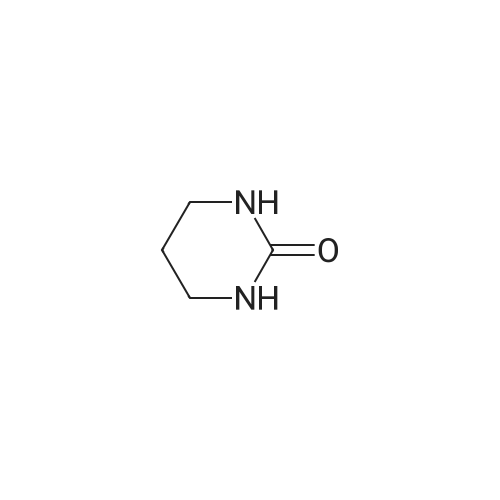
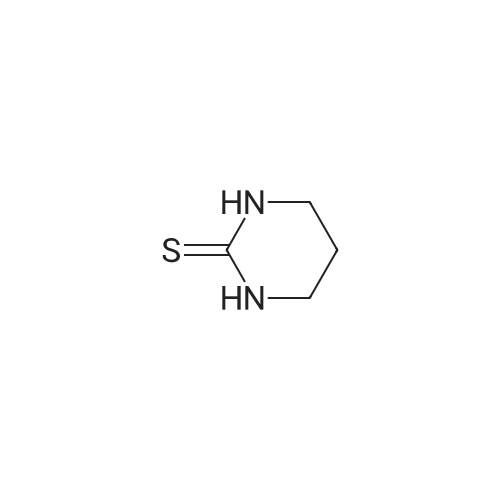
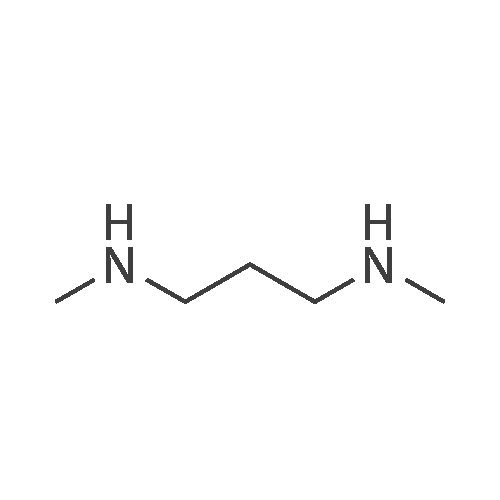
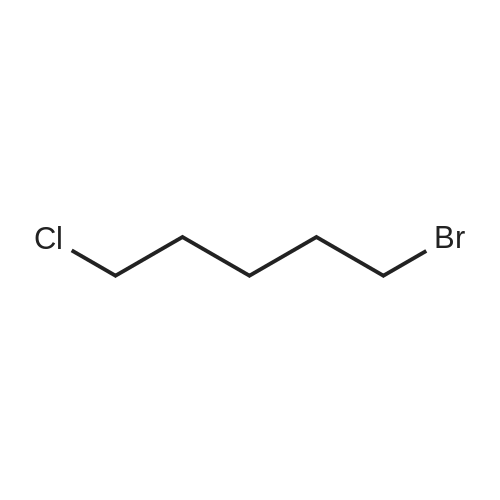
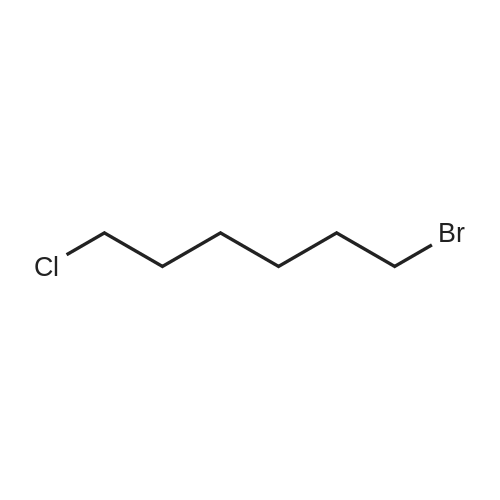
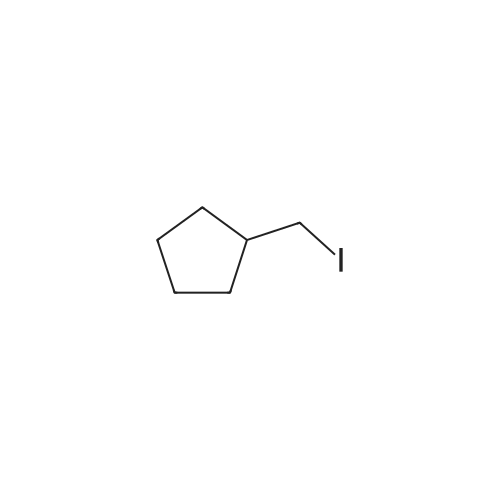
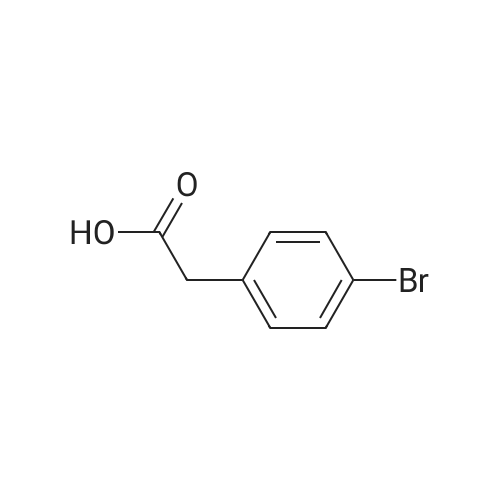
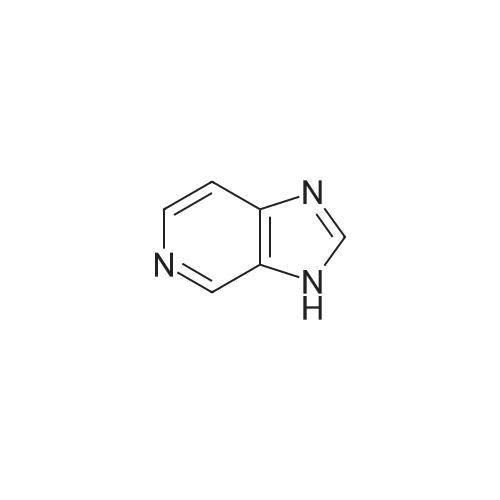
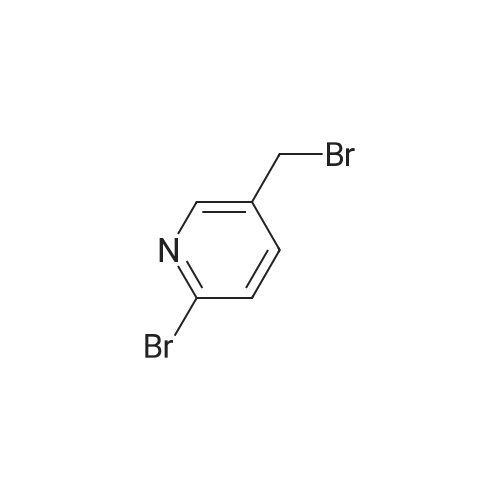
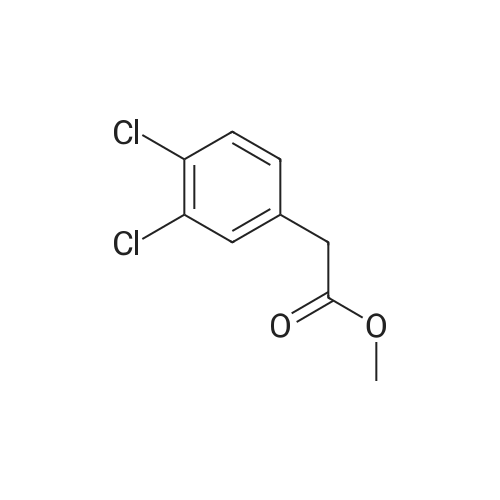
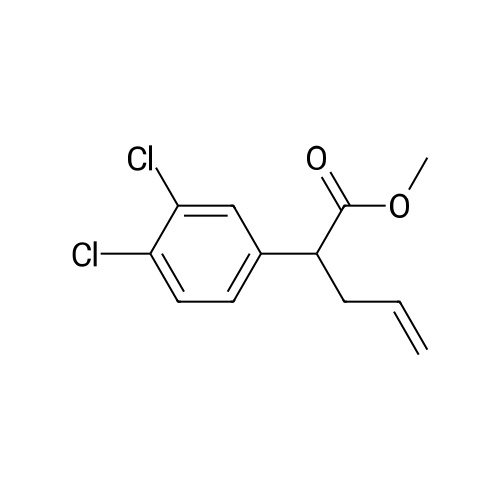
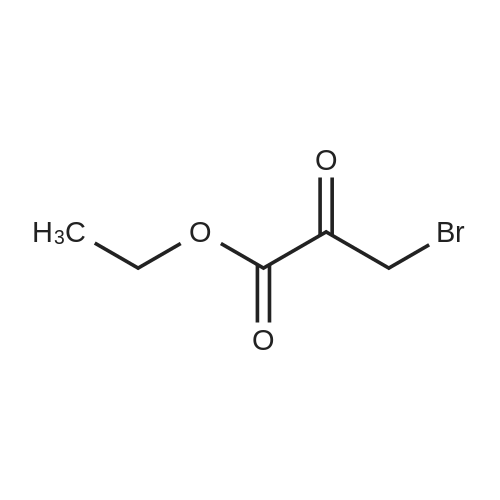
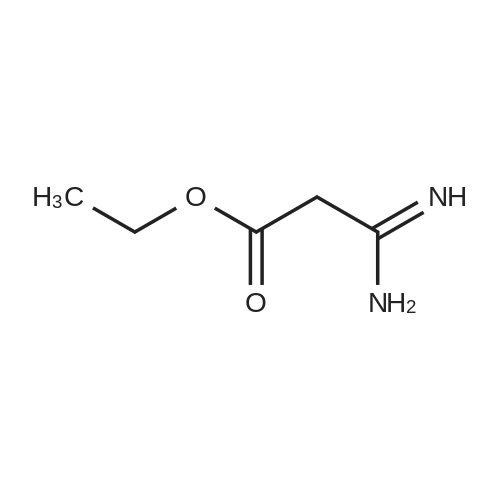
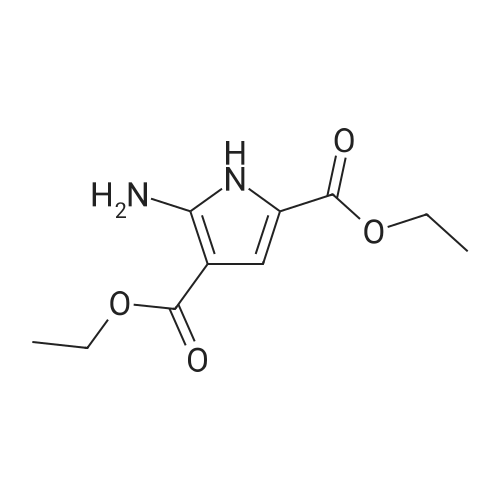
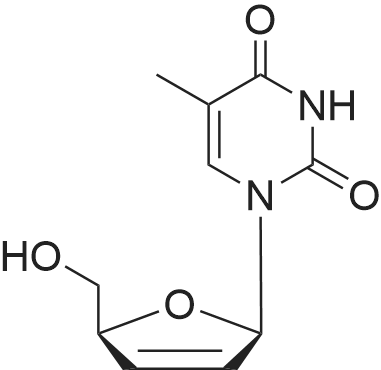
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
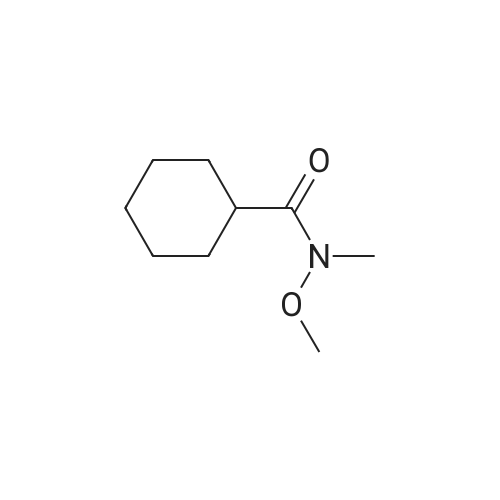
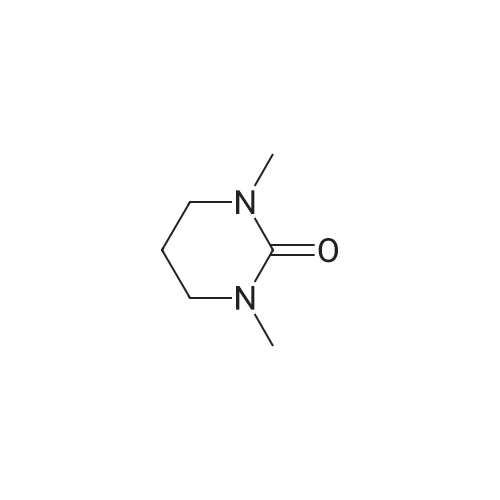
EXAMPLE 29 3-Cyclopentyl-N-pyridin-2-yl-2-(4-pyridin-4-yl-phenyl)-propionamide A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stirred at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 58.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 35 1-[3-Cyclopentyl-2-(4-naphthalen-1-yl-phenyl)-propionyl]-3-methyl Urea A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stirred at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 58.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 36 1-[3-Cyclopentyl-2-(4-pyridin-3-yl-phenyl)-propionyl]-3-methyl-urea A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stirred at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 58.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 37 1-{3-Cyclopentyl-2-[4-(1H-indol-5-yl)-phenyl]-propionyl}-3-methyl-urea A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stiffed at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 5 8.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 5 3-Cyclopentyl-2-(4-naphthalen-1-yl-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stirred at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 58.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EL-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 8 3-Cyclopentyl-2-(4-pyridin-3-yl-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stirred at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 58.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EL-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 70% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 9 3-Cyclopentyl-2-(4-pyridin-4-yl-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (17.1 mL, 122.21 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL) was cooled to -78 C. under nitrogen and then treated with a 10M solution of n-butyllithium in hexanes (12.2 mL, 122.21 mmol). The yellow reaction mixture was stirred at -78 C. for 30 min and then treated dropwise with a solution of 4-iodophenylacetic acid (15.25 g, 58.19 mmol) in dry tetrahydrofuran (55 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (18 mL). The reaction mixture turned dark in color and was allowed to stir at -78 C. for 45 min, at which time, a solution of iodomethylcyclopentane (13.45 g, 64.02 mmol) in a small amount of dry tetrahydrofuran was added dropwise. The reaction mixture was allowed to warm to 25 C. where it was stirred for 42 h. The reaction mixture was concentrated in vacuo to remove tetrahydrofuran and then quenched with a 10% aqueous hydrochloric acid solution (100 mL). The resulting aqueous layer was extracted with ethyl acetate (3*200 mL). The combined organic extracts were washed with a saturated aqueous sodium chloride solution (1*200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (13.97 g, 70%) as a cream solid: mp 121-122 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
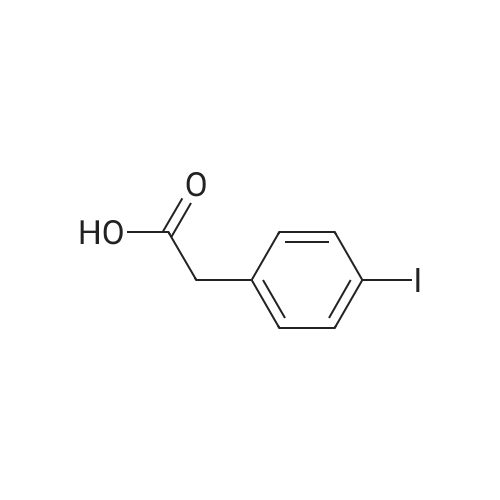
EXAMPLE 1 3-Cyclopentyl-2-(4-ethynyl-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofiran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 10 3-Cyclopentyl-2-(4-pyrimidin-5-ylethynyl-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL), a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 3 3-Cyclopentyl-2-[4-(3-methoxy-prop-1-ynyl)-phenyl]-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 4 3-Cyclopentyl-2-[4-(3-hydroxy-3-methyl-pent-1-ynyl)-phenyl]-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and was acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 5 3-Cyclopentyl-2-[4-(4-hydroxy-pent-1-ynyl)-phenyl]-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and was acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 6 3-Cyclopentyl-2-[4-(3-hydroxy-prop-1-ynyl)-phenyl]-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 7 3-Cyclopentyl-2-[4-(3-dimethylamino-prop-1-ynyl)-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS In/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 8 3-Cyclopentyl-2-[4-(3-morpholin-4-yl-prop-1-ynyl)-phenyl]-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |
| 57.8% |
With n-butyllithium; diisopropylamine; In tetrahydrofuran; |
EXAMPLE 9 3-Cyclopentyl-2-(4-pyridin-2-ylethynyl-phenyl)-N-thiazol-2-yl-propionamide A solution of diisopropylamine (11.2 mL, 80.13 mmol) in tetrahydrofuran (120 mL) cooled to -78 C. was treated with a 2.5M solution of n-butyllithium in hexanes (32 mL, 80.13 mmol). This solution was stirred at -78 C. for 30 min and then treated with a solution of (4-iodo-phenyl)-acetic acid (9.67 g, 36.9 mmol) in tetrahydrofuran (88 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (29 mL). The reaction mixture was allowed to stir at -78 C. for 1 h. At this time, the reaction was treated with iodomethylcyclopentane (8.53 g, 40.6 mmol). The reaction mixture was allowed to slowly warm to 25 C. where it was stirred at 25 C. for 18 h. At this time, the reaction mixture was quenched with water (5 mL) and then concentrated in vacuo. The residue was diluted with water (800 mL) and then acidified to pH=2 with concentrated hydrochloric acid. This solution was extracted with ethyl acetate (2*800 mL). The combined organic extracts were washed with water (1*600 mL) and a saturated aqueous sodium chloride solution (1*600 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 50/50 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-iodo-phenyl)-propionic acid (7.34 g, 57.8%) as a white solid: mp 105-107 C.; EI-HRMS m/e calcd for C14H17IO2 (M+) 344.0273, found 344.0275. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +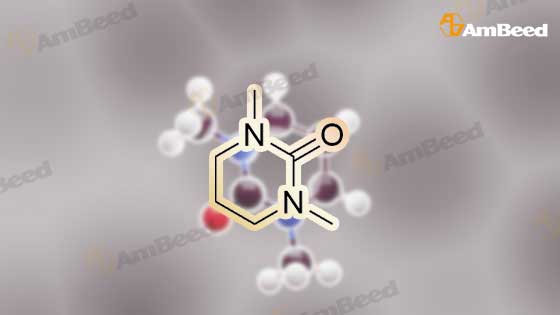

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping