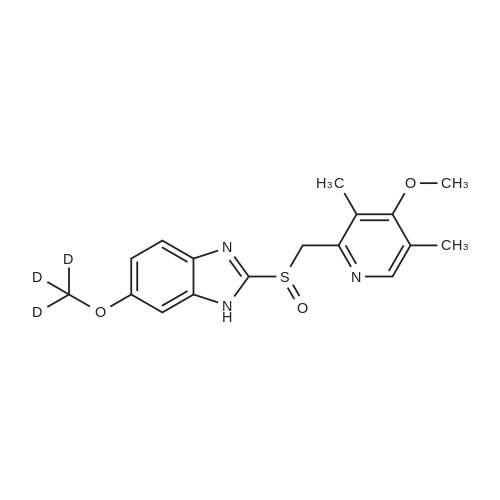
| 89% |
With ethylamine; In ethanol; water; at 20 - 55℃;Product distribution / selectivity; |
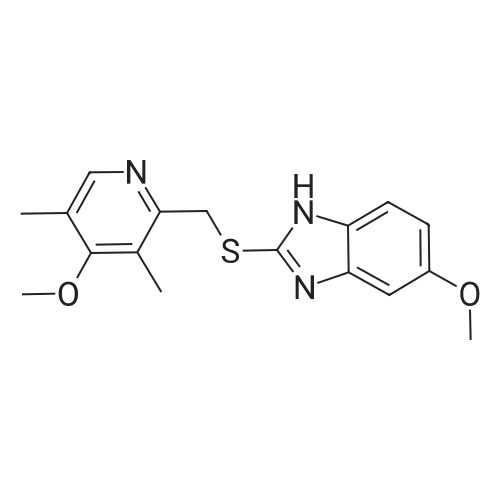
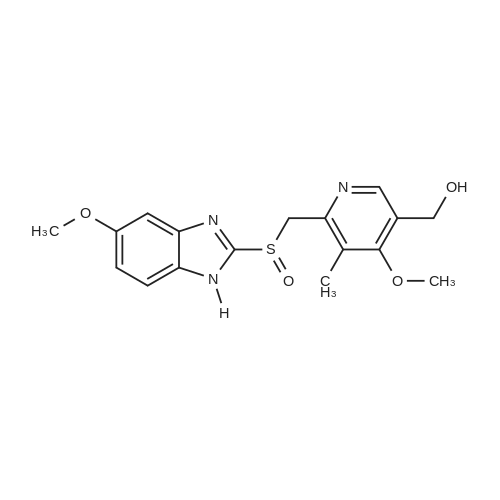
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 87.3% |
With phenol; In toluene; at 20 - 30℃;seeded with esomeprazole-BINOL;Product distribution / selectivity; |
EXAMPLE 2Preparation of Es<strong>[73590-58-6]omeprazole</strong>-BINOL Inclusion ComplexBINOL (6.22 g; 21.72 mmol) and phenol (4.09 g; 43.46 mmol) were added to toluene (70 ml) at 20-30 C. Omeprazole (10 g; 28.95 mmol) was added to the reaction mixture and seeded with es<strong>[73590-58-6]omeprazole</strong>-BINOL inclusion complex. Thereafter, the reaction mass was stirred for 2 h to crystallize the product. Then, hexane (35 ml) was added to the reaction mass and stirred for 15 h at 20-30 C. for complete crystallization of title compound, which was filtered, washed with toluene-hexane mixture and dried to obtain white solid of es<strong>[73590-58-6]omeprazole</strong>-BINOL inclusion complex. |
| 86% |
With ammonia; In water; acetone; at 50 - 60℃;Product distribution / selectivity; |
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 85% |
With triethylamine; In ethanol; water; at 20 - 60℃; for 12h;Product distribution / selectivity; |
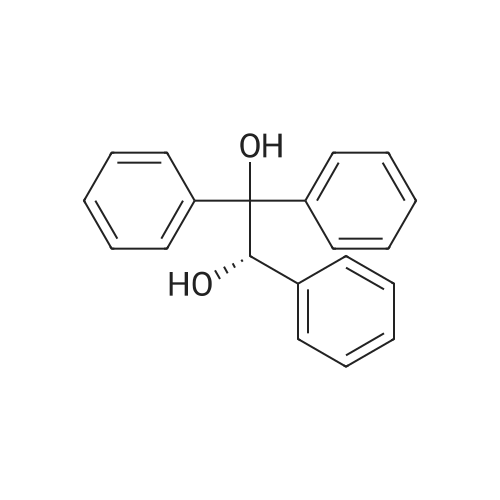
25.0 g of (S)-(-)-binol (87.3 mmol) was dissolved in a mixture of 400 ml of ethanol and 100 ml of water at 60 C, and 5.0 ml of triethylamine (35.9 mmol) and 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol) were dissolved thereto while maintaining the temperature at 50-55 C . Then, the resulting solution was slowly cooled to room temperature and stirred at that temperature for 12 hrs. The precipitated solids were filtered, washed sequentially with a mixture of 85 ml of ethanol and 15 ml of water and with 100 ml of n-hexane, and dried at 40 C to obtain 38.9 g of the white-yellow title compound (yield: 85%). M.p.: 158-160 C .Specific linear luminosity: [alpha]D20 = -146.2 (c=1, THF).Optical purity: 98.7% ee (chiral HPLC).1H-NMR (CDCl3, ppm): delta 2.24 (s, 6H), 3.73 (s, 3H), 3.87 (s, 3H), 4.65 (d, 1H), 4.78 (d, 2H), 5.50 (br. s, 2H), 6.96 (br. s, 2H), 7.18 (d, 2H), 7.40-7.28 (m, 8H), 7.70 (br. s, 1H), 7.90 (d, 2H), 7.98 (d, 2H), 8.16 (s, 1H), 11.60 (bs, 1H).IR (KBr, cm-1): 3057, 1619, 1595, 1576, 1471, 1462, 1401, 1380, 1271, 1205, 1146, 1073, 1028, 815, 570, 506, 422. |
| 85% |
With ammonia; In water; butan-1-ol; at 50 - 60℃;Product distribution / selectivity; |
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 85% |
In cyclohexane; toluene; at 0 - 55℃; for 1.5 - 1.75h; |
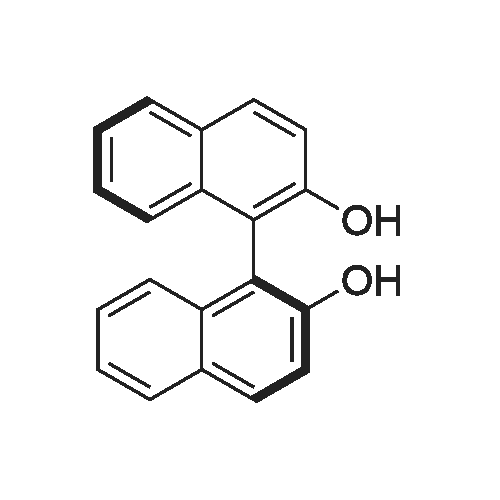
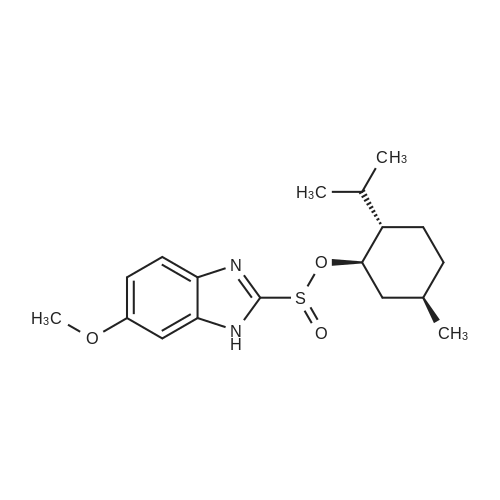
EXAMPLE 1; Preparation of (S)-<strong>[73590-58-6]omeprazole</strong>-(S)-(-)-BINOL complex; Omeprazole (100 g, 0.2898 mole) was added to a mixture of toluene (1600 ml) and cyclohexane (400 ml) in a round bottom flask kept at 25-30 0C. (S)-(-)-BINOL (124.3 g, 0.4346 mole) was added and the content warmed to about 50-55 0C with stirring for 30-45 minutes. The content of the flask was allowed to attain the ambient temperature and then cooled to 0-5 0C with Stirling for about an hour. The (S)-<strong>[73590-58-6]omeprazole</strong>-(S)-(-)-BINOL complex crystallizes out, filtered and washed with a mixture of cyclohexane/toluene (1 :4, v/v) pre-cooled to 0-5 0C. The (S)-<strong>[73590-58-6]omeprazole</strong>-(S)-(-)-BINOL complex was dried at 35- 40 0C under reduced pressure. The e.e. of (S)-<strong>[73590-58-6]omeprazole</strong> in the complex was found to be99.5%. Yield: 85 %.The IR spectrum of the complex is given in Fig. 3. The powder X-ray diffraction pattern is given in Fig. 4 |
| 83% |
With ammonia; In methanol; water; at 50 - 60℃;Product distribution / selectivity; |
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 81% |
With ammonia; In water; acetonitrile; at 50 - 60℃;Product distribution / selectivity; |
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 80 - 92% |
With ammonia; In ethanol; water; at 50 - 60℃;Product distribution / selectivity; |
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 40% |
With ammonia; In 1,4-dioxane; water; at 50 - 60℃;Product distribution / selectivity; |
Examples 3 to 14The procedure of Example 1 or 2 was repeated employing 50.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> (144.8 mmol), 500 ml of a reaction solvent and the other specifics shown in Table I5 to obtain various compounds, whose yields and optical purities are listed in Table 1.Table 1 |
| 25% |
In n-heptane; toluene; at 85℃; for 0.5h;Product distribution / selectivity; |
20.0 g of <strong>[73590-58-6]omeprazole</strong> (57.9 mmol) and 25.0 g of (S)-(-)-[1 ,1'-binaphthalen]- 2,2'-diol (86.8 mmol) were suspended in 600 ml of toluene and 150 ml of heptane. It was heated at 85 0C for 30 min. It was cooled at 0-5 0C, the suspending solid was filtered and dried in vacuo at 40 0C. S-<strong>[73590-58-6]omeprazole</strong>*(S)- [1 ,1'-Binaphthalen]-2,2'-diol inclusion complex with 1 :1 stoichiometric ratio was obtained with a 25% yield corrected by HPLC and a 94% e.e. according to HPLC. |
|
In hexane; benzene; at 0 - 110℃; for 12h;Product distribution / selectivity; |
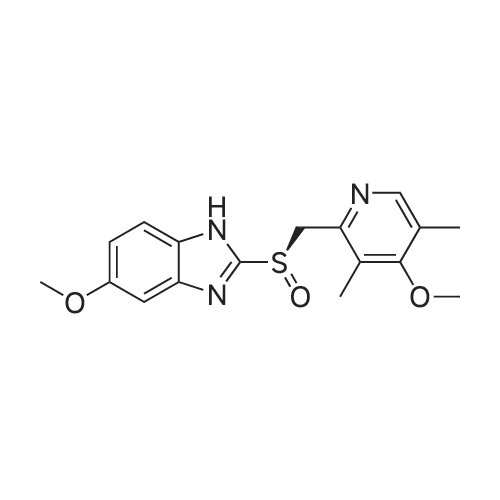
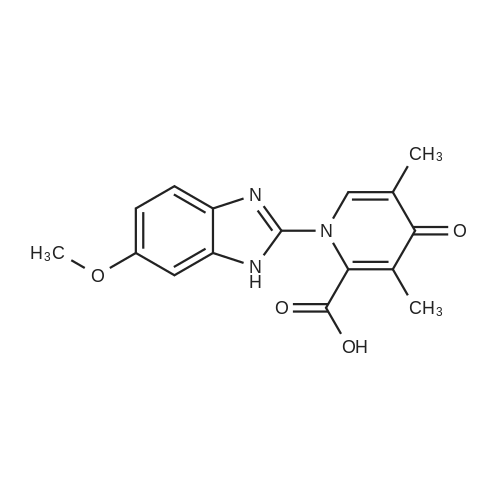
Preparation of inclusion complex of es<strong>[73590-58-6]omeprazole</strong> and (S)-(-)-binol (formula (I)) in accordance with the method disclosed in [J. Deng et al., Tetrahedron: Asymmetry, 11, 1729-1732, 2000] and [H. Cotton et al., Tetrahedron: Asymmetry, 11, 3819-3825, 2000](S)-(-)-binol and 10.0 g of the racemic form of <strong>[73590-58-6]omeprazole</strong> were dissolved in a mixture of 288 ml of benzene and 72 ml of n-hexane at 110 C . The (S)-(-)-binol was used in the amount of 0.6 mole equivalents based on <strong>[73590-58-6]omeprazole</strong>. Then, the resulting solution was slowly cooled to 0 C and stirred for 12 hrs. The precipitated solids were filtered, and washed with a mixture of benzene and hexane to obtain the title compound.Color of the product: yellow.Optical purity: 20.0 %ee (chiral HPLC). |
| 94%Chromat. |
With triethylamine; In n-heptane; toluene; at 70℃; for 0.5h;Product distribution / selectivity; |
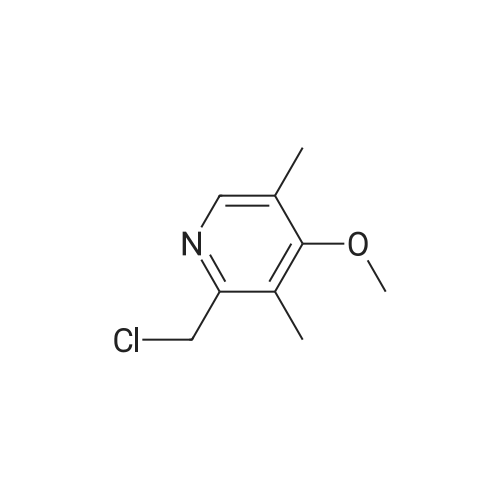
Example 1 Preparation of the S-<strong>[73590-58-6]omeprazole</strong>.(S)-[1,1'-binaphthalen]-2,2'-diol inclusion complex in toluene/heptane with triethylamine 10.0 g of <strong>[73590-58-6]omeprazole</strong> (29.0 mmol) and 12.4 g of (S)-(-)-[1,1'-binaphthalen]-2,2'-diol (43.4 mmol) were suspended in 96 ml of toluene, 24 ml of heptane and 2 ml of triethylamine. It was heated at 70 C. for 30 min. It was cooled at 0-5 C., the suspending solid was filtered and dried in vacuo at 40 C. S-<strong>[73590-58-6]omeprazole</strong>.(S)-[1,1'-binaphthalen]-2,2'-diol inclusion complex with 1:1 stoichiometric ratio was obtained with a 94% yield (corrected by HPLC) and a 97% e.e. (according to HPLC). 1H-RMN (400 MHz, CDCl3): delta 11.9 (1H, wide signal), 7.96 (1H, s), 7.86 (2H, d, J=8.9 Hz), 7.82 (2H, d, J=8.0 Hz), 7.51 (1H, wide signal), 7.32 (4H, m), 7.25 (2H, t, J=8.0 Hz), 7.14 (2H, d, J=8.3 Hz), 6.89 (1H, d, J=8.5 Hz), 6.79 (1H, wide signal), 4.70 (1H, d, J=13.6 Hz), 4.63 (1H, d, J=13.6 Hz), 3.80 (3H, s), 3.67 (3H, s), 2.17 (6H, s). |
| 89%Chromat. |
With triethylamine; In toluene; at 70℃; for 0.5h;Product distribution / selectivity; |
Example 2Preparation of the Inclusion Complex S-<strong>[73590-58-6]omeprazole</strong>.(S)-[1,1-Binaphthalen]-2,2'-diol in toluene with triethylamine10.0 g of <strong>[73590-58-6]omeprazole</strong> (29.0 mmol) and 12.4 g of (S)-(-)-[1,1'-Binaphthalen]-2,2'-diol (43.4 mmol) were suspended in 80 ml of toluene and 2 ml of triethylamine. It was heated at 70 C. for 30 min. It was cooled at 0-5 C., the suspending solid was filtered and dried in vacuo at 40 C. S-<strong>[73590-58-6]omeprazole</strong>.(S)-[1,1'-Binaphthalen]-2,2'-diol inclusion complex with 1:1 stoichiometric ratio was obtained with a 89% yield corrected by HPLC and a 97% e.e. according to HPLC. 1H-RMN (400 MHz, CDCl3): delta 11.9 (1H, wide signal), 7.96 (1H, s), 7.86 (2H, d, J=8.9 Hz), 7.82 (2H, d, J=8.0 Hz), 7.51 (1H, wide signal), 7.32 (4H, m), 7.25 (2H, t, J=8.0 Hz), 7.14 (2H, d, J=8.3 Hz), 6.89 (1H, d, J=8.5 Hz), 6.79 (1H, wide signal), 4.70 (1H, d, J=13.6 Hz), 4.63 (1H, d, J=13.6 Hz), 3.80 (3H, s), 3.67 (3H, s), 2.17 (6H, s). |
| 25%Chromat. |
In n-heptane; toluene; at 85℃; for 0.5h;Product distribution / selectivity; |
Comparative Example 2Preparation of the inclusion complex S-<strong>[73590-58-6]omeprazole</strong>.(S)-[1,1'-binaphthalen]-2,2'-diol in toluene/heptane without amine20.0 g of <strong>[73590-58-6]omeprazole</strong> (57.9 mmol) and 25.0 g of (S)-(-)-[1,1'-binaphthalen]-2,2'-diol (86.8 mmol) were suspended in 600 ml of toluene and 150 ml of heptane. It was heated at 85 C. for 30 min. It was cooled at 0-5 C., the suspending solid was filtered and dried in vacuo at 40 C. S-<strong>[73590-58-6]omeprazole</strong>.(S)-[1,1'-Binaphthalen]-2,2'-diol inclusion complex with 1:1 stoichiometric ratio was obtained with a 25% yield corrected by HPLC and a 94% e.e. according to HPLC. |
|
In dichloromethane; toluene; at 20℃; for 4.25h; |
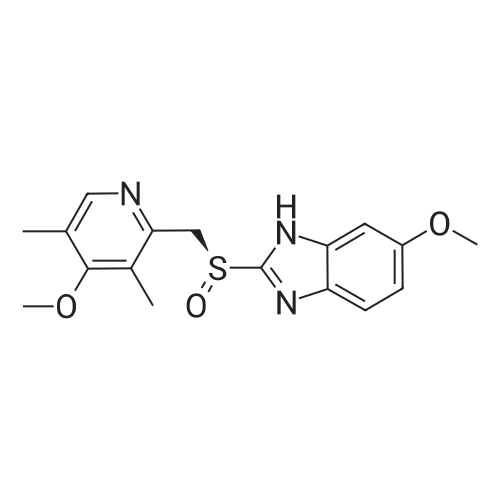
Preparation of es<strong>[73590-58-6]omeprazole</strong>-S-BINOLS-l ,l '-bi-2-naphthol (364.5 gm) was added to a solution of <strong>[73590-58-6]omeprazole</strong> (405 gm) as obtained in example 1 in methylene chloride (1000 ml) at room temperature and stirred for 15 minutes. To the reaction mixture was added toluene (6000 ml) and maintained for 4 hours at room temperature. The solid obtained was collected by filtration and dried to obtain 310 gm of es<strong>[73590-58-6]omeprazole</strong>-S-BINOL. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping