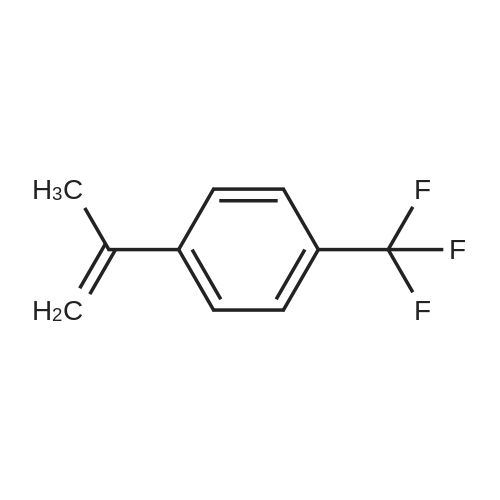
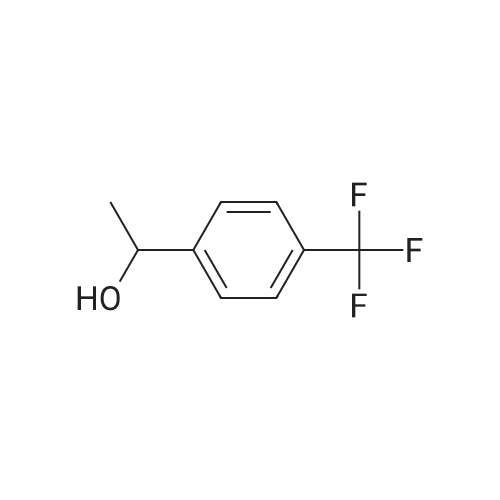
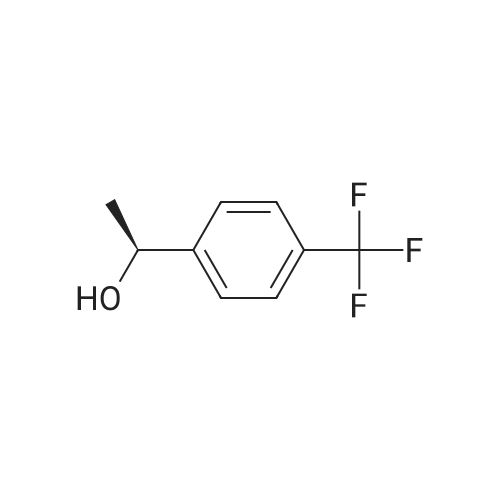
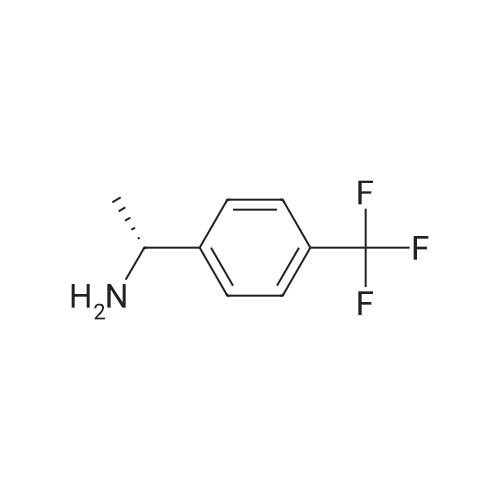
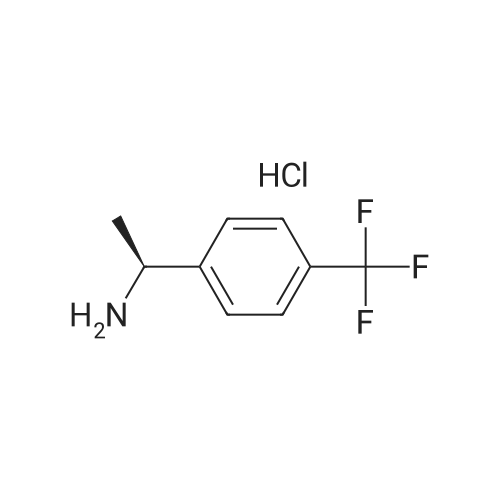
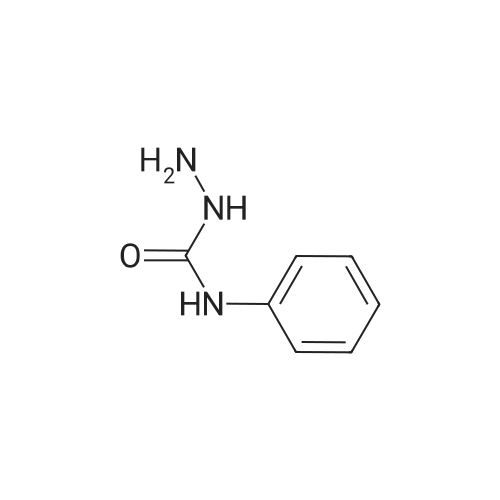
|
With (3aS)-1-methyl-3, 3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole borane complex In dichloromethane at -20℃; Yield given. Yields of byproduct given. Title compound not separated from byproducts; |
|
|
With potassium isopropoxide; (1S,3R,4R)-2-azabicyclo[2.2.1]heptane-3-(R)-methylmethanol In isopropanol at 20℃; for 1h; Title compound not separated from byproducts; |
|
|
With Rhizopus arrhizus In ethanol for 168h; Title compound not separated from byproducts; |
|
|
With (p-cymene)ruthenium(II) chloride; (S,S)-1,2-diphenyl-1,2-diaminoethane; potassium-t-butoxide In water monomer; isopropanol at 22℃; for 4h; Title compound not separated from byproducts; |
|
|
With (p-cymene)ruthenium(II) chloride; (1R,2R)-N-(p-sulfonylbenzolsulfonyl)-1,2-diaminocyclohexane; potassium-t-butoxide In water monomer; isopropanol at 22℃; for 4h; Title compound not separated from byproducts; |
|
|
With Merulius tremellosus ono991 In acetone at 28℃; for 72h; Title compound not separated from byproducts; |
|
|
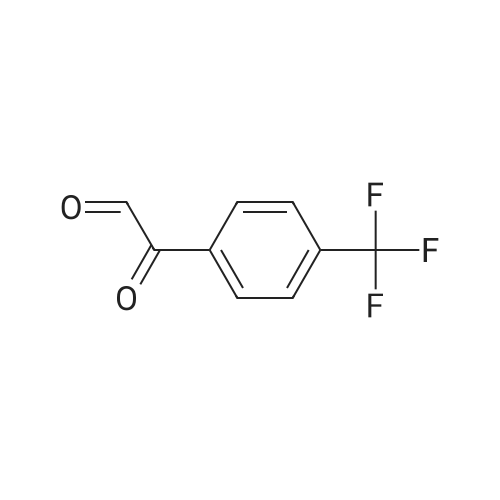
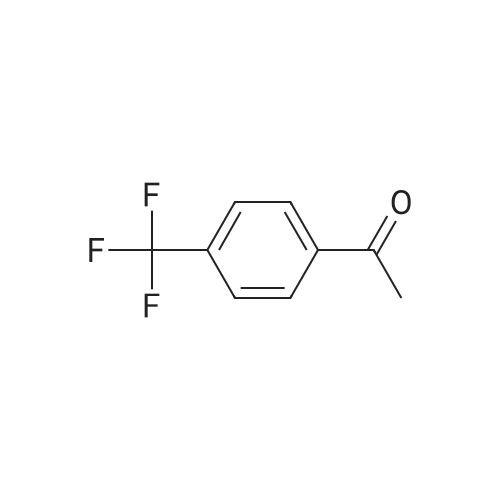
Stage #1: 1-(4-trifluoromethylphenyl)ethanone With copper difluoride; phenylsilane; (S)-(1,1'-binaphthalene)-2,2'-diylbis(diphenylphosphine) In toluene at 20℃; for 3h;
Stage #2: With hydrogenchloride Title compound not separated from byproducts; |
|
|
With dimethylsulfide borane complex; chiral deriv. of oxazaboralodinone Title compound not separated from byproducts; |
|
|
With (R)-3,5-xyl-MeO-BIPHEP; copper chloride (I) In toluene at -50℃; for 1h; Title compound not separated from byproducts; |
|
|
With copper difluoride; phenylsilane; (S)-(1,1'-binaphthalene)-2,2'-diylbis(diphenylphosphine) In toluene at 20℃; for 3h; Title compound not separated from byproducts; |
|
|
With (-)-C5Me5FeC10H10N; mesitylphenylsilane In tetrahydrofuran at 20℃; Title compound not separated from byproducts; |
|
|
With PMHS; copper chloride (I); sodium tertiary butoxide In toluene at -78 - 20℃; for 3h; Title compound not separated from byproducts; |
|
|
With chiral Zn(OTf)2*C5H5FeC5H3(CHNCPh3)(C3H3NO-i-Pr); benzo[1,3,2]dioxaborole In dichloromethane at -15℃; Title compound not separated from byproducts; |
|
|
With potassium-t-butoxide; hydrogen In isopropanol at 20℃; for 4h; Title compound not separated from byproducts; |
|
|
With potassium-t-butoxide In isopropanol at 50℃; for 24h; |
|
|
With (R)-1-[(1R,4R,5R,6S)-5,6-Me2CO2-2-aza-3-norbornyl]ethanol; potassium isopropoxide; isopropanol at 20℃; for 1h; Title compound not separated from byproducts; |
|
|
With N-(p-toluenesulfonyl)-(1R,2R)-diphenylethylenediamine; anhydrous sodium formate at 40℃; for 2h; Title compound not separated from byproducts; |
|
|
With anhydrous sodium formate at 40℃; for 2h; Title compound not separated from byproducts; |
|
|
With [(S)-BrXuPHOS]2RuCl2(S,S-DPEN); potassium-t-butoxide; hydrogen In dichloromethane; isopropanol at 20 - 22℃; for 20h; Title compound not separated from byproducts; |
|
|
With potassium-t-butoxide; isopropanol; (R)-(3-benzylamino-2-hydroxy)propyl trityl ester In toluene at 20℃; for 1h; Title compound not separated from byproducts; |
|
|
With (S)-(3-benzylamino-2-mercapto)propyl trityl ester; potassium-t-butoxide; isopropanol In toluene at 20℃; for 1h; Title compound not separated from byproducts; |
|
|
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; anhydrous sodium formate; (1R, 2S)-2-(methylamino)-1-phenylpropan-1-ol hydrochloride at 20℃; Title compound not separated from byproducts; |
|
|
With C49H47O2N2(1+)*BF4(1-); diphenylsilane; trifluoromethane sulfonic acid silver salt In tetrahydrofuran at 0℃; for 48h; Title compound not separated from byproducts; |
|
|
With anhydrous sodium formate In water monomer at 60℃; Title compound not separated from byproducts; |
|
|
With 1,2-O-isopropylidene-3-O-Ph2P-5-deoxy-5-tBuS-D-xylofuranose; bis[chlorido(η2,η2-cycloocta-1,5-diene)rhodium(I)]; diphenylsilane In toluene at -15℃; Title compound not separated from byproducts; |
|
|
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; poly(ethylene glycol)-N-pTos-1,2-diphenylethylenediamine; anhydrous sodium formate In water monomer at 40℃; for 2h; Title compound not separated from byproducts; |
|
|
With [Ru(μ-Cl)(η(5)-pentamethylcyclopentadienyl)]4; potassium-t-butoxide; hydrogen In isopropanol for 0.7h; Title compound not separated from byproducts; |
|
|
With formic acid; Ru-(R,R)-Ts-dpen; triethylamine In water monomer at 40℃; for 1.3h; Title compound not separated from byproducts; |
|
|
With formic acid; imidazolium salt unit attached to (1S,2S)-TsDPEN; triethylamine at 20℃; for 24h; Title compound not separated from byproducts; |
|
|
With potassium-t-butoxide; hydrogen In isopropanol at 25℃; for 18h; Title compound not separated from byproducts; |
|
|
With potassium hydroxide; 9-amino-9-deoxyepicinchonine In isopropanol at -20℃; for 30h; Title compound not separated from byproducts; |
|
|
With potassium-t-butoxide; hydrogen In dichloromethane; isopropanol for 8h; cooling; Title compound not separated from byproducts; |
|
|
With air; anhydrous sodium formate at 40℃; for 0.166667h; Title compound not separated from byproducts; |
|
|
With Trimethyl borate In (2)H8-toluene at 20℃; for 1h; Title compound not separated from byproducts.; |
|
|
In formic acid; triethylamine at 50℃; for 10h; |
|
|
With air; phenylsilane In toluene at -60℃; for 18h; Title compound not separated from byproducts.; |
|
|
With anhydrous sodium formate In water monomer at 40℃; for 2h; Title compound not separated from byproducts.; |
|
|
With trichlorosilane In toluene at -20℃; for 16h; Title compound not separated from byproducts.; |
|
|
With potassium-t-butoxide; hydrogen In isopropanol; <i>tert</i>-butyl alcohol at 20℃; for 6h; |
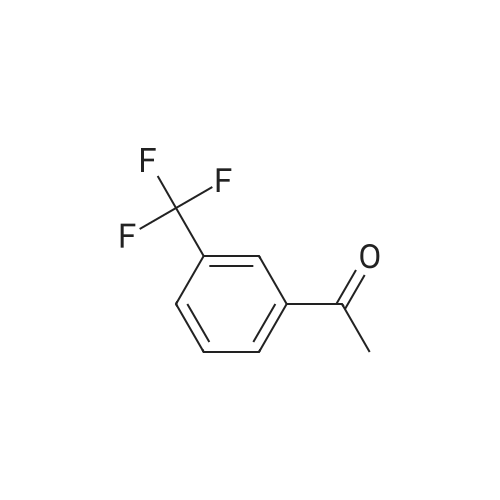
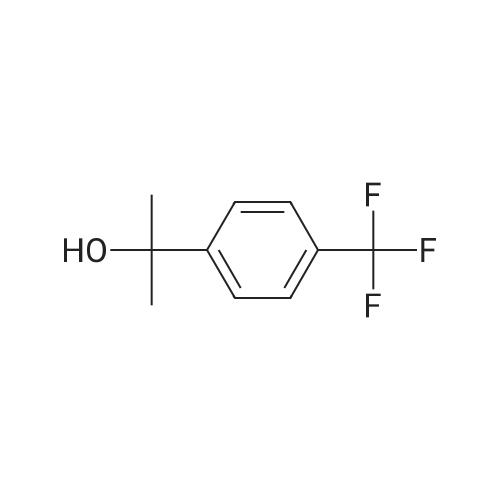
40 Hydrogenation of 4-Trifluoromethylacetophenone using Complex(R,S)-13d
Complex R,S-13d (3.0 mg; 0.0025 mmol; 0.005 equiv) was placed in a reaction vessel, which was pressurized with argon and vented five times. Argon-degassed isopropanol (2 mL) was added and the mixture was stirred for 15 min. 4-Trifluoromethylacetophenone (94 mg; 0.5 mmol) dissolved in 1 mL of argon-degassed isopropanol was added and was washed in with 1.0 mL of argon-degassed isopropanol. Potassium tert-butoxide in tert-butanol (1M; 0.05 mL; 0.05 mmol; 0.1 equiv) in 0.5 mL of argon-degassed isopropanol was added and was washed in with 0.5 mL of argon-degassed isopropanol. The reaction mixture was pressurized with argon and vented five times and then pressurized to 300 psig with hydrogen and stirred at ambient temperature for 6 h. The vessel was vented, then pressurized with argon and vented five times, and the solution was assayed by chiral GC to indicate 99.8% conversion to 1-(4-trifluoromethylphenyl)ethanol with 60.0% ee. Chiral GC [Cyclosil-B (J&W Scientific), 40° C. to 100° C. at 70° C./min, hold at 100° C. for 15 minutes, 100° C. to 170° C. at 15° C./min, hold at 170° C. for 7 min]: tR=16.7 min (4-trifluoromethylacetophenone), tR=20.7 min [1-(4-trifluoromethylphenyl)ethanol, enantiomer 1], tR=20.9 min [1-(4-trifluoromethylphenyl)ethanol, enantiomer 2]. |
|
With potassium-t-butoxide; hydrogen In isopropanol at 25℃; for 1h; Title compound not separated from byproducts.; |
|
|
With sodium isopropanolate; isopropanol at 20℃; for 2h; Title compound not separated from byproducts.; |
|
|
With potassium hydroxide; hydrogen In isopropanol at 40℃; for 3h; Title compound not separated from byproducts.; |
|
|
With (S)-2-((1-formylpyrrolidine-2-carbonyl)amino)propane-1,3-diyl diacetate; trichlorosilane In dichloromethane at -20℃; for 16h; Inert atmosphere; enantioselective reaction; |
|
|
With C58H56Cl2N2P2Ru; potassium-t-butoxide; hydrogen In isopropanol at 25℃; for 3h; Inert atmosphere; optical yield given as %ee; |
|
|
With RuCl2[(R,S)-Josiphos][(S)-Me-bimaH]; potassium-t-butoxide; hydrogen; triphenylphosphine In toluene; <i>tert</i>-butyl alcohol at 25℃; for 8h; Inert atmosphere; Autoclave; optical yield given as %ee; enantioselective reaction; |
|
| 82 % ee |
With potassium-t-butoxide; hydrogen In toluene; <i>tert</i>-butyl alcohol at 25℃; for 8h; |
22
Several aromatic ketones were reduced using the same process described in Example 19. The hydrogenation conditions and results were shown in Table 3. Table 3. Asymmetric hydrogenation of simple aryl ketones catalyzed by RuCl2[(R,5)-Josiphos)][(5)-Me-bimaH)][(λS',ιS)-8] complex. a ee (%yPI1R2 YieldEntry Substrate S/C ratio Time (h) (config (atm) (%)b1 acetophenone 1,000 8 2 100 96 (5)2 acetophenone 10,000 20 12 100 96 (5)3d acetophenone 50,000 40 10 100 97 (5)4-MeO-acetopheno 4 5,000 20 16 95 97 (S) ne4-CF3-acetophenon5 5,000 20 8 90 82 (5) e2-Me-acetophenon6 5,000 20 8 100 95 (5) e7 3 -Br-acetophenone 5,000 20 6 100 92 (5) ,oCM 1,000 8 12 92 94 (5) a Hydrogenation conditions: [ketone] = 0.3-1.9 M, [(i?5,5)-8] = 0.04-0.3 mM, [KO-^-C4H9] = 15-20 mM, [PPh3] = 1.0-3.4 mM, t = 25 0C, solvent = toluene/t-BuOH (9/1). b Determined by GC. c Absolute configuration (config) determined from [α]D. d Toluene/t-BuOH (7/3). e Yield determined by 1H NMR; ee determined by HPLC. |
|
With bis[dichlorido(η5-1,2,3,4,5-pentamethyl-cyclopentadienyl)rhodium (III)]; C21H28N2O2S; sodium isopropanolate; lithium chloride In isopropanol at 20℃; for 0.5h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
|
Stage #1: 1-(4-trifluoromethylphenyl)ethanone With phenylsilane; copper (II) acetate; (S)-4-phenyl-4,5-dihydro-3H-dinaphtho[2,1-c;1',2'-e]phosphepine In toluene at -20℃; for 5h; Inert atmosphere;
Stage #2: With methanol; N,N,N-tributylbutan-1-aminium fluoride In tetrahydrofuran for 2h; optical yield given as %ee; enantioselective reaction; |
|
|
With trans-[RuH(BH4){(R)-2,2'-bis(diphenylphosphino)-1,1'-binaphthyl}{(R,R)-1,2-diphenylethylenediamine}]; potassium-t-butoxide; hydrogen In isopropanol at 30 - 32℃; optical yield given as %ee; enantioselective reaction; |
|
|
Stage #1: 1-(4-trifluoromethylphenyl)ethanone With [(4S,5S)-1,3-bis[2,6-diisopropylphenyl]-4,5-di-tert-butylimidazolidine-2-ylidene][1,5-cyclooctadiene]-iodo-rhodium(I); diphenylsilane In tetrahydrofuran at 25℃; for 3h;
Stage #2: With sodium hydroxide In tetrahydrofuran; methanol for 0.166667h; enantioselective reaction; |
|
|
With (2S,1'S,2'R)-1-formyl-piperidine-2-carboxylic acid (2-methoxy-1,2-diphenyl-ethyl)-amide; trichlorosilane In chloroform-d1 at 20℃; for 16h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
|
With formic acid; C28H28ClN2O2RuS; triethylamine at 28℃; for 2h; optical yield given as %ee; enantioselective reaction; |
|
|
With bis[dichlorido(η5-1,2,3,4,5-pentamethyl-cyclopentadienyl)rhodium (III)]; (S)-tert-butyl 1-((1-benzyl-1H-1,2,3-triazol-4-yl)methylamino)-3-methyl-1-thioxobutan-2-ylcarbamate; sodium isopropanolate; isopropanol; lithium chloride at 20℃; for 0.166667h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
|
With dichloro(benzene)ruthenium(II) dimer; C16H23NO4; potassium-t-butoxide In isopropanol at 70℃; for 21h; Inert atmosphere; enantioselective reaction; |
|
|
With C68H78Cl2N2O6P2Ru; hydrogen; potassium hydroxide In isopropanol at 20℃; for 12h; Autoclave; optical yield given as %ee; enantioselective reaction; |
|
|
With D-glucose; D-glucose dehydrogenase; recombinant Sporobolomyces salmonicolor carbonyl reductase M242D mutant; NADPH In dimethyl sulfoxide at 20℃; aq. phosphate buffer; Enzymatic reaction; optical yield given as %ee; enantioselective reaction; |
|
|
With potassium-t-butoxide; hydrogen; copper (II) acetate; 4-(4-trifluoromethyl-phenyl)-4,5-dihydro-3<i>H</i>-4-phospha-cyclohepta[2,1-<i>a</i>;3,4-<i>a</i>']dinaphthalene In isopropanol at 10℃; for 16h; Inert atmosphere; Autoclave; optical yield given as %ee; enantioselective reaction; |
|
|
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; {2-[(S)-1-aminoethyl]phenyl}methanol; isopropanol; potassium hydroxide at -10℃; for 5h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
4.3. General procedure for the transfer hydrogenation of aromatic ketones
General procedure: A mixture of [RuCl2(p-cymene)]2 (1.5 mg, 0.0025 mmol) and (S)-4a (3.0 mg, 0.02 mmol) in 2 mL of 2-propanol was stirred at 80 °C for 30 min under argon atmosphere. After cooling to room temperature, 2-propanol (15 mL), KOH (0.6 mL, 0.1 M in 2-propanol), acetophenone (0.5 mmol, dissolved in 5 mL of 2-propanol) were added. The resulting solution was stirred at -10 °C, and the reaction was monitored by GC or HPLC. The mixture was neutralized with dilute HCl and 2-propanol was removed under reduced pressure. The residue was diluted with ethyl acetate (25 mL) and the organic solution was washed with brine (3 × 20 mL) and dried over anhydrous MgSO4. After evaporation of the solvent, the residue was subjected to short column chromatography on silica gel (hexane/ethyl acetate as eluent) for ee and conversion determination. |
|
With bis[chlorido(η2,η2-cycloocta-1,5-diene)rhodium(I)]; C7H7O3S(1-)*C9H14N3O(1+); potassium-t-butoxide; potassium hydroxide In isopropanol at 80℃; for 20h; Inert atmosphere; |
|
|
Multi-step reaction with 2 steps
1.1: ferrous acetate; 1,2-bis((2S,5S)-2,5-dimethylphospholano)benzene / tetrahydrofuran / 65 °C / Inert atmosphere
1.2: 45 h / 20 °C / Inert atmosphere
2.1: water monomer; sodium hydroxide / tetrahydrofuran; methanol / 0 - 20 °C / Inert atmosphere |
|
| 82 % ee |
With potassium-t-butoxide; hydrogen In toluene; <i>tert</i>-butyl alcohol at 25℃; for 8h; |
22
Several aromatic ketones were reduced using the same process described in Example 19. The hydrogenation conditions and results were shown in Table 3. TABLE 3 Asymmetric hydrogenation of simple aryl ketones catalyzed byRuCl2[(R,S)-Josiphos)][(S)-Me-bimaH)][(RS,S)-8] complex.a P/H2 Yieldee (%)b Entry Substrate S/C ratio (atm) Time (h)(%)b (config)c 1 acetophenone 1,000 8 2 100 96 (S) 2 acetophenone 10,000 20 12 100 96 (S) 3d acetophenone 50,000 40 10 100 97 (S) 4 4-MeO-acetophenone 5,000 20 16 95 97 (S) 54-CF3-acetophenone 5,000 20 8 90 82 (S) 6 2-Me-acetophenone 5,000 20 8 100 95 (S) 7 3-Br-acetophenone 5,000 20 6 100 92 (S) 8e 5,000 20 24 95 99 (R) 9 1,000 8 12 92 94 (S) 10 1,000 8 12 100 97 (S) aHydrogenation conditions: [ketone] = 0.3-1.9M, [(RS,S)-8] = 0.04-0.3 mM, [KO-t-C4H9] = 15-20 mM, [PPh3] = 1.0-3.4 mM, t = 25° C., solvent = toluene/t-BuOH (9/1).bDetermined by GC.cAbsolute configuration (config) determined from [α]D.dToluene/t-BuOH (7/3).eYield determined by 1H NMR; ee determined by HPLC. |
|
With formic acid; di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; triethylamine; N-((1R,2R)-2-aminocyclohexyl)-4-methylbenzenesulfonamide at 40℃; for 24h; optical yield given as %ee; enantioselective reaction; |
4.3. Asymmetric transfer hydrogenation in formic acid/triethylamine (Investigation scale)
General procedure: A suspension of the [RuCl2(arene)]2 (0.001 mmol) and ligand (0.0027 mmol) in CH2Cl2 (0.5 mL) was stirred at 20 °C for 30 min. After removal of CH2Cl2 by a stream of N2, the ketone (0.1 mmol) in a physical mixture of HCO2H/Et3N (5/2 mol ratio, 0.25 mL) was added. The reaction mixture was stirred vigorously at 40 °C for the specified number of hours. Samples were withdrawn from the reaction mixture and the solvent was removed under a stream of N2. The samples were then dissolved in the HPLC eluent, filtered through silica and analysed by HPLC for determination of conversion and ee. Note: on scale-up the pressure built-up of CO2 must be taken into account. |
|
With (R)-2,2',6,6'-tetramethoxy-4,4'-bis(di-3,5-dimethylphenylphosphine)3,3'-bipyridine; phenylsilane; Co(OAc)2.4H2O In toluene at 40℃; for 60h; Molecular sieve; Under air; optical yield given as %ee; enantioselective reaction; |
|
|
With bis[dichlorido(η5-1,2,3,4,5-pentamethyl-cyclopentadienyl)rhodium (III)]; lithium formate; L-prolinamide In water monomer at 35℃; for 18h; optical yield given as %ee; enantioselective reaction; |
|
|
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; (3R,3aR,6R,6aS)-6-(benzyloxy)-hexahydrofuro-N-(2(tosylamino)ethyl)furo[3,2-b]furan-3-amine; potassium-t-butoxide; isopropanol at 25℃; for 24h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
| 96 %Chromat. |
With stems and germinated seed of Brassica oleracea variety italica In water monomer at 20℃; for 48h; optical yield given as %ee; enantioselective reaction; |
|
|
Stage #1: 1-(4-trifluoromethylphenyl)ethanone With diethylzinc(II); 2,6-bis((R)-4-phenyl-4,5-dihydrooxazol-2-yl)pyridine In tetrahydrofuran; toluene at 20℃; for 20h; Inert atmosphere;
Stage #2: With sodium hydroxide In tetrahydrofuran; methanol; diethyl ether; toluene for 0.5h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
|
With (S)-(+)-2-(5-phenyl-4,5-dihydro-1,3-oxazol-2-yl)pyridine; trichlorosilane In chloroform at -20℃; for 16h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
|
With anhydrous sodium formate In water monomer at 40℃; enantioselective reaction; |
|
|
With RuCl2P(C6H5)3C5H5FeC5H3CHP(C6H5)2CH3COCH2CHCH(CH3)2N; potassium isopropoxide; isopropanol at 20℃; for 0.333333h; optical yield given as %ee; enantioselective reaction; |
|
|
With (S,S)-DPENDS; C36H24Cl2O18P2RuS6(6-)*6Na(1+); hydrogen; potassium hydroxide In water monomer at 30℃; for 3h; Autoclave; optical yield given as %ee; enantioselective reaction; |
4.2. Typical procedure for asymmetric hydrogenation of aromatic ketones
General procedure: To a 60 mL stainless autoclave with a glass liner and magnetic stirrer were added PEG-400, H2O, RuCl2(TPPTS)2, (S,S)-DPENDS, KOH, and reactant. Hydrogen was introduced to the desired pressure after the reaction mixture had been purged with H2 five times. The products were extracted by n-hexane and analyzed by GC-960 with a FID detector and β-DEX120 capillary column (30 m × 0.25 mm, 0.25 μm film) at 115 °C. The enantiomeric excess (ee value) was calculated from the equation: ee (%) = 100 × (R - S)/(R + S). |
| 88.2 % ee |
With [(R,R)-Teth-TsDpen RuCl]; hydrogen In methanol at 60℃; for 16h; enantioselective reaction; |
|
| 88 % ee |
With anhydrous sodium formate In water monomer at 40℃; for 12h; enantioselective reaction; |
|
|
Multi-step reaction with 2 steps
1: (Cp*RhTsDPEN)+CF3SO3-; tetrabutylammonium bromide; anhydrous sodium formate / water monomer / 20 °C / Inert atmosphere; Schlenk technique; Sonication
2: Supelco Β-Dex 120 chiral column
/ Resolution of racemate |
|
| 86 % ee |
With triruthenium dodecacarbonyl; (R,R)-4-methyl-N-{2-[(pyridin-2-ylmethyl)-amino]cyclohexyl}benzenesulfonamide In isopropanol at 80℃; for 48h; Inert atmosphere; Schlenk technique; enantioselective reaction; |
General procedure for asymmetric transfer hydrogenation of ketones:
General procedure: A mixture of catalyst (2 mol%) and Ru3 (CO)12 (0.67 mol%) in IPA (10 cm3) wasstirred at 80 °C under an inert atmosphere in a schlenk tube for 30 min. To this solution, ketone (1 mmol) was added and the resulting mixture was stirred at 80 °Cfor 48 h. The reaction mixture was filtered through a short column of silica using (EtOAc:hexane 1:1), a small amount of the filtrate was dilluted in EtOAc and then injected on the GC to determine the conversion and enantiomeric excess. |
| 46 % ee |
With 1,4-diaza-bicyclo[2.2.2]octane; [Ru(η6-p-cymene)(κ2-o-{(11bS)-3H-dinaphtho(2,1-c:1',2'-e)phosphepin-4(5H)-yl}C6H4SO3)Cl] In methanol at 60℃; for 15h; Autoclave; Overall yield = 99 %Chromat.; enantioselective reaction; |
|
| 20 % ee |
With carbonylhydridotris(triphenylphosphine)iridium(I); C32H43N4P; isopropanol; potassium hydroxide at 40℃; for 5h; Schlenk technique; Inert atmosphere; enantioselective reaction; |
ATH of aromatic ketones
General procedure: To a 50mL Schlenk tube were added Ir complex (0.005mmol) and ligand 5 (0.005mmol). Under nitrogen atmosphere, freshly distilled and degassed iPrOH (10mL) were introduced. After stirring at 40°C for 30min, an appropriate amount of KOH/iPrOH solution was then added. The mixture was continually stirred for another 15min, ketone was then introduced and the mixture was stirred at 40°C for a certain period of time. At the end of experiment, the reaction products were analyzed by GC using a chiral CP-Chiralsil-Dex CB column. |
| 85.8 % ee |
With C40H41FeNP2; tris(3,5-dimethylphenyl)phosphine; potassium-t-butoxide; hydrogen; copper (II) acetate In isopropanol at 15℃; for 12h; Inert atmosphere; Overall yield = 76 %; Overall yield = 626 mg; |
|
| 74 % ee |
With N-{(1R,2R)-2-[3-(3,5-dimethoxyphenyl)propylamino]-1,2-diphenylethyl}-4-methylbenzenesulfonamide ruthenium chloride; hydrogen In methanol at 60℃; for 16h; Inert atmosphere; Sealed tube; |
|
| 84 % ee |
With water monomer; anhydrous sodium formate at 40℃; enantioselective reaction; |
|
| 84 % ee |
With anhydrous sodium formate In water monomer at 40℃; for 4h; |
Typical catalytic procedure
General procedure: A typical procedure was as follows [32]: GH-catalyst, HCOONa (20 mg, 0.3 mmol), ketones (2.0 mmol), and 2.0 mL water were added in a 10 mL round bottom flask. The mixture was allowed to react at 40 8C for 4 h. During the reaction, it was monitored constantly by TLC. The conversion and ee value could be determined by chiral GC using a Supelco b-Dex 120 chiral column (30 m 0.25 mm (i.d.), 0.25 mm film) or HPLC analysis with UV-vis detector using Daicel OJ-H chiral column (10.46 cm*25 cm). |
| 46 % ee |
With hydrogen; 1,4-dithiothreitol In aq. phosphate buffer; dimethyl sulfoxide at 40℃; for 96h; Overall yield = 16 %Spectr.; enantioselective reaction; |
|
| 88.2 % ee |
With N-{(1R,2R)-2-[3-(3,5-dimethoxyphenyl)propylamino]-1,2-diphenylethyl}-4-methylbenzenesulfonamide ruthenium chloride; hydrogen In methanol at 60℃; for 16h; |
|
| 65 % ee |
With formic acid; [Cp*Ir(H2O)((R)-8-amino-5,6,7,8-tetrahydroquinoline)]SO4 In methanol; water monomer at 70℃; for 6h; enantioselective reaction; |
Method A
The ATH procedure when formic acid or sodium formate was used as the hydrogen donor. To a solution of the substrate (0.5mmol) in a 1:1 mixture methanol and water (2mL), [Cp Ir(H2O)(L)]SO4 (0.0025mmol) and hydrogen donor (2.5mmol, 5equiv) were added. The reaction mixture was stirred at 70°C for a fixed time (3h for 2-cyanoacetophenones, 6h for acetophenones and 24h for β-ketoesters). The reaction mixture was quenched with brine (4mL) and extracted with ethyl acetate (2×5mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. |
| 78 % ee |
With Ru/Al2O3/2ttp; hydrogen; potassium hydroxide; (2S)-N-phenylpyrrolidine-2-carboxamide In isopropanol at 30℃; for 10h; Autoclave; Overall yield = 57 %; enantioselective reaction; |
Heterogeneous enantioselective hydrogenation reaction; general procedure
General procedure: The hydrogenation was performed in a 20 mL stainless autoclave with a magnetic stirrer bar. The desired amounts of 1.0%Ru/γ-Al2O3/2tpp(20 mg), iPrOH (2 mL), KOH (0.18 mol L-1), L-proline derivative 1 and substrate (Substrate/Ru/proline derivative 1 = 300 : 1 : 27.4) were added into the autoclave and this was then sealed and purged with pure hydrogen several times (reaction pressure, PH2: 6.0 MPa). After the reactants were heated to 30 °C, the reaction timing began. After completion of the reaction (10 h), the organic phase was dried over MgSO4 and the conversion and enantiomeric excess were determined by GC analysis according to literature.17 With the exception of aromatic alcohol, no other products were detected (GC-MS and NMR). |
| 46 % ee |
With trimethylamine-N-oxide; C38H38FeO6Si2; hydrogen In water monomer; isopropanol at 70℃; for 18h; Inert atmosphere; Autoclave; Schlenk technique; enantioselective reaction; |
|
| 78 % ee |
With fruits of Ligustrum lucidum (glossy privet) In aq. phosphate buffer; dimethyl sulfoxide at 20℃; for 144h; Overall yield = 5 %; enantioselective reaction; |
|
|
With dichloro(benzene)ruthenium(II) dimer; formic acid; 1-[4-[4-[[[(1S,2S)-2-amino-1,2-diphenylethyl]amino]sulfonyl]phenoxy]butyl]-3-methyl-1H-imidazolium mono(trifluoroacetate); triethylamine at 20℃; for 24h; Inert atmosphere; Ionic liquid; Optical yield = 85 %ee; |
Typical Procedure of RCATH
General procedure: Acetophenone (120 mg, 1.0 mmol) was added to a solution of ionic ligand (0.012 mmol) and [RuCl2(benzene)]2 (2.5 mg, 0.005 mmol) in [bmim][PF6] (1.0 mL) with stirring under N2, followed by additionof a formic acid-triethylamine azeotropic mixture31) (bp 108 °C/29 mmHg, 0.5 mL). The reaction mixture was stirred at rt for 24 h. Next, n-hexane (5 mL×3) was added to the reaction mixture and the products were extracted by decantation of the upper layer, and the residual IL phase was dried in vacuo for 30 min. Acetophenone (120 mg, 1.0 mmol) and formic acid-triethylamine azeotropic mixture (0.5 mL) were added to the remaining IL solution, and the next cycle of the reaction was started. |
| 65 % ee |
With C28H36ClNOP2Ru; potassium-t-butoxide; hydrogen In ethanol at -40℃; for 16h; Autoclave; Inert atmosphere; enantioselective reaction; |
|
| 40 % ee |
With (R)-N-(3-amino-3-phenylpropyl)-4-methylbenzenesulfonamide; C10H14*Ru(1+)*Cl(1-)*C16H20N2O2S; anhydrous sodium formate In water monomer; dimethyl sulfoxide at 40℃; for 48h; Schlenk technique; enantioselective reaction; |
|
| 24 % ee |
With (S)-N-(3-amino-1-phenylpropyl)-4-methylbenzenesulfonamide; C10H14*Ru(1+)*Cl(1-)*C16H20N2O2S; anhydrous sodium formate In water monomer; dimethyl sulfoxide at 40℃; for 48h; Schlenk technique; enantioselective reaction; |
|
| 46 % ee |
With trimethylamine-N-oxide; C38H38FeO6Si2; hydrogen In water monomer; isopropanol at 70℃; for 18h; Autoclave; enantioselective reaction; |
|
| 89 % ee |
With (R)-bis[[N-(2-diphenylphosphinite-2-phenyl)ethyl]-1,1'-ferrocenylmethyldiamine(dichloro η6-p-cymene ruthenium(II))]; sodium hydroxide In isopropanol at 82℃; for 0.5h; Inert atmosphere; Schlenk technique; enantioselective reaction; |
|
| 84 % ee |
With (S)-bis[[N-(2-diphenylphosphinitepropyl)]-1,1'-ferrocenylmethyldiamine(dichloro η6-p-cymene ruthenium(II))]; sodium hydroxide In isopropanol at 82℃; for 0.33h; Inert atmosphere; Schlenk technique; enantioselective reaction; |
|
| 86 % ee |
With bis(1,5-cyclooctadiene)iridium(I) tetrafluoroborate; formic acid; anhydrous sodium formate; (1R,2R)-N<SUP>1</SUP>,N-di(naphthalen-1-yl)cyclohexane-1,2-diamine In methanol; water monomer at 70℃; for 22h; Inert atmosphere; Overall yield = 86 %; enantioselective reaction; |
2.4. Procedure for the reduction of ketone with [C16H24BF4Ir] andchiral amine ligand
General procedure: In a pressure tube, 0.5 mol% of metal precursor [C16H24BF4Ir](2.48 mg, 0.005 mol) and 1 mol% of chiral amine ligand (3.66 mg,0.01 mmol) were dissolved in 2 mL of water and methanol (ratio1:1) and stirred at room temperature for 1 h under argon atmo-sphere. Then formic acid (2.5eq, 0.1 mL), sodium formate (2.5eq,170 mg) and 1eq of ketone substrate (1 mmol) were introduced.The reaction mixture was stirred at 500 rpm and heated at 70C for22 h. After that, the tube was cooled to room temperature; and theorganic compound was extracted with either with ethyl acetate orCH2Cl2, then the solution was dried over Na2SO4, filtrated and con-centrated under reduced pressure. The crude material was purifiedby flash column chromatography on silica gel using cyclohex-ane/ethyl acetate as gradient eluent (90:10-7:3). After evaporation,alcohols were obtained as oil or solid. The products were identifiedby NMR. The conversion and the enantioselectivity were deter-mined by chiral GC or chiral HPLC analysis (Scheme 1). |
| 73 % ee |
With dichloro(o-isopropoxyphenylmethylene)(tricyclohexylphosphine)ruthenium(II); potassium-t-butoxide; (1R,2R)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine In tetrahydrofuran; isopropanol at 30℃; for 20h; Inert atmosphere; Glovebox; enantioselective reaction; |
|
| 82 % ee |
With phenylsilane; C42H30BF5S In neat (no solvent) at 20℃; for 96h; Inert atmosphere; Glovebox; Overall yield = 57 %; Overall yield = 12.8 mg; enantioselective reaction; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping