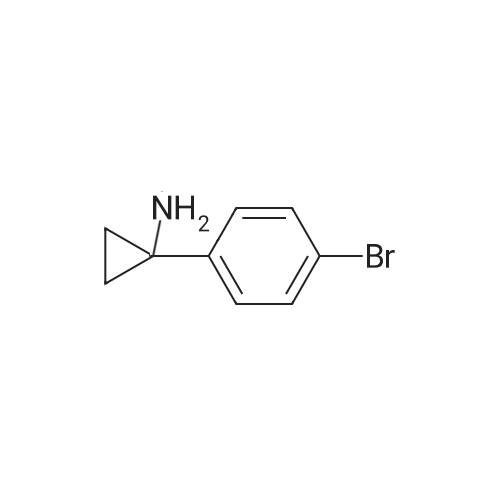
N-(1-(4-bromophenyl)cyclopropyl)-4-chlorobenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In dichloromethane; at 20℃; for 15h;
To a stirred solution of l-(4-bromophenyl)cyclopropanamine (200 mg, 0.943 mmol) in CH2CI2 (10 mL) were added 4-chlorobenzoic acid (221 mg, 1.415 mmol), TEA (0.4 mL, 2.87 mmol) and HATU (538 mg, 1.415 mmol) at RT and the reaction was stirred at RT for 15 h. The solvent was concentrated and the residue was diluted with water (20 mL) and extracted with EtOAc (30 mLx2). The organic layers were collected, washed with brine (20 mL), dried over Na2S04, filtered, and the filtrate was concentrated in vacuo. The residue was first purified by silica gel chromatography followed by additional purification using reversed phase HPLC on a GILSON 281 instrument fitted with a Waters Xbridge Prep OBD Cl 8 column (l00xl9mmx5um) using water (0.225% formic acid)-ACN as eluents. The title compound was obtained as a solid after concentration of the desired fractions. MS (ESI) m/z: 349.9 [M+H+]
With tetrakis(triphenylphosphine) palladium(0); triethylamine; In 1,4-dioxane; at 100℃; for 8h;
At room temperature, 783 mg (5 mmol) of p-chlorobenzoic acid 1b, 625 mg (6 mmol) of styrene 2a, and 721 mg (5 mmol) of <strong>[580-19-8]7-aminoquinoline</strong> 3a were added to a 25 mL round-bottomed flask, and then 578 mg (0.5 mmol) were sequentially added. Tetratriphenylphosphine palladium, 15 mL of 1,4-dioxane, and 1010 mg (10 mmol) of triethylamine were stirred at 100 C for 8 hours. After the reaction was completed, 15 mL of a saturated sodium chloride aqueous solution was added to the system, and extracted three times with 10 mL of ethyl acetate. The organic phases were combined, dried over anhydrous sodium sulfate, and the solvent was distilled off. Silica gel column chromatography of 200-300 mesh Pure 4b was obtained (1589 mg, yield 83%, pale yellow solid).
With dmap; dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;
General procedure: A mixture of 2 or 3 (0.50 mmol), the corresponding acids RCOOH (0.60 mmol),DCC (0.60 mmol), DMAP (0.1 mmol) in dry dichloromethane (15 mL) was stirred atroom temperature. When the reaction was completed, and checked by TLC, the mixturewas filtered to remove urea from the reaction, and the filtrate was diluted bydichloromethane (45 mL). Subsequently, the diluted organic phase was washed bysaturated aqueous NaHCO3 (30 mL), and brine (30 mL), dried over anhydrousNa2SO4, concentrated in vacuo, and purified by CC to give the pure 9R/S-acyloxyderivatives of cinchonidine and cinchonine 5a-j,l-o and 6a,c,e-o [17-19]. The dataof target compounds are shown as follows.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping